

Abstract
Reduced expression of CCR5 on target CD4+ cells lowers their susceptibility to infection by R5-tropic HIV-1, potentially preventing transmission of infection and delaying disease progression. Binding of the HIV-1 envelope (Env) protein gp120 with CCR5 is essential for the entry of R5 viruses into target cells. The threshold surface density of gp120-CCR5 complexes that enables HIV-1 entry remains poorly estimated. We constructed a mathematical model that mimics Env-mediated cell-cell fusion assays, where target CD4+CCR5+ cells are exposed to effector cells expressing Env in the presence of a coreceptor antagonist and the fraction of target cells fused with effector cells is measured. Our model employs a reaction network-based approach to describe protein interactions that precede viral entry coupled with the ternary complex model to quantify the allosteric interactions of the coreceptor antagonist and predicts the fraction of target cells fused. By fitting model predictions to published data of cell-cell fusion in the presence of the CCR5 antagonist vicriviroc, we estimated the threshold surface density of gp120-CCR5 complexes for cell-cell fusion as ∼20 µm −2. Model predictions with this threshold captured data from independent cell-cell fusion assays in the presence of vicriviroc and rapamycin, a drug that modulates CCR5 expression, as well as assays in the presence of maraviroc, another CCR5 antagonist, using sixteen different Env clones derived from transmitted or early founder viruses. Our estimate of the threshold surface density of gp120-CCR5 complexes necessary for HIV-1 entry thus appears robust and may have implications for optimizing treatment with coreceptor antagonists, understanding the non-pathogenic infection of non-human primates, and designing vaccines that suppress the availability of target CD4+CCR5+ cells.
Introduction
The entry of HIV-1 into target cells requires the formation of complexes between the viral envelope protein (Env) and the cellular receptor, CD4, as well as a coreceptor, either CCR5 or CXCR4. CCR5 appears to play a central role in HIV-1 transmission and disease progression to AIDS. Viruses transmitted across individuals are predominantly R5 viruses, i.e., require CCR5 for entry [1], [2]. Studies of simian immunodeficiency virus (SIV) infections of non-human primates suggest that differences in the expression level of CCR5 on target CD4+ cells may underlie the difference between the non-pathogenic infection of natural hosts, such as African green monkeys and sooty mangabeys, and the pathogenic infection of non-natural hosts, such as rhesus macaques [1]–[3]. The former have substantially lower levels of CD4+CCR5+ target cells than the latter [4]. Low CCR5 expression may imply reduced susceptibility of cells to infection [5], [6]. Consequently, the extent of viral replication at mucosal sites may be suppressed, lowering the probability of transmission. Indeed, low CCR5 expression in newborns correlated with poor SIV transmission via breast-feeding, which may underlie the negligible mother-to-child transmission of infection in natural hosts [7]. Similarly, humans homozygous for the CCR5Δ32 allele, which results in complete suppression of CCR5 expression, are extraordinarily resistant to HIV-1 infection [8]. At the same time, low CCR5 expression may control damage to the gut mucosa, suppressing microbial translocation, and also reduce T cell homing to sites of inflammation, thereby lowering immune activation and contributing to the non-pathogenic nature of infection in natural hosts [4], [9], [10]. Reducing the availability of target CD4+CCR5+ cells therefore appears to be a promising strategy for therapeutic and preventive vaccine development [1]–[3]. Indeed, the CCR5 antagonist maraviroc was found recently to protect rhesus macaques from vaginal transmission (Veazey et al., Abstract # 84LB 17th Conference on Retroviruses and Opportunistic Infections, 2010).
Env is a trimer of non-covalently attached extracellular gp120 and transmembrane gp41 glycoprotein heterodimers [11]. During viral entry, gp120 first binds to CD4, following which conformational changes expose a cryptic binding site on gp120 for CCR5 [12]. Following CCR5 binding to gp120, further conformational changes bring the viral and cell membranes into close apposition, culminating in viral entry [12]–[14]. Direct observation of the protein complexes that mediate viral entry has remained a challenge. One strategy to overcome this limitation has been to employ mathematical models to analyse viral infectivity assays and infer the stoichiometry and/or the number of complexes necessary for viral entry [14]–[21]. Following such an approach, previous studies have argued that multiple CD4 and CCR5 molecules must be bound to gp120 for viral entry [15], [16]. More recent studies using virions expressing heterotrimeric Env containing combinations of wild-type and mutant gp120 molecules, the latter incapable of mediating entry, suggested that a single Env trimer with at least two functional gp120 subunits is adequate for HIV-1 entry [17], [18]. When the latter experiments were reanalysed using more detailed mathematical models, one study estimated that 5 trimers on a virion carrying 9 trimers are necessary for entry [19], whereas another study estimated that 8 trimers (range 2–19 depending on the assumptions employed) represent the threshold for entry [20]. Further, a model of allosteric interactions between CCR5 and gp120 argued that the better adapted a viral strain is to utilize CCR5, the fewer the CCR5 molecules needed for entry, with highly adapted strains requiring a single CCR5 bound to gp120 [14]. Robust estimates of the threshold number and the stoichiometry of Env-CD4-CCR5 complexes necessary for HIV-1 entry are thus lacking.
Here, we developed a mathematical model that mimics cell-cell fusion assays widely employed to investigate HIV entry into target cells (e.g., [13], [22]–[25]). The model employs a reaction network-based approach to describe the protein interactions that precede viral entry and quantitatively predicts the influence of the CCR5 expression level on the susceptibility of target cells to Env-mediated cell-cell fusion. We applied the model to analyse data from cell-cell fusion assays in the presence of the CCR5 antagonist vicriviroc and obtained estimates of the threshold surface density of gp120-CCR5 complexes necessary for cell-cell fusion. We validated the estimate by comparison of model predictions with independent data from cell-cell fusion assays in the presence of rapamycin, which down-regulates CCR5 expression, as well as assays using different Env clones in the presence of another coreceptor antagonist, maraviroc.
Results
Model formulation
We modelled cell-cell fusion assays where target cells expressing CD4 and CCR5 are exposed to effector cells expressing Env in the presence of a coreceptor antagonist and the percentage of target cells fused with effector cells is measured (e.g., see [23], [24]). To describe these assays, we first considered a single target cell-effector cell pair in close apposition and employed reaction kinetics to determine the surface densities of different Env-CD4-CCR5 complexes formed across the pair. The reaction network and the rate equations are mentioned in Text S1. We found by solving the rate equations that reaction equilibrium was attained rapidly (∼1 s) compared to the time required for cell-cell fusion (∼min) (Fig. S1). Further, for typical CD4 and Env expression levels, CD4 appeared to be in large excess so that all gp120 monomers were bound to CD4 at equilibrium. The reaction network may therefore be simplified by ignoring the trimeric nature of Env and considering the total surface density of gp120 molecules as being available for interaction with CCR5. With this simplification, we determined the surface density of gp120-CCR5 complexes formed across a closely apposed target cell-effector cell pair as a function of the CCR5 expression level on the target cell (Methods). We postulated that a threshold surface density of gp120-CCR5 complexes must be formed for cell-cell fusion. Thus, if the surface density of gp120-CCR5 complexes formed is larger than the threshold, the target cell-effector cell pair is fused (Fig. 1). We next assumed that the CCR5 expression level on target cells in a cell-cell fusion assay follows a truncated normal distribution. Cells with smaller expression levels of CCR5 form fewer complexes and may not fuse. We thus estimated the fraction of cells that expresses CCR5 at levels larger than that required to form the threshold surface density of gp120-CCR5 complexes, which yields the fraction of cells fused in the assay.
Figure 1. Schematic of the model.

A) High CCR5 expression on a target cell allows the formation of the requisite gp120-CCR5 complexes for cell-cell fusion. B) Low CCR5 expression or C) the presence of a coreceptor antagonist reduces the surface density of gp120-CCR5 complexes and prevents fusion.
A coreceptor antagonist typically binds to an allosteric site on CCR5 and inhibits CCR5 binding to gp120 [26]. Consequently, fewer gp120-CCR5 complexes are formed between a cell-cell pair as exposure to the coreceptor antagonist increases (Fig. 1). A target cell would therefore require higher CCR5 expression to form the threshold surface density of gp120-CCR5 complexes when exposed to the coreceptor antagonist. Thus, in a cell-cell fusion assay, the fraction of cells fused decreases as the concentration of the coreceptor antagonist increases. We employed the standard ternary complex model to describe the gp120-CCR5 interaction across a cell-cell pair in the presence of a coreceptor antagonist [27]. Accordingly, we estimated the fraction of cells fused at different levels of exposure to the antagonist (Methods). We present model predictions below.
Model predictions
Distribution of CCR5 on target cells
In Fig. 2A, we present
the distribution, f(C
0), of the
CCR5 expression level, C
0, on cells, computed
using Eq. (12) (Methods), for a fixed
mean expression level 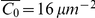 , which
corresponds to ∼5000 molecules/cell (radius∼5
µm), and different standard deviations,
, which
corresponds to ∼5000 molecules/cell (radius∼5
µm), and different standard deviations,
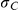 . For small values of
. For small values of 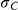 (
(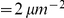 in Fig.
2A), the distribution is nearly normal and sharply peaked at
in Fig.
2A), the distribution is nearly normal and sharply peaked at
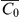 . As
. As 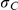 increases, the
size of the peak drops and the distribution spreads over a broader range of
values of
increases, the
size of the peak drops and the distribution spreads over a broader range of
values of 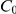 centred near
centred near 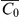 . For even
larger values of
. For even
larger values of 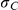 (
(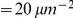 in Fig.
2A), because the distribution is truncated at
in Fig.
2A), because the distribution is truncated at
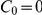 , the distribution spreads to larger values of
, the distribution spreads to larger values of
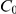 and is unevenly distributed about
and is unevenly distributed about
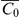 .
.
Figure 2. Model predictions of cell-cell fusion in the absence of a coreceptor antagonist.

A) Distribution, f(C
0),
of the CCR5 expression level, C
0, across
cells, predicted using Eq. (12), for the mean expression,
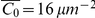 and
different values of the standard deviation,
and
different values of the standard deviation,
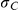 .
Inset: Fraction of cells fused, F, is the (shaded)
area under the f(C
0)
curve for
.
Inset: Fraction of cells fused, F, is the (shaded)
area under the f(C
0)
curve for 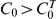 , the
threshold CCR5 expression level for fusion. B) F as
a function of GCT, the threshold surface
density of gp120-CCR5 complexes necessary for fusion. Inset: The
dependence of
, the
threshold CCR5 expression level for fusion. B) F as
a function of GCT, the threshold surface
density of gp120-CCR5 complexes necessary for fusion. Inset: The
dependence of 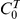 on
GCT computed using Eq. (11).
on
GCT computed using Eq. (11).
Threshold CCR5 expression and cell-cell fusion
With the above distribution of CCR5 expression and given a threshold CCR5
expression level necessary for fusion, 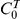 , we computed
the fraction of cells fused in a cell-cell fusion assay,
, we computed
the fraction of cells fused in a cell-cell fusion assay,
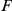 , using Eq. (13) (area of the shaded region in Fig. 2A (inset)).
, using Eq. (13) (area of the shaded region in Fig. 2A (inset)).
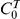 depends on the threshold surface density of
gp120-CCR5 complexes that enables entry,
depends on the threshold surface density of
gp120-CCR5 complexes that enables entry, 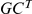 (Eq. (11)). We
therefore examined model predictions of the dependence of
(Eq. (11)). We
therefore examined model predictions of the dependence of
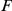 on
on 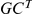 for different
values of
for different
values of 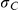 (Fig.
2B).
(Fig.
2B). 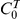 increases upon
increasing
increases upon
increasing 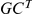 (Fig. 2B (inset)). Thus,
for a fixed
(Fig. 2B (inset)). Thus,
for a fixed 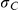 , increasing
, increasing
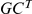 resulted in smaller F (Fig. 2B). When
resulted in smaller F (Fig. 2B). When
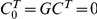 , all cells had
, all cells had 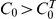 , which implied
F = 1. As
, which implied
F = 1. As
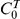 increased, a smaller fraction of cells had
expression levels
increased, a smaller fraction of cells had
expression levels 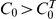 and
F decreased. With small
and
F decreased. With small 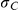 (
(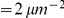 in Fig.
2), because the distribution of CCR5 expression was sharply
peaked at
in Fig.
2), because the distribution of CCR5 expression was sharply
peaked at 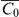 , nearly all cells had
, nearly all cells had
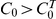 when
when 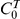 was modestly
smaller than
was modestly
smaller than 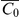 , whereas few
cells had
, whereas few
cells had 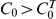 when
when 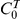 increased
modestly above
increased
modestly above 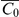 . Consequently,
F exhibited a sharp drop from 1 to 0 as
. Consequently,
F exhibited a sharp drop from 1 to 0 as
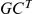 increased. The drop occurred around the value of
increased. The drop occurred around the value of
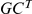 at which
at which 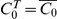 . With larger
. With larger
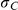 , the wider distribution of CCR5 implied that the
drop in F was gradual (
, the wider distribution of CCR5 implied that the
drop in F was gradual (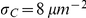 and
and
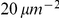 in Fig.
2). Eventually, as
in Fig.
2). Eventually, as 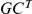 approached
approached
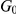 , the expression level of gp120 on effector cells,
, the expression level of gp120 on effector cells,
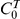 rose sharply (Fig. 2B (inset)) because of the
limitation in the availability of gp120. Correspondingly, F
dropped sharply as
rose sharply (Fig. 2B (inset)) because of the
limitation in the availability of gp120. Correspondingly, F
dropped sharply as 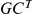 approached
approached
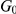 ( =
( = 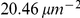 in Fig. 2B).
in Fig. 2B).
Gp120-CCR5 interactions in the presence of a coreceptor antagonist
We next predicted the equilibrium surface densities of the various reacting
species across a single target cell-effector cell pair
(Eq. 5), calculated using Eqs.
(6)–(9), as functions of the concentration of the coreceptor
antagonist, 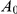 , for different
values of the cooperativity factor,
, for different
values of the cooperativity factor,  (Fig. 3).
(
(Fig. 3).
(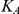 and
and 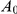 always appear
as their product in the model equations, so that changes in
always appear
as their product in the model equations, so that changes in
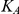 have the same effect as changes in
have the same effect as changes in
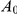 .)
.)  is the ratio
of the binding affinities of the antagonist for gp120-bound CCR5 and unbound
CCR5 (Eq. 5). For fixed
is the ratio
of the binding affinities of the antagonist for gp120-bound CCR5 and unbound
CCR5 (Eq. 5). For fixed
 , as we increased
, as we increased 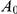 , we found that
the surface density of unbound gp120,
, we found that
the surface density of unbound gp120, 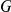 , increased
implying that fewer CCR5 molecules bound to gp120. At the same time, the
surface densities of free CCR5 and gp120-CCR5 complexes,
, increased
implying that fewer CCR5 molecules bound to gp120. At the same time, the
surface densities of free CCR5 and gp120-CCR5 complexes,
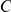 and
and 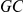 , respectively,
decreased, whereas the surface densities of their antagonist bound
counterparts,
, respectively,
decreased, whereas the surface densities of their antagonist bound
counterparts, 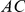 and
and
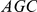 , increased, indicating greater binding of the
antagonist to CCR5. Higher values of
, increased, indicating greater binding of the
antagonist to CCR5. Higher values of  imply greater
affinity of the antagonist for gp120-bound CCR5. Accordingly, for fixed
imply greater
affinity of the antagonist for gp120-bound CCR5. Accordingly, for fixed
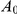 , increasing
, increasing  resulted in
increased
resulted in
increased 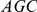 , and decreased surface densities of all the other
species. Thus, increasing
, and decreased surface densities of all the other
species. Thus, increasing 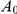 or decreasing
or decreasing
 resulted in fewer CCR5 molecules binding gp120.
resulted in fewer CCR5 molecules binding gp120.
Figure 3. Predictions of the ternary complex model.

Equilibrium surface densities of A) unbound gp120, G, B) unbound CCR5, C, C) gp120-CCR5 complexes, GC, D) CCR5-coreceptor antagonist complexes, AC and E) gp120-CCR5-coreceptor antagonist complexes, AGC, as functions of the concentration of the coreceptor antagonist, A 0, for different values of the cooperativity factor, α, calculated by solving Eqs. (6)–(9).
Cell-cell fusion in the presence of a coreceptor antagonist
In Fig. 4, we present the
fraction of target cells fused, 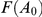 , and the
inhibition of fusion,
, and the
inhibition of fusion, 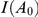 , calculated
using Eqs. (3)–(16), as
functions of
, calculated
using Eqs. (3)–(16), as
functions of 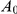 , for different
values of
, for different
values of 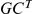 and
and  . For fixed
. For fixed
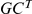 and
and  , we found
that, as expected, increasing
, we found
that, as expected, increasing 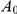 lowered
lowered
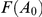 and increased
and increased 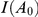 . When
. When
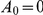 ,
, 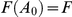 , predicted
above in the absence of the drug (Fig. 2B). Increasing
, predicted
above in the absence of the drug (Fig. 2B). Increasing
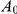 resulted in fewer gp120-CCR5 complexes, which
lowered
resulted in fewer gp120-CCR5 complexes, which
lowered 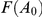 and increased
and increased 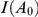 . For very
large values of
. For very
large values of 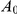 , all CCR5
molecules were bound to the antagonist. Yet, because, according to the
ternary complex model, the antagonist when bound to CCR5 lowers but does not
annihilate the ability of CCR5 to bind gp120, gp120-CCR5 complexes formed
even when all the CCR5 molecules were bound to the antagonist. Further,
because we assumed that antagonist-bound CCR5 may also trigger fusion when
bound to gp120, a fraction of cells, with sufficiently high expression
levels of CCR5, fused even when
, all CCR5
molecules were bound to the antagonist. Yet, because, according to the
ternary complex model, the antagonist when bound to CCR5 lowers but does not
annihilate the ability of CCR5 to bind gp120, gp120-CCR5 complexes formed
even when all the CCR5 molecules were bound to the antagonist. Further,
because we assumed that antagonist-bound CCR5 may also trigger fusion when
bound to gp120, a fraction of cells, with sufficiently high expression
levels of CCR5, fused even when 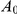 was very
large. For given
was very
large. For given 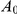 and
and
 , increasing
, increasing 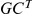 resulted in
lower
resulted in
lower 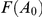 because the threshold expression level
because the threshold expression level
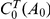 increased with
increased with 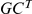 and fewer
cells had expression levels
and fewer
cells had expression levels 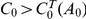 . Accordingly,
. Accordingly,
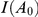 also increased with
also increased with 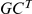 . For fixed
. For fixed
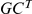 and
and 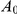 , as
, as
 increased, greater binding of gp120 to
antagonist-bound CCR5 increased
increased, greater binding of gp120 to
antagonist-bound CCR5 increased 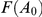 and
consequently decreased
and
consequently decreased 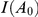 .
.
Figure 4. Model predictions of cell-cell fusion in the presence of a coreceptor antagonist.

The fraction of cells fused,
F(A
0), as a
function of the concentration of the coreceptor antagonist for A)
different values of GCT with
α = 0.01 and B) different values of α
with
GCT = 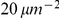 computed using Eqs (1)–(15). C) and D) The corresponding
inhibition of fusion due to the coreceptor antagonist calculated
using Eq. (16). The standard deviation of the CCR5 expression level,
computed using Eqs (1)–(15). C) and D) The corresponding
inhibition of fusion due to the coreceptor antagonist calculated
using Eq. (16). The standard deviation of the CCR5 expression level,
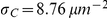 .
.
Our model thus describes the outcome of a cell-cell fusion assay in the presence of a coreceptor antagonist. Below, we present comparisons of our predictions with experiments.
Comparisons with experiments
Estimation of the threshold surface density of gp120-CCR5 complexes
Recently, Heredia et al.
[24]
performed cell-cell fusion assays to examine the antiviral activity of
vicriviroc, a CCR5 antagonist in phase III trials (Gathe et al., Abstract #
45LB 17th Conference on Retroviruses and Opportunistic Infections, 2010).
They performed the assays with and without rapamycin, a drug that lowers the
expression level of CCR5 on cells [6] (Methods). We fit our prediction of
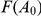 to their data of the percentage of cells fused as a
function of vicriviroc concentration in the absence of rapamycin using three
adjustable parameters,
to their data of the percentage of cells fused as a
function of vicriviroc concentration in the absence of rapamycin using three
adjustable parameters, 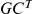 ,
,
 , and
, and 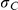 . Our model
provided excellent fits to the data (Fig. 5A). The best-fit parameter
estimates (95% confidence intervals) were
. Our model
provided excellent fits to the data (Fig. 5A). The best-fit parameter
estimates (95% confidence intervals) were
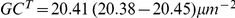 ,
, 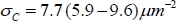 , and
, and
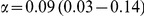 . The best-fit estimate of
. The best-fit estimate of
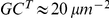 gives the minimum surface density of gp120-CCR5
complexes that must be formed between apposed cells for the cells to fuse.
Further, using the best-fit value of
gives the minimum surface density of gp120-CCR5
complexes that must be formed between apposed cells for the cells to fuse.
Further, using the best-fit value of 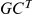 in Eq. (11),
we obtained
in Eq. (11),
we obtained 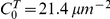 , which yields
the minimum expression level of CCR5 on target cells expressing excess CD4
that can fuse with effector cells with the gp120 expression level
, which yields
the minimum expression level of CCR5 on target cells expressing excess CD4
that can fuse with effector cells with the gp120 expression level
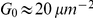 . The latter minimum expression level corresponds to
∼6700 CCR5 molecules/cell (radius∼5 µm).
. The latter minimum expression level corresponds to
∼6700 CCR5 molecules/cell (radius∼5 µm).
Figure 5. Comparisons of model predictions with experiments.
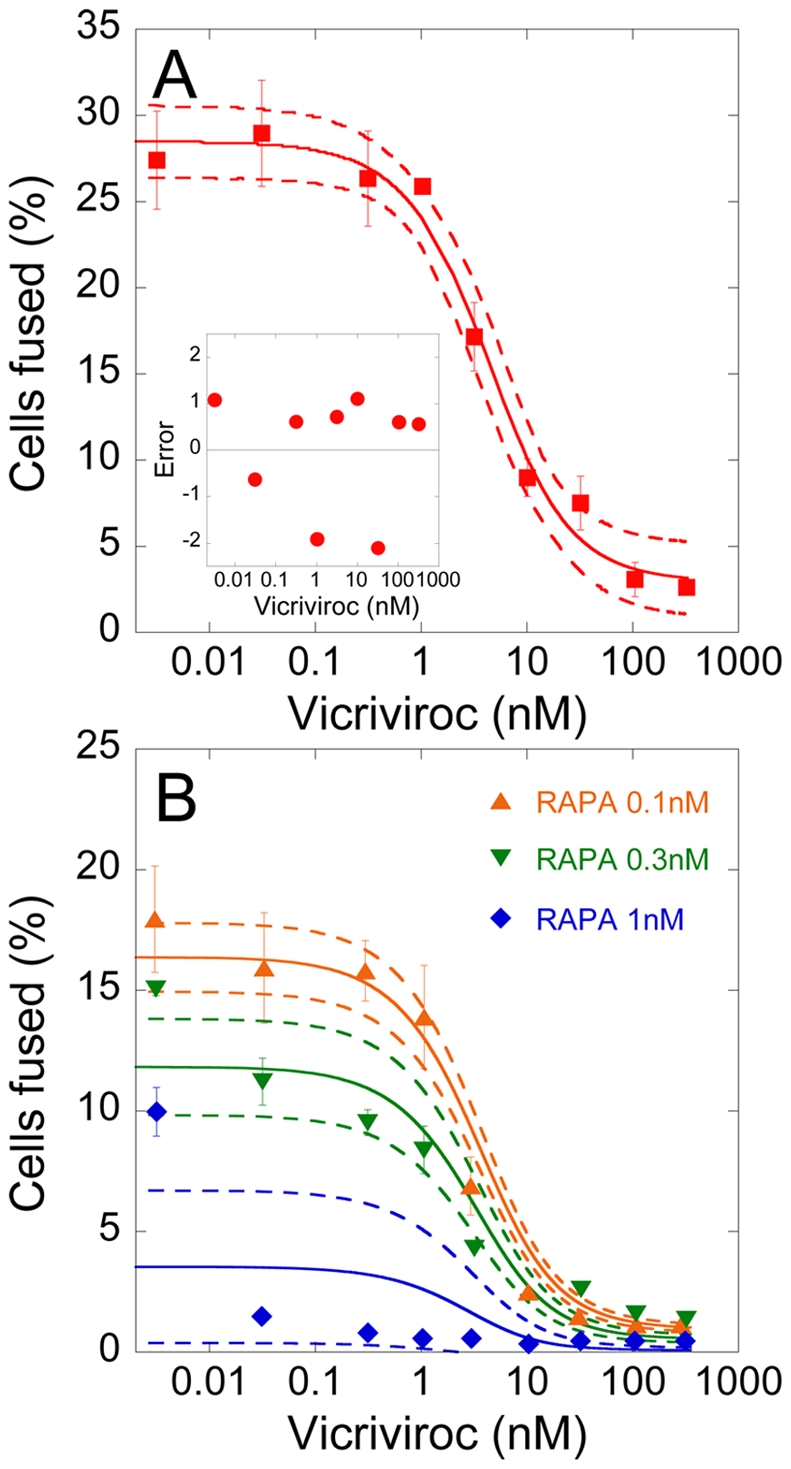
A) Fit of model predictions of
F(A
0) (line) to
published experimental data [24] (symbols) of
the fraction of cells fused as a function of vicriviroc
concentration using 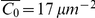 ( = 5318 molecules/cell [24]) and with
( = 5318 molecules/cell [24]) and with
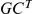 ,
,
 , and
, and
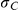 as
adjustable parameters. The other parameters are mentioned in Methods. The dashed lines are
95% confidence limits on the predictions. Inset: Difference
between model predictions and the experimental data; the mean error
is 0.002 (in units of the percentage of cells fused) and is not
significantly different from zero
(P = 0.996 using a two-tailed
t-test). B) Fits of model predictions of
F(A
0) (lines) to
data [24] (symbols) of the fraction of cells fused
as a function of vicriciroc concentration in the presence of
different concentrations of rapamycin (RAPA) using
as
adjustable parameters. The other parameters are mentioned in Methods. The dashed lines are
95% confidence limits on the predictions. Inset: Difference
between model predictions and the experimental data; the mean error
is 0.002 (in units of the percentage of cells fused) and is not
significantly different from zero
(P = 0.996 using a two-tailed
t-test). B) Fits of model predictions of
F(A
0) (lines) to
data [24] (symbols) of the fraction of cells fused
as a function of vicriciroc concentration in the presence of
different concentrations of rapamycin (RAPA) using
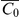 as an
adjustable parameter. The other parameters are the same as in A).
The best-fit parameter estimates are mentioned in the text.
as an
adjustable parameter. The other parameters are the same as in A).
The best-fit parameter estimates are mentioned in the text.
Validation of best-fit parameter estimates
To validate our parameter estimates, we compared our model predictions with
independent data of cell-cell fusion as a function of vicriviroc
concentration in the presence of rapamycin, reported by Heredia et
al.
[24].
Rapamycin lowers the mean expression level of CCR5 on target cells,
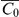 , in a dose-dependent manner [6]. Using the above
best-fit parameter estimates, we therefore fit model predictions to the data
of Heredia et al.
[24]
using
, in a dose-dependent manner [6]. Using the above
best-fit parameter estimates, we therefore fit model predictions to the data
of Heredia et al.
[24]
using 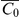 as an adjustable parameter. Our model provided good
fits to the data (Fig.
5B) and yielded estimates of
as an adjustable parameter. Our model provided good
fits to the data (Fig.
5B) and yielded estimates of 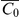 that are in
close agreement with measurements, presenting a successful validation of our
model and parameter estimates. Thus, for rapamycin levels of 0.1, 0.3, and 1
nM, we obtained
that are in
close agreement with measurements, presenting a successful validation of our
model and parameter estimates. Thus, for rapamycin levels of 0.1, 0.3, and 1
nM, we obtained 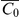 (95%
CI) as 4300 (4100–4400), 3700 (3500–4000), and 2100
(1000–3200) molecules/cell, whereas experimental measurements of the
mean CCR5 expression levels under the same conditions were 3900, 3535, and
2791, respectively [24].
(95%
CI) as 4300 (4100–4400), 3700 (3500–4000), and 2100
(1000–3200) molecules/cell, whereas experimental measurements of the
mean CCR5 expression levels under the same conditions were 3900, 3535, and
2791, respectively [24].
Robustness of best-fit parameter estimates
The above experiments have employed the HIV-1 JRFL Env. Also, rapamycin is
known to have a cytostatic effect on cells [6], the influence of which
on cell-cell fusion remains unknown. To test the robustness of our parameter
estimates, we therefore examined additional experiments on cell-cell fusion
reported by Hu et al. [28] that employed a wide variety of Env clones
derived from transmitted or are early founder viral genomes [29]. The
experiments were performed in the presence of maraviroc, a coreceptor
antagonist approved for clinical use, and the extent of cell-cell fusion
relative to that in the absence of maraviroc was reported (Methods). We compared our predictions of
the relative extent of cell-cell fusion, 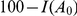 , as a function
of maraviroc concentration with the observations of Hu et al. [28] for 16
different Env clones. To describe this data, we employed the affinity of
maraviroc for CCR5,
KA = 1.15
nM−1
[30], and
let the cooperativity factor,
, as a function
of maraviroc concentration with the observations of Hu et al. [28] for 16
different Env clones. To describe this data, we employed the affinity of
maraviroc for CCR5,
KA = 1.15
nM−1
[30], and
let the cooperativity factor,  , which is
unknown for maraviroc, be an adjustable parameter. We also employed the
threshold surface density of gp120-CCR5 complexes,
, which is
unknown for maraviroc, be an adjustable parameter. We also employed the
threshold surface density of gp120-CCR5 complexes,
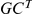 , as an adjustable parameter to examine the
dependence of this threshold on variations in the HIV-1 Env. Our model
provided good fits to all the 16 data sets (Fig. 6). The best-fit values of
, as an adjustable parameter to examine the
dependence of this threshold on variations in the HIV-1 Env. Our model
provided good fits to all the 16 data sets (Fig. 6). The best-fit values of
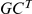 and
and  are presented
in Table 1. We found
that
are presented
in Table 1. We found
that  varied substantially across the different Env clones
(range: 0.002–0.13) indicating varying degrees of sensitivity to
maraviroc, in agreement with the conclusions of Hu et al. [28]. The
varying sensitivity was also evident from the corresponding values of
IC50 (range: approximately 14–1300
nM), which we obtained for each clone as the maraviroc concentration at
which the relative extent of fusion was 50% (Table 1). Interestingly,
varied substantially across the different Env clones
(range: 0.002–0.13) indicating varying degrees of sensitivity to
maraviroc, in agreement with the conclusions of Hu et al. [28]. The
varying sensitivity was also evident from the corresponding values of
IC50 (range: approximately 14–1300
nM), which we obtained for each clone as the maraviroc concentration at
which the relative extent of fusion was 50% (Table 1). Interestingly,
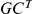 appeared to be nearly constant across the clones
(
appeared to be nearly constant across the clones
(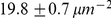 ) and close to the value estimated above
(
) and close to the value estimated above
(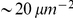 ) indicating the robustness of the latter estimate.
That nearly the same value of
) indicating the robustness of the latter estimate.
That nearly the same value of 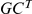 captured
multiple experimental data sets with different HIV-1 Env clones in the
presence of two different coreceptor antagonists and an agent that altered
CCR5 expression levels suggests that our model captures the cell-cell fusion
assays accurately and gives us confidence in our estimate of the threshold
surface density of gp120-CCR5 complexes necessary for cell-cell fusion.
captured
multiple experimental data sets with different HIV-1 Env clones in the
presence of two different coreceptor antagonists and an agent that altered
CCR5 expression levels suggests that our model captures the cell-cell fusion
assays accurately and gives us confidence in our estimate of the threshold
surface density of gp120-CCR5 complexes necessary for cell-cell fusion.
Figure 6. Robustness of model predictions.

Fits of model predictions (lines) of the relative extent of cell-cell
fusion,
100−I(A
0), as a
function of maraviroc concentration to published experimental
observations [28] (symbols) using
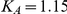 nM−1 and with
nM−1 and with  and
and
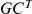 as
adjustable parameters. The other parameters are mentioned in Methods. The different panels
represent data from experiments using different Env clones
(legends). The best fits (solid lines) and the corresponding
95% confidence limits (dashed lines) are shown. The best-fit
parameter estimates are mentioned in Table 1.
as
adjustable parameters. The other parameters are mentioned in Methods. The different panels
represent data from experiments using different Env clones
(legends). The best fits (solid lines) and the corresponding
95% confidence limits (dashed lines) are shown. The best-fit
parameter estimates are mentioned in Table 1.
Table 1. Threshold surface density of gp120-CCR5 complexes for different Env clones.
| Env clone | Threshold complex surface density, GCT (µm−2) | Cooperativity factor, α | IC50 (nM) |
| RHPA.A19.2000 | 19.9 (19.5–20.2) | 0.011 (0.005–0.031) | 79.5 |
| PRB958_06.TB1.4305 | 19.6 (18.6–20.1) | 0.009 (0.002–0.026) | 242.1 |
| PRB926_04.A9.4237 | 20.2 (20.1–20.3) | 0.021 (0.012–0.033) | 29.0 |
| 6244_13.B5.4576 | 20.0 (18.7–20.4) | 0.020 (0.003–0.130) | 143.3 |
| 1054.TC4.1499 | 20.1 (19.3–20.3) | 0.020 (0.005–0.058) | 100.9 |
| 62357_14.D3.4589 | 20.1 (20.0–20.2) | 0.015 (0.009–0.021) | 44.4 |
| 9021_14.B2.4571 | 20.2 (19.3–20.4) | 0.028 (0.005–0.100) | 71.4 |
| 1006_11.C3.1601 | 20.1 (19.7–20.3) | 0.013 (0.005–0.030) | 46.0 |
| 6240_08.TA5.4622 | 19.7 (18.5–20.2) | 0.013 (0.004–0.040) | 798.5 |
| SC05.8C112344 | 20.3 (18.2–20.5) | 0.082 (0.003–0.741) | 1287.0 |
| 700010058.A4.4375 | 19.4 (16.4–20.2) | 0.005 (0.001–0.030) | 201.1 |
| REJO.D12.1972 | 20.4 (20.2–20.5) | 0.128 (0.032–0.473) | 17.6 |
| PRB931_06.TC3.4930 | 18.3 (17.4–19.0) | 0.002 (0.001–0.004) | 469.8 |
| SC45.4B5.2631 | 17.8 (16.7–18.5) | 0.002 (0.001–0.003) | 699.3 |
| 62615_03.P4.3964 | 20.3 (19.5–20.4) | 0.042 (0.007–0.200) | 43.2 |
| 700010040.C9.4520 | 20.4 (20.3–20.4) | 0.061 (0.038–0.096) | 13.9 |
| Mean | 19.8 | 0.030 | 267.9 |
Estimates of the threshold surface density of gp120-CCR5 complexes, GCT, and the cooperativity factor, α, for 16 different transmitted or early founder viruses determined from fits of model predictions with experimental data [28] of cell-cell fusion in the presence of maraviroc (Fig. 6). 95% confidence intervals for the best-fit parameter estimates are shown in brackets. The corresponding IC50 values of maraviroc are also listed.
Discussion
The role of CCR5 in mediating HIV-1 entry has important implications for HIV-1
transmission and disease progression to AIDS as well as for strategies of
intervention [1]–[3]. Yet, the threshold surface density of CCR5 molecules that
must interact with gp120 to facilitate HIV-1 entry remains poorly estimated. Here,
we constructed a mathematical model to analyse data from cell-cell fusion assays and
estimated the threshold surface density of gp120-CCR5 complexes that enables HIV-1
Env-mediated cell-cell fusion. We found the threshold surface density of gp120-CCR5
complexes to be 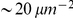 . The corresponding minimum expression level of CCR5 on
target cells that allows cell-cell fusion given the gp120 expression level on
effector cells employed in our analysis and when CD4 is not limiting is
. The corresponding minimum expression level of CCR5 on
target cells that allows cell-cell fusion given the gp120 expression level on
effector cells employed in our analysis and when CD4 is not limiting is
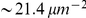 , equivalent to ∼6700 molecules/cell (radius ∼5
µm). To validate our estimate, we analysed data from
independent cell-cell fusion experiments performed in the presence of vicriviroc and
maraviroc, both CCR5 antagonists, and rapamycin, a drug that down-regulated CCR5
expression, as well as with sixteen different Env clones derived from transmitted or
early founder viruses. Our model provided good fits to the data and yielded an
estimate of the threshold surface density of gp120-CCR5 complexes that remained
nearly constant at
, equivalent to ∼6700 molecules/cell (radius ∼5
µm). To validate our estimate, we analysed data from
independent cell-cell fusion experiments performed in the presence of vicriviroc and
maraviroc, both CCR5 antagonists, and rapamycin, a drug that down-regulated CCR5
expression, as well as with sixteen different Env clones derived from transmitted or
early founder viruses. Our model provided good fits to the data and yielded an
estimate of the threshold surface density of gp120-CCR5 complexes that remained
nearly constant at 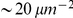 across these
experiments, indicating the robustness of our estimate.
across these
experiments, indicating the robustness of our estimate.
Our estimate of the threshold surface density of gp120-CCR5 complexes necessary for
HIV-1 entry may facilitate optimal utilization of coreceptor antagonists for
preventive and therapeutic intervention. For instance, the estimate suggests that a
potent coreceptor antagonist must reduce the surface density of gp120-CCR5 complexes
to below 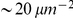 . CCR5 expression levels vary substantially across
individuals, with mean levels in the range ∼1000 to ∼10000 molecules/cell
[31]. The
variations may be due at least in part to variations in the CCL3L1 gene copy number,
which was recently observed to be correlated with the susceptibility of individuals
to HIV-1 infection [32]. Individuals with larger mean CCR5 expression levels
would require greater drug exposure to achieve the same level of inhibition [33]. Our model may
be applied to estimate the necessary drug exposure and may thus serve to personalize
the usage of coreceptor antagonists based on the CCR5 expression level and/or the
CCL3L1 gene dose in patients. Maraviroc was recently found to prevent transmission
in rhesus macaques in a dose dependent manner when employed as a vaginal microbicide
(Veazey et al., Abstract # 84LB 17th Conference on Retroviruses and Opportunistic
Infections, 2010). CCR5 expression levels on target CD4+ T cells in
mucosal regions may be higher than in peripheral blood [34]. Consequently, greater exposure
to a coreceptor antagonist in mucosal regions may be necessary to prevent
transmission than is necessary in plasma during treatment. Our model may again serve
to quantify this greater exposure. Similarly, for vaccine strategies that aim to
reduce the availability of target CD4+CCR5+ cells at
sites of transmission [1], [2], our study suggests that CCR5 expression must be lowered
to a level that restricts the formation of gp120-CCR5 complexes to below
. CCR5 expression levels vary substantially across
individuals, with mean levels in the range ∼1000 to ∼10000 molecules/cell
[31]. The
variations may be due at least in part to variations in the CCL3L1 gene copy number,
which was recently observed to be correlated with the susceptibility of individuals
to HIV-1 infection [32]. Individuals with larger mean CCR5 expression levels
would require greater drug exposure to achieve the same level of inhibition [33]. Our model may
be applied to estimate the necessary drug exposure and may thus serve to personalize
the usage of coreceptor antagonists based on the CCR5 expression level and/or the
CCL3L1 gene dose in patients. Maraviroc was recently found to prevent transmission
in rhesus macaques in a dose dependent manner when employed as a vaginal microbicide
(Veazey et al., Abstract # 84LB 17th Conference on Retroviruses and Opportunistic
Infections, 2010). CCR5 expression levels on target CD4+ T cells in
mucosal regions may be higher than in peripheral blood [34]. Consequently, greater exposure
to a coreceptor antagonist in mucosal regions may be necessary to prevent
transmission than is necessary in plasma during treatment. Our model may again serve
to quantify this greater exposure. Similarly, for vaccine strategies that aim to
reduce the availability of target CD4+CCR5+ cells at
sites of transmission [1], [2], our study suggests that CCR5 expression must be lowered
to a level that restricts the formation of gp120-CCR5 complexes to below
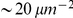 in order to prevent infection of target cells.
in order to prevent infection of target cells.
Our study may also inform the substantial ongoing efforts to elucidate the origins of the differences between SIV infection of natural and non-natural hosts (reviewed in [1]–[3]). An intriguing hypothesis explaining the non-progressive infection of natural hosts despite high plasma viral loads hinges on the reduced susceptibility of the central memory cell compartment in these animals due to low CCR5 expression [2]. In contrast, in non-natural hosts, the central memory compartment may be depleted more rapidly because of higher CCR5 expression levels. In both natural and non-natural hosts, the activated and effector memory cells have high CCR5 levels and are responsible for high plasma viral loads. The threshold expression level of CCR5 that renders target cells susceptible to SIV infection remains unknown. Our model may be applied to analyse data from SIV-Env-mediated cell-cell fusion assays and estimate the corresponding threshold CCR5 expression level for SIV infection, which may serve to elucidate the differences between natural and non-natural hosts of SIV. Indeed, more generally, our model provides a framework for analysing cell-cell fusion assays, widely employed to investigate HIV entry and related intervention strategies.
Recent studies have argued that the mechanism of viral entry into cells may be distinct from cell-cell fusion: while cell-cell fusion involves membrane fusion at the cell surface, HIV-1 entry appears to involve receptor and coreceptor mediated endocytosis [35], [36]. Following endocytosis, however, the viral membrane fuses with the endosomal membrane facilitating the release of viral contents into the cytoplasm leading to productive infection [35], [36]. Thus, if the membrane fusion processes are similar in cell-cell fusion and viral-endosomal fusion, which remains to be ascertained, the same threshold surface density of gp120-CCR5 complexes may underlie both cell-cell fusion and viral infection of target cells.
We recognize approximations in our model that hold for cell-cell fusion but may not apply to viral entry in vivo. First, our model describes the protein interactions that precede viral entry using a continuum, mass action-based approach. Such a continuum approximation is expected to be accurate for cell-cell fusion, where the number of protein molecules per cell is large (>103). The advantage of the continuum approach is the simplicity of the resulting model equations and their facile application to data analysis. With virus-cell interaction, however, because virions express far fewer gp120 molecules (14±7 Env trimers per virion [37]), a stochastic description may be more appropriate (see, e.g., [38]). Second, our model considers the distribution of CCR5 expression levels across cells, but not of CD4 and gp120 levels. This approximation is reasonable for analysis of cell-cell fusion assays in the presence of coreceptor antagonists, which effectively lower the availability of CCR5 for binding gp120 and render CD4 and gp120 not limiting. With viral entry, however, a distribution of Env trimers is observed [37] and may have to be accounted for to accurately describe the susceptibility of target cells in vivo [20]. Further, where CD4 is down-modulated, as in African green monkeys [39], the assumption that all gp120 molecules are bound to CD4 and therefore accessible to CCR5 may not hold and the complete network of Env-CD4-CCR5 interactions (Text S1) may have to be considered. Indeed, cells with low CD4 expression have been suggested to require high CCR5 for infection [40]. Nonetheless, because the above approximations are expected to hold for the cell-cell fusion assays we analysed, they may not confound our estimate of the threshold surface density of gp120-CCR5 complexes necessary for viral entry.
The spatial distribution of CCR5 across the surface of a target cell and its role on viral entry remains to be established. While one study suggests that CCR5 molecules are localized within lipid rafts [41], another finds CCR5 molecules in non-raft regions [42]. Several studies suggest that CCR5 is colocalized and/or associated with CD4 [41], [43], [44]. Although CD4 is preferentially localized within rafts, such localization may not be essential for viral entry [45]. At the same time, membrane cholesterol depletion, which is known to affect raft formation and may also influence CCR5 mobility [46], inhibits viral entry when the receptors are not expressed in excess [41], [42], [47]. Current studies thus leave unclear the spatial distribution of CCR5 and its role in HIV-1 entry. Here, as an approximation, therefore, we have assumed CCR5 to be randomly distributed on the target cell surface. Further, effector cells have been suggested to recruit CCR5 to regions of cell-cell contact [41], the mechanisms underlying which remain unknown. Only recently have studies begun to unravel the local organization of protein complexes in the virus-cell contact region, which may be important for viral entry [38], [48], [49]. A quantitative assessment of the impact of the latter phenomena on estimates of the threshold surface density of gp120-CCR5 complexes necessary for viral entry awaits further studies that would establish the spatial distribution of CCR5 on target cells and of the mechanisms that underlie receptor migration and recruitment following virus-cell contact.
Finally, we note that our model assumed that each gp120 monomer in an Env trimer is independently accessible to CCR5. In contrast, steric constraints may result in increasingly hindered successive binding of CCR5 to the second and third gp120 monomers of an Env trimer. Conversely, cooperative binding may render successive binding easier [14]. Besides, recent single-molecule studies suggest that a complex energy landscape underlies gp120-CD4-CCR5 interactions [50], [51], the implications of which for viral entry remain to be fully understood. Further, heterogeneity in the CCR5 molecules, arising, for instance, due to post-translational modifications [52], may introduce additional variations in the affinity of CCR5 for gp120. Nonetheless, by assuming that all gp120 monomers are equally accessible to CCR5, our model ignores the association of gp120 into trimers and precludes identification of the stoichiometry of the Env-CD4-CCR5 complexes that renders the complexes fusion competent. At the threshold surface density, gp120-CCR5 complexes would be distributed such that some Env trimers are bound to 3 CCR5 molecules, some to 2, some to 1 and some to none (Fig. S1). It is possible that only Env trimers bound to 2 or more CCR5 molecules, for instance, mediate entry. While our model can be extended to predict this latter distribution (Text S1), currently available data does not allow establishment of the stoichiometry of CCR5 binding to Env that enables entry [20].
Methods
Data
We have analysed experimental data of HIV-1-Env mediated cell-cell fusion published recently by Heredia et al. [24] and Hu et al. [28]. In the cell-cell fusion assays reported by Heredia et al. [24], lymphocytes expressing CD4 and CCR5 (target cells) were treated with different concentrations of rapamycin and coincubated with 293T cells transfected with HIV-1 JRFL Env (effector cells) for 2.5 h at 37°C in the presence of known concentrations of vicriviroc. The two cell types were stained with different fluorescent dyes. Flow cytometry was used to detect cell-cell fusion: cells that were positive for both dyes indicated a fusion event. The fraction of target cells that eventually fused was reported as a function of vicriviroc concentration for different levels of exposure to rapamycin.
In the experiments performed by Hu et al. [28], QT6 cells transfected with CD4 and CCR5 (target cells) were exposed to QT6 cells transfected with HIV-1 Env expression constructs (effector cells) in the presence of different concentrations of maraviroc. The target cells were also transfected with a luciferase construct under the transcriptional control of T7 promoter. Luciferase activity was measured ∼8 h following co-incubation and reported as a percentage of the activity in the absence of maraviroc, thus representing the extent of inhibition of cell-cell fusion due to maraviroc. The experiments were performed using different Env clones that were derived from transmitted or early founder viral strains [29]. Hu et al. reported cell-cell fusion data for 18 different clones of which two were found to be highly resistant to maraviroc [28]. Here, we analysed data for the remaining 16 clones.
Mathematical model
Single target cell-effector cell pair
We considered first the interactions between proteins across a single target cell-effector cell pair in the absence of a coreceptor antagonist (Fig. 1). Typically, CD4 molecules are in excess and rapidly bind all available gp120 molecules in the contact region on an apposed effector cell, as shown by our detailed reaction kinetics calculations (Text S1) and by independent simulations of virus-cell interactions [49]. We therefore considered the interactions between gp120 and CCR5:
| (1) |
Here, G, C, and GC are the surface densities (numbers per unit area) of gp120, CCR5, and gp120-CCR5 complexes, respectively. Eq. (1) assumes that each gp120 molecule in an Env trimer is bound to CD4 and is independently accessible to CCR5. The reaction in Eq. (1) attains equilibrium rapidly compared to fusion; equilibrium is attained within seconds (Fig. S1), whereas the lag time for fusion is in minutes [22]. At equilibrium, the surface densities of the reacting species obey
| (2) |
where 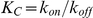 is the
equilibrium association constant of CCR5 with gp120. If the effector cell
expresses
is the
equilibrium association constant of CCR5 with gp120. If the effector cell
expresses 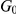 gp120 molecules per unit area
(i.e.,
gp120 molecules per unit area
(i.e., 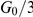 Env trimers
per unit area) and the target cell
Env trimers
per unit area) and the target cell 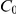 CCR5 molecules
per unit area, then species balance implies
CCR5 molecules
per unit area, then species balance implies
| (3) |
where changes in the protein surface densities due to protein diffusion in and out of the cell-cell contact region are assumed negligible (see Discussion). Combining Eqs. (2) and (3) resulted in a quadratic equation in GC, solving which we obtained
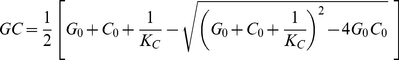 |
(4) |
Eq. (4) yields the surface density of CCR5 bound to gp120 between a single target cell-effector cell pair.
Threshold CCR5 binding for cell-cell fusion
We defined GCT as the minimum surface density of gp120-CCR5 complexes that must be formed between a target cell-effector cell pair for the cells to fuse. Thus, the cell pair fuses if GC≥GCT.
Single target cell-effector cell pair in the presence of a coreceptor antagonist
We next considered a single target cell-effector cell pair in the presence of
a CCR5 antagonist, 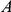 , at
concentration
, at
concentration 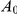 . We employed
the standard ternary complex model to describe the resulting allosteric
interactions [27]:
. We employed
the standard ternary complex model to describe the resulting allosteric
interactions [27]:
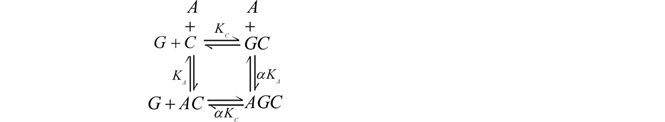 |
(5) |
Here, gp120 can bind to a complex of CCR5 and A, denoted
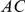 , with an altered binding affinity
, with an altered binding affinity
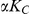 , where
, where  is the
cooperativity factor. Similarly,
is the
cooperativity factor. Similarly, 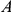 can bind to
can bind to
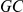 with affinity
with affinity 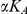 , where
, where
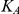 is the affinity of the antagonist for CCR5. At
equilibrium, the ternary complex model yields
is the affinity of the antagonist for CCR5. At
equilibrium, the ternary complex model yields
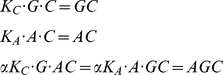 |
(6) |
along with the species balance equations
| (7) |
We assumed that the concentration of 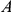 does not
decrease substantially below
does not
decrease substantially below 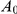 . Combining
Eqs. (6) and (7) yielded a quadratic equation in
. Combining
Eqs. (6) and (7) yielded a quadratic equation in
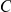 ,
, 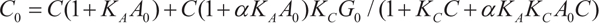 , which we
solved to obtain
, which we
solved to obtain
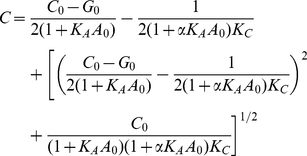 |
(8) |
Eqs. (6) and (7) also imply
| (9) |
using which in Eq. (6) along with Eq. (8), we obtained the surface densities of all the reacting species in the ternary complex model. In particular, the surface density of CCR5 bound to gp120,
| (10) |
For fusion, this latter surface
density must be larger than the threshold surface density,
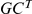 .
.
Cell-cell fusion assay
In a cell-cell fusion assay, target cells with different expression levels of
CCR5 form different surface densities of gp120-CCR5 complexes at
equilibrium. We defined 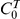 as that
expression level of CCR5 that would result in the formation of the threshold
surface density,
as that
expression level of CCR5 that would result in the formation of the threshold
surface density, 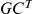 , of complexes.
In the absence of a coreceptor antagonist, from Eqs. (2) and (3), it follows
that
, of complexes.
In the absence of a coreceptor antagonist, from Eqs. (2) and (3), it follows
that
| (11) |
Because
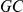 increases with
increases with 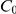 (Eq. (4)), all
cells with
(Eq. (4)), all
cells with 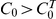 will fuse with
effector cells. On the other hand, all cells with
will fuse with
effector cells. On the other hand, all cells with
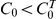 will be unable to fuse. We assumed next that the
expression level of CCR5 on cells follows a truncated normal distribution
with mean
will be unable to fuse. We assumed next that the
expression level of CCR5 on cells follows a truncated normal distribution
with mean 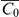 and standard deviation
and standard deviation
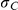 ,
,
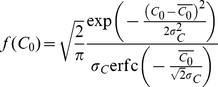 |
(12) |
where
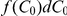 represents the fraction of cells with the CCR5
expression level within a small range
represents the fraction of cells with the CCR5
expression level within a small range 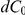 near
near
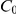 , and
, and 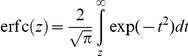 is the
complementary error function. Note that
is the
complementary error function. Note that 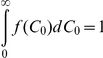 . The fraction
of target cells that fuses,
. The fraction
of target cells that fuses, 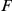 , is then the
fraction of cells with
, is then the
fraction of cells with 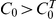 ,
i.e.,
,
i.e., 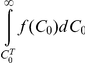 , which upon
substituting for
, which upon
substituting for 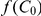 from Eq. (12)
yielded
from Eq. (12)
yielded
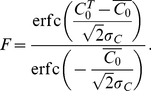 |
(13) |
Equation (13) predicts the fraction of cells fused in a cell-cell fusion assay in the absence of a coreceptor antagonist.
Cell-cell fusion assay in the presence of a coreceptor antagonist
In the presence of the coreceptor antagonist, the expression level of CCR5
that results in the formation of complexes at the surface density
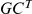 would be higher than
would be higher than 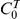 because the
coreceptor antagonist inhibits the binding of CCR5 with gp120. We defined
because the
coreceptor antagonist inhibits the binding of CCR5 with gp120. We defined
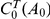 as that expression level of CCR5 that would result
in the formation of the threshold surface density,
as that expression level of CCR5 that would result
in the formation of the threshold surface density,
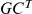 , of complexes in the presence of the coreceptor
antagonist at concentration
, of complexes in the presence of the coreceptor
antagonist at concentration 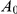 . We obtained
. We obtained
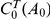 as that value of
as that value of 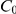 that
satisfies
that
satisfies
| (14) |
The fraction of cells that
fuses, 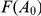 , is then the fraction of cells with
, is then the fraction of cells with
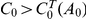 , i.e.,
, i.e.,
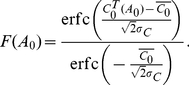 |
(15) |
Equation (15) predicts the fraction
of target cells fused in a cell-cell fusion assay in the presence of a
coreceptor antagonist. Note that Eq. (15) reduces to Eq. (13) when
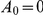 . The percentage of inhibition of cell-cell fusion
due to the coreceptor antagonist is then
. The percentage of inhibition of cell-cell fusion
due to the coreceptor antagonist is then
| (16) |
Parameter estimates
We performed model calculations using parameter estimates representative of the
cell-cell fusion assays we considered [24] unless mentioned
otherwise. Target cells employed in the assays were lymphocytes from donors,
with CCR5 expression levels in the range 2000–7000 molecules/cell [24]. We
therefore assumed the mean CCR5 expression level on cells,
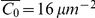 , corresponding to ∼5000 molecules/target cell
(radius∼5 µm). We set the expression level of gp120
on the effector cells,
, corresponding to ∼5000 molecules/target cell
(radius∼5 µm). We set the expression level of gp120
on the effector cells, 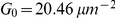 , equivalent to
∼10000 Env trimers/effector cell (radius∼10 µm),
following observations of ∼2 ng of gp120 on
∼106 effector cells [22]. The equilibrium
dissociation constant of gp120 binding to CCR5 is ∼4 nM [53]. The
corresponding affinity when both gp120 and CCR5 are restricted to membranes,
following the analysis of Bell [54] and assuming an encounter radius of 0.75
nm, is
, equivalent to
∼10000 Env trimers/effector cell (radius∼10 µm),
following observations of ∼2 ng of gp120 on
∼106 effector cells [22]. The equilibrium
dissociation constant of gp120 binding to CCR5 is ∼4 nM [53]. The
corresponding affinity when both gp120 and CCR5 are restricted to membranes,
following the analysis of Bell [54] and assuming an encounter radius of 0.75
nm, is 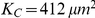 (Text S1).
The affinity of vicriviroc for CCR5,
(Text S1).
The affinity of vicriviroc for CCR5, 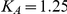 nM−1
[55]. The
cooperativity factor,
nM−1
[55]. The
cooperativity factor,  , and the standard
deviation of the CCR5 expression level on cells,
, and the standard
deviation of the CCR5 expression level on cells, 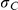 , are not known and
we estimated them along with the threshold surface density of complexes,
, are not known and
we estimated them along with the threshold surface density of complexes,
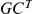 , by fitting model predictions to data. The parameters
employed are summarized in Table
2.
, by fitting model predictions to data. The parameters
employed are summarized in Table
2.
Table 2. Summary of model parameters and their values employed.
| Parameter | Description | Valuea | Source |
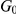
|
Surface density of gp120 on effector cells |
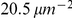
|
[22], see text |
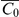
|
Mean surface density of CCR5 on target cells |
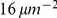
|
[24], see text |
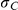
|
Standard deviation of the surface density of CCR5 across cells |
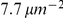
|
Best-fit (Fig. 5A) |
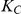
|
Equilibrium association constant of gp120 with CCR5 |
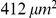
|
[53], see text |
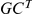
|
Threshold surface density of gp120-CCR5 complexes |
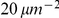
|
Best-fits (Figs. 5 and 6, Table 1) |

|
Cooperativity factor in the ternary complex model Eq.(5) | 0.03 | Best-fits (Fig. 6, Table 1) |
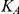
|
Equilibrium association constant of a coreceptor antagonist with CCR5 |
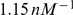
|
[30] |
Typical values; variations are indicated in the text and in figure legends.
Model calculations and comparisons with experiments
We solved the above equations and fit model predictions to data using a computer program written in MATLAB. We employed the inbuilt routine NLINFIT, which uses the Levenberg-Margquardt algorithm for nonlinear least squares, for fitting model predictions to data and for obtaining 95% confidence intervals. For some of the data sets of transmitted/founder Env-mediated cell-cell fusion in the presence of maraviroc, NLINFIT yielded confidence intervals that included negative parameter values. We therefore determined 95% confidence intervals on the best-fit parameter values for the transmitted/founder Env-mediated cell-cell fusion data sets by performing 200 bootstrap replicates each, again in MATLAB.
Supporting Information
Time-evolution of the surface densities of the species in the reaction network. Surface densities of the species in the network (Eq. (1) in Text S1), namely, (A) unbound Env and Env molecules bound to single CD4 molecules, (B) Env bound to 2 CD4 molecules, (C) Env bound to 3 CD4 molecules, and (D) unbound CD4 and CCR5, obtained by solving Eq. (2) in Text S1 (solid lines), and of the equilibrium surface densities of CCR5 and the gp120-CCR5 complexes in the simplified network (Eq. (5) in Text S1) (dashed lines), obtained by solving Eqs. (6)–(9) in Text S1. Parameter values and initial conditions are mentioned in Text S1. All surface densities are normalized with the initial Env surface density.
(TIF)
Detailed kinetics of Env-CD4-CCR5 binding. A detailed description of the kinetics of the Env-CD4-CCR5 binding across a target cell-effector cell pair is presented.
(DOC)
Acknowledgments
We thank Alonso Heredia for providing details of their experimental observations and Pradeep Nagaraja and Prithwiraj M. Mukherjee for help with parameter estimation.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work was supported in part by the National Institutes of Health grant AI065334. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Sodora DL, Allan JS, Apetrei C, Brenchley JM, Douek DC, et al. Toward an AIDS vaccine: lessons from natural simian immunodeficiency virus infections of African nonhuman primate hosts. Nat Med. 2009;15:861–865. doi: 10.1038/nm.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brenchley JM, Silvestri G, Douek DC. Nonprogressive and progressive primate immunodeficiency lentivirus infections. Immunity. 2010;32:737–742. doi: 10.1016/j.immuni.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pandrea I, Apetrei C. Where the wild things are: Pathogenesis of SIV infection in African nonhuman primate hosts. Curr HIV/AIDS Rep. 2010;7:28–36. doi: 10.1007/s11904-009-0034-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pandrea I, Apetrei C, Gordon S, Barbercheck J, Dufour J, et al. Paucity of CD4+CCR5+ T cells is a typical feature of natural SIV hosts. Blood. 2007;109:1069–1076. doi: 10.1182/blood-2006-05-024364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin Y-L, Mettling C, Portales P, Reynes J, Clot J, et al. Cell surface CCR5 density determines the postentry efficiency of R5 HIV-1 infection. Proc Natl Acad Sci U S A. 2002;99:15590–15595. doi: 10.1073/pnas.242134499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heredia A, Amoroso A, Davis C, Le N, Reardon E, et al. Rapamycin causes down-regulation of CCR5 and accumulation of anti-HIV beta-chemokines: An approach to suppress R5 strains of HIV-1. Proc Natl Acad Sci U S A. 2003;100:10411–10416. doi: 10.1073/pnas.1834278100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pandrea I, Onanga R, Souquiere S, Mouinga-Ondeme A, Bourry O, et al. Paucity of CD4+ CCR5+ T cells may prevent transmission of simian immunodeficiency virus in natural nonhuman primate hosts by breast-feeding. J Virol. 2008;82:5501–5509. doi: 10.1128/JVI.02555-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, et al. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell. 1996;86:367–377. doi: 10.1016/s0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- 9.Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12:1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 10.Estes JD, Harris LD, Klatt NR, Tabb B, Pittaluga S, et al. Damaged intestinal epithelial integrity linked to microbial translocation in pathogenic simian immunodeficiency virus infections. PLoS Pathog. 2010;6:e1001052. doi: 10.1371/journal.ppat.1001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu J, Bartesaghi A, Borgnia MJ, Sapiro G, Subramaniam S. Molecular architecture of native HIV-1 gp120 trimers. Nature. 2008;455:109–113. doi: 10.1038/nature07159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gallo SA, Finnegan CM, Viard M, Raviv Y, Dimitrov A, et al. The HIV Env-mediated fusion reaction. BBA Biomembranes. 2003;1614:36–50. doi: 10.1016/s0005-2736(03)00161-5. [DOI] [PubMed] [Google Scholar]
- 13.Melikyan GB, Markosyan RM, Hemmati H, Delmedico MK, Lambert DM, et al. Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. The Journal of Cell Biology. 2000;151:413–424. doi: 10.1083/jcb.151.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Platt EJ, Durnin JP, Shinde U, Kabat D. An allosteric rheostat in HIV-1 gp120 reduces CCR5 stoichiometry required for membrane fusion and overcomes diverse entry limitations. J Mol Biol. 2007;374:64–79. doi: 10.1016/j.jmb.2007.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Layne SP, Merges MJ, Dembo M, Spouge JL, Nara PL. HIV requires multiple gp120 molecules for CD4-mediated infection. Nature. 1990;346:277–279. doi: 10.1038/346277a0. [DOI] [PubMed] [Google Scholar]
- 16.Kuhmann SE, Platt EJ, Kozak SL, Kabat D. Cooperation of multiple CCR5 coreceptors is required for infections by human immunodeficiency virus type 1. J Virol. 2000;74:7005–7015. doi: 10.1128/jvi.74.15.7005-7015.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang X, Kurteva S, Ren X, Lee S, Sodroski J. Stoichiometry of envelope glycoprotein trimers in the entry of human immunodeficiency virus type 1. J Virol. 2005;79:12132–12147. doi: 10.1128/JVI.79.19.12132-12147.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang X, Kurteva S, Ren X, Lee S, Sodroski J. Subunit stoichiometry of human immunodeficiency virus type 1 envelope glycoprotein trimers during virus entry into host cells. J Virol. 2006;80:4388–4395. doi: 10.1128/JVI.80.9.4388-4395.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klasse PJ. Modeling how many envelope glycoprotein trimers per virion participate in human immunodeficiency virus infectivity and its neutralization by antibody. Virology. 2007;369:245–262. doi: 10.1016/j.virol.2007.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Magnus C, Rusert P, Bonhoeffer S, Trkola A, Regoes RR. Estimating the stoichiometry of human immunodeficiency virus entry. J Virol. 2009;83:1523–1531. doi: 10.1128/JVI.01764-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Magnus C, Regoes RR. Estimating the stoichiometry of HIV neutralization. PLoS Comput Biol. 2010;6:e1000713. doi: 10.1371/journal.pcbi.1000713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lineberger JE, Danzeisen R, Hazuda DJ, Simon AJ, Miller MD. Altering expression levels of human immunodeficiency virus type 1 gp120-gp41 affects efficiency but not kinetics of cell-cell fusion. J Virol. 2002;76:3522–3533. doi: 10.1128/JVI.76.7.3522-3533.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dorr P, Westby M, Dobbs S, Griffin P, Irvine B, et al. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob Agents Chemother. 2005;49:4721–4732. doi: 10.1128/AAC.49.11.4721-4732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heredia A, Latinovic O, Gallo RC, Melikyan G, Reitz M, et al. Reduction of CCR5 with low-dose rapamycin enhances the antiviral activity of vicriviroc against both sensitive and drug-resistant HIV-1. Proc Natl Acad Sci U S A. 2008;105:20476–20481. doi: 10.1073/pnas.0810843106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Markosyan RM, Leung MY, Cohen FS. The six-helix bundle of human immunodeficiency virus env controls pore formation and enlargement and is initiated at residues proximal to the hairpin turn. J Virol. 2009;83:10048–10057. doi: 10.1128/JVI.00316-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kondru R, Zhang J, Ji C, Mirzadegan T, Rotstein D, et al. Molecular interactions of CCR5 with major classes of small-molecule anti-HIV CCR5 antagonists. Mol Pharm. 2008;73:789–800. doi: 10.1124/mol.107.042101. [DOI] [PubMed] [Google Scholar]
- 27.Christopoulos A. Allosteric binding sites on cell-surface receptors: novel targets for drug discovery. Nat Rev Drug Discov. 2002;1:198–210. doi: 10.1038/nrd746. [DOI] [PubMed] [Google Scholar]
- 28.Hu Q, Huang X, Shattock RJ. C-C chemokine receptor type 5 (CCR5) utilization of transmitted and early founder human immunodeficiency virus type 1 envelopes and sensitivity to small-molecule CCR5 inhibitors. J Gen Virol. 2010;91:2965–2973. doi: 10.1099/vir.0.025270-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, Pham KT, et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Natl Acad Sci U S A. 2008;105:7552–7557. doi: 10.1073/pnas.0802203105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Napier C, Sale H, Mosley M, Rickett G, Dorr P, et al. Molecular cloning and radioligand binding characterization of the chemokine receptor CCR5 from rhesus macaque and human. Biochem Pharm. 2005;71:163–172. doi: 10.1016/j.bcp.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 31.Hladik F, Liu H, Speelmon E, Livingston-Rosanoff D, Wilson S, et al. Combined effect of CCR5-delta32 heterozygosity and the CCR5 promoter polymorphism -2459 A/G on CCR5 expression and resistance to human immunodeficiency virus type 1 transmission. J Virol. 2005;79:11677–11684. doi: 10.1128/JVI.79.18.11677-11684.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gonzalez E, Kulkarni H, Bolivar H, Mangano A, Sanchez R, et al. The Influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science. 2005;307:1434–1440. doi: 10.1126/science.1101160. [DOI] [PubMed] [Google Scholar]
- 33.Ketas TJ, Kuhmann SE, Palmer A, Zurita J, He W, et al. Cell surface expression of CCR5 and other host factors influence the inhibition of HIV-1 infection of human lymphocytes by CCR5 ligands. Virology. 2007;364:281–290. doi: 10.1016/j.virol.2007.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hladik F, Sakchalathorn P, Ballweber L, Lentz G, Fialkow M, et al. Initial events in establishing vaginal entry and infection by human immunodeficiency virus type-1. Immunity. 2007;26:257–270. doi: 10.1016/j.immuni.2007.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miyauchi K, Kim Y, Latinovic O, Morozov V, Melikyan GB. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell. 2009;137:433–444. doi: 10.1016/j.cell.2009.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miyauchi K, Kozlov MM, Melikyan GB. Early steps of HIV-1 fusion define the sensitivity to inhibitory peptides that block 6-helix bundle formation. PLoS Pathog. 2009;5:e1000585. doi: 10.1371/journal.ppat.1000585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu P, Liu J, Bess J, Jr, Chertova E, Lifson JD, et al. Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature. 2006;441:847–852. doi: 10.1038/nature04817. [DOI] [PubMed] [Google Scholar]
- 38.Dobrowsky TM, Daniels BR, Siliciano RF, Sun SX, Wirtz D. Organization of cellular receptors into a nanoscale junction during HIV-1 adhesion. PLoS Comput Biol. 2010;6:e1000855. doi: 10.1371/journal.pcbi.1000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beaumier CM, Harris LD, Goldstein S, Klatt NR, Whitted S, et al. CD4 downregulation by memory CD4+ T cells in vivo renders African green monkeys resistant to progressive SIVagm infection. Nat Med. 2009;15:879–885. doi: 10.1038/nm.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, Kabat D. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J Virol. 1998;72:2855–2864. doi: 10.1128/jvi.72.4.2855-2864.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Popik W, Alce TM, Au W-C. Human immunodeficiency virus type 1 uses lipid raft-colocalized CD4 and chemokine receptors for productive entry into CD4+ T cells. J Virol. 2002;76:4709–4722. doi: 10.1128/JVI.76.10.4709-4722.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Percherancier Y, Lagane B, Planchenault T, Staropoli I, Altmeyer R, et al. HIV-1 entry into T-cells is not dependent on CD4 and CCR5 localization to sphingolipid-enriched, detergent-resistant, raft membrane domains. J Biol Chem. 2003;278:3153–3161. doi: 10.1074/jbc.M207371200. [DOI] [PubMed] [Google Scholar]
- 43.Steffens CM, Hope TJ. Localization of CD4 and CCR5 in living cells. J Virol. 2003;77:4985–4991. doi: 10.1128/JVI.77.8.4985-4991.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baker AM, Sauliere A, Gaibelet G, Lagane B, Mazeres S, et al. CD4 interacts constitutively with multiple CCR5 at the plasma membrane of living cells: A fluorescence recovery after photobleaching at variable radii approach. J Biol Chem. 2007;282:35163–35168. doi: 10.1074/jbc.M705617200. [DOI] [PubMed] [Google Scholar]
- 45.Popik W, Alce TM. CD4 Receptor localized to non-raft membrane microdomains supports HIV-1 entry. J Biol Chem. 2004;279:704–712. doi: 10.1074/jbc.M306380200. [DOI] [PubMed] [Google Scholar]
- 46.Steffens CM, Hope TJ. Mobility of the human immunodeficiency virus (HIV) receptor CD4 and coreceptor CCR5 in living cells: Implications for HIV fusion and entry events. J Virol. 2004;78:9573–9578. doi: 10.1128/JVI.78.17.9573-9578.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Viard M, Parolini I, Sargiacomo M, Fecchi K, Ramoni C, et al. Role of cholesterol in human immunodeficiency virus type 1 envelope protein-mediated fusion with host cells. J Virol. 2002;76:11584–11595. doi: 10.1128/JVI.76.22.11584-11595.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sougrat R, Bartesaghi A, Lifson JD, Bennett AE, Bess JW, et al. Electron tomography of the contact between T cells and SIV/HIV-1: Implications for viral entry. PLoS Pathog. 2007;3:e63. doi: 10.1371/journal.ppat.0030063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trister AD, Hammer DA. Trimerization on HIV binding elucidated with Brownian adhesive dynamics. Biophys J. 2008;95:40–53. doi: 10.1529/biophysj.107.118430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chang MI, Panorchan P, Dobrowsky TM, Tseng Y, Wirtz D. Single-molecule analysis of human immunodeficiency virus type 1 gp120-receptor interactions in living cells. J Virol. 2005;79:14748–14755. doi: 10.1128/JVI.79.23.14748-14755.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dobrowsky TM, Zhou Y, Sun SX, Siliciano RF, Wirtz D. Monitoring early fusion dynamics of human immunodeficiency virus type 1 at single-molecule resolution. J Virol. 2008;82:7022–7033. doi: 10.1128/JVI.00053-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zaitseva M, Peden K, Golding H. HIV coreceptors: role of structure, posttranslational modifications, and internalization in viral-cell fusion and as targets for entry inhibitors. BBA Biomembranes. 2003;1614:51–61. doi: 10.1016/s0005-2736(03)00162-7. [DOI] [PubMed] [Google Scholar]
- 53.Doranz BJ, Baik SSW, Doms RW. Use of a gp120 binding assay to dissect the requirements and kinetics of human immunodeficiency virus fusion events. J Virol. 1999;73:10346–10358. doi: 10.1128/jvi.73.12.10346-10358.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bell GI. Models for the specific adhesion of cells to cells. Science. 1978;200:618–627. doi: 10.1126/science.347575. [DOI] [PubMed] [Google Scholar]
- 55.Strizki JM, Tremblay C, Xu S, Wojcik L, Wagner N, et al. Discovery and characterization of vicriviroc (SCH 417690), a CCR5 antagonist with potent activity against human immunodeficiency virus type 1. Antimicrob Agents Chemother. 2005;49:4911–4919. doi: 10.1128/AAC.49.12.4911-4919.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Time-evolution of the surface densities of the species in the reaction network. Surface densities of the species in the network (Eq. (1) in Text S1), namely, (A) unbound Env and Env molecules bound to single CD4 molecules, (B) Env bound to 2 CD4 molecules, (C) Env bound to 3 CD4 molecules, and (D) unbound CD4 and CCR5, obtained by solving Eq. (2) in Text S1 (solid lines), and of the equilibrium surface densities of CCR5 and the gp120-CCR5 complexes in the simplified network (Eq. (5) in Text S1) (dashed lines), obtained by solving Eqs. (6)–(9) in Text S1. Parameter values and initial conditions are mentioned in Text S1. All surface densities are normalized with the initial Env surface density.
(TIF)
Detailed kinetics of Env-CD4-CCR5 binding. A detailed description of the kinetics of the Env-CD4-CCR5 binding across a target cell-effector cell pair is presented.
(DOC)