

Abstract
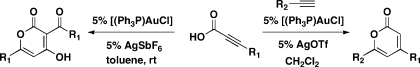
Sequential alkyne activation of terminal alkynes and propiolic acids by gold(I) catalysts yields compounds having α-pyrone skeletons. Novel cascade reactions involving propiolic acids are reported that give rise to α-pyrones with different substitution patterns.
In efforts to synthesize compounds having properties that facilitate small-molecule probe and drug discovery,(1) we have developed multicomponent coupling reactions that use gold(I) catalysts and yield, among others, complex α-pyrones.(2) Activation of the electron-deficient alkyne in propargyl propiolate 1 by a cationic gold(I) catalyst results in allenyl propiolate 2, which undergoes a 6-endo-dig cyclization(3) to oxocarbenium intermediate A (Figure 1). In order to generate diverse and previously inaccessible α-pyrones,(4) we investigated the possibility of generating the vinyl propiolate 5. We imagined this intermediate undergoing a similar 6-endo cyclization to afford oxocarbenium intermediate B and then α-pyrone 6 after deprotonation and proto-demetalation. Intermediate 5 would result from an intermolecular coupling of propiolic acid 3 and alkyne 4 catalyzed by the same gold(I) catalyst.(5) Herein, we describe a new gold(I)-catalyzed cascade reaction based on the concept of sequential alkyne activation,2,6 synthesizing substituted α-pyrones in one step from readily available propiolic acids.
Figure 1.

Syntheses of α-pyrones via gold(I)-catalyzed cascade reactions.
We initiated our investigation using commercially available propiolic acid 3a and terminal alkyne 4a. The counterion of the cationic gold(I) catalyst was determined to have a significant effect on the product distribution (Table 1, entries 1–4). When AgOTf or AgPF6 was used, we isolated α-pyrone 6a in modest yields (entries 1 and 2), presumably via the vinyl propiolate 5a resulting from the gold-catalyzed Markovnikov addition of the carboxylic acid to the terminal alkyne.(5) However, AgSbF6 led to α-pyrone 7a as the predominant product (entry 3; structure determined by X-ray crystallography),(7) whereas AgNTf2 gave both α-pyrones (entry 4). The reaction was also sensitive to the identity of the solvent. With [(Ph3P)AuCl]/AgPF6 as the catalyst, switching the solvent from toluene to dichloromethane significantly lowered the yield of 6a with substantial starting material recovery (Table 1, entry 5). In contrast, with [(Ph3P)AuCl]/AgOTf as the catalyst, dichloromethane afforded 6a in higher yield than that afforded by toluene (entry 6). The more electron-donating ligand tricyclohexylphosphine and less electron-donating ligand tris(para-trifluoromethylphenyl)phosphine had minimal effects on the reaction (entries 7 and 8). AuCl or AgOTf alone failed to catalyze the cascade reaction (entries 9 and 10). The best result was obtained by increasing the amount of alkyne 4a to 5 equiv and using the catalyst [(Ph3P)AuCl]/AgOTf in dichloromethane, which gave rise to 6a in 83% yield (entry 11). Unexpectedly, increasing the amount of alkyne 4a and using the catalyst [(Ph3P)AuCl]/AgPF6 in toluene (entry 12) resulted in the isolation of the vinyl propiolate 5a, which was further subjected to the optimized reaction conditions to give 6a in excellent yield (Figure 2).
Table 1. Optimization of Reaction Conditions for the Synthesis of 6a.

| yield%b |
||||
|---|---|---|---|---|
| entry | catalyst | conditionsa | 6a | 7a |
| 1 | [(Ph3P)AuCl]/AgOTf | toluene, rt | 43 | <5 |
| 2 | [(Ph3P)AuCl]/AgPF6 | toluene, rt | 52 | <5 |
| 3 | [(Ph3P)AuCl]/AgSbF6 | toluene, rt | <5 | 84 |
| 4 | [(Ph3P)AuCl]/AgNTf2 | toluene, rt | 20 | 60 |
| 5 | [(Ph3P)AuCl]/AgPF6 | CH2Cl2, rt | 12 | <5c |
| 6 | [(Ph3P)AuCl]/AgOTf | CH2Cl2, rt | 74 | <5 |
| 7 | [(Cy3P)AuCl]/AgOTf | CH2Cl2, rt | 75 | <5 |
| 8 | [(p-CF3C6H4)3P]AuCl/AgOTf | CH2Cl2, rt | 68 | <5 |
| 9 | AuCl | CH2Cl2, rt | N.R. | |
| 10 | AgOTf | CH2Cl2, rt | N.R. | |
| 11 | [(Ph3P)AuCl]/AgOTf | CH2Cl2, rt | 83d | <5 |
| 12 | [(Ph3P)AuCl]/AgPF6 | toluene, rt | 35d | <5e |
[3a] = 0.2 M, 1.5 equiv of 4a.
Isolated yields after column chromatography.
39% of 3a was recovered.
5 equiv of 4a were employed.
5a was isolated in 29% yield.
Figure 2.

Cyclization of vinyl propiolate 5a into α-pyrone 6a.
While we do not understand why different counterions provide different product distributions (6a vs 7a), a proposed mechanism of the serendipitously discovered propiolic acid dimerization is offered in Figure 3. The addition of the carboxylic acid to the β-position of the propiolic acid yields vinyl ester D. Further activation of D by cationic gold(I) generates oxocarbenium E, where the acyl group is transferred to the C–Au bond with concomitant regeneration of the gold(I) catalyst.(8) The 6-endo-dig cyclization of carboxylic acid G onto the activated alkyne followed by enolization affords 4-hydroxy α-pyrone 7 as the final product.
Figure 3.

Dimerization of propiolic acid leading to 4-hydroxy α-pyrone.
The scope of the cascade reaction was explored with a variety of propiolic acids and alkynes (Scheme 1). Generally, moderate to excellent yields were obtained with different terminal alkynes and propiolic acids. More sterically hindered alkynes gave lower yields (6g). Ether (6h and 6o), ester (6i and 6j), halide (6k), and alkyne (6m, 6n, and 6o) functional groups are compatible with the reaction conditions. The structure of α-pyrone 6p was verified by X-ray analysis.(7) Notably, α-pyrone 6t is a natural product with antibiotic and antifungal activity,(9) which recently has been synthesized using a gold(I)-catalyzed cycloisomerization of β-alkynylpropiolactone.(10) Using the new method reported here, 6t was synthesized in one step from the commercially available propiolic acid and 1-heptyne. Pyrone 6u was synthesized in 77% yield from 3-bromopropiolic acid. The bromide functionality provides a handle to introduce other groups at the C-4 position via transition-metal-catalyzed cross-coupling reactions. Gratifyingly, in separate reactions using 0.5 g of phenylpropiolic acid, pyrones 6c and 6l were obtained in good yields. Unfortunately, neither phenylacetylene nor the internal alkyne 2-butyne reacts at room temperature in CH2Cl2. Only a low yield of the corresponding α-pyrone was obtained using elevated reaction conditions (6v and 6w). When 4-methoxy-4-oxobut-2-ynoic acid was used, only a 1,2-addition of the acid to 3-phenyl-1-propyne took place, yielding 5b in 59% yield. A higher reaction temperature (50 °C) gave a similar result, presumably because the capacity of the triple bond of 4-methoxy-4-oxobut-2-ynoic acid to coordinate gold is diminished by the existing ester group.
Scheme 1. Gold(I)-Catalyzed Syntheses of α-Pyrones from Propiolic Acids and Alkynes.

Reaction conditions: propiolic acid (0.2–0.7 mmol, 0.2 M), alkyne (5–6 equiv), [(Ph3P)AuCl]/AgOTf (5 mol %), CH2Cl2, rt, 12 h; bphenylpropiolic acid (3.4 mmol), alkyne (5 equiv), [(Ph3P)AuCl]/AgOTf (5 mol %), CH2Cl2, rt, 24 h; cCH2Cl2, 50 °C, 12 h; d3-(naphthalene-2-yl)propiolic acid (0.1 M); ephenylacetylene (6 equiv), toluene, 60 °C, slow addition of acid (over 2 h), 12 h; f2-butyne (10 equiv), toluene, 60 °C, 12 h.
The method described herein provides an efficient and simple route to multiply substituted α-pyrones. The generality observed thus far suggests that it will find many future applications.
Acknowledgments
The NIGMS-sponsored Center of Excellence in Chemical Methodology and Library Development (P50-GM069721) sponsored this research. S.L.S. is an investigator with the Howard Hughes Medical Institute.
Supporting Information Available
Experimental procedures and full spectroscopic data for all new compounds and CIFs for 7a and 6p. The material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Nielsen T. E.; Schreiber S. L. Angew. Chem., Int. Ed. 2008, 47, 48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Luo T.; Schreiber S. L. Angew. Chem., Int. Ed. 2007, 46, 8250–8253. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Luo T.; Schreiber S. L. J. Am. Chem. Soc. 2009, 131, 5667–5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For the application of gold-catalyzed 6-endo-dig cyclization, see: ; a Sherry B. D.; Maus L.; Laforteza B. N.; Toste F. D. J. Am. Chem. Soc. 2006, 128, 8132–8133. [DOI] [PubMed] [Google Scholar]; b Minnihan E. C.; Colletti S. L.; Toste F. D.; Shen H. C. J. Org. Chem. 2007, 72, 6287–6289. [DOI] [PubMed] [Google Scholar]; c Imase H.; Noguchi K.; Hirano M.; Tanaka K. Org. Lett. 2008, 10, 3563–3566. [DOI] [PubMed] [Google Scholar]; d Barabé F.; Bétournay G.; Bellavance G.; Barriault L. Org. Lett. 2009, 11, 4236–4238. [DOI] [PubMed] [Google Scholar]; e Menon R. S.; Findlay A. D.; Bissember A. C.; Banwell M. G. J. Org. Chem. 2009, 74, 8901–8903. [DOI] [PubMed] [Google Scholar]; f Jiang C.; Xu M.; Wang S.; Wang H.; Yao Z. J. J. Org. Chem. 2010, 75, 4323–4325. [DOI] [PubMed] [Google Scholar]; g Liu Y.; Xu W.; Wang X. Org. Lett. 2010, 12, 1448–1451. [DOI] [PubMed] [Google Scholar]
- a Dickinson J. M. Nat. Prod. Rep. 1993, 10, 71–98. [DOI] [PubMed] [Google Scholar]; b van Raaij M. J.; Abrahams J. P.; Leslie A. G.; Walker J. E. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 6913–6917. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Steyn P. S.; van Heerden F. R. Nat. Prod. Rep. 1998, 15, 397–413. [DOI] [PubMed] [Google Scholar]; d Salomon C. E.; Magarvey N. A.; Sherman D. H. Nat. Prod. Rep. 2004, 21, 105–121. [DOI] [PubMed] [Google Scholar]; e McGlacken G. P.; Fairlamb I. J. Nat. Prod. Rep. 2005, 22, 369–385. [DOI] [PubMed] [Google Scholar]; f Sunazuka T.; O̅mura S. Chem. Rev. 2005, 105, 4559–4580. [DOI] [PubMed] [Google Scholar]
- a Roembke P.; Schmidbaur H.; Cronje S.; Raubenheimer H. J. Mol. Catal. A 2004, 212, 35–42. [Google Scholar]; b Cui D.-M.; Meng Q.; Zheng J.-Z.; Zhang C. Chem. Commun. 2009, 1577–1579. [DOI] [PubMed] [Google Scholar]; c Chary B. C.; Kim S. J. Org. Chem. 2010, 75, 7928–7931. [DOI] [PubMed] [Google Scholar]; d Lee P. H.; Kim S.; Park A.; Chary B. C.; Kim S. Angew. Chem., Int. Ed. 2010, 49, 6806–6809. [DOI] [PubMed] [Google Scholar]
- a Hashmi A. S. K.; Schwarz L.; Choi J.-H.; Frost T. M. Angew. Chem., Int. Ed. 2000, 39, 2285–2288. [DOI] [PubMed] [Google Scholar]; b Zhao J.; Hughes C. O.; Toste F. D. J. Am. Chem. Soc. 2006, 128, 7436–7437. [DOI] [PubMed] [Google Scholar]; c Liu X.-Y.; Che C.-M. Angew. Chem., Int. Ed. 2008, 47, 3805–3810. [DOI] [PubMed] [Google Scholar]; d Sperger C.; Fiksdahl A. Org. Lett. 2009, 11, 2449–2452. [DOI] [PubMed] [Google Scholar]; e Sperger C. A.; Fiksdahl A. J. Org. Chem. 2010, 75, 4542–4553. [DOI] [PubMed] [Google Scholar]
- Crystallographic Information Files (CIFs) for 7a and 6p are available at the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. See Supporting Information for further details.
- For an example of a related acyl transfer involving a gold(III) intermediate, see: Wang S.; Zhang L. J. Am. Chem. Soc. 2006, 128, 8414–8415. [DOI] [PubMed] [Google Scholar]
- Evidente A.; Cabras A.; Maddau L.; Serra S.; Andolfi A.; Motta A. J. Agric. Food Chem. 2003, 51, 6957–6960. [DOI] [PubMed] [Google Scholar]
- Dombray T.; Blanc A.; Weibel J.-M.; Pale P. Org. Lett. 2010, 12, 5362–5365. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.