

Abstract
DAXX is a scaffold protein with diverse roles that often depend upon binding SUMO via its N- and/or C-terminal SUMO-interacting motifs (SIM-N and SIM-C). Using NMR spectroscopy, we characterized the in vitro binding properties of peptide models of SIM-N and SIM-C to SUMO-1 and SUMO-2. In each case, binding was mediated by hydrophobic and electrostatic interactions and weakened with increasing ionic strength. Neither isolated SIM showed any significant paralog specificity, and the measured μm range KD values of SIM-N toward both SUMO-1 and SUMO-2 were ∼4-fold lower than those of SIM-C. Furthermore, SIM-N bound SUMO-1 predominantly in a parallel orientation, whereas SIM-C interconverted between parallel and antiparallel binding modes on an ms to μs time scale. The differences in affinities and binding modes are attributed to the differences in charged residues that flank the otherwise identical hydrophobic core sequences of the two SIMs. In addition, within its native context, SIM-N bound intramolecularly to the adjacent N-terminal helical bundle domain of DAXX, thus reducing its apparent affinity for SUMO. This behavior suggests a possible autoregulatory mechanism for DAXX. The interaction of a C-terminal fragment of DAXX with an N-terminal fragment of the sumoylated Ets1 transcription factor was mediated by SIM-C. Importantly, this interaction did not involve any direct contacts between DAXX and Ets1, but rather was derived from the non-covalent binding of SIM-C to SUMO-1, which in turn was covalently linked to the unstructured N-terminal segment of Ets1. These results provide insights into the binding mechanisms and hence biological roles of the DAXX SUMO-interacting motifs.
Keywords: Biophysics, Ets Family Transcription Factor, Protein-Protein Interactions, Spectroscopy, Sumoylation, DAXX, NMR Spectroscopy
Introduction
DAXX is an enigmatic protein with diverse and often controversial roles. Although first discovered through a screen for association with the cytoplasmic Fas death domain (1), DAXX is now recognized to function primarily in the nucleus and to often be localized within subnuclear structures known as promyelocytic leukemia nuclear bodies (PML-NBs)3 (2, 3). Perhaps the best established role of DAXX lies with the regulation of transcription factors. Although some of these factors may be activated by DAXX (4, 5), in most cases, DAXX is linked with transcriptional repression (3). DAXX also associates with histone deacetylases as well as other chromatin-remodeling systems, thereby providing plausible routes to the repression of gene expression (6–10). The key to the localization and function of DAXX is its ability to bind the ubiquitin-like protein SUMO (3, 11).
The human genome encodes four SUMO paralogs (for reviews, see Refs. 12–16). SUMO-2 and SUMO-3 are virtually identical and are generally referred to as SUMO-2/3. SUMO-1 and SUMO-2/3 have ∼50% sequence identity and are expressed in all cells, whereas the function of SUMO-4 remains unclear. Using a common enzymatic pathway, many substrates can be covalently modified by both SUMO-1 and SUMO-2/3. However, the latter can also form chains on substrate proteins, and the levels of SUMO-2/3 conjugation increase with cellular stress (17). Dissecting the regulation and consequences of sumoylation, including paralog specificity, remains a very active area of research. Of the many possible functions of sumoylation, also prevalent is the repression of transcription (18). The recruitment of chromatin-remodeling complexes and histone-modifying enzymes by sumoylated transcription factors is thought to be mediated by scaffold proteins, such as DAXX (19–21).
DAXX interacts with SUMO via its conserved N-terminal (SIM-N) and C-terminal (SIM-C) SUMO-interacting motifs (22–24). SIMs are short sequences typically characterized by a hydrophobic HXHH or HHXH core (where H is Ile, Leu, or Val and X is any amino acid) flanked by negatively charges residues. These peptide motifs non-covalently bind the SUMO paralogs in an extended parallel or antiparallel conformation along a conserved groove between an α-helix and β-strand (16, 25–33). SIM-N and SIM-C play a crucial role in the recruitment of DAXX into PML-NBs via recognition of the sumoylated PML (6, 9, 23, 24). DAXX recruitment into PML-NBs has been proposed to regulate its function (11, 34). In one possible model, PML-NBs serve as “storage depots” sequestering DAXX in an inactive state. Release of DAXX from the nuclear bodies, for example in response to desumoylation of PML, allows it to bridge sumoylated transcription factors with transcriptional co-repressors and thereby inhibit gene expression. In support of this model, DAXX-mediated repression of several transcription factors is reduced by the increased expression or sumoylation of PML (6, 23, 35).
To provide a biophysical foundation for understanding the interplay of DAXX and the SUMO paralogs, we characterized the interactions of peptide models of SIM-N and SIM-C with SUMO-1 and SUMO-2 using NMR spectroscopy. These studies confirmed that both SIMs bind both SUMO paralogs along a common interface via hydrophobic and complementary electrostatic interactions. Neither SIM exhibited any significant paralog specificity, and SIM-N bound both SUMOs with ∼4-fold higher affinity than did SIM-C. Interestingly, SIM-N bound SUMO-1 with one predominant orientation, whereas SIM-C adopted multiple, interconverting binding modes. The latter conclusion was confirmed through specialized treatment of paramagnetic relaxation enhancement (PRE) data for molecular docking calculations. In addition, we found that within its native context SIM-N binds intramolecularly to the adjacent N-terminal DAXX helical bundle (DHB) domain (36), thereby reducing its apparent affinity for SUMO-1. This could constitute an autoregulatory mechanism for DAXX. Finally, we demonstrated that SIM-C mediates the interaction of a C-terminal fragment of DAXX with the sumoylated Ets1 transcription factor. Importantly, there are no direct contacts between DAXX and Ets1, but rather the two are bridged in a “beads-on-a-string” fashion by SIM-C recognizing SUMO-1 that in turn is covalently attached via an isopeptide bond to Lys15 in an unstructured N-terminal segment of Ets1 (37). These results provide insights into the binding mechanisms and hence biological roles of the DAXX SUMO-interacting motifs.
EXPERIMENTAL PROCEDURES
Expression Vectors
Genes encoding residues 1–97 of human SUMO-1 (National Center for Biotechnology information database: NP_003343), 1–93 of human SUMO-2 (P61956), 566–739 and 566–726 of murine DAXX (NP_031855), 1–144 and 55–144 of human DAXX (CAG33366), and 1–138 of murine Ets1 (AAN38317) were cloned via PCR methods into the pET28a expression vector (Novagen) using NdeI and XhoI restriction enzyme sites. The resulting constructs contained an N-terminal His6 tag with a thrombin cleavage site such that a Gly-Ser-His extension remained after proteolytic processing. A tryptophan residue was also introduced between this extension and Met1 of SUMO-1 and SUMO-2 to allow quantification via UV absorbance.
Genes encoding residues 1–19 (SIM-N) of human DAXX, 1–19 with 7IIVL10 mutated to 7GSGS10 (SIM-NGSGS), 718–739 of murine DAXX (SIM-C), 718–739 of murine DAXX with the C728A and S738C mutations (SIM-CS738C), and 718–739 of murine DAXX with the C728A mutation and an additional C-terminal cysteine residue (SIM-C740C) were cloned via PCR methods into pGEX-2T expression vector (GE Healthcare) using BamHI and EcoRI restriction enzyme sites. For the titrations where a KD value was calculated, a tryptophan residue was added at the C terminus of DAXX1–19 and at the N terminus of DAXX718–739 to facilitate quantification. In each case, the NMR signals of the tryptophan were not perturbed during any titrations, confirming that this residue does not influence the SIM/SUMO interactions. All of the peptide constructs also contained an N-terminal GST tag with an intervening thrombin cleavage site such that a Gly-Ser extension remained after proteolytic processing.
A gene encoding residues 1–97 of SUMO-1 fused directly to residues 16–138 of Ets1 (SUMO1–97-Ets16–138) was cloned by PCR methods into the pET28a vector. The resulting SUMO1–97-Ets116–138 chimera served as a model of Ets1 with Lys15 (within its unstructured N-terminal segment) linked via an isopeptide bond to the C-terminal Gly97 of SUMO-1. All clones were verified by DNA sequencing.
Protein Expression and Purification
Freshly transformed Escherichia coli BL21 (λDE3) cells were grown overnight in 25 ml of LB medium, collected by centrifugation, and transferred to 1 liter of minimal M9 medium enriched with 1 g of 15NH4Cl or 1 g of 15NH4Cl and 3 g of [13C6]glucose (Spectral Stable Isotopes) for uniform 15N labeling or 13C/15N labeling, respectively, or to 1 liter of LB medium (for unlabeled proteins). When cells reached an A600 of ∼0.6, protein expression was induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside (final concentration). Cells were then grown overnight (for labeled proteins) or for 5 h (for unlabeled proteins) at 30 °C, harvested by centrifugation, and frozen at −70 °C prior to further purification. Thawed E. coli pellets were resuspended in binding buffer (5 mm imidazole, 50 mm HEPES, 500 mm NaCl, 5% (v/v) glycerol, pH 7.5) with a protease inhibitor mixture tablet (Roche Applied Science) and lysed by passage through a French press (1000 p.s.i.) followed by sonication (duty cycle, ∼50%; 10 min). After centrifugation at 15,000 rpm in a Sorvall SS32 rotor for 30 min, the supernatant was passed through a 0.80-μm filter (Millex).
For pET28a-encoded proteins, the lysate was applied to a 5-ml His-Trap metal affinity column connected to an ÄKTA chromatography system (GE Healthcare). The column was washed with 200 ml of washing buffer (binding buffer plus 60 mm imidazole), and the His6-tagged proteins were eluted with a gradient to 500 mm imidazole. Fractions containing the protein, as detected by absorption at 280 nm, were pooled and exchanged with a 3000 molecular weight cutoff Amicon concentrator into thrombin cleavage buffer (20 mm Tris-HCl, 150 mm NaCl, 2.5 mm CaCl2, pH 8.4). The His6 tag was removed by incubation with thrombin (∼1.4 units; Novagen) at room temperature for ∼5 h. Proteolysis was monitored by MALDI-TOF MS. The reaction was terminated by incubation with 200 μl of p-aminobenzamidine beads (Sigma) for 1 h. The supernatant was then diluted 10-fold in buffer without salt, applied to a HiPrep Q FF 16/10 ion exchange column (GE Healthcare), and eluted with a salt gradient (20 mm Tris, pH 8.0, 2 mm DTT, 0–1 m NaCl). Fractions absorbing at 280 nm were evaluated by SDS-PAGE, and those containing the desired sample were concentrated using a 3000 molecular weight cutoff Amicon Centricon device (Millipore). Sumoylated Ets1 was prepared and purified as described previously (37).
For GST-tagged peptides, the lysate was applied to four 5-ml GSTrap HP affinity columns connected in series to an ÄKTA chromatography system (GE Healthcare). The lysate was kept on ice-cold water to prevent proteolytic cleavage and slowly loaded to the room temperature column at 0.7 ml/min. Once loaded, the column was washed with 100 ml of PBS/EDTA buffer (137 mm NaCl, 2.7 mm KCl, 4.3 mm Na2HPO4, 1.4 mm KH2PO4, 5 mm EDTA, pH 7.3), and the GST-tagged peptides were eluted with freshly prepared glutathione buffer (10 mm Tris, 20 mm glutathione, 5 mm DTT, pH 8.0). Fractions containing the GST-tagged peptides, as detected by absorption at 280 nm, were pooled and buffer-exchanged with a 10,000 molecular weight cutoff Amicon concentrator into thrombin cleavage buffer. The GST was removed by incubation with thrombin (∼1.4 units; Novagen) at room temperature for ∼5 h. Proteolysis was monitored by MALDI-TOF MS. The reaction was terminated by incubation with 200 μl of p-aminobenzamidine beads (Sigma) for 1 h. The supernatant was passed through the 10,000 molecular weight cutoff Amicon filter, which retained the GST and uncleaved GST-peptide in the concentrate. The cleaved peptides were collected in the flow-through. The flow-through was injected into a reverse phase C18 HPLC column, washed thoroughly with 0.1% TFA in water, and eluted with an H2O:acetonitrile gradient (0.1% TFA). Fractions absorbing at 280 nm were evaluated by MALDI-TOF MS, and those containing the desired peptide were lyophilized. A peptide containing residues 729–740 of human DAXX was also synthesized and HPLC-purified by EZBiolab (Westfield, IN).
Protein concentrations were determined by UV light absorption spectroscopy using predicted ϵ278 values of 9970 m−1 cm−1 for SUMO-1, 6990 m−1 cm−1 for SUMO-2 and the SIM-C peptides, 5500 m−1 cm−1 for SIM-N, 6210 m−1 cm−1 for DAXX1–144, 5960 m−1 cm−1 for DAXX55–144, 23,000 m−1 cm−1 for Ets1–138, and 26,720 for SUMO1–97-Ets116–138 chimera. In each case, the sample purity was judged to be >95% by SDS-PAGE, and the protein or peptide identity was confirmed by MALDI-TOF MS.
NMR Spectroscopy
NMR spectra were recorded using a Varian Unity 500-MHz spectrometer and a cryoprobe-equipped Inova 600-MHz spectrometer, processed with NMRpipe (38), and analyzed with Sparky (39) and NMRview (40). Spectra were assigned using HNCACB and CBCA(CO)NH experiments combined with data for SUMO-1, SUMO-2, and sumoylated-Ets1 deposited in the BioMagResBank. Annotated 15N HSQC spectra are provided in supplemental Fig. S1.
NMR-monitored Titrations
All NMR-monitored titrations were carried out using sensitivity-enhanced 15N HSQC spectra (41) recorded at 25 °C with a 600-MHz spectrometer. Samples were in an NMR buffer composed of ∼5% D2O, 10 mm potassium phosphate, 0.1 mm EDTA, 10 mm DTT, pH 6.5 with 100 mm KCl unless otherwise stated. Proteins were exchanged into this buffer by concentrating/diluting 1:10 with a 3000 molecular weight cutoff Amicon Centricon device at least three times. Lyophilized peptides were dissolved in NMR buffer with small amounts of NaOH added to adjust the pH value to 6.5. Samples (500 μl) of the 15N-labeled species were titrated with increasing amounts of the concentrated unlabeled species as summarized in supplemental Table S1. Combined amide chemical shift changes were calculated as Δδ = {(Δδ1H)2 + (0.2Δδ15N)2}1/2. Equilibrium dissociation constants (KD values) for the 1:1 complexes with 1HN and 15N signals titrating in the fast exchange regime were obtained from non-linear least square fitting of chemical shift changes as a function of total ligand concentration using Sigma Plot (42).
PRE Measurements
To ensure full reduction of all Cys residues, 20 mm DTT was maintained throughout the purification of SIM-C, SIM-C738C, and SIM-C740C. After HPLC purification, the peptides were lyophilized, resuspended in DTT-free 50 mm phosphate buffer, pH 7.4, and subsequently incubated for 24 h in the dark at room temperature with (1-oxyl-2,2,5,5-tetramethyl-Δ3-pyrroline-3-methyl)methanethiosulfonate (MTSL; Toronto Research Chemicals), previously dissolved in acetone, at a peptide:MTSL ratio of at least 1:5. MALDI-TOF MS showed the reactions to be ∼90% complete. Peptides were repurified by HPLC to remove any unreacted material and side products. The resulting spin-labeled peptides are denoted as NOSIM-C, NOSIM-C738C, and NOSIM-C740C, respectively. 13C/15N-labeled SUMO-1 was dialyzed into non-reducing NMR buffer with 100 mm KCl, and the spin-labeled peptide was titrated to saturation. A series of relaxation experiments was carried out with a 600-MHz spectrometer to determine amide 1HN R2 values (43). Ascorbic acid (2 mm final concentration from a stock solution prepared in NMR buffer) for NOSIM-C and NOSIM-C738C and DTT (10 mm) for NOSIM-C740C were then added to reduce the spin label, and the same measurements were repeated to obtain site-specific ΔR2 PRE values.
The previously determined average global correlation time τc of 8.6 ns for SUMO-1 (44) was used to estimate the effective distance r from the amide proton to the paramagnetic center according to the following equation (45).
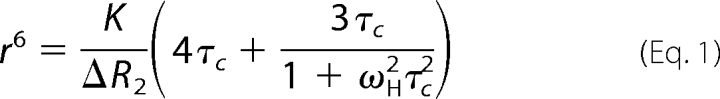 |
K is 1.23 × 10−32 cm6 s−2 for a nitroxide radical, and ωH is the Larmor frequency of the proton (3.8 × 109 radians s−1). Because of the sixth power in this equation, the calculated distances are relatively insensitive to the exact choice of τc (42).
NMR-based Docking
A model of the SIM-N·SUMO-1 complex was generated with HADDOCK version 2.1 (46, 47) using the web server interface (48). Chemical shift perturbation and NOE information were provided as experimental restraints. The initial structures for SUMO-1 and SIM-N were Protein Data Bank code 2BF8 chain B (49) and a homology model of Protein Data Bank code 2ASQ chain B (26), respectively. Intermolecular NOEs between SIM-N with SUMO-1 were obtained from a 13C/15N-filtered edited NOESY-HSQC spectrum (0.15 s mixing time) of 13C/15N-SUMO-1 complexed with unlabeled SIM-N. Signals from the peptide residues taking part in the interaction were assigned partially from two-dimensional 13C/15N-filtered NOESY of the complex, whereas partial protein assignments were obtained from 15N HSQC, HNCACB, CC(CO)NH-TOCSY, and H(CC)(CO)NH-TOCSY spectra (50). Protons displaying intermolecular NOEs were restrained to be <5.5 Å apart. Solvent-exposed SUMO-1 residues with combined chemical shift changes greater than 0.1 ppm upon SIM-N binding (residues 21, 23, 35–37, 39, 43–46, 50, 74, and 85) were defined as active. Solvent-exposed SUMO-1 residues surrounding these active residues (residues 18–20, 25, 33, 41, 42, 49, 53, 54, 73, 75, 84, and 86) were defined as passive. For the peptide, the active residues were 7–10 and 12, passive residues were automatically selected, and full flexibility was allowed.
Ensemble-averaged models of the SIM-C·SUMO-1 complex were generated using a modified version of the multibody web interface of HADDOCK (51) with chemical shift perturbation and PRE information as experimental restraints. PRE data were used to derive unambiguous distance restraints for a hypothetical combined Cys727-Cys738-Cys740 peptide modeled in an extended conformation. Because the N-terminal residues of SIM-C do not interact with SUMO-1, the peptide was truncated to residues 725–740, and its N terminus was defined as uncharged. Amide protons with signals broadened beyond detection due to the spin label were defined as <12.5 Å from the corresponding sulfur atom. Otherwise the ΔR2-calculated distances ±3 Å were used. Solvent-exposed SUMO-1 residues in Protein Data Bank code 2BF8 chain B with combined chemical shift changes greater than 0.1 ppm upon SIM-C binding (residues 37–39, 42, 43, 45, 46, and 50) were defined as active. Solvent-exposed SUMO-1 residues surrounding these active residues (residues 20, 21, 35, 36, 41, 53, and 54) were defined as passive. The peptide was fully flexible and active. A three-body docking was performed consisting of SUMO-1 and two copies of the peptide. The HADDOCK protocol was modified by turning off the intermolecular interactions between the peptides to allow ensemble docking. Ambiguous interaction restraints based on chemical shift perturbation data were defined separately for each peptide, whereas the PRE restraints were calculated as the average over the two peptides. The starting orientations of all three molecules were randomized in the usual manner to not introduce any bias toward parallel or antiparallel orientations. Restraints were not randomly removed, and the number of structures for the rigid body stage was increased to 2000. The weighting of distance restraints was increased from the default values of 0.01 or 0.1 to 1.0 for all stages. All other parameters were left to their default values. This modified HADDOCK protocol was first developed and tested using synthetic ensemble-averaged PRE data (data not shown).
A model of the SIM-N·DHB complex was also generated with the HADDOCK server using only chemical shift perturbation restraints. The initial structures were Protein Data Bank code 2KZS (36) for the DHB domain and the above SIM-N model. Solvent-exposed DHB domain residues with combined chemical shift changes greater than 0.045 ppm (residues 75, 78–80, 87, 88, 92, 98, 124, 128, 129, 132, 136, 137, 140, and 141) were defined as active, and surrounding passive residues were defined automatically. SIM-N residues 7–19 were defined as active with automatic selection of passive residues and full flexibility. Statistics for all HADDOCK models are provided in supplemental Table S3.
RESULTS
NMR-monitored Titrations Confirm DAXX SIM/SUMO Binding
The interactions of the DAXX SIM peptides and the SUMO paralogs were monitored by following perturbations in the 15N HSQC spectra of each 15N-labeled species upon titration with the unlabeled species. As evident from spectral changes in complementary titrations (Figs. 1 and 2), both isolated SIM peptides bound both SUMOs in vitro. Chemical shift perturbation mapping (Fig. 3 and supplemental Fig. S2) confirmed that the binding of SIM-N and SIM-C occurred along the cleft between strand β2 and helix α1 of SUMO-1 and SUMO-2. Conversely, the Ile-Ile-Val-Leu hydrophobic cores of SIM-N and SIM-C as well as the flanking negatively charged aspartate and glutamate residues showed the greatest chemical shift perturbations upon binding to either SUMO paralog. This is in agreement with mutational studies of the DAXX/SUMO interactions (23, 24) and the reported structures of SUMO·SIM complexes (33). Details of these titrations are provided in the following sections.
FIGURE 1.

NMR-monitored titrations demonstrated that SIM-N binds both SUMO-1 and SUMO-2 in ionic strength-dependent manner. A and B, superimposed sections of the 15N HSQC spectra of 15N-labeled SUMO-1 or SUMO-2 in the absence and presence of increasing amounts of unlabeled SIM-N in sample buffer with 0 mm KCl. C–F, superimposed sections of the 15N HSQC spectra of 15N-labeled SIM-N in the absence and presence of increasing amounts of unlabeled SUMO-1 and SUMO-2 in sample buffer with 100 or 200 mm KCl. The arrows indicate the changes of the 1HN-15N signals from selected residues over the course of the titrations. Labeled peaks not reappearing upon saturation are underlined. See supplemental Table S1 for experimental details. Note that the behavior of signals depends upon the effective rate constant for association and dissociation (kex = kon[ligand] + koff), the chemical shift difference between the free and bound states (Δω), and the relaxation behavior in both states (69). In the fast exchange limit (kex > Δω), peaks shift progressively with added ligand, whereas in the slow exchange limit (kex < Δω), the relative intensities of distinct peaks from the free and bound states change progressively. In the intermediate exchange limit, severe signal broadening occurs. For a residue with a given Δω, as binding weakens and kex increases, there is generally a shift from slow to intermediate to fast exchange behavior. As discussed in the text, conformational exchange within the bound state adds further complications.
FIGURE 2.

NMR-monitored titrations demonstrated that SIM-C binds both SUMO-1 and SUMO-2 in ionic strength-dependent manner. Superimposed sections of the 15N HSQC spectra of 15N-labeled SIM-C in the absence and presence of increasing amounts of unlabeled SUMO-1 (A, C, and E) and SUMO-2 (B, D, and F) in sample buffer with 0 (A and B), 100 (C and D), or 200 mm KCl (E and F) are shown. The arrows indicate the changes of the 1HN-15N signals from selected residues over the course of the titration. Labeled peaks not reappearing upon saturation are underlined. See supplemental Table S1 for experimental details.
FIGURE 3.

Mapping binding interfaces of SUMO-1 and SUMO-2 with SIM-N and SIM-C. A, modular organization of murine and human DAXX, including SIM-N, SIM-C, the DHB domain, an uncharacterized helical domain, an “acidic” region, which contains 80% Glu/Asp residues, and segments rich in Ser/Pro/Glu residues (SPE) and in Ser/Pro/Thr (SPT) residues (36). The latter three regions as well as the linkers flanking the helical domains are predicted to be intrinsically disordered. B, residues of the human SIM-N- and murine SIM-C-containing peptides exhibiting combined amide chemical shift changes greater than 0.2 ppm (or disappearing) upon binding SUMO-1 and SUMO-2 are highlighted in black. Although differing in residue numbering by 1, the sequences of murine and human SIM-C that interact with SUMO are identical. C, residues with chemical shift changes greater than 0.2 ppm upon binding SIM-N and SIM-C are identified in black on the schematic structures of SUMO-1 (Protein Data Bank code 1A5R) and SUMO-2 (Protein Data Bank code 1WM2). The flexible terminal residues of these proteins have been removed for clarity. Histograms showing the chemical shift changes for all of these SIM/SUMO titrations are provided as supplemental Fig. S2, and titration conditions are summarized in supplemental Table S1.
SIM-N Binding to SUMO-1 and SUMO-2
Upon titration with unlabeled SIM-N, the 1HN-15N signals from many amides in 15N-labeled SUMO-1 and SUMO-2 showed small shift changes and a progressive loss of intensity along with the concomitant appearance of new peaks arising from the resulting complex (Fig. 1, A and B). This behavior is indicative of moderately high affinity binding at the edge of the slow exchange regime on the chemical shift time scale (kex < Δω). For those amides showing fast exchange behavior due to small Δω values between free and bound states (i.e. progressive chemical shift changes with added ligand), plots of Δδ versus SIM-N concentration indicated essentially stoichiometric binding under the experimental conditions and hence a KD value <1 μm (Table 1 and supplemental Fig. S3). To confirm the predicted role of electrostatic interactions in the binding of SIM-N to SUMO-1 and SUMO-2, titrations were also carried out as a function of sample ionic strength. Consistent with this hypothesis, as the KCl concentration was increased from 0 to 200 mm, binding progressively weakened to a KD value of ∼40 μm (Fig. 1, Table 1, and supplemental Fig. S3). Finally, inspection of Table 1 reveals that SIM-N bound to SUMO-1 and SUMO-2 with similar affinities at all tested salt concentrations. Thus, the isolated SIM-N peptide does not display any significant paralog specificity.
TABLE 1.
Dissociation constants for complexes of DAXX SIMs with the SUMO paralogs and the DHB domain
Reported KD values and errors correspond to the mean ± S.D. of the individually fit titration curves of at least three residues as shown in supplemental Fig. S3.
| SUMO-1 KD |
SUMO-2 KD |
DHB domainbKD, |
|||||
|---|---|---|---|---|---|---|---|
| 0 mm KCla | 100 mm KCla | 200 mm KCla | 0 mm KCla | 100 mm KCla | 200 mm KCla | 100 mm KCla | |
| μm | μm | μm | |||||
| SIM-N | <1c | 8.1 ± 1.7 | 36 ± 3 | <1c | 8.7 ± 0.9 | 38 ± 6 | 170 ± 60 |
| SIM-NGSGS | 440 ± 30 | ||||||
| DAXX1–144 | 1200 ± 500 | ||||||
| SIM-C | 4.5 ± 1.4 | 30 ± 7 | 110 ± 14 | 16 ± 7 | 24 ± 6 | 170 ± 24 | |
a Plus 10 mm K2HPO4, 0.1 mm EDTA, 10 mm DTT, ∼5% D2O, pH 6.5, 25 °C.
b The DHB domain is residues 55–144 of DAXX.
c An upper limit as binding is essentially stoichiometric under the experimental conditions.
SIM-C Binding to SUMO-1 and SUMO-2
The NMR-monitored titrations of SIM-C with SUMO-1 and SUMO-2 showed complex behavior (Fig. 2). In addition to spectral changes at low ionic strength, resulting from slow-to-intermediate exchange between free and bound states, signals from the Ile-Ile-Val-Leu hydrophobic core in SIM-C and Arg39 in SUMO-1 were not detected even under saturating conditions. A comparable pattern of weak or absent peaks was also reported recently for a similar complex of SIM-C and SUMO-1 (52). As explained below, this is attributed to conformational exchange within the bound state. Using residues showing fast exchange behavior, the KD values for of SIM-C with SUMO-1 and SUMO-2 as a function of added KCl were determined (Table 1 and supplemental Fig. S3). As observed with SIM-N, binding progressively weakened with increasing ionic strength, thus confirming the role of electrostatic interactions in complex formation. Under the same conditions, SIM-C showed ∼4-fold weaker binding to both SUMO-1 and SUMO-2 than observed with SIM-N (Table 1). Furthermore, in the absence of added KCl, SIM-C exhibited ∼3.5-fold higher affinity toward SUMO-1 than SUMO-2. This very modest paralog preference was reduced at 100 and 200 mm salt concentrations, indicating that electrostatic interactions play a role in determining the relative affinity of SIM-C for the two SUMOs.
DAXX SIM-N Binds SUMO-1 in a Parallel Orientation
In contrast to SIM-C, SIM-N bound SUMO-1 and SUMO-2 without exchange broadening effects under saturating conditions at low ionic strength. We hypothesized that, due to its asymmetric charge distribution, SIM-N interacts with both SUMO paralogs in a preferred orientation to form well defined complexes. To determine this orientation, 13C/15N-filtered edited NOESY-HSQC experiments were used to measure intermolecular NOEs between unlabeled SIM-N and labeled SUMO-1 (Fig. 4A and supplemental Table S2). The intermolecular NOEs were then mapped on the structure of SUMO-1 and the sequence of SIM-N. For example, Ile8 showed NOE interactions with Arg54, Ile34, His35, and Phe36, whereas Asp11 yielded NOEs to Thr42, His43, and Lys39 (Fig. 4B). This clearly demonstrated that the SIM-N peptide binds predominantly in a parallel conformation relative to strand β2.
FIGURE 4.

SIM-N binds SUMO-1 in a parallel orientation. A, 1H-1H strips from a three-dimensional 13C/15N-filtered edited NOESY-HSQC spectrum showing intermolecular NOEs between unlabeled SIM-N (annotated on the vertical axes) and three residues from strand β2 and three residues from helix α1 in 13C/15N-labeled SUMO-1 (horizontal axes). B, mapping of intermolecular NOEs on the structure of SUMO-1 (Protein Data Bank code 1A5R; red, negatively charged; blue, positively charged; green, hydrophobic; gray, neutral polar). Strand β2 and the helix are also shown as isolated elements with residues that receive intermolecular NOEs from SIM-N highlighted in magenta. Each line represents the presence of at least one intermolecular NOE (supplemental Table S2). C, HADDOCK model of SIM-N in stick format (yellow, carbon; red, oxygen; blue, nitrogen) docked on SUMO-1 with its orientation and transparent surface coloring to match the schematic of B. Residue labels have the same color coding.
To better define the physicochemical basis for this binding orientation, we generated a low resolution model of SIM-N·SUMO-1 complex using NOE interactions and chemical shift perturbations as experimental restraints for the data-driven docking program HADDOCK (Fig. 4C). Similar to previously described SIM·SUMO complexes, SIM-N adopted a short β-strand-like conformation, pairing with strand β2 of SUMO-1 to form an intermolecular β-sheet. The second (Ile8) and fourth (Leu10) residues of the SIM-N core packed against a hydrophobic interface of SUMO-1 that is located between strand β2 and helix αl and formed primarily by the side chains of Ile34, Phe36, Val38, Leu44, Leu47, and Tyr52. These nonpolar contacts were augmented by potential electrostatic interactions between Asp11, Asp12, Asp14, and Asp15 of SIM-N and Lys39, His43, Lys46, and Lys49 near the “bottom” of SUMO-1 as drawn in Fig. 4C. In contrast to this favorable juxtapositioning of oppositely charged groups, an antiparallel binding orientation would position the aspartates and glutamates of SIM-N near a negatively charged surface on the “top” of SUMO-1.
If the charges of the juxtaposing glutamate and aspartate residues in SIM-N are responsible for establishing the binding mode of SIM-N on SUMO-1, then increasing the ionic strength of the solution should not only weaken the electrostatic contribution of those residues as noted above (Table 1) but could also lead to multiple binding modes. Indeed, 1HN-15N signals from several amides in the hydrophobic core of SIM-N were broadened beyond detection in the 15N HSQC spectra of the SUMO-1 complex recorded in the presence of 200 mm KCl (i.e. Ile8, Val9, and Leu10; Fig. 1E). This behavior is similar to that exhibited by SIM-C with SUMO-1 at 100 mm KCl (Fig. 2C) and suggestive of conformational exchange within the bound complex.
Although flanking charged residues appear to dictate the orientation of binding, the closest SIM-N and SUMO-1 contacts, as detected by intermolecular NOE interactions, involve the hydrophobic residues of the peptide. Accordingly, we hypothesized that mutation of these residues would severely disrupt binding. To test this, a SIM-N variant with the hydrophobic Ile-Ile-Val-Leu core mutated to Gly-Ser-Gly-Ser (SIM-NGSGS) was produced. Surprisingly, the mutant was still able to bind the same interface of SUMO-1 at low ionic strength, although weakly and in the fast exchange limit (supplemental Fig. S4). The dissociation constant of the mutant was determined to be 440 ± 30 μm, which is at least 400-fold weaker than the value of <1 μm for the wild type (Table 1). Collectively, the disruptive effects of mutations and elevated ionic strength highlight the contribution of both the hydrophobic and charged residues of SIM-N in binding SUMO-1.
DAXX SIM-C Binds SUMO-1 and SUMO-2 in Both Parallel and Antiparallel Orientations
Several amide signals in the 15N HSQC spectrum of 15N-labeled SIM-C were broadened beyond detection upon reaching saturation with unlabeled SUMO-1 (Fig. 2). Similarly, the 1HN-15N signal of Arg39 in SUMO-1 was no longer detected in the spectrum of the SIM-C complex (supplemental Fig. S2). We hypothesized that ms to μs time scale exchange between two or more conformations of the SIM-C·SUMO-1 complex led to this observed spectral behavior (53). To test this, PRE measurements using a nitroxide spin label placed on one end or the other of the SIM-C peptide were undertaken (54). A wild-type cysteine residue (Cys727) was used to covalently link a spin label N-terminal to the hydrophobic core of SIM-C. To attach the spin label near the C-terminal end of SIM-C and closer to the hydrophobic core, this residue was mutated to an alanine, and a new single cysteine was introduced at either position 738 or 740 (Fig. 5A).
FIGURE 5.

SIM-C binds SUMO-1 and SUMO-2 in both parallel and antiparallel orientations. A, three variants of SIM-C with a single cysteine (red) at different positions were nitroxide spin-labeled. B, 15N HSQC spectrum of SUMO-2 saturated with NOSIM-C740C (left). Upon reduction of the spin label, peaks that were broadened beyond detection due to paramagnetic relaxation reappeared (right, red boxes). Similar results were obtained with the three SIM-C variants and SUMO-1 (not shown). C, SUMO-1 and SUMO-2 residues that displayed large changes in 1HN transverse relaxation rates, ΔR2 (>20 s−1; blue) or that reappeared (ΔR2 > 100 s−1; red) upon reduction of the NOSIM-CS738C or NOSIM-C740C spin label are located along the binding surface, including both ends. This cannot be rationalized with a single binding orientation. In the case of NOSIM-C, the PRE effects are smaller due to the greater separation of Cys727 from the hydrophobic core. D, HADDOCK models of a theoretical SIM-C peptide (residues 725–740; yellow ribbon) with all three cysteine residues docked on SUMO-1 using ensemble-averaged PRE distance restraints. Four representative low energy ensemble-averaged models are shown. Although shown separately for clarity, the antiparallel (top) and parallel (bottom) orientations are the result of simultaneous ensemble-averaged docking of the two peptides onto SUMO-1, and both orientations are required to satisfy the PRE data. The cysteines and hydrophobic core residues are indicated as color-coded balls.
PREs were measured qualitatively from 15N HSQC spectra of 15N-labeled SUMO-1 titrated separately with the three SIM-C derivatives in their paramagnetic nitroxide forms and then subsequently reduced to their diamagnetic hydroxylamine forms. Similar measurements were carried out for 15N-labeled SUMO-2 with the NOSIM-C740C derivative. As expected, upon reaching saturation, a number of peaks in the spectra of SUMO-1 and SUMO-2 were paramagnetically broadened, often beyond detection, due to the bound nitroxide yet reappeared upon addition of reducing agent (Fig. 5B). However, recall that several amides were undetectable in the unmodified SIM-C·SUMO-1 complex, thus limiting the analysis of these spectra. Nevertheless, in all four cases, amides experiencing 15N HSQC intensity changes due to nitroxide-enhanced relaxation are at both ends as well as the middle of the SIM-C binding site. This indicates that the peptide is indeed bound in both parallel and antiparallel orientations with respect to strand β2 (Fig. 5C). We attribute the presence of both orientations to the roughly symmetrical distribution of negatively charged residues on both sides of this core motif. Importantly, an analysis of the chemical shift perturbations accompanying these titrations confirmed that the presence of MTSL-modified cysteines did not significantly alter the binding mode or affinity of SIM-C for the SUMOs (not shown).
We also used HADDOCK to generate models of the SIM-C·SUMO-1 complexes consistent with ensemble-averaged PRE effects (Fig. 5D). Distance restraints for each peptide were quantitated from the differences in transverse relaxation rates, ΔR2, of corresponding amide 1HN nuclei in the presence of the MTSL group in its paramagnetic versus diamagnetic states. The HADDOCK algorithm was modified to allow the simultaneous docking of two peptides in orientations 1 and 2 to SUMO-1. We made use of the multibody docking ability of HADDOCK, turning off the interaction terms between the two peptides and refining against effective distance restraints calculated as the average for the two peptides (reff = {(0.5/r16) + (0.5/r26)}1/6). Although this approach assumes that the two orientations occur in an equal ratio, because of the limited accuracy of the PRE data, their relative populations could differ substantially. Nevertheless, the HADDOCK modeling confirmed that SIM-C binds SUMO-1 in both parallel and antiparallel modes. Importantly, this result was obtained without a priori assumptions because the orientations of the peptides were randomized prior to docking.
Intramolecular Binding of SIM-N to the DAXX Helical Bundle Domain Competes with SUMO
With the goal of investigating the behavior of SIM-N in a more native context, NMR spectroscopy was used to characterize the fragment DAXX1–144, which contains both SIM-N and the recently discovered DHB domain (residues 55–144) (36). Surprisingly, the 15N HSQC spectra of this species at low ionic strength indicated extensive aggregation (supplemental Fig. S5). This behavior was not alleviated with addition of excess DTT to ensure complete reduction of all four cysteine residues. However, progressively increasing the sample ionic strength to 500 mm KCl yielded progressively sharper 15N HSQC signals, diagnostic of monomeric DAXX1–144. Given that both DAXX1–56 and the DHB domain (DAXX55–144) are soluble and monomeric under all of these conditions, a simple explanation for this behavior is an ionic strength-dependent self-association of DAXX1–144 due to intermolecular interactions between its flexible N-terminal residues and the adjacent structured domain.
To test for such an interaction, 15N-labeled DAXX1–56 was produced and changes in its 15N HSQC spectra upon addition of unlabeled DAXX55–144 were monitored. As hypothesized, binding occurred, and the residues of 15N-labeled DAXX1–56 most affected corresponded to those of the hydrophobic core and flanking aspartates and glutamates of SIM-N (supplemental Fig. S6A). Because at pH 6.5, SIM-N is negatively charged and DAXX55–144 is net positive, this is consistent with an electrostatically driven interaction. This interaction was confirmed through the reverse titration of 15N-labeled DAXX55–144 with both unlabeled DAXX1–56 (supplemental Fig. S6B) and SIM-N (not shown). Following the peaks in the fast exchange regime, the dissociation constant was determined to be 170 ± 60 μm (100 mm KCl; Table 1 and supplemental Fig. S6C). However, in the context of DAXX1–144 or the full-length protein, the free energy of intramolecular association will likely be more favorable due to the covalent linkage of SIM-N and the DHB domain.
Mapping the amide chemical shift perturbations due to SIM-N binding on the structure of the DHB domain (Fig. 6A) as well as use of these perturbations as restraints to generate a HADOCK model of the resulting complex (Fig. 6B) shows that the peptide interacts along a cleft previously identified as the association interface for peptides from Rassf1C, p53, and Mdm2 (36). Similar to SIM-N, these peptides also have a hydrophobic core with flanking negatively charged residues. This implies that the intermolecular association of SIM-N and the DHB domain will compete with the interaction of DAXX and its partner proteins as well as with SUMO. To test this idea, we repeated the NMR-monitored titration of SUMO-1 with DAXX1–144. In contrast to the relatively high affinity binding of the isolated SIM-N, DAXX1–144 bound the same interface on SUMO-1 with an ∼150-fold weaker apparent KD value of only 1.2 ± 0.5 mm (Fig. 6C and Table 1). Thus, in its native context, SIM-N is likely sequestered from SUMO (Fig. 6D).
FIGURE 6.

SIM-N binds the DHB domain and is sequestered from SUMO-1 when present within DAXX1–144. A, amides in the DHB domain of DAXX55–144 undergoing the greatest combined amide shift perturbations (magenta; Δδ > 0.045 ppm) upon binding DAXX1–56 cluster along helices H2 and H5. This same region is the binding interface for DAXX partners, including Rassf1C, p53, and Mdm2 (36). See supplemental Fig. S6 for the titration data. B, model of the resulting complex of SIM-N (stick format: yellow, carbon; red, oxygen; blue, nitrogen) with the DHB domain (transparent surface: red, negatively charged; blue, positively charged; green, hydrophobic; gray, neutral polar) generated using chemical shift perturbations as experimental restraints for HADDOCK. C, titration curves for 15N-labeled SIM-N with unlabeled SUMO-1 (red) and 15N-labeled SUMO-1 with unlabeled DAXX1–144 (black) in NMR buffer with 100 mm KCl. The reported values are the mean ± S.D. of five individually fit residues. D, schematic model of a potential autoregulatory mechanism of DAXX involving an equilibrium between inactive and active states in which SIM-N and the DHB domain are sequestered through their intramolecular association or exposed to bind both SUMO and partner proteins, respectively.
DAXX SIM-C Binds Only the SUMO-1 Moiety of Sumoylated Ets1
A prevalent function of DAXX is to repress transcription factors. One such factor is Ets1, which contains a DNA-binding ETS domain (residues 331–415) as well as a helical bundle PNT domain (residues 42–135) (55). The latter mediates MAPK docking and binding to the general transcriptional co-activator CBP. The unstructured N-terminal segment of Ets1 (residues 1–42) is the target for regulation via sumoylation (Lys15) and phosphorylation (Thr38 and Ser41) (37, 56).
Ets1 was reported to interact with and be repressed by DAXX as evidence by a yeast two-hybrid screen and a luciferase assay, respectively (57). This interaction was mapped to the C-terminal 173 residues of human DAXX and the N-terminal 139 residues of Ets1. Our attempts to reproduce the association of Ets1 and DAXX in vitro were unsuccessful as the 15N HSQC spectrum of 15N-labeled Ets1–138 did not exhibit any perturbations upon addition of excess unlabeled murine DAXX566–739 (not shown). Thus, the effect of DAXX566–739 on Ets1–138 sumoylated at Lys15 was examined. The latter was prepared initially from unlabeled SUMO-1 and 15N-labeled Ets1–138 using an in vitro sumoylation system (37). Addition of DAXX566–739 did not cause any spectral changes for the 15N-labeled Ets11–138, showing that the binding of its SIM-C to the invisible SUMO-1 did not perturb the covalently linked Ets11–138 (Fig. 7A). To confirm that only the SUMO moiety is involved in the interaction, we titrated a genetically fused 15N-labeled chimera, SUMO-11–97-Ets116–138, with unlabeled DAXX566–739. Indeed, only residues from SUMO-1, but not Ets1, were perturbed (Fig. 7B). Control experiments verified that these same residues showed spectral changes upon titration of 15N-labeled SUMO-1 with unlabeled DAXX566–739 (Fig. 7C) but not with unlabeled DAXX566–727 (not shown). The latter lacks SIM-C. These results clearly demonstrated that DAXX566–739 interacts via SIM-C exclusively with the SUMO-1 moiety of sumoylated Ets11–138 (Fig. 7D).
FIGURE 7.

SIM-C of DAXX566–739 binds only the SUMO-1 moiety of sumoylated Ets1. A, superimposed 15N HSQC spectra of sumoylated 15N-labeled Ets11–138 in the absence (red) and presence (black) of unlabeled DAXX566–739. The lack of any detectable shift or intensity perturbations in the amide signals of Ets11–138 demonstrates that the Ets1 and DAXX fragments did not interact directly with any appreciable affinity. Note that the SUMO moiety of this covalently linked species was unlabeled and hence invisible. B, superimposed 15N HSQC spectra of a 15N-labeled SUMO-11–97-Ets116–138 chimera in the absence (290 μm; red) and presence (black) of 1.9 eq of unlabeled DAXX566–739. Only residues from the SUMO-1 moiety (italicized) showed severe signal broadening due to the higher molecular mass of the resulting complex. In contrast, peaks arising from amides in Ets116–138 (underlined) were unperturbed. C, superimposed 15N HSQC spectra of 15N-labeled SUMO-1 in the absence (120 μm; red) and presence (black) of 9 eq of unlabeled DAXX566–739. The same residues as those that disappeared in the spectrum of the chimera showed intermediate exchange broadening, thus confirming that SUMO-1 functions as an independent tag on Ets1. D, beads-on-a-string model for the non-covalent binding of DAXX566–739 via SIM-C to sumoylated Ets11–138, emphasizing the lack of any direct interaction between the PNT domain-containing fragment of Ets1 and the intrinsically disordered C-terminal segment of DAXX.
DISCUSSION
Using NMR spectroscopy, we characterized the interaction of peptide models of the two DAXX SIMs with two SUMO paralogs. The low μm range KD values for the DAXX SIM·SUMO complexes were comparable with those reported for the interactions of these SUMOs with other SIM-containing peptides (supplemental Table S4). In each case, SIM-N and SIM-C bound SUMO-1 or SUMO-2 along a common interface between helix α1 and strand β2 of the protein via both hydrophobic and electrostatic interactions. Evidence for the former is provided by a >400-fold increase in the KD value of SIM-N for SUMO-1 upon mutation of the core Ile-Ile-Val-Leu motif to Gly-Ser-Gly-Ser. Evidence for the latter is provided both by the ionic strength dependence of the SIM/SUMO interactions and the observation that SIM-NGSGS still weakly bound SUMO-1. Also, because both DAXX SIMs have identical hydrophobic core motifs, the ∼4-fold higher affinity of SIM-N versus SIM-C for both SUMO paralogs likely reflects the greater number of flanking aspartate and glutamate residues present in SIM-N relative to SIM-C (i.e. seven versus five).
SIM-C Binds SUMO-1 with Multiple Conformations
To date, structures of SIMs from PIASx (25, 26), RanBP2 (28), thymine-DNA glycosylase (27), and DAXX SIM-C (52) with SUMO-1 and thymine-DNA glycosylase (29) and MBD1-containing chromatin-associated factor MCAF1 (32) with SUMO-3 have been reported. In each case, the hydrophobic core of the SIM adopts an extended conformation, pairing with strand β2 of the SUMO. However, a detailed comparison of the complex structures reveals a high degree of plasticity in the intermolecular contacts between the various SIM peptides and SUMO proteins. Most striking of course is that the pairing can be in either a parallel or antiparallel orientation.
It has been suggested that the juxtaposition of charged groups relative to the hydrophobic core is responsible for determining the binding mode of a given SIM (33). Consistent with this hypothesis, NOE measurements verified that SIM-N binds SUMO-1 predominantly in a parallel orientation, thereby positioning up to seven negatively charge residues (11Asp-Asp-Asp-Asp-Glu-Asp-Glu17) of the peptide near a lysine-rich surface of the protein (Fig. 4). In contrast, SIM-C contains two or three negatively charge residues on each side of a rather symmetrical hydrophobic core and thus binds SUMO-1 and SUMO-2 in both parallel and antiparallel orientations. These binding modes were verified by PRE effects from three separate nitroxide spin-labeled SIM-C variants to amides on both ends of the binding clefts of the SUMO paralogs (Fig. 5). Data-driven HADDOCK docking using a single peptide could not explain fully the observed PRE effects. Only when two peptides were docked in an ensemble-averaged manner could all PRE restraints measured for the three SIM-C derivatives be satisfied simultaneously (supplemental Table S3). These results also indicate that the absence of detectable 1HN-15N signals from amides in the hydrophobic core of SIM-C when bound to SUMO-1 likely results from interconversion between these two orientations on a ms to μs time scale. This interconversion appears to be faster for SIM-C when bound to SUMO-2 as peaks did reappear upon saturation in an HSQC-monitored titration (Fig. 2). Unfortunately, due to the nature of ensemble-averaged PRE effects, we were unable to determine the relative populations of the two orientations. Indeed, the recently released NOE-derived structural coordinates of a complex of SIM-C bound in a parallel orientation with SUMO-1 (Protein Data Bank code 2KQS)4 suggest that this particular binding mode is predominant. Similarly, it is possible that a small fraction of SIM-N also binds SUMO-1 in an antiparallel manner not detectable by NOE measurements (which are a less sensitive indicator of alternative conformations than are PREs). This hypothesis is supported by the loss of some 1HN-15N signals from the SIM-N·SUMO-1 complex at elevated ionic strength.
Although important for understanding the structural and thermodynamic mechanisms underlying SIM/SUMO interactions, the biological significance of the exact orientations of the resulting DAXX complexes is unclear. That is, SIM-N and SIM-C are within intrinsically unstructured regions of DAXX (36), thus allowing a high degree of conformational freedom between a bound SUMO and other functional moieties of this protein.
DAXX SIM Peptides Do Not Show SUMO Paralog Specificity
Using NMR-monitored titrations, we found that peptide models of the DAXX SIMs show no significant paralog specificity. As summarized in Table 1, the SIM-N peptide bound SUMO-1 and SUMO-2 with the same affinity in the presence of 0 or 200 mm KCl. Although at low ionic strength the isolated SIM-C peptide bound SUMO-1 with ∼3.5-fold higher affinity than SUMO-2, this very modest effect was absent under more physiological salt concentrations. Using yeast two-hybrid screens, Santiago et al. (24) reported that full-length DAXX binds SUMO-1, -2, and -3 with comparable affinity, whereas Lin et al. (23) detected a stronger interaction with SUMO-1 than with SUMO-2. Although the biological implications of these results remain to be established, it is noteworthy that endogenous DAXX in unstressed HeLa cells is predominantly modified by SUMO-1 (23), whereas heat shock significantly increases its modification by SUMO-2 (17). Although this specificity could result from the many regulatory steps along the sumoylation/desumoylation pathway (16), it is interesting that SIM-C is required for the covalent modification of DAXX in vivo presumably through the non-covalent recruitment of a thiol ester-linked SUMO-Ubc9 conjugate (23). Thus, small differences in affinities measured in vitro might translate to larger effects in a cellular context.
Several studies using qualitative pulldown or yeast two-hybrid assays have demonstrated that some SIMs do show specificity for SUMO-1 versus SUMO-2/3, whereas others bind both paralogs with comparable affinity (25, 30, 32, 58–61). However, the largest quantitative effect reported to date is only an ∼10-fold lower KD value of the isolated MCAF1 SIM for SUMO-3 versus SUMO-1 (supplemental Table S3) (32). Furthermore, the molecular basis for any potential paralog specificity is not readily apparent from published SUMO·SIM complex structures. SUMO-1 and SUMO-2/3 share ∼50% sequence identity and adopt highly similar ubiquitin-like folds. Not surprisingly, each utilizes an analogous cleft between strand β2 and helix α1 to bind SIMs. This cleft consists mostly of hydrophobic residues surrounded by positively charged histidine, lysine, and arginine side chains and thus matches the complementary negative charges from aspartate and glutamate residues flanking the core hydrophobic motifs of SIMs. However, the number and positions of these ionizable residues do differ between SUMO-1 and SUMO-2/3 as well as between various SIMs, suggesting that electrostatic interactions could lead to a degree of preferential binding (30, 32, 62). Further detailed in vitro pH- and ionic strength-dependent thermodynamic and structural studies of wild-type and mutant SIMs and SUMOs combined with in vivo cellular assays will be required to resolve the mechanisms and biological significance of any paralog specificity.
SIM-N Binds Both SUMO and DHB Domain
We have recently identified the N-terminal DHB domain of DAXX and structurally characterized it in complex with a peptide model from the tumor suppressor Rassf1C (36). This peptide undergoes a coil-to-helix transition upon binding the DHB domain along a shallow groove formed by two parallel helices. Binding is driven by both hydrophobic interactions involving one side of the amphipathic helix of Rassf1C and flanking negatively charged residues (KD ∼ 60 μm). Peptides corresponding to segments of p53 and Mdm2 also associate in a similar manner with the DHB domain.
We have now discovered that SIM-N binds the same interface of the DHB domain. Although at first unexpected, as to date no SIM has been reported to have binding partners other than SUMO paralogs or SUMO-like domains, this result can be rationalized by the similar physicochemical properties of the SIM-N and Rassf1C peptides. This raises the question of whether the DHB domain and SIM-N also interact in vivo within the context of full-length DAXX. If so, then an autoregulatory mechanism could result with the intramolecular association of the DHB domain and SIM-N sequestering both regions of DAXX from their respective binding partners (i.e. Rassf1C and SUMO; Fig. 6D). Indeed, SIM-N within DAXX1–144 bound SUMO-1 ∼150-fold weaker than as an isolated peptide. According to this linked equilibrium model, we speculate that binding of SIM-N by SUMO would also free the DHB domain for binding to Rassf1C (or other partners) and vice versa. Cells could control such a competitive/cooperative mechanism by regulating the available concentrations of each interacting species or via post-translational modifications of either DAXX or its partners. For example, Lys122 has been reported recently to be a ubiquitylation site in DAXX (63). A ubiquitin at this position might sterically hinder Rassf1C or SIM-N binding.
SUMO Functions as Independent Tag to Bridge Ets1 and DAXX
In 2000, Li et al. (57) reported that the transcriptional activity of Ets1 is repressed by DAXX. This effect was mapped to an apparent interaction between the N-terminal 139 residues of Ets1 and the C-terminal 173 residues of DAXX. Subsequently, several groups discovered that Ets1 is sumoylated at Lys15 and Lys227 (37, 64–66). Furthermore, we demonstrated via NMR methods that Lys15-sumoylated Ets11–138 behaves as two independent beads-on-a-string with the Ets1 and SUMO-1 components simply linked via a flexible isopeptide bond to an unstructured region of the transcription factor (37). We now show that the C-terminal portion of DAXX binds only the SUMO component of sumoylated Ets11–138 via SIM-C. Thus, the initial identification of DAXX as an Ets1 partner using a two-hybrid screen in yeast (57) was likely mediated by an unrecognized SUMO attached to the transcription factor. This extends the beads-on-a-string model to three components with the intrinsically disordered C terminus of DAXX non-covalently bound to a SUMO moiety that in turn is covalently connected to Ets1 via a flexible linker (Fig. 7D). Although an isolated SIM-N peptide will certainly bind sumoylated Ets1, the fact that N-terminal fragments of DAXX were not identified in the original interaction screens with Ets1 (57) could reflect the sequestration of SIM-N by it intramolecular interaction with the adjacent DHB domain.
We speculate that SUMO frequently serves as an independent tag to bridge sumoylated transcription factors, such as Ets1, with DAXX, which in turn recruits co-repressors, including chromatin-remodeling histone deacetylases, to inhibit gene expression (3, 21). However, this leads to a conundrum for understanding the necessary specificity of SUMO-mediated transcriptional regulation. One plausible explanation is that a combinatorial network of additional interactions between components of the transcriptional machinery with both DAXX and the transcription factors is required for a regulated biological response. For example, the two sumoylation sites in Ets1 were originally identified as “synergy control motifs” (64). As implied by this name, the presence of both motifs leads to transcriptional repression of only reporter genes with promoters containing multiple Ets1 binding sites. This phenomenon is suggestive of higher order, multivalent interactions involving transcriptional regulators.
Biological Significance
In this study, the interactions of the two SIMs in DAXX with SUMO-1 and SUMO-2 were characterized in vitro. There is solid evidence that SIM-C is also active in vivo, mediating the sumoylation and localization of DAXX to both PML-NBs and chromatin as well repression of sumoylated transcription factors, including the glucocorticoid receptor and c-Jun (23, 24). A new paradigm for regulating SIM interactions has emerged very recently. Casein kinase 2, an essential kinase in numerous signaling pathways, has been shown to phosphorylate serine residues adjacent to the hydrophobic core of SIMs (67). All known DAXX orthologs contain an exact target site for casein kinase 2 at the C-terminal end of their SIM-C sequence (736SDSD739 by murine numbering). We have demonstrated the importance of electrostatic interactions in SUMO/SIM interactions, and thus, it is expected that phosphorylation at either serine would change the ability of DAXX to recognize sumoylated proteins for several reasons. First of all, SIM-C binds SUMO-1 in multiple orientations due to the presence of negatively charged residues on both sides of its hydrophobic core. Phosphorylation of the acceptor serines might induce a single binding mode by changing this charge distribution. Second, SIM-C phosphorylation will almost certainly increase its net affinity for SUMO. On the other hand, the role of SIM-N is less clear. In particular, only DAXX1–740, but not DAXX1–732 (which lacks SIM-C but not SIM-N), was reported to repress the activity of Smad4, androgen receptor, and CBP (23). This occurs despite the fact that isolated SIM-N bound SUMO-1 and SUMO-2 with ∼4-fold higher affinity than did SIM-C. Again, one possible explanation is that the accessibility of SIM-N is limited by its intramolecular association with the adjacent DHB domain.
It is striking that DAXX contains a short SIM at both of its termini. Given their large separation by ∼700 residues, it is likely that each functions independently and not in synchrony as is the case for RNF4, which binds polySUMO-2/3 chains via adjacent SIMs (68). However, both SIMs may be simultaneously involved in the formation of higher order multiprotein complexes or could interact with transcription factors, such as Ets1, that are sumoylated at multiple positions.
In closing, DAXX is an essential scaffold protein with key roles in diverse cellular processes spanning transcription to apoptosis and mitosis. Despite its importance, the molecular bases for its scaffolding properties and regulation are still largely unknown (3). Characterizing in vitro the interactions of its N- and C-terminal SIMs with SUMO-1 and SUMO-2 is a needed stepping stone toward understanding the in vivo roles of this enigmatic protein.
Supplementary Material
Acknowledgments
We thank Pitter Huesgen, Alexander Ishov, Lewis Kay, and Cameron Mackereth for advice and helpful ideas. Instrument support was provided by the Canadian Institutes for Health Research, the Canadian Foundation for Innovation, the British Columbia Knowledge Development Fund, the University of British Columbia Blusson Fund, and the Michael Smith Foundation for Health Research.
Note Added in Proof
A complementary study of DAXX-SUMO interactions has been published by Chang et al. (C.-C. Chang, M. T. Naik, Y.-S. Huang, J.-C. Jeng, P.-H. Lian, H.-Y. Kuo, C.-C. Ho, Y.-L. Hsieh, C.-H. Lin, N.-J. Huang, M. M. Naik, C. C.-H. Kung, S.-Y. Lin, R.-H. Chen, K.-S. Chang, T.-H. Huang, H.-M. Shih (2011) Mol. Cell 42, 62–74).
This work was supported in part by the Canadian Cancer Society (Grant 017308 to L. P. M.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S6 and Tables S1–S4.
M. T. Naik, T. Huang, and H. Shih, unpublished data.
- PML-NB
- promyelocytic leukemia nuclear body
- SUMO
- small ubiquitin-like modifier
- SIM-N
- N-terminal SUMO-interacting motif
- SIM-C
- C-terminal SUMO-interacting motif
- PRE
- paramagnetic relaxation enhancement
- DHB
- DAXX helix bundle
- HSQC
- heteronuclear single quantum correlation
- MTSL
- (1-oxyl-2,2,5,5-tetramethyl-Δ3-pyrroline-3-methyl)methanethiosulfonate
- CBP
- cAMP-response element-binding protein (CREB)-binding protein
- MS
- mass spectrometry.
REFERENCES
- 1. Yang X., Khosravi-Far R., Chang H. Y., Baltimore D. (1997) Cell 89, 1067–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bernardi R., Pandolfi P. P. (2007) Nat. Rev. Mol. Cell Biol. 8, 1006–1016 [DOI] [PubMed] [Google Scholar]
- 3. Lindsay C. R., Morozov V. M., Ishov A. M. (2008) Front. Biosci. 13, 7132–7142 [DOI] [PubMed] [Google Scholar]
- 4. Emelyanov A. V., Kovac C. R., Sepulveda M. A., Birshtein B. K. (2002) J. Biol. Chem. 277, 11156–11164 [DOI] [PubMed] [Google Scholar]
- 5. Huang Y. S., Shih H. M. (2009) Biochem. Biophys. Res. Commun. 386, 762–768 [DOI] [PubMed] [Google Scholar]
- 6. Li H., Leo C., Zhu J., Wu X., O'Neil J., Park E. J., Chen J. D. (2000) Mol. Cell. Biol. 20, 1784–1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hollenbach A. D., McPherson C. J., Mientjes E. J., Iyengar R., Grosveld G. (2002) J. Cell Sci. 115, 3319–3330 [DOI] [PubMed] [Google Scholar]
- 8. Muromoto R., Sugiyama K., Takachi A., Imoto S., Sato N., Yamamoto T., Oritani K., Shimoda K., Matsuda T. (2004) J. Immunol. 172, 2985–2993 [DOI] [PubMed] [Google Scholar]
- 9. Ishov A. M., Vladimirova O. V., Maul G. G. (2004) J. Cell Sci. 117, 3807–3820 [DOI] [PubMed] [Google Scholar]
- 10. Puto L. A., Reed J. C. (2008) Genes Dev. 22, 998–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shih H. M., Chang C. C., Kuo H. Y., Lin D. Y. (2007) Biochem. Soc. Trans. 35, 1397–1400 [DOI] [PubMed] [Google Scholar]
- 12. Johnson E. S. (2004) Annu. Rev. Biochem. 73, 355–382 [DOI] [PubMed] [Google Scholar]
- 13. Hay R. T. (2005) Mol. Cell 18, 1–12 [DOI] [PubMed] [Google Scholar]
- 14. Geiss-Friedlander R., Melchior F. (2007) Nat. Rev. Mol. Cell Biol. 8, 947–956 [DOI] [PubMed] [Google Scholar]
- 15. Wilkinson K. A., Henley J. M. (2010) Biochem. J. 428, 133–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gareau J. R., Lima C. D. (2010) Nat. Rev. Mol. Cell Biol. 11, 861–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Golebiowski F., Matic I., Tatham M. H., Cole C., Yin Y., Nakamura A., Cox J., Barton G. J., Mann M., Hay R. T. (2009) Sci. Signal. 2, ra24. [DOI] [PubMed] [Google Scholar]
- 18. Gill G. (2005) Curr. Opin. Genet. Dev. 15, 536–541 [DOI] [PubMed] [Google Scholar]
- 19. Lin D. Y., Fang H. I., Ma A. H., Huang Y. S., Pu Y. S., Jenster G., Kung H. J., Shih H. M. (2004) Mol. Cell. Biol. 24, 10529–10541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kuo H. Y., Chang C. C., Jeng J. C., Hu H. M., Lin D. Y., Maul G. G., Kwok R. P., Shih H. M. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 16973–16978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Garcia-Dominguez M., Reyes J. C. (2009) Biochim. Biophys. Acta 1789, 451–459 [DOI] [PubMed] [Google Scholar]
- 22. Ryu S. W., Chae S. K., Kim E. (2000) Biochem. Biophys. Res. Commun. 279, 6–10 [DOI] [PubMed] [Google Scholar]
- 23. Lin D. Y., Huang Y. S., Jeng J. C., Kuo H. Y., Chang C. C., Chao T. T., Ho C. C., Chen Y. C., Lin T. P., Fang H. I., Hung C. C., Suen C. S., Hwang M. J., Chang K. S., Maul G. G., Shih H. M. (2006) Mol. Cell 24, 341–354 [DOI] [PubMed] [Google Scholar]
- 24. Santiago A., Godsey A. C., Hossain J., Zhao L. Y., Liao D. (2009) Cell Cycle 8, 76–87 [DOI] [PubMed] [Google Scholar]
- 25. Song J., Durrin L. K., Wilkinson T. A., Krontiris T. G., Chen Y. A. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 14373–14378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Song J., Zhang Z., Hu W., Chen Y. (2005) J. Biol. Chem. 280, 40122–40129 [DOI] [PubMed] [Google Scholar]
- 27. Baba D., Maita N., Jee J. G., Uchimura Y., Saitoh H., Sugasawa K., Hanaoka F., Tochio H., Hiroaki H., Shirakawa M. (2005) Nature 435, 979–982 [DOI] [PubMed] [Google Scholar]
- 28. Reverter D., Lima C. D. (2005) Nature 435, 687–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Baba D., Maita N., Jee J. G., Uchimura Y., Saitoh H., Sugasawa K., Hanaoka F., Tochio H., Hiroaki H., Shirakawa M. (2006) J. Mol. Biol. 359, 137–147 [DOI] [PubMed] [Google Scholar]
- 30. Hecker C. M., Rabiller M., Haglund K., Bayer P., Dikic I. (2006) J. Biol. Chem. 281, 16117–16127 [DOI] [PubMed] [Google Scholar]
- 31. Duda D. M., van Waardenburg R. C., Borg L. A., McGarity S., Nourse A., Waddell M. B., Bjornsti M. A., Schulman B. A. (2007) J. Mol. Biol. 369, 619–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sekiyama N., Ikegami T., Yamane T., Ikeguchi M., Uchimura Y., Baba D., Ariyoshi M., Tochio H., Saitoh H., Shirakawa M. (2008) J. Biol. Chem. 283, 35966–35975 [DOI] [PubMed] [Google Scholar]
- 33. Kerscher O. (2007) EMBO Rep. 8, 550–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Matunis M. J., Zhang X. D., Ellis N. A. (2006) Dev. Cell 11, 596–597 [DOI] [PubMed] [Google Scholar]
- 35. Lehembre F., Müller S., Pandolfi P. P., Dejean A. (2001) Oncogene 20, 1–9 [DOI] [PubMed] [Google Scholar]
- 36. Escobar-Cabrera E., Lau D. K., Giovinazzi S., Ishov A. M., McIntosh L. P. (2010) Structure 18, 1642–1653 [DOI] [PubMed] [Google Scholar]
- 37. Macauley M. S., Errington W. J., Schärpf M., Mackereth C. D., Blaszczak A. G., Graves B. J., McIntosh L. P. (2006) J. Biol. Chem. 281, 4164–4172 [DOI] [PubMed] [Google Scholar]
- 38. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 39. Goddard T. D., Kneller D. G. (2004) Sparky 3, Version 3.11, University of California, San Francisco [Google Scholar]
- 40. Johnson B. A., Blevins R. A. (1994) J. Biomol. NMR 4, 603–614 [DOI] [PubMed] [Google Scholar]
- 41. Kay L. E., Keifer P., Saarinen T. (1992) J. Am. Chem. Soc. 114, 10663–10665 [Google Scholar]
- 42. Johnson P. E., Tomme P., Joshi M. D., McIntosh L. P. (1996) Biochemistry 35, 13895–13906 [DOI] [PubMed] [Google Scholar]
- 43. Donaldson L. W., Skrynnikov N. R., Choy W. Y., Muhandiram D. R., Sarkar B., Forman-Kay J. D., Kay L. E. (2001) J. Am. Chem. Soc. 123, 9843–9847 [DOI] [PubMed] [Google Scholar]
- 44. Macauley M. S., Errington W. J., Okon M., Schärpf M., Mackereth C. D., Schulman B. A., McIntosh L. P. (2004) J. Biol. Chem. 279, 49131–49137 [DOI] [PubMed] [Google Scholar]
- 45. Kosen P. A. (1989) Methods Enzymol. 177, 86–121 [DOI] [PubMed] [Google Scholar]
- 46. Dominguez C., Boelens R., Bonvin A. M. J. J. (2003) J. Am. Chem. Soc. 125, 1731–1737 [DOI] [PubMed] [Google Scholar]
- 47. de Vries S. J., van Dijk A. D., Krzeminski M., van Dijk M., Thureau A., Hsu V., Wassenaar T., Bonvin A. M. (2007) Proteins 69, 726–733 [DOI] [PubMed] [Google Scholar]
- 48. de Vries S. J., van Dijk M., Bonvin A. M. (2010) Nat. Protoc. 5, 883–897 [DOI] [PubMed] [Google Scholar]
- 49. Pichler A., Knipscheer P., Oberhofer E., van Dijk W. J., Körner R., Olsen J. V., Jentsch S., Melchior F., Sixma T. K. (2005) Nat. Struct. Mol. Biol. 12, 264–269 [DOI] [PubMed] [Google Scholar]
- 50. Sattler M., Schleucher J., Griesinger C. (1999) Prog. Nucl. Magn. Reson. Spectrosc. 34, 93–158 [Google Scholar]
- 51. Karaca E., Melquiond A. S., de Vries S. J., Kastritis P. L., Bonvin A. M. J. J. (2010) Mol. Cell. Proteomics 9, 1784–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Naik M. T., Chang C. C., Naik N. M., Kung C. C., Shih H. M., Huang T. H. (2011) Biomol. NMR Assign. 5, 75–77 [DOI] [PubMed] [Google Scholar]
- 53. Reibarkh M., Malia T. J., Wagner G. (2006) J. Am. Chem. Soc. 128, 2160–2161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Su X. C., Otting G. (2010) J. Biomol. NMR 46, 101–112 [DOI] [PubMed] [Google Scholar]
- 55. Hollenhorst P., McIntosh L. P., Graves B. J. (2011) Annu. Rev. Biochem., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nelson M. L., Kang H. S., Lee G. M., Blaszczak A. G., Lau D. K., McIntosh L. P., Graves B. J. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 10026–10031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Li R., Pei H., Watson D. K., Papas T. S. (2000) Oncogene 19, 745–753 [DOI] [PubMed] [Google Scholar]
- 58. Tatham M. H., Kim S., Jaffray E., Song J., Chen Y., Hay R. T. (2005) Nat. Struct. Mol. Biol. 12, 67–74 [DOI] [PubMed] [Google Scholar]
- 59. Zhu J., Zhu S., Guzzo C. M., Ellis N. A., Sung K. S., Choi C. Y., Matunis M. J. (2008) J. Biol. Chem. 283, 29405–29415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Meulmeester E., Kunze M., Hsiao H. H., Urlaub H., Melchior F. (2008) Mol. Cell 30, 610–619 [DOI] [PubMed] [Google Scholar]
- 61. Ouyang J., Shi Y., Valin A., Xuan Y., Gill G. (2009) Mol. Cell 34, 145–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chupreta S., Holmstrom S., Subramanian L., Iñiguez-Lluhí J. A. (2005) Mol. Cell. Biol. 25, 4272–4282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fukuyo Y., Kitamura T., Inoue M., Horikoshi N. T., Higashikubo R., Hunt C. R., Usheva A., Horikoshi N. (2009) Cancer Res. 69, 7512–7517 [DOI] [PubMed] [Google Scholar]
- 64. Iñiguez-Lluhí J. A., Pearce D. (2000) Mol. Cell. Biol. 20, 6040–6050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Nishida T., Terashima M., Fukami K. (2006) Biochem. Biophys. Res. Commun. 345, 1536–1546 [DOI] [PubMed] [Google Scholar]
- 66. Ji Z., Degerny C., Vintonenko N., Deheuninck J., Foveau B., Leroy C., Coll J., Tulasne D., Baert J. L., Fafeur V. (2007) Oncogene 26, 395–406 [DOI] [PubMed] [Google Scholar]
- 67. Stehmeier P., Muller S. (2009) Mol. Cell 33, 400–409 [DOI] [PubMed] [Google Scholar]
- 68. Tatham M. H., Geoffroy M. C., Shen L., Plechanovova A., Hattersley N., Jaffray E. G., Palvimo J. J., Hay R. T. (2008) Nat. Cell Biol. 10, 538–546 [DOI] [PubMed] [Google Scholar]
- 69. Cavanagh J., Fairbrother W. J., Palmer A. G., Rance M., Skelton N. J. (2007) Protein NMR Spectroscopy: Principles and Practices, Academic Press, New York [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.