

Abstract
The involvement of exosite I in α-thrombin (FIIa) binding to platelet glycoprotein Ibα (GPIbα), which could influence interactions with other substrates, remains undefined. To address the problem, we generated the GPIbα amino terminal domain (GPIbα-N) fully sulfated on three tyrosine residues and solved the structure of its complex with FIIa. We found that sulfotyrosine (Tys) 278 enhances the interaction mainly by establishing contacts with exosite I. We then evaluated how substituting tyrosine with phenylalanine, which cannot be sulfated, affects FIIa binding to soluble or surface-immobilized GPIbα-N. Mutating Tyr276, which mostly contacts exosite II residues, markedly reduced FIIa interaction with both soluble and immobilized GPIbα-N; mutating Tyr278 or Tyr279, which mostly contact exosite I residues, reduced FIIa complexing in solution by 0–20% but affinity for immobilized GPIbα-N 2 to 6-fold, respectively. Moreover, three exosite I ligands—aptamer HD1, hirugen, and lepirudin—did not interfere with soluble FIIa complexing to GPIbα-N, excluding that their binding caused allosteric effects influencing the interaction; nonetheless, all impaired FIIa binding to immobilized GPIbα-N and platelet GPIb nearly as much as aptamer HD22 and heparin, both exosite II ligands. Bound HD1 and hirugen alter Trp148 orientation in a loop near exosite I preventing contacts with the sulfate oxygen atoms of Tys279. These results support a mechanism in which binding occurs when the two exosites of one FIIa molecule independently interact with two immobilized GPIbα molecules. Through exosite engagement, GPIbα may influence FIIa-dependent processes relevant to hemostasis and thrombosis.
Keywords: tyrosine sulfation, tyrosylprotein sulfotransferase-2, protease-activated receptor, platelet activation, platelet aggregation
The disulfide-linked α and β chains of glycoprotein (GP) Ib associate with GPIX and GPV in a noncovalent hetero-oligomeric platelet membrane complex that binds proteins involved in hemostasis, thrombosis, and inflammation (1, 2). The best-characterized interactions are with von Willebrand factor (VWF) (3), essential for platelet adhesion and aggregation under conditions of rapid blood flow (4, 5), and α-thrombin (FIIa) (6). The latter has been known for decades (7) and may support platelet aggregation (8, 9), but its biological significance is not fully understood. Crystal structures of GPIbα amino terminal fragments (GPIbα-N) in complex with VWF-A1 domain (10, 11) or FIIa (12, 13) have been solved. The GPIbα sequence beyond residue Asp269 is not involved in contacts with VWF-A1 and appears disordered in the corresponding complex. This region contains three Tyr residues—positions 276, 278, 279—that are sulfated (sulfotyrosine = Tys) in mammalian cells (14, 15). The sulfate groups on Tys276 and Tys279 are directly involved in close contacts with FIIa and essential for binding (12, 13). Of note, Tyr278 is not sulfated in the known structures of the complex with FIIa (12, 13), indicating that the posttranslational modification of these residues may vary. Tyr O-sulfation occurs in proteins that enter the secretory pathway and is mediated by tyrosylprotein sulfotransferase (TPST). Two enzyme isoforms, TPST-1 and TPST-2, have been cloned and are broadly coexpressed in animal cells. One exception is Drosophila melanogaster, which has a single TPST gene (16). TPST-2 is distinctly required for sulfation of specific proteins (17), thus we wondered whether variation in TPST expression could influence Tyr278 sulfation and, consequently, function in GPIbα-N. Here we show that coexpression of human GPIbα-N and TPST-2 in D. melanogaster cells leads to a marked increase of Tyr278 sulfation, which allowed us to solve the crystal structure of GPIbα-N with three Tys residues in complex with FIIa. Moreover, because Tys sulfate moieties may interact with two distinct FIIa sites (12, 13), and the significance of GPIbα-N interaction with the site involving exosite I (site I) is still disputed (18), we integrated structural information with site-directed mutagenesis and functional studies to characterize the individual role of each FIIa contact site in binding to GPIbα. Our findings delineate a mode of interaction in which FIIa site I, while not required for forming a soluble bimolecular complex, is necessary for binding to surface-immobilized GPIbα-N and membrane-anchored platelet GPIbα. The involvement of both exosites in the interaction with platelets may influence FIIa cleavage of other substrates and/or inactivation of the protease, with consequences on hemostasis and thrombosis.
Results
TPST-2 and Tys Content in GPIbα-N.
Human GPIbα-N (residues -2 to 290) was homogeneous by SDS-PAGE analysis but not by ion exchange chromatography. Drosophila cells cotransfected with human TPST-2 (TPST2+) secreted a greater proportion of GPIbα-N eluting at higher NaCl concentration than noncotransfected (TPST2-) cells (Fig. 1A). Four species with decreasing isoelectric point (Fig. 1B) were isolated and designated peaks 0 to 3 to indicate the presumed content of negatively charged sulfate per molecule. Digestion of peak 3 with aryl-sulfate sulfohydrolase (abalone sulfatase) progressively shortened the retention time (Fig. 1C) in agreement with sequential sulfate loss. Phenylalanine is identical to tyrosine except for lacking the hydroxyl group required for sulfation. The longest retention time (indicating the highest sulfate content) of mutants with single (Y276F, Y278F, or Y279F), double (Y276/8F or Y278/9F) or triple (Y276/8/9F) Tyr → Phe substitutions expressed in TPST2+ cells corresponded to that of wild type peaks 2, 1, and 0, respectively. Digestion by abalone sulfatase progressively shortened the retention time of single and double, but not triple mutants (Fig. 1D), confirming that three Tyr residues in the GPIbα amino terminal domain can be variably sulfated.
Fig. 1.

Characterization of GPIbα-N sulfation. (A). Culture medium of TPST2- or TPST2+ cells secreting GPIbα-N was subjected to ion exchange chromatography; then, a constant volume of fractions eluted at increasing NaCl concentration was analyzed by nonreducing 4–20% SDS-PAGE. The intensity of Coomassie Blue staining reflects protein concentration. (B). Isoelectric focusing with 5% polyacrylamide-2% ampholytes (pH 3–10; Invitrogen) of four GPIbα-N peaks (0–3; 5 μg each) separated by ion exchange chromatography. After applying 100 V for 1 h, 200 V for 1 h and 500 V for 30 min, gels were fixed and proteins stained with Coomassie Blue. Peaks 2a and 2b were from TPST2+ or TPST2− cells, respectively. (C). Ion exchange chromatography after digestion of GPIbα-N peak 3 with abalone sulfatase at 37 °C, showing the sequential generation of species eluting at lower NaCl concentration (shorter retention time). Top, time 0; center, after 75 min; bottom, after 18 h. A dotted line indicates the elution position of purified peaks 0, 1, and 2. (D). Top. Ion exchange chromatography of purified GPIbα-N with the indicated Tyr → Phe mutations. Each mutant was expressed in TPST2+ cells, and the species with the longest retention time (highest sulfate content) is shown. Bottom. After digestion with abalone sulfatase for 75 min at 37 °C, two new species with shorter retention time are generated from the single mutant (Y276F), one from the double mutant (Y278/9F), but none from the triple mutant (Y276/8/9F). In C and D, diagonal lines indicate the 0 to 1 M NaCl gradient. The deduced number of Tys residues is indicated next to each peak.
Crystal Structure of Sulfated GPIbα-N with Bound P-FIIa.
In structures known to date, Tyr278 in GPIbα-N is not sulfated (12, 13). Thus, we generated crystals (Table 1) of FIIa with the active site blocked by D-Phe-Pro-Arg chloromethylketone (P-FIIa) bound to GPIbα-N secreted by TPST2+ cells. Electron density maps confirmed that most Tyr279 and part of Tyr276 and Tyr278 residues were modified (Fig. 2A). The overall structure of the complex was like in Celikel et al., who used the same crystallization conditions at pH 7.0 (12), and differed from Dumas et al., who obtained crystals at pH 6.0 (13). We confirmed the 1∶1 stoichiometry of the complex, with each molecule related by crystallographic symmetry to two binding partners each defining a distinct interaction site (Fig. 2B). We confirmed also the atomic interactions of Tys276 and Tys279 (Table S1), while the side chain of Tys278 was repositioned to form salt bridges not seen with the unmodified residue (Fig. 2C). In the previous structure (12), the hydroxyl group of Tyr278 was hydrogen bonded to Nζ of Lys236 in exosite II. Instead, the sulfate oxygen atoms of Tys278 form a salt bridge with Lys236 in exosite II and six tight (< 4 Å) contacts with Arg35 and Pro37 at the FIIa site I interface, including two hydrophilic interactions with the former (Table S1). The two residues are located within the Phe34-Leu41 loop, which is part of FIIa exosite I and together with the Leu59-Asn62 insertion loop borders one side of the active-site cleft. On the opposite side is the Leu144-Gly150 loop (a.k.a. the 149 insertion or autolysis loop) in which Thr147 and Trp148 form 10 close contacts, including two hydrophilic interactions, with the sulfate oxygen atoms of Tys279 (Table S1). Thus, Tys278 and Tys279, but not unmodified tyrosine, together anchor the Asp269-Asp287 anionic loop of GPIbα to a positively charged FIIa region including exosite I; the detailed structure of FIIa exosite I has been described elsewhere (19).
Table 1.
Data collection parameters and refinement statistics
| Shell Å | I/σ(I) | Rsym | Completeness |
| 50.00-6.89 | 60.9 | 0.034 | 92.2 |
| 6.89-5.47 | 33.8 | 0.079 | 93.9 |
| 5.47-4.78 | 32.0 | 0.086 | 92.3 |
| 4.78-4.34 | 29.8 | 0.098 | 93.4 |
| 4.34-4.03 | 25.4 | 0.119 | 93.6 |
| 4.03-3.79 | 20.3 | 0.153 | 94.5 |
| 3.79-3.60 | 16.9 | 0.187 | 94.3 |
| 3.60-3.45 | 12.8 | 0.242 | 96.3 |
| 3.45-3.31 | 9.5 | 0.306 | 95.9 |
| 3.31-3.20 | 7.4 | 0.397 | 95.7 |
| All reflections | 26.4 | 0.104 | 94.2 |
| Refinement | ||
| Protein atoms (1 GPIbα-N, 1 thrombin) | 4,648 | |
| Polysaccharide atoms | 81 | |
| Cl atom | 1 | |
| Resolution range for refinements: | 15–3.2 Å | |
| Number of reflections in the working set: | 11,467 | |
| Number of reflections in the test set: | 611 | |
| R factor = 21.9 % | ||
| Rfree = 27.0% | ||
| rms Deviations | ||
| Bond lengths | 0.009 Å | |
| Bond Angles | 1.6° | |
| Ramachandran plot | ||
| Residues in the most favored regions | 73.0% | |
| Residues in additionally allowed regions | 23.9% | |
| Residues in generously allowed regions | 2.4% | |
| Residues in disallowed regions | 0.6% |
Space group P43212. Unit cell constants: a = 67.657 Å, c = 329.730 Å Solvent content = 55%
Data collection: Stanford Synchrotron Radiation Laboratory, Beam Line 9-2
Wavelength = 1.03317 Å
Detector: Quantum-315 CCD
Data reduction: HKL2000
Number of observations = 84,700. Unique reflections = 12,914 Redundancy = 6.6
Fig. 2.

Details of the crystal structure of the complex between fully sulfated GPIbα-N and P-FIIa. (A). Stereo representation of electron density maps computed at 3.2 Å resolution. Top. Initial omit map showing electron densities for the sulfate moieties on Y276, Y278, and Y279. The map was calculated using the previously published structure (12) and deleting the sulfate moieties from Y276 and Y279, but otherwise keeping the geometry unaltered. This goal was achieved by performing a rigid body refinement followed by a B-factor refinement prior to map calculation. Contours are drawn at 2σ level. Center. Final 2Fo-Fc electron density map of the new structure with fully sulfated GPIbα-N. Contours are drawn at 1σ level. Bottom. Final 2Fo-Fc map of the previously published structure (12) with sulfation on Y276 and Y279 but not on Y278, shown for comparison. Contours are drawn at 1σ level. (B). Orientation of the side chains of Tys residues 276, 278, and 279. A close-up view is shown in the inset on the left. (C). Comparison of the interatomic interactions established by Tys, Tyr, or Phe residues at position 278 in GPIbα-N.
Tyr Sulfation and P-FIIa Binding to GPIbα-N.
To evaluate the effects of Tyr sulfation on P-FIIa binding, we used surface plasmon resonance (SPR) with immobilized GPIbα-N. The off-rate was slower and maximal binding greater when GPIbα-N had three rather than two sulfate groups; binding was minimal with one sulfate (Fig. 3). The P-FIIa binding affinity of triple-sulfated GPIbα-N secreted by TPST2+ or TPST2− cells was similar, but that of partially sulfated GPIbα-N varied with TPST-2 coexpression (Table 2). Species with one sulfate group had ∼2-fold lower KD when secreted by TPST2+ as compared to TPST2− cells; in contrast, species with two sulfate groups had ∼2-fold lower KD when secreted by TPST2- as compared to TPST2+ cells (Table 2). These results suggested that TPST-2 might influence which Tyr residues are sulfated.
Fig. 3.

Effect of tyrosine sulfation on P-FIIa binding to immobilized GPIbα-N. (A–C). Kinetic analysis by surface plasmon resonance of P-FIIa (25, 50, 100 nM) binding to wild type GPIbα-N with three Tys residues (A) or the mutants Y279F (B), and Y278/279F (C) with two and one Tys residues, respectively. Results are expressed as difference in Biacore Units (ΔRU) on surfaces coated or not with GPIbα-N. Note the different scale on the ordinate. (D). Dose-response of P-FIIa binding to fully sulfated GPIbα-N (3) or the single (2) and double (1) Tyr → Phe mutants. Binding is reported as mole of P-FIIa bound per mole of immobilized GPIbα-N.
Table 2.
Kinetic parameters and affinity of P-FIIa binding to wild type and Tyr to Phe GPIbα-N mutants with different sulfate content
| GPIbα-N | Tys Residue(s) | TPST-2 | n | KD(10-9 M) | Bmax | ka(105 M-1 s-1) | kd (s-1) | ||
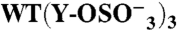 |
276 | 278 | 279 | + | (3) | 76 ± 24 | 0.89 ± 0.09 | 12.4 ± 2.9 | 0.11 ± 0.03 |
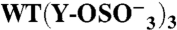 |
276 | 278 | 279 | − | (2) | 81 ± 20 | 0.96 ± 0.08 | 23.4 ± 3.6 | 0.20 ± 0.02 |
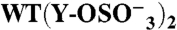 |
(276) | (278) | + | (4) | 393 ± 11 | 0.45 ± 0.04 | 9.8 ± 3.3 | 0.59 ± 0.10 | |
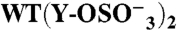 |
(276) | (279) | − | (3) | 164 ± 11 | 0.83 ± 0.01 | 11.9 ± 1.8 | 0.24 ± 0.18 | |
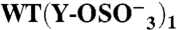 |
(276) | + | (2) | 556 ± 47 | 0.49 ± 0.02 | 6.8 ± 1.7 | 0.57 ± 0.06 | ||
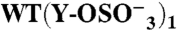 |
(279) | − | (2) | 961 ± 421 | 0.68 ± 0.04 | 4.8 ± 0.3 | 0.61 ± 0.09 | ||
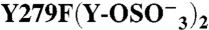 |
276 | 278 | + | (2) | 498 ± 106 | 0.71 ± 0.05 | 10.2 ± 3.2 | 0.53 ± 0.06 | |
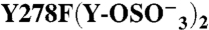 |
276 | 279 | + | (2) | 211 ± 45 | 0.76 ± 0.06 | 9.8 ± 0.9 | 0.36 ± 0.08 | |
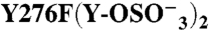 |
278 | 279 | + | (3) | 1612 ± 514 | 0.61 ± 0.15 | 3.1 ± 0.3 | 0.66 ± 0.1 | |
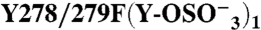 |
276 | + | (3) | 978 ± 539 | 0.26 ± 0.08 | 7.9 ± 1.3 | 1.31 ± 0.07 | ||
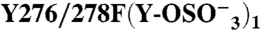 |
279 | + | (2) | 1548 ± 412 | 0.83 ± 0.18 | 4.3 ± 0.3 | 0.76 ± 0.03 | ||
Binding was measured by SPR at 25 °C. Association and dissociation rate constants (ka and kd, respectively), equilibrium dissociation constant (kD = kd/ka) and Bmax, expressed as molar ratio relative to the amount of GPIbα-N immobilized onto the SPR chip, were calculated as described in Methods. A subscript indicates the number of sulfate groups ( ) present in each GPIbα-N molecule tested. Assignment of Tys residues is indicated by residue number; hypothetical assignments are shown in italics and in parentheses. Results are expressed as mean ± SEM of the indicated number of experiments (n). Note that there was no measurable P-FIIa interaction with
) present in each GPIbα-N molecule tested. Assignment of Tys residues is indicated by residue number; hypothetical assignments are shown in italics and in parentheses. Results are expressed as mean ± SEM of the indicated number of experiments (n). Note that there was no measurable P-FIIa interaction with 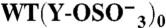 and Y276/8/9F GPIbαN, thus no binding parameters are reported for these species.
and Y276/8/9F GPIbαN, thus no binding parameters are reported for these species.
To test this hypothesis, we compared P-FIIa binding to fully sulfated Tyr → Phe mutants with known identity of Tys residues and wild type GPIbα-N with the same sulfate content secreted by TPST2+ or TPST2− cells. P-FIIa failed to interact with nonsulfated wild type GPIbα-N or the Y276/278/279F triple mutant, and bound with low affinity (KD in the μM range) to double mutants with one Tys residue or wild type GPIbα-N with one sulfate (Table 2). The KD of the Y278/279F mutant with Tys276 was ∼2-fold lower than that of the Y276/278F mutant with Tys279; and the KD of wild type GPIbα-N with one sulfate was ∼2-fold lower when secreted by TPST2+ than TPST2− cells (Table 2). Both wild type species had relatively better affinity for P-FIIa than the double mutants, likely because of contacts mediated by the hydroxyl group of tyrosine not supported by phenylalanine (see Fig. 2C). Among the single mutants, Y276F had P-FIIa binding affinity similar to that of the Y276/278F double mutant, highlighting the key functional role of Tys276; in contrast, the mutant lacking Tyr279 had a KD similar to wild type GPIbα-N with two Tys residues secreted by TPST2+ cells, and the mutant lacking Tyr278 to the corresponding species secreted by TPST-2- cells (Table 2). Of note, the latter was indeed shown by crystallography to be sulfated on Tyr276/Tyr279, but not Tyr278 (12). Therefore, it appears that sulfation follows the order Tyr279 → Tyr276 in the absence of TPST-2 or Tyr276 → Tyr278 in its presence. The latter intermediate may lead to modification of the third tyrosine more efficiently than the former, explaining the enrichment of fully sulfated GPIbα-N in TPST2+ cells.
Two Distinct FIIa Sites Mediate Binding to Immobilized GPIbα-N.
In the P-FIIa/GPIbα-N complex, Tys276 sulfate oxygen atoms establish close contacts (< 4 Å distance) with 13 FIIa residues in exosite II (site II) or 3 within or near exosite I (site I); in contrast, Tys279 and Tys278 sulfate oxygen atoms contact more residues in site I (10 and 6) than site II (3 and 1, respectively; Table S1). Substituting different Tyr residues with Phe, which cannot be sulfated, variably reduced P-FIIa binding to GPIbα-N. Replacing Tyr276 had a greater effect than replacing Tyr279 or Tyr278, with ∼70% vs. 20% or 0% reduction of P-FIIa incorporation into soluble complex (Fig. 4 A and B) and ∼20- vs. 6-or 2-fold increase in the KD of P-FIIa binding to immobilized GPIbα-N (Table 2), respectively. Thus, preventing exosite II contacts with Tys276 disrupted FIIa interaction with GPIbα-N regardless of its physical state. In contrast, immobilized GPIbα-N missing Tys278 or Tys279 showed, respectively, a 2- to 6-fold decrease in FIIa binding affinity that may be more severe functionally than the corresponding 0 to 20% loss of soluble complex formation. Because both residues have more contacts with FIIa site I than II, this finding may indicate a greater involvement of the former in binding to immobilized rather than soluble GPIbα-N. Such a conclusion is tempered by the consideration that all three GPIbα Tys residues interact with both FIIa exosites.
Fig. 4.

Effect of sulfate content, Tyr mutations, and aptamers selectively inhibiting FIIa exosites on P-FIIa binding to soluble GPIbα-N. (A). P-FIIa mixed in a 1∶3 mass ratio with wild type GPIbα-N (left) forms a complex of larger mass than free P-FIIa, as shown by the earlier elution from a GPC column. In contrast, P-FIIa mixed with the Y276F mutant GPIbα-N (center) elutes in the same fractions as free P-FIIa, indicating impaired formation of a stable complex. The Y279F mutant GPIbα-N (right) forms a complex with P-FIIa but a small residual amount of free P-FIIa indicates decreased stability. (B). Quantification of P-FIIa incorporation into a soluble complex with wild type GPIbα-N (black bar); or different Tyr → Phe GPIbα-N mutants (white bars); or wild type GPIbα-N in the presence of a 9-fold molar excess of aptamers (gray bars) blocking exosite I (HD1), exosite II (HD22), or nonspecific for P-FIIa (HD23). Results are expressed as percent P-FIIa in fractions not overlapping with free P-FIIa after GPC (mean and SD of at least two determinations). (C). Elution time from a GPC column of exosite I-binding aptamer HD1 (peak 1), P-FIIa (peak 2), or wild type GPIbα-N (peak 3) analyzed separately; or of the mixtures HD1/P-FIIa (peak 4), P-FIIa/GPIbα-N (peak 5), and HD1/P-FIIa/GPIbα-N (peak 6). Note the similar residual amount of free HD1 (added at a 9-fold molar excess) in the mixtures with P-FIIa or P-FIIa/GPIbα-N, and the small but reproducible shift of peak 6 (HD1/P-FIIa/GPIbα-N) as compared to peak 5 (P-FIIa/GPIbα-N), indicating that P-FIIa with bound HD1 interacts with GPIbα-N in solution.
To define the mechanism of FIIa binding to GPIbα-N we then used selective exosite ligands, namely the DNA aptamer HD1 (20) or hirudin-related hirugen and lepirudin, all of which form structurally defined complexes with FIIa (21–23), to block exosite I; and the DNA aptamer HD22 (24, 25) or heparin to block exosite II. HD22 interfered with FIIa binding to GPIbα-N in solution but HD1 did not (Fig. 4B), even though the latter (Fig. 4C), as well as hirugen and lepirudin (Fig. S1) remained bound to FIIa in complex with GPIbα-N. The lack of effect by exosite I ligands demonstrated that allosteric changes caused by their binding (26) had no influence on exosite II function in this interaction. Nonetheless, HD1 inhibited FIIa binding to immobilized GPIbα-N, as measured by SPR, nearly as well as HD22 (Fig. 5A, B); the same was observed with hirugen and heparin to block exosite I and II, respectively (Fig. S2 A and B). Altogether, these results indicate that both FIIa exosites must interact with GPIbα-N—each with a distinct molecule, for structural reasons (12)—in order to bind to receptor anchored onto a surface. Of note, maximal FIIa binding and KD were not substantially different whether 50 or 400 RU (Biacore Units) of GPIbα-N, i.e., 0.93 or 7.48•109 molecules/mm2, was immobilized onto the SPR chip.
Fig. 5.

Inhibition of P-FIIa binding to surface-anchored GPIbα by aptamers blocking specifically exosite I or II. (A, B). Biacore analysis of aptamer HD1 (exosite I ligand; A) or HD22 (exosite II ligand; B) inhibitory effect on P-FIIa (used at 80 nM, i.e., approximately KD value) binding to GPIbα-N peak 3 linked to a surface-immobilized antibody. The corresponding IC50 and Ki values (nM; mean ± standard error of the mean; n = 2), respectively, were: 154.6 ± 1.08 and 77.3 ± 0.54 for HD1; 120.9 ± 1.04 and 60.45 ± 0.52 for HD22. (C, D). Flow cytometric analysis of B-P- FIIa (used at 800 nM, i.e., in excess of saturating concentration for wild type GPIbα) binding to human platelets in the presence of aptamer HD1 (C) or HD22 (D); mixtures were incubated for 20 min before adding S-PE and measuring binding. Results are expressed as percent binding relative to a control without inhibitor. The calculated Ki values (nM; mean ± standard error of the mean; n = 6) were 12.25 ± 1.09 for HD1 and 19.85 ± 1.04 for HD22.
The mode of HD1 inhibition was competitive (Fig. S3), indicating a mutually exclusive FIIa interaction with exosite I ligands or immobilized, but not soluble (Fig. 4C) GPIbα-N. Inhibition of binding by HD1 and hirugen was incomplete, and the slope of dose-response curves covered a 2-log concentration range; in contrast, exosite II ligands inhibited completely and the slope of dose-response curves covered a 1-log range. These differences could signify the possibility of binding mediated exclusively by exosite II and/or cooperative interactions. In this regard, the crystal structure of Celikel et al. shows close contacts between two FIIa molecules bound to the same GPIbα-N receptor through exosite I or II, respectively, with a 7.6% surface involvement (12). Accordingly, the distinct effects of exosite-specific blockers are compatible with a binding mechanism in which one FIIa molecule interacting first through exosite II independently contributes to the energy of interaction of a second molecule bound through exosite I.
To rule out that GPIbα-N linkage to the immobilized capturing antibody recognizing an epitope in the N-terminal domain (27) could interfere with FIIa binding when its exosite I is occupied, we used a chimeric GPIbα-N with a 168-residue C-terminal extension (GPIbαN-Long). A monoclonal antibody binding to this extended sequence allowed a different receptor orientation on the surface as compared to GPIbα-N. Immobilized GPIbαN-Long bound P-FIIa (Fig. S4A) with KD of 53.4 nM comparable to that of GPIbα-N (Table 2) and the interaction was blocked by HD1, which exhibited a relatively greater effect than seen with the latter (compare Fig. S4B and Fig. 5A). This difference may indicate that conformational changes in the FIIa binding region—from a flexible C-terminal loop in GPIbα-N to a more constrained structure in GPIbαN-Long owing to the presence, as in native GPIbα, of additional C-terminal domains (28)—may influence the interaction with FIIa exosite I and the consequences of its blockade.
Role of Exosites I and II in FIIa Binding to Platelets.
To assess the physiologic relevance of our findings, we evaluated the role of FIIa exosite I and II in binding to native GPIbα in the platelet GPIb-IX-V complex. FIIa tagged with biotin/PPACK (B-P-FIIa) and coupled to streptavidin (SP) labeled with phycoerythrin (S-PE) or Alexa Fluor 488 bound to platelets rapidly (Fig. S5A) and at equilibrium (Fig. S5B); binding was saturable (KD = 26.0 nM; 95% confidence interval: 14.6–37.3 nM; n = 3) and fully blocked by LJ-Ib10 (Fig. S5C), an anti-GPIbα antibody known to recognize a sulfotyrosine-containing epitope (15, 27). B-P-FIIa binding to platelets was inhibited equally, completely, and independently by HD1 and HD22 (Fig. 5 C and D) as well as by hirugen and heparin (Fig. S5D). To validate these results, we verified that the fluorescent signal of SP bound to B-P-FIIa was not decreased when exosite I was occupied by HD1 (Fig. S6). Therefore, FIIa binding to platelet GPIb, like binding to immobilized but not soluble GPIbα-N, depends on the necessary contribution of both exosite I and II.
Discussion
We show here that distinct interactions involving residues in FIIa exosite II (site II) and within or near exosite I (site I) are concurrently required, but individually insufficient, for binding to platelet GPIbα (28). Our findings confirm the role of exosite II (29–34) and define that of exosite I (35), proving its necessary involvement in binding to immobilized but not soluble receptor. Although hirugen and HD1, exosite I ligands, lack inhibitory effect on FIIa interaction with GPIbα fragments linked to agarose (31) or plastic (30), we found that both inhibit binding to GPIbα fragments linked to immobilized antibodies and to membrane-anchored platelet GPIb, the biologically relevant receptor.
The structure of sulfated GPIbα-N can explain the mechanism of FIIa binding and inhibition by exosite I ligands. Analogous to Tys276 with respect to exosite II, the Tys278/Tys279 sulfate groups form multiple contacts with two loops within or bordering exosite I (Table S1). Bound HD1 (21) or hirudin (36) cause steric hindrance as well as conformational changes that prevent these contacts, in particular between Tys279 and Trp148 (Fig. S7), which justifies why substituting GPIbα Tyr278/Tyr279 or blocking FIIa exosite I similarly inhibit binding. In the Dumas et al. structure (13), a 180° rotation of the site I-bound FIIa prevents any interaction with Tys278/Tys279 and is associated with a reorientation of the GPIbα Asp269-Asp287 anionic region past Pro265 (28), which moves the site II-bound FIIa close to the convex surface of the leucine-rich GPIbα region (13). Such rotation may result from the loss of interaction between the two FIIa molecules bound to the same GPIbα-N, possibly a consequence of the acidic pH used by Dumas et al. during crystal growth. We suggest that the latter structure may represent the soluble FIIa complex with truncated GPIbα-N fragments, while the structure shown here, which accounts for the distinct consequences of Tyr → Phe substitutions and functional inhibition by exosite I ligands, may reflect FIIa bound to surface-anchored GPIbα.
Experimental results indicate that two contact interfaces participate in FIIa binding to platelet GPIb and immobilized GPIbα-N. Predominantly electrostatic exosite II-mediated contacts, effective over a broad range of angles and distances, may be established first and lead to subsequent site I-mediated contacts that involve hydrogen bonds and may stabilize the association. This model implies that one FIIa interacts with two GPIbα molecules; moreover, the 1∶1 stoichiometry in the crystal of the complex, verified by binding to immobilized GPIbα-N (Table 2), implies that each GPIbα interacts with two FIIa molecules. On platelets, the suggested subunit composition of the GPIb-IX-V complex, with four GPIbα chains (37), is compatible with such a functional arrangement (Fig. S8). Moreover, for binding to occur according to this mechanism, GPIbα surface density should not be less than required to maintain a distance between two molecules compatible with the concurrent binding of one FIIa molecule. Based on crystal structure dimensions and predicted 1 pg/mm2 bound protein per RU measured in SPR, such critical density may be as high as 4•109 GPIbα-N molecules/mm2; however, functionally efficient clustering appears to occur even at 1/4th such density, likely explained by the known fluidity and mobility of the dextran surface of SPR chips to which molecules are anchored (38).
In conclusion, TPST-2 can modulate GPIbα sulfation influencing the proportion of species with higher affinity for FIIa; thus, individual differences in its megakaryocytic expression may alter the platelet potential for binding the enzyme. Because the corresponding mechanism of interaction involves both FIIa exosites, the functional consequences could vary. For example, competition for exosite I that mediates binding to many substrates may limit proteolytic activity; in the case of fibrinogen, generation of the fibrin clot would be reduced and/or delayed with increasing FIIa binding to platelets. Blockade of exosite II, in turn, could affect factor VIII activation (39), with anticoagulant effects, but also protect from inhibition by serpins (30). It remains to be explored how these GPIb-dependent events may change the local balance of FIIa procoagulant and anticoagulant functions and influence hemostasis and thrombosis.
Methods
Expression, Purification, and Characterization of GPIbα-N.
The human GPIbα fragment -2 to 290 with Cys65 substituted to Ala to prevent dimerization was expressed in D. melanogaster as reported (12), without or with cotransfection with human TPST-2 (40). We also expressed GPIbα-N with Tyr to Phe substitutions at positions 276, 278, and 279; three mutants had single, two double (Y276/278F and Y278/279F), and one triple substitutions. All GPIbα-N species were purified by ion exchange and gel permeation chromatography as previously described (12). To demonstrate differences in sulfate content, purified GPIbα-N was digested with abalone sulfatase (type VIII; Sigma-Aldrich Co.).
Crystallization of the Complex of Fully Sulfated GPIbα-N with FIIa.
Human FIIa (Haematologic Technologies Inc.) blocked in the active site with PPACK was crystallized in complex with fully sulfated GPIbα-N by the hanging drop technique as previously described (12).
Evaluation of FIIa Binding to GPIbα-N in Solution and by SPR.
The soluble complex formed by GPIbα-N and P-FIIa was analyzed by gel permeation chromatography and reversed-phase high-performance liquid chromatography (HPLC). SPR sensorgrams were generated with a Biacore 3000 (GE Healthcare) using a CM5 sensor chip to which the anti-GPIbα monoclonal antibody, LJ-P3 (27), was covalently bound; this antibody interacts with the NH2-terminal region of GPIbα without affecting FIIa binding (15). Binding at saturation (Bmax) and equilibrium dissociation constant (KD, binding affinity) were obtained by nonlinear fitting of specific binding (ΔRU, the difference in P-FIIa RU on the surface with and without immobilized GPIbα-N at equilibrium) as a function of P-FIIa concentration. A one-site model of interaction (GraphPad Prism version 5.0; GraphPad Software) yielded consistently the best fitting, indicating that the required concurrent involvement of two distinct contact interfaces represents a functional unit. Bmax was reported as molar ratio of bound P-FIIa and GPIbα-N immobilized onto the chip, the latter calculated with the assumption that 1 RU measured by SPR corresponds to 1 pg of protein per mm2 of sensor chip surface. Details on the method used to derive the kinetics rate constants (off-rate, kd; on-rate, ka) are reported as SI Text. Goodness of fitting was evaluated by the value of R2 (> 0.95 for all assays). To probe the role of FIIa exosites in binding to GPIbα-N we used single-stranded DNA oligonucleotides: HD1 (G15D), a 15-mer exosite I ligand (20); HD22 (60-18), a 29-mer exosite II ligand (24); and HD23, a scrambled specificity control (41); all were from IDT. See SI Text for other reagents used and for the methods used to calculate IC50 and Ki of inhibitors.
FIIa Binding to Platelets.
For these assays, FIIa was blocked in the active site with biotin-PPACK (B-P-FIIa; Haematologic Technologies). B-P-FIIa and phycoerythrin-conjugated streptavidin (S-PE; Invitrogen) were added into aliquots of washed platelets and incubated at room temperature (22–25 °C) for the indicated time. When needed, the anti-GPIbα monoclonal antibody, LJ-Ib10 (27), which specifically inhibits FIIa binding (8), was added to the platelet suspension 5 min before the addition of S-PE/B-P-FIIa. Binding was measured by flow-cytometry without sample dilution. Results were expressed as geometric mean of the fluorescence intensity of 10,000 events and analyzed with GraphPad Prism version 5.0.
Additional methodological details are reported as SI Text.
Supplementary Material
Acknowledgments.
This work is supported by National Institutes of Health (NIH) grants HL42846 (to Z.M.R.), HL55375 (to K.I.V.) and HD056022 (to K.L.M.), and by a grant from CSL Behring (Prof. Heimburger Award to G.L.M.). We thank the Stanford Synchrotron Radiation Laboratory for providing beam time for data collection.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank www.pdb.org (RCSB ID code rcsb062553 and PDB ID code 3PMH).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1017042108/-/DCSupplemental.
References
- 1.Berndt MC, Shen Y, Dopheide SM, Gardiner EE, Andrews RK. The vascular biology of the glycoprotein Ib-IX-V complex. Thromb Haemostasis. 2001;86:178–188. [PubMed] [Google Scholar]
- 2.Du X. Signaling and regulation of the platelet glycoprotein Ib-IX-V complex. Curr Opin Hematol. 2007;14:262–269. doi: 10.1097/MOH.0b013e3280dce51a. [DOI] [PubMed] [Google Scholar]
- 3.Ruggeri ZM. Von Willebrand factor. Curr Opin Hematol. 2003;10:142–149. doi: 10.1097/00062752-200303000-00008. [DOI] [PubMed] [Google Scholar]
- 4.Ruggeri ZM, Mendolicchio GL. Adhesion mechanisms in platelet function. Circ Res. 2007;100:1673–1685. doi: 10.1161/01.RES.0000267878.97021.ab. [DOI] [PubMed] [Google Scholar]
- 5.Ruggeri ZM. Platelet adhesion under flow. Microcirculation. 2009;16:58–83. doi: 10.1080/10739680802651477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Marco L, Mazzucato M, Masotti A, Ruggeri ZM. Localization and characterization of an α-thrombin-binding site on platelet glycoprotein Ibα. J Biol Chem. 1994;269:6478–6484. [PubMed] [Google Scholar]
- 7.Okumura T, Hasitz M, Jamieson GA. Platelet glycocalicin. Interaction with thrombin and role as thrombin receptor of the platelet surface. J Biol Chem. 1978;253:3435–3443. [PubMed] [Google Scholar]
- 8.De Marco L, Mazzucato M, Masotti A, Fenton JW, II, Ruggeri ZM. Function of glycoprotein Ibα in platelet activation induced by α-thrombin. J Biol Chem. 1991;266:23776–23783. [PubMed] [Google Scholar]
- 9.Guerrero JA, et al. In vivo relevance for platelet glycoprotein Ibα residue tyr276 in thrombus formation. J Thromb Haemost. 2008;6:684–691. doi: 10.1111/j.1538-7836.2008.02916.x. [DOI] [PubMed] [Google Scholar]
- 10.Huizinga EG, et al. Structures of glycoprotein Ibα and its complex with von Willebrand factor A1 domain. Science. 2002;297:1176–1179. doi: 10.1126/science.107355. [DOI] [PubMed] [Google Scholar]
- 11.Dumas JJ, et al. Crystal structure of the wild-type von Willebrand factor A1-glycoprotein Ibα complex reveals conformation differences with a complex bearing von Willebrand disease mutations. J Biol Chem. 2004;279:23327–22334. doi: 10.1074/jbc.M401659200. [DOI] [PubMed] [Google Scholar]
- 12.Celikel R, et al. Modulation of α-thrombin function by distinct interactions with platelet glycoprotein Ibα. Science. 2003;301:218–221. doi: 10.1126/science.1084183. [DOI] [PubMed] [Google Scholar]
- 13.Dumas JJ, Kumar R, Seehra J, Somers WS, Mosyak L. Crystal structure of the gpibα-thrombin complex essential for platelet aggregation. Science. 2003;301:222–226. doi: 10.1126/science.1083917. [DOI] [PubMed] [Google Scholar]
- 14.Dong JF, Li CQ, Lopez JA. Tyrosine sulfation of the glycoprotein Ib-IX complex: Identification of sulfated residues and effect on ligand binding. Biochemistry. 1994;33:13946–13953. doi: 10.1021/bi00250a050. [DOI] [PubMed] [Google Scholar]
- 15.Marchese P, et al. Identification of three tyrosine residues of glycoprotein Ibα with distinct roles in von Willebrand factor and α-thrombin binding. J Biol Chem. 1995;270:9571–9578. doi: 10.1074/jbc.270.16.9571. [DOI] [PubMed] [Google Scholar]
- 16.Moore KL. The biology and enzymology of protein tyrosine o-sulfation. J Biol Chem. 2003;278:24243–24246. doi: 10.1074/jbc.R300008200. [DOI] [PubMed] [Google Scholar]
- 17.Hoffhines AJ, Jen CH, Leary JA, Moore KL. Tyrosylprotein sulfotransferase-2 expression is required for sulfation of rnase 9 and mfge8 in vivo. J Biol Chem. 2009;284:3096–3105. doi: 10.1074/jbc.M808434200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vanhoorelbeke K, Ulrichts H, Romijn RA, Huizinga EG, Deckmyn H. The gpibalpha-thrombin interaction: far from crystal clear. Trends Mol Med. 2004;10:33–39. doi: 10.1016/j.molmed.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 19.Bode W, Turk D, Karshikov A. The refined 1.9-å X-ray crystal structure of d-Phe-Pro-Arg chloromethylketone-inhibited human α-thrombin: Structure analysis, overall structure, electrostatic properties, detailed active-site geometry, and structure-function relationships. Protein Sci. 1992;1:426–471. doi: 10.1002/pro.5560010402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bock LC, Griffin LC, Latham JA, Vermaas EH, Toole JJ. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature. 1992;355:564–566. doi: 10.1038/355564a0. [DOI] [PubMed] [Google Scholar]
- 21.Padmanabhan K, Padmanabhan KP, Ferrara JD, Sadler JE, Tulinsky A. The structure of α-thrombin inhibited by a 15-mer single-stranded DNA aptamer. J Biol Chem. 1993;268:17651–17654. doi: 10.2210/pdb1hut/pdb. [DOI] [PubMed] [Google Scholar]
- 22.Skrzypczak-Jankun E, et al. Structure of the hirugen and hirulog 1 complexes of α-thrombin. J Mol Biol. 1991;221:1379–1393. [PubMed] [Google Scholar]
- 23.Rydel TJ, et al. The structure of a complex of recombinant hirudin and human α-thrombin. Science. 1990;249:277–280. doi: 10.1126/science.2374926. [DOI] [PubMed] [Google Scholar]
- 24.Tasset DM, Kubik MF, Steiner W. Oligonucleotide inhibitors of human thrombin that bind distinct epitopes. J Mol Biol. 1997;272:688–698. doi: 10.1006/jmbi.1997.1275. [DOI] [PubMed] [Google Scholar]
- 25.Muller J, Freitag D, Mayer G, Potzsch B. Anticoagulant characteristics of hd1-22, a bivalent aptamer that specifically inhibits thrombin and prothrombinase. J Thromb Haemost. 2008;6:2105–2112. doi: 10.1111/j.1538-7836.2008.03162.x. [DOI] [PubMed] [Google Scholar]
- 26.Petrera NS, et al. Long range communication between exosites 1 and 2 modulates thrombin function. J Biol Chem. 2009;284:25620–25629. doi: 10.1074/jbc.M109.000042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Handa M, Titani K, Holland LZ, Roberts JR, Ruggeri ZM. The von Willebrand factor-binding domain of platelet membrane glycoprotein Ib. Characterization by monoclonal antibodies and partial amino acid sequence analysis of proteolytic fragments. J Biol Chem. 1986;261:12579–12585. [PubMed] [Google Scholar]
- 28.Ruggeri ZM, et al. Unravelling the mechanism and significance of thrombin binding to platelet glycoprotein Ib. Thromb Haemostasis. 2010;104:894–902. doi: 10.1160/TH10-09-0578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Candia E, et al. Thrombin interaction with platelet GPIb: role of the heparin binding domain. Thromb Haemostasis. 1997;77:735–740. [PubMed] [Google Scholar]
- 30.De Cristofaro R, De Candia E, Rutella S, Weitz JI. The Asp(272)-Glu(282) region of platelet glycoprotein Ibα interacts with the heparin-binding site of α-thrombin and protects the enzyme from the heparin-catalyzed inhibition by antithrombin III. J Biol Chem. 2000;275:3887–3895. doi: 10.1074/jbc.275.6.3887. [DOI] [PubMed] [Google Scholar]
- 31.Li CQ, Vindigni A, Sadler JE, Wardell MR. Platelet gylcoprotein Ibα binds to thrombin anion-binding exosite II inducing allosteric changes in the activity of thrombin. J Biol Chem. 2001;276:6161–6168. doi: 10.1074/jbc.M004164200. [DOI] [PubMed] [Google Scholar]
- 32.De Cristofaro R, De Candia E, Landolfi R, Rutella S, Hall SW. Structural and functional mapping of the thrombin domain involved in the binding to the platelet glycoprotein Ib. Biochemistry. 2001;40:13268–13273. doi: 10.1021/bi010491f. [DOI] [PubMed] [Google Scholar]
- 33.Sabo TM, Maurer MC. Biophysical investigation of gpibα binding to thrombin anion binding exosite II. Biochemistry. 2009;48:7110–7122. doi: 10.1021/bi900745b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Richardson JL, Fuentes-Prior P, Sadler JE, Huber R, Bode W. Characterization of the residues involved in the human α-thrombin-haemadin complex: an exosite II-binding inhibitor. Biochemistry. 2002;41:2535–2542. doi: 10.1021/bi011605q. [DOI] [PubMed] [Google Scholar]
- 35.Jandrot-Perrus M, Huisse MG, Krstenansky JL, Bezeaud A, Guillin MC. Effect of the hirudin carboxy-terminal peptide 54–65 on the interaction of thrombin with platelets. Thromb Haemostasis. 1991;66:300–305. [PubMed] [Google Scholar]
- 36.Rydel TJ, Tulinsky A, Bode W, Huber R. Refined structure of the hirudin-thrombin complex. J Mol Biol. 1991;221:583–601. doi: 10.1016/0022-2836(91)80074-5. [DOI] [PubMed] [Google Scholar]
- 37.Lopez JA, Smith DR, Dong J-F. A unique receptor for a unique function: The glycoprotein Ib-IX-V complex in platelet adhesion and activation. In: Berndt MC, editor. Platelets, thrombosis and the vessel wall, Advances in vascular biology. Boca Raton, Florida, USA: CRC Press; 2000. pp. 65–82. [Google Scholar]
- 38.Rich RL, Myszka DG. Survey of the year 2006 commercial optical biosensor literature. J Mol Recognit. 2007;20:300–366. doi: 10.1002/jmr.862. [DOI] [PubMed] [Google Scholar]
- 39.De Cristofaro R, De Filippis V. Interaction of the 268–282 region of glycoprotein Ibα with the heparin-binding site of thrombin inhibits the enzyme activation of factor VIII. Biochem J. 2003;373:593–601. doi: 10.1042/BJ20030167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ouyang YB, Moore KL. Molecular cloning and expression of human and mouse tyrosylprotein sulfotransferase-2 and a tyrosylprotein sulfotransferase homologue in caenorhabditis elegans. J Biol Chem. 1998;273:24770–24774. doi: 10.1074/jbc.273.38.24770. [DOI] [PubMed] [Google Scholar]
- 41.Kretz CA, Stafford AR, Fredenburgh JC, Weitz JI. Hd1, a thrombin-directed aptamer, binds exosite 1 on prothrombin with high affinity and inhibits its activation by prothrombinase. J Biol Chem. 2006;281:37477–37485. doi: 10.1074/jbc.M607359200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.