

Abstract
Mutations in the A-type lamin (LMNA) gene are associated with age-associated degenerative disorders of mesenchymal tissues, such as dilated cardiomyopathy, Emery-Dreifuss muscular dystrophy, and limb-girdle muscular dystrophy. The molecular mechanisms that connect mutations in LMNA with different human diseases are poorly understood. Here, we report the identification of a Muscle-enriched A-type Lamin-interacting Protein, MLIP (C6orf142 and 2310046A06rik), a unique single copy gene that is an innovation of amniotes (reptiles, birds, and mammals). MLIP encodes alternatively spliced variants (23–57 kDa) and possesses several novel structural motifs not found in other proteins. MLIP is expressed ubiquitously and most abundantly in heart, skeletal, and smooth muscle. MLIP interacts directly and co-localizes with lamin A and C in the nuclear envelope. MLIP also co-localizes with promyelocytic leukemia (PML) bodies within the nucleus. PML, like MLIP, is only found in amniotes, suggesting that a functional link between the nuclear envelope and PML bodies may exist through MLIP. Down-regulation of lamin A/C expression by shRNA results in the up-regulation and mislocalization of MLIP. Given that MLIP is expressed most highly in striated and smooth muscle, it is likely to contribute to the mesenchymal phenotypes of laminopathies.
Keywords: Aging, Cell Differentiation, Differentiation, Genetic Diseases, Lamin
Introduction
Lamins are intermediate filament proteins of the nuclear lamina that underlies the inner nuclear membrane of all multicellular eukaryotes. They are fundamentally important for nuclear architecture, chromatin organization, and transcriptional regulation of gene expression (1–3). A-type lamin expression is closely associated with cellular differentiation during development (4) and stem cell differentiation (5). However, beyond this, the molecular mechanisms responsible for transcriptional specificity and cellular commitment and differentiation are largely unknown.
More than 400 different mutations in the lamin A/C (LMNA)3 gene have been described and result in a heterogeneous group of disorders that are collectively defined as laminopathies (6). The diseases caused by the wide spectrum of LMNA gene mutations are characterized by a diverse variability of clinical phenotypes; ranging from cardiac and skeletal myopathies to partial lipodystrophy, peripheral neuropathy, and premature aging. No clear genotype-phenotype correlation has been identified because mutations in the same codon can cause different diseases in unrelated families (7–9) and even among family members (10). A complete loss of A-type lamins resulted in a postnatal appearance of abnormal nuclear structures that manifests as muscular dystrophy and dilated cardiomyopathy (11, 12). A recent study used hierarchical cluster analysis for assembling laminopathies into classes based on organ system involvement. They uncovered a nonrandom relationship between the class of laminopathy and the mutation (13).
Several hypotheses have been proposed for the pathogenesis of laminopathies, and most research has been focused on the “mechanical stress” and “altered gene regulation” hypotheses. The structural integrity of the nucleus may be affected by the expression of mutant A-type lamins because mice that lack LMNA have varied nuclear morphologies with a significant redistribution of nuclear envelope proteins (12, 14). The fragility of the nuclear envelope is believed to contribute (in part) to pathologies in tissues subjected to mechanical stresses, such as skeletal and cardiac muscle. The complete loss of A-type lamins supports this hypothesis. The effect of autosomal dominant missense mutations on the structural integrity of the nucleus remains to be determined.
Evidence in support of the altered gene regulation hypothesis include many of the proteins that are involved in chromatin organization, transcription, and binding to DNA are either directly or indirectly associated with the nuclear envelope. Chromatin organization and transcriptional regulation of gene expression are therefore affected in specific ways due to the disruption of the nuclear envelope (15). We know very little about the molecular pathogenesis from mutations in the LMNA gene to heart phenotypes. Therefore, the question is how these different pathologies arise from alterations in the same gene (LMNA) that is almost ubiquitously expressed in adult cells.
To address this question we probed the first 230 amino acids of A-type lamin (rich in mutations associated with skeletal and cardiac muscle laminopathies) for new interactors. A muscle-enriched A-type lamin-interacting protein (MLIP, C6orf142) was identified in our laboratory. The MLIP gene encodes at least seven, alternatively spliced, LMNA-interacting factors. Phylogenetic analysis revealed that MLIP is a unique, single copy gene that is found only in the genomes of amniotes. MLIP may represent an innovation of amniotes.
EXPERIMENTAL PROCEDURES
Yeast Two-hybrid
The bait-vector was constructed by subcloning human LMNA fragment (660 bp corresponding to the 1–230 aa) into EcoRI/BamHI sites of pGBKT7 vector (Clontech). The bait-vector was transformed into yeast (AH109). Yeast toxicity, autotranscription activation, and bait expression were all assessed. The bait-vector-transformed yeast were then mated with the pretransformed human heart library (MatchmakerTM, Clontech) and screened according to manufacture's protocol (BD Biosciences Clontech).
Bacterial Protein Expression, Purification, and in Vitro Binding Assay
Bacterial expression plasmids for MLIP proteins and lamin A were constructed by subcloning MLIP cDNA into the His tag fusion vector pET100D (Invitrogen) and lamin A (1–230 aa) into the GST fusion vector pGEX-2T (GE Healthcare). Plasmids were transformed independently into the bacterial strain BL21 DE3(pLysS) and induced with 250 μm isopropyl-1-thio-d-galactopyranoside for 2 h during exponential growth phase. Cells were lysed in 50 mm Tris-HCl (pH 7.5), 500 mm NaCl, and 0.1% Triton X-100 in the presence of a mixture of protease (Sigma) inhibitors, and then centrifuged at 100,000 × g for 45 min. The His6-MLIP supernatant was loaded on to a Ni2+-nitrilotriacetic acid column (GE Healthcare), washed with 50 mm Tris-HCl (pH 7.5), 500 mm NaCl, and 25 mm imidazole buffer, and eluted with a linear gradient from 25 to 250 mm imidazole in the presences of 50 mm Tris-HCl (pH 7.5), 500 mm NaCl. The GST-lamin bacterial supernantants were run across a GSTrap 4B column (GE Healthcare) and washed, and the GST-lamin was eluted with 50 mm Tris-HCl + 20 mm reduced glutathione, pH 8.0. The fractions containing the His6-tagged MLIP proteins or GST-lamin were then dialyzed against 10 mm phosphate buffer (pH 7.4), 50 mm NaCl, and 0.05% Triton X-100. The purity of each fusion protein was 95%, as determined by Coomassie Blue-stained protein gels. Various combinations of His6-MLIP and GST-lamin recombinant proteins were mixed together (as indicated) and incubated at room temperature for 30 min in 10 mm phosphate buffer (pH 7.4), 50 mm NaCl, 0.05%Triton X-100. Ni2+-nitrilotriacetic acid-Sepharose beads were added to each reaction mixture, and complexes were isolated and washed by centrifugation in phosphate buffer (pH 7.4), 50 mm NaCl, 0.05% Triton X-100. Complexes were eluted by the addition of SDS-PAGE loading buffer, boiled, and resolved by SDS-PAGE. Western blot analysis was performed using anti-GST (Cell Signaling) and anti-MLIP polyclonal antibodies. A 1:10 dilution of the total starting material was run on the same gel. The assay was repeated two additional times with similar results.
Cell Culture
Mouse HL1 cell line were cultured at 37 °C in an atmosphere of 5% CO2 in growth medium consisting of Claycomb medium supplemented with 10% fetal bovine serum (FBS), 1% v/v penicillin-streptomycin, and 2 mm l-glutamine. C2C12 mouse myoblasts were cultured at 37 °C in an atmosphere of 10% CO2 in growth medium consisting of DMEM supplemented with 20% FBS, 1% v/v penicillin-streptomycin, and 2 mm l-glutamine. For immunofluorescence, cells were grown on sterile coverslips coated with gelatin in 12-well cell culture-treated dishes.
Northern Blot Analyses
MLIP cDNA fragment was radiolabeled with [γ-32P]dCTP and hybridized to the human (Clontech) or mouse (Zyagen, San Diego, CA) multitissue Northern blot. A human β-actin cDNA fragment was random hexamer-radiolabeled with [γ-32P]dCTP and used to probe the blots as an internal loading control.
MLIP-specific Antibody
MLIP antibodies were raised in a rabbit against two synthetic peptides NH2-MEFGKHEPGSSLKRNKKNL-COOH and NH2-LRKDEEVYEPNPFSKYL-COOH (21st Century Biochemicals).
Whole Cell Extract Preparation, Western Blotting, and Co-immunoprecipitations
Proteins were extracted using lysis buffer containing 50 mm Tris-HCl (pH = 8.0), 200 mm NaCl, 20 mm NaF, 20 mm β-glycerophosphate, 0.5% Nonidet P-40, 0.1 mm Na3VO4, 1 mm dithiothreitol, 1× protease inhibitor mixture (1 tablet/7.0 ml; Roche), and phosphatase inhibitor cocktails (0.1 ml/7.0 ml; Sigma). Cells were scraped from dishes and lysates pipetted into 1.5-ml centrifuge tubes. Lysates were incubated on ice for 20 min and then cleared by centrifugation at 10,000 × g for 10 min at 4 °C. Supernatants were collected, and protein concentrations were determined using the Bradford protein assay (Bio-Rad). For co-immunprecipitation experiments affinity purified (against MLIP COOH-terminal peptide NH2-LRKDEEVYEPNPFSKYL-COOH) was covalently coupled to an agarose bead according to the manufacturer's instructions (co-immunoprecipitation kit; Pierce). 20 μl of a 50% slurry of anti-MLIP resin or 20 μl of MLIP-peptide neutralized anti-MLIP resin (control) was added to 100 μg of precleared total mouse heart lysates and incubated overnight at 4 °C. Primary antibody-protein complexes were isolated and eluted from the antibody-resins according to the manufacturer's instructions (co-immunoprecipitation kit). Eluted protein complexes were resolved on a 15% 0.75-mm-thick SDS-Tris-glycine-polyacrylamide gel (200 V) and transferred to PVDF membranes (Millipore) for 1 h at 100 V at 4 °C. Membranes were blocked in 5% nonfat milk dissolved in Tris-buffered saline + 0.05% Tween 20. The MLIP primary antibody concentration used was 1:100,000. Membranes were then incubated with HRP-conjugated anti-rabbit and anti-mouse IgG secondary antibody (Santa Cruz Biotechnology). Immunoblot signals were detected with a SuperSignal West Pico Chemiluminescent kit (Thermo Scientific) and visualized on x-ray film (Sigma).
RNA Isolation, RT-PCR, RACE, and Cloning of MLIP from Mouse Hearts
Total RNA was extracted from adult mouse hearts using TRIzol Reagent (Invitrogen). RNA samples were DNase-treated prior to RT-PCR (Promega). RNA samples were assessed for quality using the Bioanalyzer Nano chips (Agilent Technologies). cDNA was reverse-transcribed using AffinityScript Multiple Temperature Reverse Transcriptase (Stratagene) from 1 μg of RNA with an oligo(dT) primer. MLIP DNA was amplified with primers for MLIP-5′-UTR (5′-ACC TCT CTC TCA TTC TTT CAC CAT G-3′) and mMLIP-3′-UTR (5′-CAC TTC CAC TCC AGC TTC C-3′). Gene amplification was carried out as follows: 95 °C for 5 min followed by 34 cycles in 3 steps, 95 °C for 30 s, 55 °C for 30 s, 72 °C for 3 min, and 72 °C for 5 min using Taq DNA polymerase (NEB). PCR products were subsequently TA-cloned into pCRII-TOPO vector (Invitrogen). MLIP clones were then sequenced using the BigDye Terminator v3.1 cycle sequencing kit (Applied Biosystems). 5′/3′-RACE was performed on cDNA isolated from mouse hearts as described in the 5′/3′-RACE kit, second generation (Roche Applied Sciences) using primers specific to MLIP exons 1, 3, and 9.
Immunofluorescence
HL1 and C2C12 cells were grown on gelatin/fibronectin-coated coverslips in 12-well cell culture-treated plates (Fisher Scientific). Cells were fixed using precooled methanol (−20 °C) for 20 min at −20 °C and were washed twice with phosphate-buffered saline (PBS). Cells were further permeabilized with 0.1% Nonidet P-40 in PBS + 5%FBS for 3 min at room temperature and then washed three times with PBS (5 min each). To reduce nonspecific binding, cells were then blocked with PBS supplemented with 5% FBS for 30 min at room temperature with gentle shaking. Cells were then incubated with anti-MLIP (1:5,000), anti-lamin A/C (1:1,000), and anti-PML (1:500) (Santa Cruz Biotechnology) in 1.5% FBS/PBS overnight at 4 °C. Primary antibodies were detected by fluorescent-conjugated secondary antibody (1:1,000) Alexa Fluor 488 donkey anti-rabbit IgG or Alexa Fluor 594 donkey anti-goat IgG (Invitrogen). Nuclei were stained with DAPI (1 μg/ml) and mounted with fluorescent mounting medium (Dako). Images were then visualized using fluorescent microscopy (Carl Zeiss AxioImager Z1 Microscope). Bleed-through control experiments were performed while acquiring images. There is no evidence of bleed-through under the conditions in which we acquired all of our immunofluorescence images (data not shown).
Immunohistochemistry
Paraffin-embedded mouse tissue sections (5–7 μm thick) were stained for MLIP (1:1,000 dilution) or peroxiredoxin 3 (1:5,000; Abcam) using a Zyagen rabbit peroxidase detection kit according to the manufacturer's instructions. All slides were counterstained with hematoxylin.
LMNA Knockdown C2C12 Myoblasts
Mouse LMNA-shRNAmir sequences in the pLKO vector were purchased from Open Biosystems. Sequences included TRCN0000089848 mature-sense CTTTCCTTGTATCTGCCTTAA. Lentiviral particles were generated using the TransLenti Viral Packaging Mix (Open Biosystems). C2C12 myoblasts were transduced at a multiplicity of infection of 0.5. Transduced cells were analyzed after 72 h for lamin A and MLIP expression by Western blotting and immunofluorescence.
RESULTS
Identification and Cloning of a Novel Amniote-specific Lamin A/C-interacting Protein
To identify specific proteins that interact with the first 230 amino acids of lamins A and C that includes the N-terminal globular head domain (1–30 aa), coil 1A (31–66 aa), linker 1 (67–77 aa), coil 1B (78–220 aa), and part of linker 12 (221–239 aa) (16) a yeast two-hybrid interaction screen was utilized. A total of 232 positive interacting cDNA clones were isolated from the 3.5 × 106 independent clones screened in a human heart cDNA library. MLIP was identified in six independent clones of the initial 232 positive clones (Fig. 1A), and it was found to be homologous to C6orf142 (NM_138569) that maps to human chromosome 6p12.1 and spans 247 kb. To further validate and partially define the interaction domain between MLIP and lamin A/C, the original lamin A/C bait was mutagenized to generate a lamin-1a bait that encompasses residues 1–130 that includes the N-terminal globular head domain, coil 1A, linker L1, and the amino-terminal portion of coil 1B and a lamin-1b bait (residues 120–230) that includes coil 1B and part of linker 12. The ability of each of these lamin A/C baits to interact with MLIP was tested. The lamin-1a bait presented a positive interaction with MLIP whereas the lamin-1b bait failed to interact (Fig. 1B). These results indicate that the MLIP interaction domain is between amino acids 1 and 130 of lamin A/C, which includes the amino-terminal globular head domain plus coil 1A, linker 1, and the first 43 amino acids of coil 1B.
FIGURE 1.

Interaction of LMNA with MLIP. A, yeast two-hybrid interaction assay that identified six independent MLIP clones from a human heart cDNA library (Clontech). B, yeast-two hybrid assay of the LMNA1–230, LMNA1–130 and LMNA120–230, and MLIP clone 1 and MLIP clone 3 interactions. Twenty microliters of the culture was plated on His+ (left) and His− (right) plates at a cell density of 1 × 106/ml.
Phylogenetic analysis through comparisons of the deduced 895 amino acid of human MLIP protein or the nucleic acid sequence against the GenBank database revealed no substantial similarities with any other human or mouse gene. No MLIP homologs were identified in anamniotes (amphibians or fish), nor were they found in invertebrates. Homologous forms of MLIP in non-human, vertebrate genomes or expressed sequence tag databases were identified and were only observed in amniotes (mammals, reptiles, turtles, and birds) (Fig. 2A).
FIGURE 2.

MLIP is a single copy, evolutionarily conserved amniota gene. Phylogenetic and gene synteny analysis centered on (A) human MLIP (C6orf142) gene and (B) human LMNA gene was performed by Genomicus v58.01 genome browser (50).
The human MLIP gene maps to human chromosome 6p12.1 and is flanked by LRRC1 and TINAG with this genomic organization conserved in amniotes (Fig. 2A). The LRRC1 gene is conserved in euteleostomi (bony vertebrates including chimpanzees, dogs, cows, mice, rats, chickens, and zebra fish) whereas TINAG is conserved in both Drosophila and Caenorhabditis elegans. By contrast, LMNA is observed in tetrapoda (amphibians, reptiles, birds, and mammals) and euteleostomi (Fig. 2B) The significance of this observation is that there are no other reported examples of a nonhomologous, single copy, amniote gene. This suggests that MLIP is an amniote acquisition that may play an important regulatory or functional role that is unique to amniotes.
Protein alignment of predicted full-length human, mouse, and chicken (Fig. 3A) MLIP defined at least five highly conserved MLIP domains. Domain 1 encompasses the lamin interaction domain (amino acids 1–147) and a putative type I sumoylation site (amino acids 123–129). Domains 2 and 3 have an unknown function (amino acids 380–397 and amino acids 448–467). Domain 4 includes a putative nuclear localization sequence (amino acids 725–775). Domain 5 is also of unknown function (amino acids 803–895) and contains a highly conserved putative tyrosine-phosphorylation site (amino acids 869–876) (Fig. 3B). None of these domains is present in any other gene or protein within anamniotes or invertebrates.
FIGURE 3.

Alignment and predicted secondary structure for human MLIP. A, the deduced amino acid sequence of human MLIP and other selected species (mouse and chicken) were aligned by ClustalW algorithm. MLIP-specific polyclonal antibodies were raised against the two highlighted (yellow) mouse MLIP peptides. The highlighted red sequence is primarily β-strand. The consensus sequence is denoted by asterisks (*) as identical, periods as conserved substitutions, and colons as semiconservative substitutions. Numbers indicate the position of the last amino acid of each line. B, the predicted secondary structure for human MLIP is represented by red, α-helical; green, β-strand; black, coil and turn. Hydrophobicity (by Kyte-Doolittle plot) of human MLIP is represented below the secondary amino acid structure of human MLIP.
The lamin-binding domain within MLIP was inferred to be within the 6 overlapping sequence of the MLIP clones that were identified in the original yeast-two hybrid screen (Fig. 1). All 6 clones included the N-terminal portion of MLIP, and we took the shortest positive clone, 120 bp, as containing the lamin-binding domain (Fig. 1A). This putative lamin-binding domain spans across exons 1 and 2 of the MLIP gene. This region of MLIP appears to interact with residues within the first 120 amino acids of lamin A/C (Fig. 1B). This region of lamin A/C includes the N-terminal globular head domain, coil 1A, linker 1, and the first 43 amino acids of coil 1B. Ongoing experiments are being performed to define further the specific interaction domains through which A-type lamins and MLIP interact. Preliminary experiments suggest that this interaction domain may comprise multiple sequence motifs whose tertiary structure may only be resolved by co-crystallization of the MLIP-lamin A/C proteins complex.
The predicted secondary structure of human MLIP shows that 46% of the sequence is β-strand and 27% α-helices. The majority of the α-helices are located in the amino and carboxyl termini of MLIP. In contrast, the majority of β-strand is located between aa 425 and 706 where 62% is β-strand with only 4% α-helical (Fig. 3B). MLIP contains a highly conserved nuclear localization sequence (PENKKSKQ from amino acid 721 to 728 in humans and PENKKPKQ from amino acid 689 to 696 in the mouse) that matches the consensus of nuclear localization sequences found in the nuclear proteins v-myb myeloblastosis viral oncogene homolog, and CCAAT/enhancer binding protein ζ. Within the N-terminal homology domain of MLIP is a highly conserved putative type I sumoylation motif (ΨKXE) DLFKAE that is perfectly conserved among humans, mice, and chickens. The hydrophobic profile of MLIP, as determined by the Kyte-Doolittle algorithm, shows it to be primarily hydrophilic with no predicted transmembrane domains (Fig. 3B).
MLIP Interacts Directly with Lamin A/C
To confirm that the MLIP-lamin A/C interaction identified by the yeast two-hybrid assay was direct, in vitro co-precipitation assays were then performed. Two major splice variants of MLIP were initially cloned from mouse hearts and fused in-frame with a His6 epitope tag. Various combinations of purified His6-MLIP and GST-lamin (residues 1–230) recombinant proteins were incubated together at room temperature. MLIP was co-precipitated with lamin, as detected by immunoblotting for the GST tag fused to lamin (Fig. 4A). Lamin did not pellet in the absence of MLIP (Fig. 4A), nor was GST alone able to be precipitated by MLIP (data not shown).
FIGURE 4.
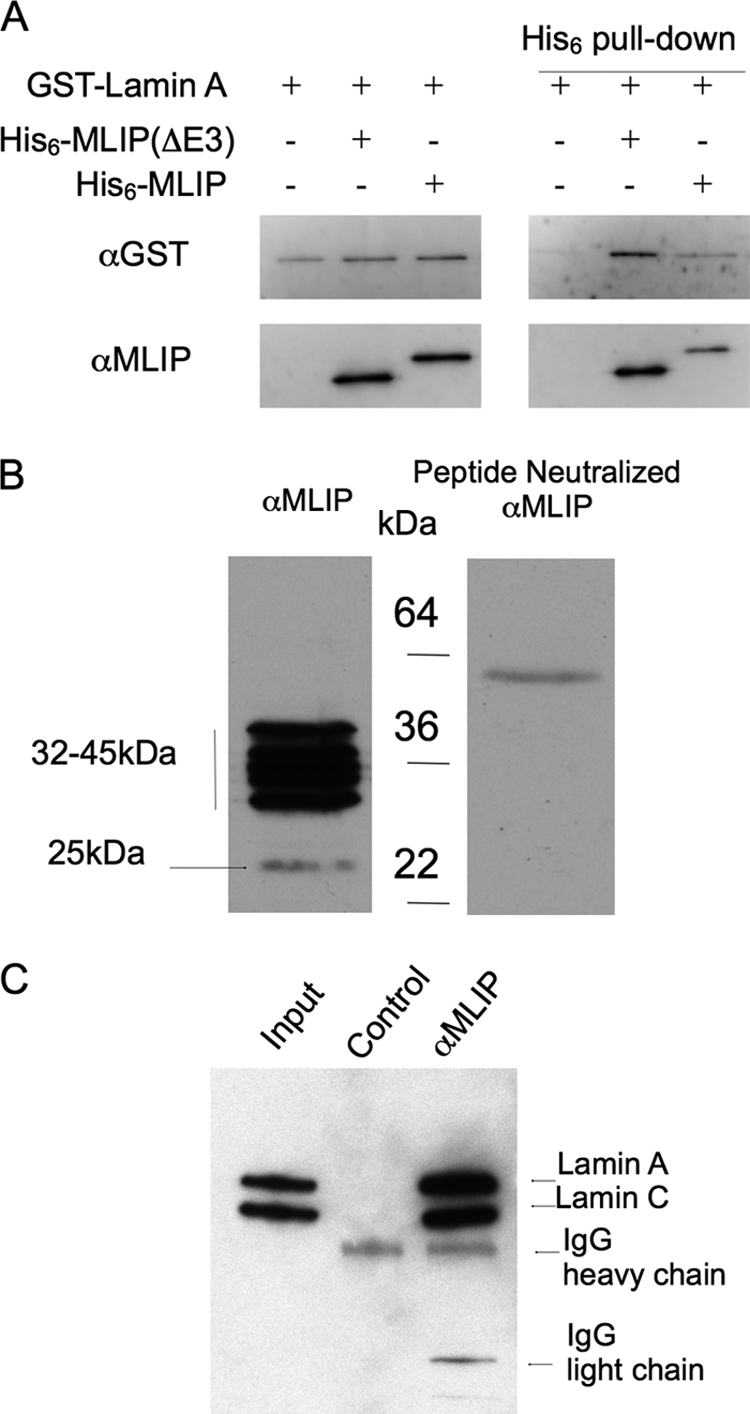
MLIP specifically binds lamin A/C. A, purified recombinant MLIP binds directly within the first 230 amino acids of recombinant lamins A and C in an in vitro immunoprecipitation assay. Purified recombinant His6-MLIP and GST-lamin A were mixed together (as indicated) with complexes precipitated through the His tag. Western blot analysis was performed using anti-GST (Cell Signaling) and anti-MLIP polyclonal antibodies. A 1:10 dilution of the total starting material was run on the same gel (left panels). The assay was repeated two additional times with similar results. B, MLIP peptide neutralization of the anti-MLIP serum demonstrates the specificity of the MLIP antibodies in Western blot analysis. C, MLIP co-immunoprecipitates with lamin A/C from adult mouse hearts. Total mouse heart lysates were divided 45%:45%:10% with 45% incubated with either anti-MLIP serum or MLIP peptide neutralized anti-MLIP serum (Control). The input control for the immunoprecipitation represents ∼2% of total heart lysate.
To investigate endogenous MLIP protein further we generated anti-peptide antibodies to peptide sequences derived from amino and carboxyl termini of the mouse MLIP amino acid sequence as underlined (Fig. 3A). Western blot analysis of mouse heart lysate indicated that the anti-peptide antibodies recognized five distinct polypeptides clustered around molecular sizes of 32–45 kDa and one polypeptide migrating at ∼25 kDa (Fig. 4B, left panel). Neutralization of the antibodies with the peptide revealed that all six polypepties were MLIP-related because all immunostaining was totally abolished in the presence of peptide (Fig. 4B, right panel).
Because we identified MLIP through its interaction with lamin A/C in the heart, we further explored this interaction in the mouse heart by performing co-immunoprecipitation experiments. Proteins were immunoprecipitated from total mouse heart lysates with the anti-MLIP peptide antibody and analyzed by Western blotting with polyclonal antibodies specific to lamin A/C. The anti-lamin A/C recognized polypeptides of ∼70 kDa (lamin A) and ∼65 kDa (lamin C) in heart lysate (Fig. 4C, Input lane) which were immunoprecipitated by anti-MLIP antibodies (Fig. 4C, α-MLIP lane) but not control Ig (Fig. 4C, Control lane).
MLIP Is Distributed in the Cytosol and Nucleus Where It Co-localizes with A-type Lamins and PML Bodies
Lamin A/C is primarily localized to the nuclear envelope as well as within the nucleus as part of branching intra- and trans-nuclear tubular structures (17). The specificity of the MLIP antibody in immunocytochemistry and immunohistochemically was determined by peptide neutralization experiments where we observed a complete loss of signal in the nucleus as well as in the cytosol (supplemental Figs. 1 and 2). Endogenous MLIP is localized to the nucleus and cytosol of C2C12 cells (Fig. 5, C, D, G, and H). MLIP specifically co-localizes with lamin A/C in the nuclear envelope and was revealed by co-staining for MLIP and A-type lamins and visualized by sequential confocal scanning (Fig. 5, B–D). In addition, MLIP was observed within the nucleus in a punctate pattern (Fig. 5, C and G) that is reminiscent of PML bodies (18, 19). Co-staining with anti-MLIP-specific antibody and anti-PML antibody revealed co-localization of MLIP within PML bodies in confocal images shown (Fig. 5, F–H). MILP did not co-localize with all PML-positive regions. The majority of MLIP is localized to the nuclear envelope and within the nucleus itself.
FIGURE 5.

MLIP co-localizes with lamin A/C and PML bodies in mouse C2C12 myoblasts. A–D, mouse C2C12 myoblasts were analyzed by indirect immunofluorescence microscopy (Carl Zeiss AxioImager Z1 Microscope) with antibodies against MLIP and lamin A/C. DNA was stained with DAPI (A), lamin A/C (B), and MLIP (C). D, merged images of DAPI (blue), lamin A/C (red), and MLIP (green) staining from A–C. Arrow indicates nuclear envelope and co-localization of MLIP with lamin A/C. E–H, C2C12 myoblasts were analyzed by indirect immunofluorescence and sequential scanning confocal microscopy with antibodies against MLIP and PML. DNA was stained with DAPI (E), PML (F), and MLIP (G). The arrows indicate co-localization of MLIP with PML. H, merged images of DAPI (blue), PML (red), and MLIP (green) staining from E–G. Scale bar, 10 μm.
MLIP Is Enriched in Various Muscle Types
To gain further insight into MLIP biology, MLIP mRNA expression by Northern blot analysis was performed. Two major MLIP RNA transcripts (3.5 and 1–2 kb in the mouse and 3.8 kb and 1.35–2.4 in the human) were observed in both mouse and human tissues (Fig. 6, A and B). MLIP transcripts were highly expressed in the heart and skeletal muscle from the mouse (Fig. 6A). A lower levels of MLIP expression were also observed in the lung and thymus (Fig. 6A). MLIP mRNA expression in human tissue was restricted primarily to the heart and the skeletal muscle with a detectable signal for MLIP also observed in the liver (Fig. 6B). Overall, the MLIP mRNA tissue distribution is very similar between human and mouse both in abundance and mRNA transcript size.
FIGURE 6.

MLIP tissue expression profile. Expression of MLIP mRNA in mouse (A) and human (B) tissue by Northern analysis revealed two transcripts. 10 μg of poly(A)-enriched RNA was loaded per lane. A human β-actin cDNA fragment was used to probe the blots as an internal loading control. The β-actin probe cross-hybridizes with cardiac α-actin and skeletal α-actin, which show up in the heart and skeletal muscle lanes. C, endogenous tissue distribution of MLIP protein by Western analysis. 5 μg of total soluble protein lysates from each tissue was loaded per lane as represented by Coomassie Blue staining of total protein, lower panel. D, normalized tissue distribution of MLIP expression in adult mouse tissue (n = 3, mean ± S.D.) as determined by quantitative PCR.
Western blot analysis of endogenous MLIP protein expression across a variety of mouse tissues revealed at least seven MLIP-related polypeptides that range in size from 23 to 57 kDa (Fig. 6C). The distribution of these MLIP polypeptides differed between tissues (Fig. 6C). The heart has as many as seven forms of MLIP (26.6, 30.2, 32.5, 33.6, 34.9, 40.9, and 44.7 kDa), whereas the lung and liver express one dominant form of MLIP (23.7 and 44.7 kDa, respectively). Skeletal muscle expresses two major forms of MLIP (41 and 44.7 kDa) that are similar in size to the two larger forms found in the heart (Fig. 6C).
Although Northern blots showed very little MLIP transcript expression in whole brain in mouse or human, Western blots showed that the brain expresses four forms of MLIP (27.7, 42.7, 47.9, and 56.9 kDa) that are not observed in any other tissue examined (Fig. 6C).
Quantitative PCR of MLIP from RNA isolated from various adult mouse tissues was performed to quantify and verify our Northern and Western blot results in the mouse. A similar distribution of MLIP expression was observed in the mouse with smooth muscle > skeletal muscle > heart > kidney > brain (Fig. 6D).
To study the expression of MLIP proteins in mouse tissues, we analyzed mouse brain, heart, lung, skeletal muscle, and liver by histology. Using the polyclonal antibody that we raised in our laboratory, MLIP was observed to be restricted to a subpopulation of brain cells within the hippocampus and cortex (Fig. 7, A–C). Within the dentate gyrus the subcellular localization of MLIP is observed within the cell body as well as in the periphery of a subpopulation cells (Fig. 7C, arrow). Similar to the brain, the heart (ventricle and atria) MLIP expression is observed within the cell body and nucleus both within the nucleus and nuclear envelope and cytosol of cardiomyocytes (Fig. 7F). MLIP also appears in smooth muscle cells of the vasculature associated with the atria (Fig. 7I) and lung (Fig. 7R). Atrial cardiomyocytes are also positive for MLIP protein expression (Fig. 7, G and H). MLIP protein expression in skeletal soleus muscle displays a mosaic pattern (Fig. 7J) that may be indicative of fiber type. To test this, we stained serial sections of mouse soleus muscle for MLIP and peroxiredoxin-3, a thioredoxin-dependent peroxide reductase localized in the mitochondria (Fig. 7, S and T). Both MLIP and peroxiredoxin-3 co-stained the same fiber type, suggesting the MLIP-positive mosaic pattern to be fast oxidative muscle fibers, possibly type 2A. The signal was specific for MLIP because preincubation of the antibody with the corresponding immunogenic peptide eliminated the labeling.
FIGURE 7.

MLIP mouse tissue expression by indirect immunohistochemistry. A–C, brain; D–E, heart ventricles; G–I, atria; J–L, skeletal muscle; M–O, liver; and P–R, lung. F and I, magnified image of heart (E and H, respectively) as indicated by box. Nucleus is identified by arrow. MLIP is expressed primarily in fast oxidative muscle fibers of soleus muscle. MLIP (S) and peroxiredoxin-3 (PRDX3) (T) expression profiles in serial soleus sections. The arrows mark similar fibers in S and T. Magnification is indicated for each image.
Based on the data presented in Figs. 5–7, MLIP is expressed primarily in striated and smooth muscle of humans and mice and co-localizes with lamin A/C at the nuclear envelope and within PML bodies of the nucleus. Furthermore, the observed variable cellular localization of MLIP (Fig. 5 and 7) may be associated with MLIP heterogeneity of isoform expression that appear to be expressed differentially among tissues (Fig. 6).
MLIP Is an Alternatively Spliced Gene
To determine whether the source of the different size MLIP isoforms observed in the Western blot analysis (Fig. 6C) was due to proteolysis or alternative splicing of the MLIP, we cloned and sequenced MLIP transcripts from mouse hearts. Comparative sequence analysis of mouse and human expressed sequence tag databases with assembled genome revealed that the putative 5′-UTR and 3′-UTR of MLIP are separated by ∼245 kbp of genomic DNA. We designed oligonucleotide primers to the 5′-UTR and 3′-UTR of MLIP to amplify mouse MLIP cDNA by RT-PCR from total heart RNA. At least eight MLIP splice forms, ranging in size from ∼762 to 1,212 bp, were identified by TA cloning and sequencing of the MLIP RT-PCR products (Fig. 8A). The putative translated protein products for each of the eight identified cDNA clones ranged in size from 27.9 to 44.4 kDa (Fig. 8A) and were very similar to the molecular size of polypeptide observed in the heart by Western blotting (Figs. 6C and 8A).
FIGURE 8.

MLIP is an alternatively spiced gene. A, RT-PCR was performed on mRNA isolated from mouse hearts using primers targeted to the 5′- and 3′-UTR of MLIP. The RT-PCR product was TA cloned into pCR-II plasmid (Invitrogen) and transformed into bacteria. Direct PCR was performed with primers targeting flanking regions of the MLIP insertion site amplified with each PCR product sequenced. Cloned MLIP cDNA length and predicted protein mass are identified for each of the bands (left and center columns). Calculated protein mass based on migration rates for each of the MLIP bands is presented in Fig. 6C (right column). B, based on these data, an alternative splice map of MLIP was constructed. The numbers below each splice map reflect the transcript size and predicted translated protein size.
Based on our cloning data and the assembled Mouse Genome (Build 37), the mouse MLIP (2310046A06Rik) gene is composed of 11 exons (Fig. 8). All of the putative introns of the MLIP gene contain a GU at the 5′-splice site and an AG at the 3′-splice site, which is the recognition motif for the major spliceosome (20, 21). Rapid amplification of 5′ and 3′ complementary DNA ends (5′- and 3′-RACE) further validated this result. Only a single 5′- and 3′-UTR sequence was identified in the mouse heart cDNA. MLIP isoforms cloned to date from the mouse heart all contain exons 1, 3, 9, 10, and 11. The two bands observed in the Northern of the mouse heart (∼3.5 kbp and 0.5–1.0 kbp) were in agreement with the predicted sizes of the mouse MLIP clones identified in mouse hearts (Fig. 6A). The observed difference in band size is due to the presence or absence of exon 4. Furthermore, the predicted protein sizes for each of the splice variants are in good agreement with those observed by Western blotting (Fig. 6C). These data suggest that the different MLIP protein bands observed by Western analysis are the products of alternative splicing of MLIP.
Lamin A/C Regulates MLIP Expression and Cellular Localization
Because we discovered MLIP through its specific interaction with lamin A/C (Fig. 1), we hypothesized that lamin A/C might regulate MLIP gene expression and/or cellular localization. A previous study has demonstrated that the localization of nuclear envelope-associated proteins (both lamin C and Emerin) are dependent on lamin A expression (22). We therefore used a shRNA specific for lamin A to down-regulate lamin A in C2C12 myoblasts and examine MLIP expression levels and cellular localization (Fig. 9). The LMNA shRNA treatment was effective because lamin A protein levels decreased by >75% in LMNA down-regulated cells (Fig. 9, A, α-LMNA, and B). We also observed a reduced but variable level of lamin C expression by Western analysis (Fig. 9, A and B). This observed decrease in lamin A protein was due to 85% of the treated cells lacking lamin A and lamin C protein (supplemental Fig. 3). This was not a surprising result because both lamin C expression and localization to the nuclear envelope are dependent upon lamin A expression (22). The lamin A/C antibody used in this study was unable to discriminate between A-type and C-type lamins by immunofluorescence analysis (Fig. 9, H–K).
FIGURE 9.

Lamin A is required for normal MLIP expression and cellular localization. A, Western blot of whole cell lysates showing reduced lamin A protein. C2C12 myoblasts down-regulated for lamin A by shRNA treatment or control cells infected with nonspecific shRNA were analyzed by Western blotting 48 h after infection using antibodies against lamin A/C, MLIP, or GAPDH. B and C, quantification of lamin A/C down-regulation (B) and MLIP isoform expression by LMNA shRNAi (C) was compared with nonspecific shRNAi. Each lamin A/C or MLIP isoform was normalized to the GAPDH internal loading control, and the data presented in B and C are the means ± S.D. of three independent experiments. D–G, control C2C12 myoblasts infected with nonspecific shRNAi were analyzed by indirect immunofluorescence microscopy with antibodies against MLIP and LMNA. DNA was stained with DAPI (D), MLIP (E), and lamin A/C (F). G, merged images of DAPI (blue), MLIP (green), and lamin A/C (red) staining from D–F are shown. H–K, C2C12 myoblasts infected with LMNA-specific shRNAi were analyzed by indirect immunofluorescence microscopy (Carl Zeiss AxioImager Z1 Microscope) with antibodies against MLIP and LMNA. DNA was stained with DAPI (H), MLIP (I), and lamin A/C (J). K, Merged images of DAPI (blue), MLIP (green) and lamin A/C (red) staining from H–J are shown. Scale bar, 10 μm.
Knockdown of LMNA resulted in a general up-regulation of MLIP protein with a significant (p < 0.01) 5–7-fold increase in MLIP isoforms 2, 3, and 4 (Fig. 9, A, α-MLIP, and C). GAPDH levels were unaffected in the LMNA-down-regulated cells (Fig. 9, A, α-MLIP and α-GAPDH). Within the cells that lack lamin A/C protein, MLIP is no longer localized to the periphery of the nucleus (Fig. 9, H–K), suggesting that MLIP localization to the nuclear envelope, like C-type lamins, is dependent upon lamin A proteins. Interestingly, MLIP protein levels were up-regulated in the LMNA down-regulated cells (Fig. 9A, α-MLIP), suggesting that lamin A is not only required for normal MLIP localization to the nuclear envelope but also for normal MLIP protein expression.
DISCUSSION
Here, we present the identity of C6orf142, a unique, single copy gene found only in amniotes that encodes a novel nuclear Lamin A/C interacting protein that we named MLIP. No previously known structural motif is present in MLIP and is ubiquitously expressed and enriched in muscle. The emergence of MLIP in amniotes suggests that MLIP might assume a novel biological function unique to amniotes. MLIP is 1 of the 1,021 genes that are unique to amniotes. Unlike MLIP, the majority of these amniote-specific genes have at least one paralogous homolog.
The MLIP gene encodes alternatively spliced variants ranging in size from 23 to 57 kDa that exhibit tissue-specific expression. The MLIP isoforms possess several novel structural motifs not found in other proteins. MLIP was found to distribute in the cytosol and the nucleus, and this may be reflective of isoform specific localization. A recent study focused on alternative splicing of 11,769 full-length human cDNAs identified MLIP (C6orf142) to be 1 of 261 genes with multiple variable first exons (23). MLIP was found to be highly expressed in the human heart with an abundant alternative transcriptional start site found only in both fetal and adult brains (23). The transcriptional start site identified in human hearts by Wakamatsu et al. (23) was homologous to the transcriptional start site we identified in the mouse heart. This raises many intriguing questions as to the functional role MLIP plays in both heart and brain.
Of significance is the finding that MLIP interacts and co-localizes with lamin A/C, a major component of nuclear lamina found between the inner nuclear membrane and the peripheral chromatin. Our data suggest that MLIP expression and localization to the nuclear rim are dependent on lamin A (Fig. 9), further supporting our original observation that MLIP interacts with lamin A/C. Thus, by association, MILP may serve important biological roles in regulating lamin function that includes important roles in maintaining nuclear structure, chromatin organization, DNA replication, cell cycle regulation, and apoptosis (24).
A large number of proteins that interact with lamin A/C proteins have been previously reported and include actin (LMNA461–536aa and LMNA563–646) (25, 26), PKCα (LMNA500–664) (27, 28), 12(S)-LOX (LMNA463–664) (29), histones (LMNA396–430) (30), SREBP1 (LMNA389–664) (31), Narf (LMNA389–664) (32), Emerin (LMNA384–566) (33–35), LAP2α (LMNA319–566) (36, 37), retinoblastoma (LMNA247–355) (37–40), and MOK2 (LMNA243–387) (41, 42). In none of these previously reported cases did the lamin A/C interaction occur within the first 230 amino acids of lamin A/C that includes the amino-terminal globular head domain (1–30 aa), coil 1A (31–66 aa), linker 1 (67–77 aa), coil 1B (78–220 aa) and part of linker 12 (221–239 aa). Thus, MLIP is the first protein to be described that interacts with this portion of lamin A/C. As a unique amniote gene, MLIP also provides new protein structural motifs that confer A-type lamin binding.
A wide range of human diseases, encompassing more than 10 different clinical syndromes, have been associated with mutations in the gene that encodes A-type lamins (1–3, 6, 43, 44). The molecular mechanism of the relationship between lamin A/C mutations with the pathogenesis of complex degenerative disorders and aging remains poorly defined. Furthermore, undifferentiated embryonic stem cells of both human and mouse origin lack lamin A/C expression (5) whereas partially differentiated cells express lamin A/C (45). There is increasing evidence that lamin A/C plays a critical role in stem cell regulation, differentiation, and cellular aging (4, 5). MLIP is only expressed in amniotes, but lamin mutations cause muscular and aging phenotypes in non-amniotes Drosophila (46) and C. elegans (47). Drosophila have two lamin genes, lamC and lamDm0, that are expressed in a pattern similar to mammalian lamin B and lamin A/C, respectively (48). C. elegans only has one lamin gene, suggesting that this singular gene is sufficient for providing the necessary functions of the different lamins genes in other species. What we find particularly intriguing about this discrepancy is that not only does MLIP co-localize with lamin A/C in the nuclear envelope but MLIP was also observed to be co-localized with PML protein in the nucleus of cells in a punctate pattern (Fig. 5J). A wide variety of biological processes have been associated within PML bodies that include protein posttranslational modifications, transcriptional regulation, DNA damage responses, and apoptosis (18, 19). PML, like MLIP, is only expressed in amniotes, suggesting that a functional link between the nuclear envelope and PML bodies may exist through MLIP.
The precise function of MLIP remains undefined. MLIP may function as a tethering protein linking LMNA to unidentified factors either within the nuclear envelope or within PML bodies. In support of this, a significant reduction in lamin A/C expression in C2C12 myoblasts (Fig. 9) resulted in the mislocalization of MLIP away from the nuclear envelope with an up-regulation in MLIP protein levels and isoform type. This dysregulation of MLIP localization and expression may (in part) be associated with skeletal and cardiac muscle laminopathies. However, a reduction of MLIP was reported in a microarray screen of the hearts of LMNA H222P mutant mouse model of autosomal dominant Emery-Dreifuss muscular dystrophy (6) (wild type versus homozygous mutant p = 0.0035 and wild type versus heterozygous mutant p = 0.0399; NCBI GEO profile GDS2746 record). MLIP has also been reported to be 1 of 36 genes differentially expressed in the placentas of women destined to develop preeclampsia (49). MLIP was observed to be down-regulated in the affected placentas and because we report here that MLIP is expressed in smooth muscle, this suggests that MLIP may have a role in maintaining normal smooth muscle function and vascular tone. It remains to be determined whether MLIP has a direct signaling or mechanistic role in A-type lamin-mediated biology.
A-type lamins have been implicated in the maintenance of cellular commitment and differentiation. Like lamin A/C, MLIP is ubiquitously expressed with tissue-specific isoforms, which may suggest that MLIP possibly plays a role in A-type lamin associated maintenance of cellular commitment and differentiation. The tissue-specific MLIP isoform patterns we observe may be necessary for the maintenance of that tissue differentiation. In addition, because we observe MLIP within PML bodies, we can infer that MLIP may have alternative functions beyond merely binding to lamin A/C. The identification of MLIP as an amniote lamin-interacting factor provides a new direction to study the mechanisms of lamin A/C during amniote development, aging, and disease.
Supplementary Material
Acknowledgments
We thank Balwant Tuana (University of Ottawa) for critical assessment of this manuscript. B. Vulesevic (University of Ottawa Heart Institute) for technical assistance, and W. Claycomb (Louisiana State University) for HL1 cells.
This work was supported by Canadian Institute of Health Research Operating Grants MOP77682 (to A. F. R. S.) and MOP82748 (to P. G. B.), an Ontario Graduate Student Science and Technology stipend (to E. A.), and a Canadian Institute of Health Research stipend (to L. K.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–3.
- LMNA
- lamin A/C
- aa
- amino acid
- MLIP
- muscle-enriched A-type lamin-interacting protein
- PML
- promyelocytic leukemia
- RACE
- rapid amplification of cDNA ends.
REFERENCES
- 1. Cohen M., Lee K. K., Wilson K. L., Gruenbaum Y. (2001) Trends Biochem. Sci. 26, 41–47 [DOI] [PubMed] [Google Scholar]
- 2. Mounkes L. C., Kozlov S., Hernandez L., Sullivan T., Stewart C. L. (2003) Nature 423, 298–301 [DOI] [PubMed] [Google Scholar]
- 3. Prokocimer M., Davidovich M., Nissim-Rafinia M., Wiesel-Motiuk N., Bar D. Z., Barkan R., Meshorer E., Gruenbaum Y. (2009) J. Cell. Mol. Med. 13, 1059–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Röber R. A., Weber K., Osborn M. (1989) Development 105, 365–378 [DOI] [PubMed] [Google Scholar]
- 5. Constantinescu D., Gray H. L., Sammak P. J., Schatten G. P., Csoka A. B. (2006) Stem Cells 24, 177–185 [DOI] [PubMed] [Google Scholar]
- 6. Muchir A., Pavlidis P., Decostre V., Herron A. J., Arimura T., Bonne G., Worman H. J. (2007) J. Clin. Invest. 117, 1282–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Charniot J. C., Pascal C., Bouchier C., Sébillon P., Salama J., Duboscq-Bidot L., Peuchmaurd M., Desnos M., Artigou J. Y., Komajda M. (2003) Hum. Mutat. 21, 473–481 [DOI] [PubMed] [Google Scholar]
- 8. Muchir A., Bonne G., van der Kooi A. J., van Meegen M., Baas F., Bolhuis P. A., de Visser M., Schwartz K. (2000) Hum. Mol. Genet 9, 1453–1459 [DOI] [PubMed] [Google Scholar]
- 9. Taylor M. R., Fain P. R., Sinagra G., Robinson M. L., Robertson A. D., Carniel E., Di Lenarda A., Bohlmeyer T. J., Ferguson D. A., Brodsky G. L., Boucek M. M., Lascor J., Moss A. C., Li W. L., Stetler G. L., Muntoni F., Bristow M. R., Mestroni L. (2003) J. Am. Coll. Cardiol. 41, 771–780 [DOI] [PubMed] [Google Scholar]
- 10. Bonne G., Mercuri E., Muchir A., Urtizberea A., Bécane H. M., Recan D., Merlini L., Wehnert M., Boor R., Reuner U., Vorgerd M., Wicklein E. M., Eymard B., Duboc D., Penisson-Besnier I., Cuisset J. M., Ferrer X., Desguerre I., Lacombe D., Bushby K., Pollitt C., Toniolo D., Fardeau M., Schwartz K., Muntoni F. (2000) Ann. Neurol. 48, 170–180 [PubMed] [Google Scholar]
- 11. Nikolova V., Leimena C., McMahon A. C., Tan J. C., Chandar S., Jogia D., Kesteven S. H., Michalicek J., Otway R., Verheyen F., Rainer S., Stewart C. L., Martin D., Feneley M. P., Fatkin D. (2004) J. Clin. Invest. 113, 357–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sullivan T., Escalante-Alcalde D., Bhatt H., Anver M., Bhat N., Nagashima K., Stewart C. L., Burke B. (1999) J. Cell Biol. 147, 913–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hegele R. (2005) Clin. Genet 68, 31–34 [DOI] [PubMed] [Google Scholar]
- 14. Raharjo W. H., Enarson P., Sullivan T., Stewart C. L., Burke B. (2001) J. Cell Sci. 114, 4447–4457 [DOI] [PubMed] [Google Scholar]
- 15. Cockell M., Gasser S. M. (1999) Curr. Opin. Genet. Dev. 9, 199–205 [DOI] [PubMed] [Google Scholar]
- 16. Herrmann H., Aebi U. (2004) Annu. Rev. Biochem. 73, 749–789 [DOI] [PubMed] [Google Scholar]
- 17. Broers J. L., Machiels B. M., van Eys G. J., Kuijpers H. J., Manders E. M., van Driel R., Ramaekers F. C. (1999) J. Cell Sci. 112, 3463–3475 [DOI] [PubMed] [Google Scholar]
- 18. Bernardi R., Pandolfi P. P. (2007) Nat. Rev. Mol. Cell Biol. 8, 1006–1016 [DOI] [PubMed] [Google Scholar]
- 19. Dellaire G., Bazett-Jones D. P. (2004) Bioessays 26, 963–977 [DOI] [PubMed] [Google Scholar]
- 20. Black D. L. (2003) Annu. Rev. Biochem. 72, 291–336 [DOI] [PubMed] [Google Scholar]
- 21. Matlin A. J., Clark F., Smith C. W. (2005) Nat. Rev. Mol. Cell Biol. 6, 386–398 [DOI] [PubMed] [Google Scholar]
- 22. Vaughan A., Alvarez-Reyes M., Bridger J. M., Broers J. L., Ramaekers F. C., Wehnert M., Morris G. E., Whitfield W. G. F., Hutchison C. J. (2001) J. Cell Sci. 114, 2577–2590 [DOI] [PubMed] [Google Scholar]
- 23. Wakamatsu A., Kimura K., Yamamoto J., Nishikawa T., Nomura N., Sugano S., Isogai T. (2009) DNA Res. 16, 371–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Holmer L., Worman H. J. (2001) Cell Mol. Life Sci. 58, 1741–1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lattanzi G., Cenni V., Marmiroli S., Capanni C., Mattioli E., Merlini L., Squarzoni S., Maraldi N. M. (2003) Biochem. Biophys. Res. Commun. 303, 764–770 [DOI] [PubMed] [Google Scholar]
- 26. Sasseville A. M., Langelier Y. (1998) FEBS Lett. 425, 485–489 [DOI] [PubMed] [Google Scholar]
- 27. Martelli A. M., Bortul R., Tabellini G., Faenza I., Cappellini A., Bareggi R., Manzoli L., Cocco L. (2002) J. Cell. Biochem. 86, 320–330 [DOI] [PubMed] [Google Scholar]
- 28. Martelli A. M., Tabellini G., Bortul R., Manzoli L., Bareggi R., Baldini G., Grill V., Zweyer M., Narducci P., Cocco L. (2000) Cancer Res. 60, 815–821 [PubMed] [Google Scholar]
- 29. Tang K., Finley R. L., Jr., Nie D., Honn K. V. (2000) Biochemistry 39, 3185–3191 [DOI] [PubMed] [Google Scholar]
- 30. Taniura H., Glass C., Gerace L. (1995) J. Cell Biol. 131, 33–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lloyd D. J., Trembath R. C., Shackleton S. (2002) Hum. Mol. Genet 11, 769–777 [DOI] [PubMed] [Google Scholar]
- 32. Barton R. M., Worman H. J. (1999) J. Biol. Chem. 274, 30008–30018 [DOI] [PubMed] [Google Scholar]
- 33. Fairley E. A., Kendrick-Jones J., Ellis J. A. (1999) J. Cell Sci. 112, 2571–2582 [DOI] [PubMed] [Google Scholar]
- 34. Sakaki M., Koike H., Takahashi N., Sasagawa N., Tomioka S., Arahata K., Ishiura S. (2001) J. Biochem. 129, 321–327 [DOI] [PubMed] [Google Scholar]
- 35. Wilkinson F. L., Holaska J. M., Zhang Z., Sharma A., Manilal S., Holt I., Stamm S., Wilson K. L., Morris G. E. (2003) Eur. J. Biochem. 270, 2459–2466 [DOI] [PubMed] [Google Scholar]
- 36. Dechat T., Korbei B., Vaughan O. A., Vlcek S., Hutchison C. J., Foisner R. (2000) J. Cell Sci. 113, 3473–3484 [DOI] [PubMed] [Google Scholar]
- 37. Markiewicz E., Dechat T., Foisner R., Quinlan R. A., Hutchison C. J. (2002) Mol. Biol. Cell 13, 4401–4413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Johnson B. R., Nitta R. T., Frock R. L., Mounkes L., Barbie D. A., Stewart C. L., Harlow E., Kennedy B. K. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 9677–9682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Markiewicz E., Ledran M., Hutchison C. J. (2005) J. Cell Sci. 118, 409–420 [DOI] [PubMed] [Google Scholar]
- 40. Van Berlo J. H., Voncken J. W., Kubben N., Broers J. L., Duisters R., van Leeuwen R. E., Crijns H. J., Ramaekers F. C., Hutchison C. J., Pinto Y. M. (2005) Hum. Mol. Genet. 14, 2839–2849 [DOI] [PubMed] [Google Scholar]
- 41. Dreuillet C., Tillit J., Kress M., Ernoult-Lange M. (2002) Nucleic Acids Res. 30, 4634–4642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Harper M., Tillit J., Kress M., Ernoult-Lange M. (2009) FEBS J. 276, 3137–3147 [DOI] [PubMed] [Google Scholar]
- 43. De Sandre-Giovannoli A., Bernard R., Cau P., Navarro C., Amiel J., Boccaccio I., Lyonnet S., Stewart C. L., Munnich A., Le Merrer M., Lévy N. (2003) Science 300, 2055. [DOI] [PubMed] [Google Scholar]
- 44. Eriksson M., Brown W. T., Gordon L. B., Glynn M. W., Singer J., Scott L., Erdos M. R., Robbins C. M., Moses T. Y., Berglund P., Dutra A., Pak E., Durkin S., Csoka A. B., Boehnke M., Glover T. W., Collins F. S. (2003) Nature 423, 293–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Meshorer E., Misteli T. (2006) Nat. Rev. Mol. Cell Biol. 7, 540–546 [DOI] [PubMed] [Google Scholar]
- 46. Muñoz-Alarcón A., Pavlovic M., Wismar J., Schmitt B., Eriksson M., Kylsten P., Dushay M. S. (2007) PLoS One 2, e532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Haithcock E., Dayani Y., Neufeld E., Zahand A. J., Feinstein N., Mattout A., Gruenbaum Y., Liu J. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 16690–16695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gruenbaum Y., Goldman R. D., Meyuhas R., Mills E., Margalit A., Fridkin A., Dayani Y., Prokocimer M., Enosh A. (2003) Int. Rev. Cytol. 226, 1–62 [DOI] [PubMed] [Google Scholar]
- 49. Founds S. A., Conley Y. P., Lyons-Weiler J. F., Jeyabalan A., Hogge W. A., Conrad K. P. (2009) Placenta 30, 15–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Muffato M., Louis A., Poisnel C. E., Roest Crollius H. (2010) Bioinformatics 26, 1119–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.