

Abstract
The purpose of this study was to compare the cellular uptake and cytotoxicity of targeted and nontargeted doxorubicin (DOX)-loaded poly(d,l-lactide co-glycolide) (PLGA) nanoparticle (NP) drug delivery systems in drug-resistant ovarian (SKOV-3) and uterine (MES-SA/Dx5) cancer cell lines. The cellular uptakes of DOX from nonconjugated DOX-loaded NPs (DNPs) and from HER-2 antibody–conjugated DOX-loaded NPs (ADNPs) in MES-SA/Dx5 cancer cells were higher compared to free DOX. Results also showed higher uptake of DOX from ADNPs in SKOV-3 cells compared with both free DOX and DNPs treatment. Cytotoxicity results at 10 μM extracellular DOX concentration were consistent with the cellular uptake results. Our study concludes that cellular uptake and cytotoxicity of DOX can be improved in MES-SA/Dx5 cells by loading DOX into PLGA NPs. DNPs targeted to membrane receptors may enhance cellular uptake and cytotoxicity in SKOV-3 cells.
Keywords: Doxorubicin, PLGA, Cytotoxicity, HER-2 antibody, Apoptosis, P-glycoprotein
Doxorubicin (DOX) is an anthracycline antibiotic that is widely used against several solid tumors, such as ovarian cancers.1 One important disadvantage of DOX is the development of multi-drug resistance (MDR) in cancer cells due to overexpression of P-glycoprotein (P-gp), which can significantly influence the therapeutic effect of DOX.2 This MDR cannot be circumvented by increasing DOX dosage because of toxic dose-dependent side effects, such as irreversible cardiotoxicity when DOX is accumulated in mitochondria.3
Drug carriers at the nanoscale may be able to help overcome the undesirable effects of traditional chemotherapeutic agents by maximizing their availability at the target tumor site and minimizing noxious effects on healthy tissue. Nanoparticulate drug delivery systems can exploit the unique vasculature characteristics of tumors and enhance drug delivery to the targeted cancer.4 The loose interconnections and intercellular openings among tumor vasculature endothelial cells, ranging in size between 100 and 780 nm, can be easily extravasated by drug-loaded nanoparticles (NPs).5,6 This phenomenon of localizing NPs in the leaky vasculature of tumor tissues is an example of passive targeting. The drug carrier also prevents the recognition of drug molecules by cellular efflux pumps such as P-gp and hence helps overcome MDR in such cell lines.7
The therapeutic potential of nanocarriers can be further magnified by tagging them with appropriate ligands that selectively interact with tumor cell membrane receptors.8,9 This method of tagging the drug delivery vehicle with a ligand and allowing it to specifically sequester in the targeted tumor is an example of active targeting. Among the wide repertoire of ligands, such as peptides, carbohydrates, and polymers, monoclonal antibodies are most widely investigated for selectively targeting nanoparticulate drug delivery systems to tumors.10 Monoclonal antibodies were initially conjugated with drugs to form immunoconjugates that conferred selectivity to the drug.11,12 However, such conjugation methods adversely affect the pharmacological action of the drug, as well as its in vivo fate,13 and the conjugation methods allow few drug molecules to be attached to the antibody.14 Nanoparticulate drug delivery systems with bound tumor-selective ligands confer greater selectivity and could potentially carry more drug compared with drug molecules directly conjugated to an antibody.15
Poly(d,l-lactide co-glycolide) (PLGA) is a U.S. Food and Drug Administration–approved biodegradable and biocompatible polymer widely used for NP preparation because of its ability to carry both hydrophobic and hydrophilic drugs.7,16 Some of the advantages include sustained and controlled drug delivery to the tumor site, easy extravasation into the tumor vasculature, higher ability to cross physiological barriers, and the possibility of targeted delivery by NP surface decoration.7,17
Several studies on DOX-loaded and targeted DOX-loaded PLGA NPs have been published.18-20 In this study we compared the cellular uptake and cytotoxicity of targeted and nontargeted DOX-loaded NPs in drug-resistant uterine and ovarian cancer cells (MES-SA/Dx5 and SKOV-3). Drug-sensitive uterine cancer cells (MES-SA) were used as a negative control. MES-SA/Dx5 shows chemoresistance through overexpression of P-gp, which can extrude significant amounts of DOX from the cells. The drug resistance of SKOV-3, on the other hand, is not associated with efflux. SKOV-3 cells cannot activate either caspase 3 or caspase 9, and this along with the defective activity of apoptotic protease-activating factor 1 makes them resistant to p53-mediated apoptosis and seriously compromises their sensitivity to DOX.21,22
Methods
Drugs and chemicals
PLGA (lactide-to-glycolide ratio 50:50, molecular weight (MW) 40,000–75,000 Da), doxorubicin hydrochloride (DOX HCl; MW 579.95), dimethylsulfoxide (DMSO; >99.9% reagent grade), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), Micro bicinchonic acid (BCA) protein assay kits, Tween-80, N-hydroxy succinimide (NHS), sodium dodecyl sulfate (SDS), monoclonal antibody to human epidermal growth factor receptor 2 (HER-2), phosphate-buffered saline (PBS), and polyvinyl alcohol (PVA; 87–89% hydrolyzed; MW 13,000–23,000) were purchased from Sigma-Aldrich (St. Louis, Missouri). Dichloromethane (DCM) was purchased from Honeywell Burdick & Jackson (Morristown, New Jersey). Sodium hydroxide (NaOH) was purchased from Fisher Scientific (Pittsburgh, Pennsylvania).
Preparation of DOX-PLGA NPs (DNPs)
PLGA NPs with encapsulated DOX were formulated by the oil-in-water emulsification solvent evaporation method23 with a slight modification using a procedure described by our group.24 Briefly, PLGA (40 mg), DOX HCl (2 mg), and triethylamine (20 μL) were dissolved in methanol (1 mL) and DCM (3 mL). This organic phase was emulsified with 8 mL of aqueous phase containing 3% PVA. The mixture was emulsified using an ultrasonicator at 21 W for 4 minutes (Cole Palmer, Vernon Hills, Illinois). The organic phase was removed through solvent evaporation of DCM in a Rotavapor (Buchi, New Castle, Delaware) at 40°C. After solvent evaporation the NPs were centrifuged at 12,200 rpm for 30 minutes. The pellets were washed four times with equal amounts of deionized water and centrifuged again for 30 minutes. This procedure was aimed at removing excess drug and PVA. Finally the washed particles were freeze-dried and lyophilized for 48 hours. Void PLGA NPs were prepared in a similar manner as mentioned above except without DOX.
Preparation of antibody-conjugated DOX-PLGA NPs (ADNPs)
Antibody-conjugated NPs were prepared using the method explained by Yang et al.23 The lyophilized NPs were resuspended in PBS (0.1 M, pH 7.4, 1 mg/mL), and 50 μL (100 μg) of IgG antibody were added to the mixture. Next, 1 mL of 2.0 mM NHS and 2.0 mM of EDC were added to the above mixture and incubated for 4 hours under shaking conditions at 4°C. After the incubation period the mixture was centrifuged at 12,200 rpm at 6°C for 30 minutes. The pellet formed was washed in PBS (0.1 M, pH 7.4, equal volume of supernatant) twice and centrifuged again at 12,200 rpm at 6°C for 30 minutes. The above procedure was repeated twice to remove the excess unreacted antibody and reaction reagents. The particles were then freeze-dried and lyophilized.
Characterization of NPs
Size and size distribution
NP size was measured by dynamic light scattering (DLS) using a Malvern Zetasizer (Malvern Instruments, Worcestershire, United Kingdom). Size measurements were taken in triplicate at 25°C using a 1:100 (vol/vol) dilution of the NP suspension in distilled water. The particle size was estimated from cumulant analysis once the correlation was determined. The polydispersity index was used as a measure of particle size distribution, scaled from 0 to 1.
Zeta potential
Zeta potential (surface charge) of the NPs dispersed in DI water was measured by the Zetasizer using the Smoluchowski fitting model. The data obtained amounted to an average of measurements of three samples (n = 3).
Drug loading
The NPs were dissolved in DMSO (3 mL), and the fluorescence spectrum of the samples was evaluated using a Fluorolog-3 spectrofluorometer (Horiba Jobin Yvon, Edison, New Jersey). Serial dilutions of the sample were done to reach the linear range (1:2, 1:4, 1:8, 1:16, 1:32 dilutions). The maximum peak intensities after DMSO blank subtraction were plotted and fitted to a linear model. Then, the concentration of DOX in the NPs was determined using a standard calibration curve of DOX in DMSO using the same spectrofluorometer. NP drug entrapment was calculated using the following formula:
| (1) |
DNPs release kinetics
DOX release kinetics from PLGA NPs was estimated using the method described by Misra and Sahoo.25 Briefly, 10 mg of DOX-loaded PLGA NPs were resuspended in 3 mL of 0.01 M PBS supplemented with 0.1% Tween-80. After this the sample was placed into Eppendorf tubes, which were then shaken at 80 rpm while maintaining at 37°C in a Cellstar incubator (Queue Systems, Asheville, North Carolina). The tubes were removed from the incubator every hour up to the first 6 hours, and then every 24 hours after that. Each time the samples were centrifuged at 14,000 rpm for 20 minutes. Following this, the supernatant was collected in 4.5-mL polystyrene cuvettes and the DOX content was estimated using a spectrofluorometer (Horiba Jobin Yvon). The NPs were again suspended in fresh PBS solution and incubated for later time release measurements. This process was repeated at regular time intervals, and the release study was done for a period of 30 days.
Antibody conjugation efficiency
We estimated conjugation efficiency using a method reported by Mo and Lim.26 In this method, 1 mg of lyophilized particles was dissolved in 0.5 mL of DMSO, and 1 mL of 0.5% (wt/vol) SDS/0.05 M NaOH (salinated SDS) was added to the above mixture followed by incubation for 4 hours. Next, 1 mL of the solution described above was mixed with equal volumes of the BCA working reagent and incubated at 60°C for 60 minutes. The absorbance of this mixture was measured using a Cary spectrophotometer (Varian, Palo Alto, California) at 562 nm. The spectrophotometer was calibrated with standard 0.5- to 40-μg/mL protein solutions (bovine serum albumin standard) in the same proportion of DMSO and salinated SDS. Solutions of unconjugated NPs served as the blank. Conjugation efficiency was calculated as the weight of protein (micrograms antibody) associated with unit weight (milligrams) of lyophilized NPs.
Cell culture
Human ovarian carcinoma SKOV-3 cells, human uterine sarcoma MES-SA cells, and their MDR (P-gp–overexpressing) derivative MES-SA/Dx5 (Dx5) cells, McCoy’s 5A medium, and fetal bovine serum were purchased from American Type Culture Collection (Manassas, Virginia). 24-well tissue culture plates, d-poly coverslips, and formalin were purchased from Fisher Scientific. Penicillin was purchased from Sigma-Aldrich. All the cells were cultured in McCoy’s 5A medium added with 1% penicillin and 10% fetal bovine serum, and kept in a 37°C cell incubator with a humidified atmosphere of 5% CO2 and 95% air.
Cellular uptake experiments
Three cell lines (MES-SA, Dx5, and SKOV-3) were used to study the cellular uptake of unencapsulated DOX (designated as free DOX), DNPs, and ADNPs. On the first day the cells were seeded into 24-well plates at cell density of 100,000 to 200,000 cells per well. On the second day the cell medium was removed, and free DOX, DNPs, or ADNPs in growth medium was added to the plates at a normalized DOX concentration of 10 μM (5.8 μg/mL) and placed in a cell incubator at 37°C for 24 hours. The control group means no drug was added. After 24 hours the cell medium was removed, and the cells were washed with ice-cold PBS (pH 7.0) four times and then lysed with 1 mL of DMSO. The supernatants were centrifuged at 140,000 rpm for 10 minutes and collected to obtain cell lysates. The DOX fluorescence intensity of cell lysates was measured by a spectrofluorometer (Horiba Jobin Yvon) at λex = 496 nm, λem = 592 nm to determine DOX concentration. To adjust the background fluorescence from cellular components, a DOX calibration curve was created by dissolving DOX, DNPs, or ADNPs in DMSO and adding the solution to untreated cells. The supernatants were then collected and measured so as to prepare different calibration curves for DOX, DNPs, and ADNPs in the presence of untreated cell lysates. The protein content in the cell lysates was measured using a micro BCA protein assay kit, and the absorption data were acquired at 562 nm with a spectrophotometer (Varian). Cellular uptake of DOX for different treatments was expressed by normalizing the DOX amount to the protein amount. An average value was obtained from three wells in each treatment for each experiment, and an average (± SD) intracellular uptake of DOX from three experiments was plotted.
Subcellular localization
To study the intracellular localization of free DOX, DNPs, and ADNPs, cells were seeded with a density of 4 × 104 cells per well on poly-d-lysine–pre-coated glass coverslips, placed inside wells of a 24-well tissue culture plate, and incubated overnight and allowed to reach confluence. On the second day, cell medium was removed and then replaced with 0.5 mL of 10 μM DOX, DNPs, or ADNPs. The plates were then kept in a 37°C incubator for 24 hours and protected from light exposure. After incubation, cells were washed with PBS three times and fixed with 4% (vol/vol) formaldehyde, followed by washing three times with PBS. Then, the specimens were observed by confocal microscopy (Nikon, Melville, New York) with a 40× objective. The confocal microscope settings were kept the same between samples. The fluorescence signals were converted to pseudo color and merged by the software EZ-C1 (Nikon, Melville, New York).
Cytotoxicity assessment
Cell viability was measured with the sulforhodamine B (SRB) assay (Invitrogen, Carlsbad, California), which colorimetrically measures cellular protein.27 In this study, the cytotoxicity of three different treatments (free DOX, DNPs, and ADNPs) was investigated. The detailed procedure was the same as we have described before.28 Briefly, cells were seeded in a 96-well plate on the first day, and after overnight incubation they were exposed to different treatments. The cytotoxicity was measured with SRB assay 24 hours post treatment. Tested DOX concentrations ranged from 0 to 10 μM, where a drug concentration equal to zero means that only PBS and no drug was added to the cells. We also tested the cytotoxicity of void PLGA NPs at much higher concentrations (5.3 mg/mL) than those used in the experiment (0.25 mg/mL).
An average value was obtained from four wells in each treatment for each experiment. An average (± SD) “cell growth” from three experiments was plotted against increasing DOX concentrations. Cell growth values were generated by normalizing the data from each treatment to the control values, which did not receive DOX or NPs.
Statistical significance
Statistical significance was identified by one-way analysis of variance (ANOVA; SPSS, Chicago, Illinois) for the difference among treatment groups at the same DOX concentration. A P value <0.05 was considered to be statistically significant.
Results
Characterization of NPs
Size, size distribution, zeta potential, and drug loading
The size, polydispersity, zeta potential, and drug loading for DNPs and ADNPs are shown in Table 1. The mean diameters of ADNPs and DNPs were 213.0 ± 3.5 nm and 162.7 ± 2.1 nm, respectively. Data represent mean ± SD obtained from size measurements of three samples prepared on three different days (n = 3). Figure 1 shows DLS measurement of void NPs, DNPs, and ADNPs. The zeta potentials of the DOX-loaded NPs with and without antibody (IgG) conjugation were −1.3 ± 3.8 mV and −13.2 ± 2.3 mV, respectively. NP (i.e., ADNPs and DNPs) drug loading was determined using Eq 1 as described in the Methods section. Drug loading values (wt/wt %) were determined and tabulated as mean ± SD (n = 3) in Table 1. The drug loading of the conjugated particles was lower than for their nonconjugated counterparts by 14% (P < 0.05).
Table 1.
Mean size, zeta potential, polydispersity, and percent entrapment efficiencies for void PLGA NPs, ADNPs, and DNPs (n = 3)*
| Formulation | Size (nm) | Polydispersity | Zeta potential (mV) | Drug loading (wt/wt %) |
|---|---|---|---|---|
| Void NPs | 163.0 ± 3.2 | 0.070 ± 0.018 | −15.7 ± 3.5 | N/A |
| ADNPs | 213.0 ± 3.5 | 0.193 ± 0.010 | −1.3 ± 3.8 | 2.3 ± 0.1 |
| DNPs | 162.7 ± 2.1 | 0.068 ± 0.016 | −13.2 ± 2.3 | 2.7 ± 0.1 |
Data represent mean ± SD.
Figure 1.
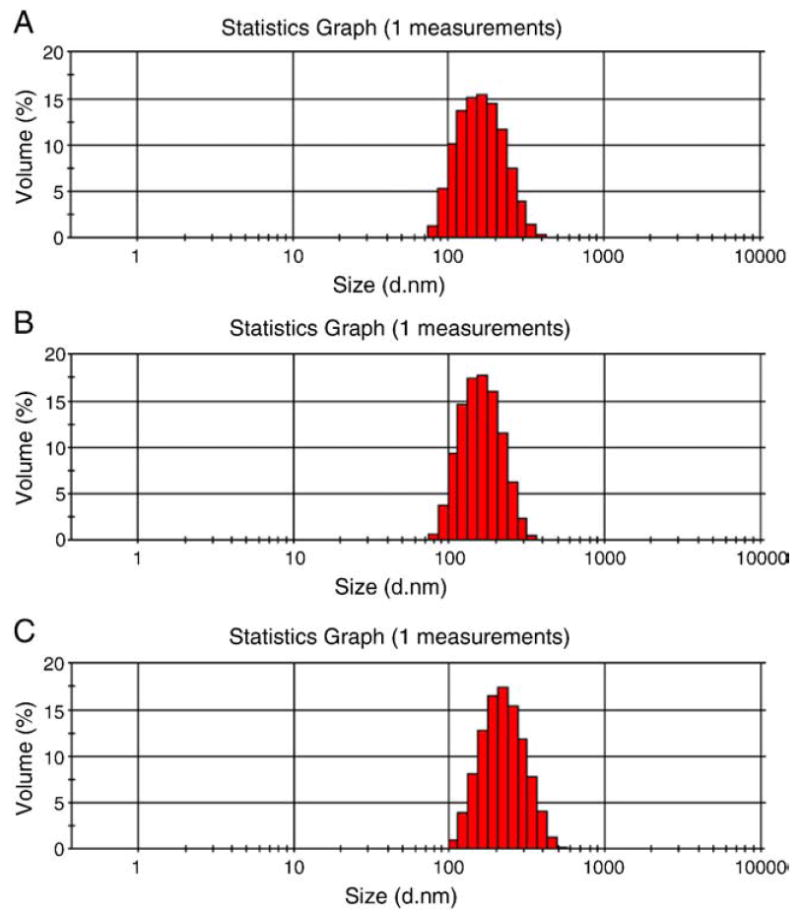
DLS size histograms of (A) void PLGA NPs, (B) DNPs, and (C) ADNPs.
In vitro release kinetics profile
Cumulative percentage of DOX released from DNPs is shown in Figure 2, which reveals a biphasic DOX release profile. There is an initial burst release of 23.42 ± 4.55% in the first 6 hours, followed by a much slower release over a period of 30 days, for a total of 47.09 ± 7.11% of DOX released in 30 days.
Figure 2.
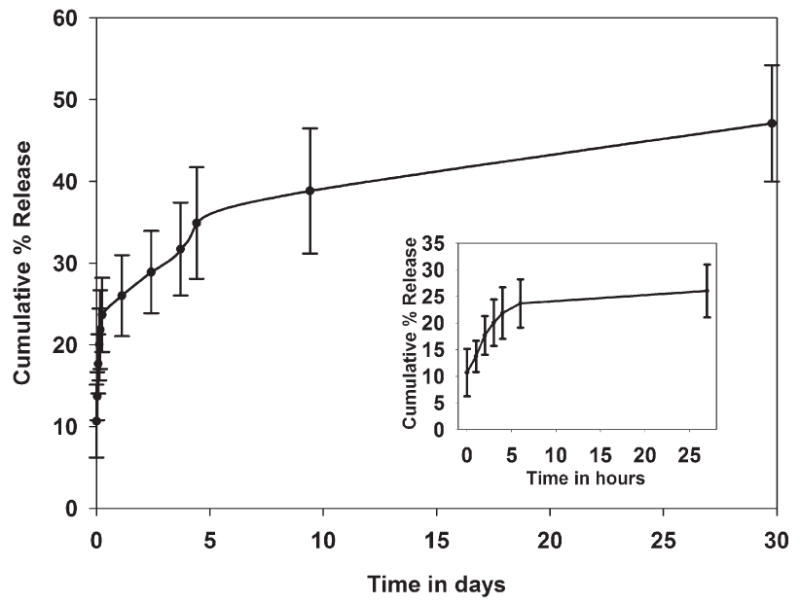
DOX release kinetics profile of DNPs.
Antibody conjugation efficiency
The conjugation efficiency of DOX-loaded NPs was measured as described in the Methods section. Our results showed that the amount of antibody conjugated to the NPs was 9.29 ± 0.66 μg antibody per milligram of NPs.
Cell culture studies
Cellular uptake of DOX
Figure 3 shows that loading DOX into PLGA NPs can significantly increase DOX uptake by Dx5 cells by approximately sevenfold as compared with free DOX. However, the NPs did not improve the uptake of DOX by MES-SA cells. In SKOV-3 cells the DOX uptake with DNPs (without antibody) was moderately higher compared with free DOX but did not reach statistical significance. We observed significantly higher DOX uptake by SKOV-3 cells after exposure to ADNPs compared with free DOX and DNPs (P < 0.05, by ANOVA). MES-SA and Dx5 cells were used as negative controls for ADNPs, and the results showed comparable DOX uptake by both cell lines after treatment with ADNPs or DNPs. This experiment indicates that a higher uptake of DOX by SKOV-3 cells can be achieved by conjugating DNPs with antibodies targeting HER-2 receptors.
Figure 3.
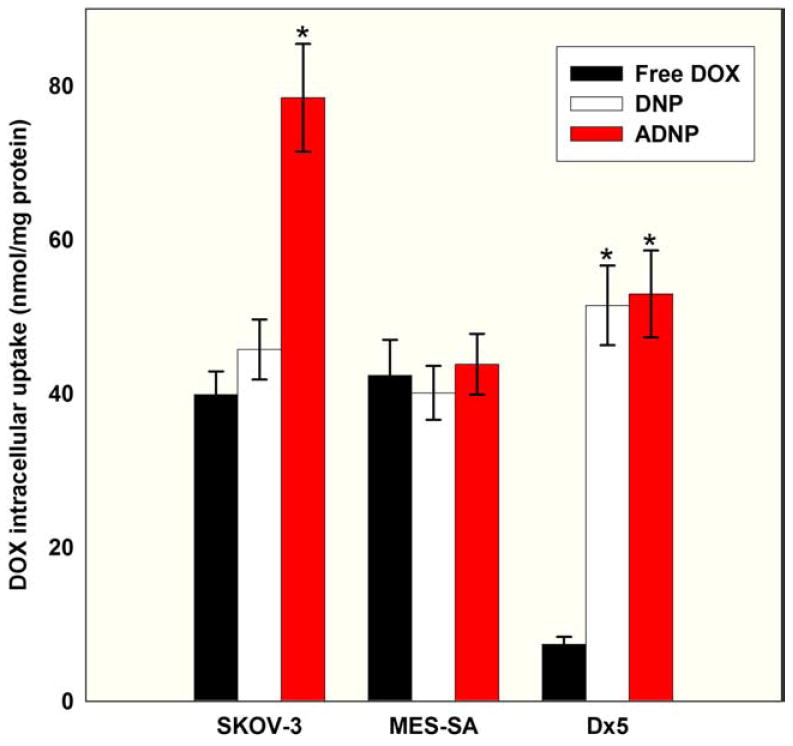
24-hour intracellular DOX uptake data in SKOV-3, MES-SA, and Dx5 cells (n = 3 experiments, three wells per treatment). *P < 0.05 (by ANOVA) in DNPs compared to free DOX and ADNPs, and ADNPs compared to free DOX and DNPs.
Subcellular localization of DOX
Figure 4 shows confocal laser microscopy images demonstrating subcellular localization for free DOX, DNPs, and ADNPs in SKOV-3, MES-SA, and Dx5 cell lines. Void PLGA NPs and void antibody-PLGA NPs produced negligible fluorescence (images not shown), so the fluorescence images obtained can be assumed to be generated by DOX only. With free DOX treatment, most DOX localized in the cell nucleus of MES-SA and SKOV-3, whereas only a small amount of DOX can be maintained in nucleus of Dx5 cells as a result of the overexpression of P-gp protein on the cell membrane. In DNPs and ADNPs treatments, we observed that some DOX molecules were still entrapped in NPs and distributed in the cytoplasm, whereas some DOX was released from NPs and delivered into the nucleus. Stronger fluorescence intensity from ADNPs treatment was observed compared with DNPs and free DOX treatment in SKOV-3 cells, indicating that a higher cellular uptake of DOX was achieved in SKOV-3 cells when DNPs were conjugated with HER-2 antibody compared with free DOX and their unconjugated counterparts. In MES-SA and Dx5 cells, similar uptake patterns were observed for ADNPs and DNPs due to a lack of HER-2 expression. In MES-SA cells, the subcellular distribution of DNPs and ADNPs is similar to that of free DOX, except that NP treatment seems to result in formation of some nuclear aggregates of DOX released from the NPs. In Dx5 cells the NPs seem to enhance nuclear delivery of DOX as compared with free DOX. We also observed stronger fluorescence intensity for DNPs and ADNPs treatment in Dx5 cells compared with free DOX treatment, indicating a higher DOX uptake by Dx5 cells when DOX was delivered through PLGA NPs. This is consistent with our cellular uptake data (Figure 3), suggesting that NP drug carrier systems can overcome MDR and lead to higher cellular uptake and nuclear delivery of DOX in cancer cell lines overexpressing P-gp.
Figure 4.
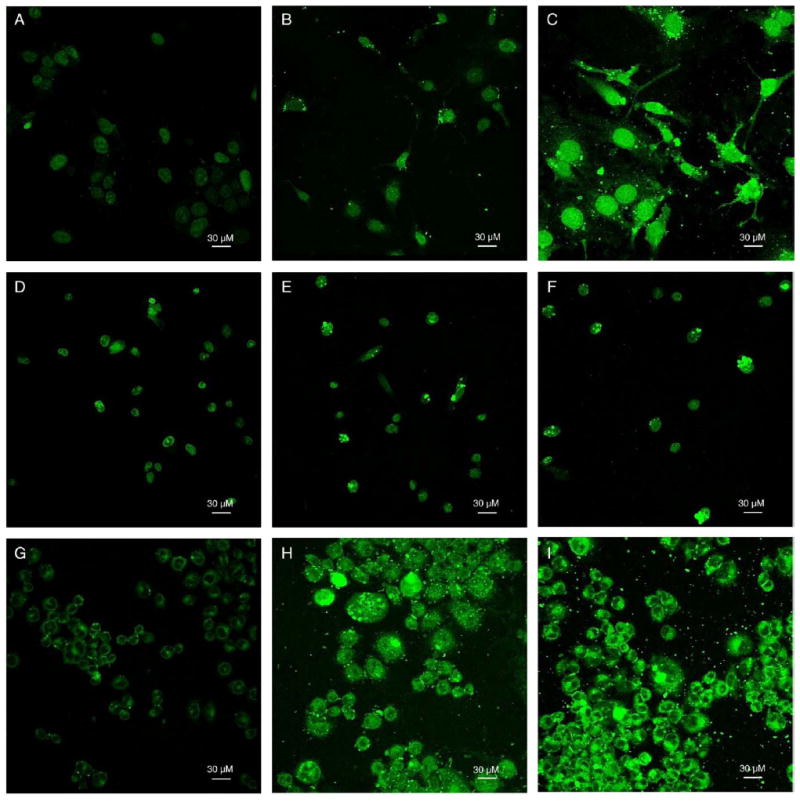
(A–I) Cell images obtained with a Nikon confocal microscope and a 488-nm argon laser at 40× objective magnification. DOX fluorescence images were recorded with software-added green pseudo color. (A–C) 10 μM free DOX, DNPs, or ADNPs were incubated with SKOV-3 cells for 24 hours, respectively. Photomultiplier tube (PMT) was set at gain 7. (D–F) 10 μM free DOX, DNPs, or ADNPs were incubated with MES-SA cells for 24 hours, respectively. PMT was set at gain 6. (G–I) 10 μM free DOX, DNPs, or ADNPs were incubated with Dx5 cells for 24 hours, respectively. PMT was set at gain 7.
Cytotoxicity of different NP formulations
PLGA is considered safe and has been widely used as a biomaterial. Cell proliferation following three different treatments is shown in Figure 5, which shows the cytotoxicity of the different DOX formulations in MES-SA and Dx5 cells, respectively (Figure 5, A and B). The expectation was that because neither MES-SA nor Dx5 cells overexpress HER-2 receptors, DNPs and ADNPs treatments should have similar cytotoxicity in both cell lines. On the other hand, given that Dx5 cells overexpress the drug efflux pump P-gp, we expected that the two NP formulations should have higher cytotoxicity than free DOX in Dx5 cells. The results confirmed our expectations. In the uptake study (10 μM DOX concentration), ADNPs showed the highest uptake by SKOV-3 cells compared with free DOX and DNPs, whereas accumulation of DOX from DNPs and free DOX was similar. This result is not surprising, because SKOV-3 cells overexpress HER-2 receptors but do not over-express P-gp. Therefore, we expect the same pattern in the cytotoxicity data. As shown in Figure 5, C, ADNPs are the most toxic among the three different DOX formulations at 10 μM DOX concentration in SKOV-3 cells, although the difference did not reach statistical significance, whereas free DOX and DNPs showed cell growth inhibition capability more comparable to each other. (See Supplementary Material in the online version of this article.)
Figure 5.
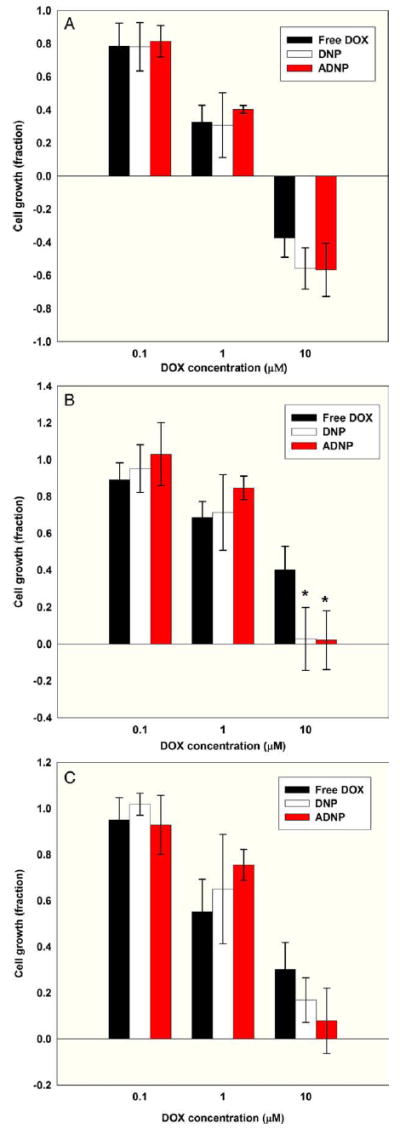
Cytotoxicity of different NP formulations. (A) MES-SA cell growth for different drug formulations (n = 3 experiments, four wells per treatment). (B) Dx5 cell growth for different drug formulations (n = 3 experiments, four wells per treatment). *P < 0.05 (by ANOVA) DNPs and ADNPs compared to free DOX, indicating significant uptake due to NP encapsulation. (C) SKOV-3 cell growth for different drug formulations (n = 3 experiments, four wells per treatment).
Discussion
DOX-encapsulated PLGA NPs were prepared by the oil-in-water emulsification solvent evaporation method, and further conjugated with antibody using the EDC–NHS carbodiimide reaction. Particles in the 100- to 200-nm range avoid premature clearance by the reticuloendothelial system. DNPs measured ~160 nm, and antibody conjugation led to an increase in particle size of ~50 nm. It also affected the polydispersity of the particles after conjugation. Similar results have been reported by other researchers.26 We observed that increasing PVA concentration increases the zeta potential of PLGA NPs toward positive as a result of the capping of negative charges of the PLGA polymer by the PVA. This observation is consistent with results from other groups.29-31 Stolnik et al also reported a zeta potential of −45 mV for PLGA particles prepared without PVA in neutral buffer.32 The observed increase in zeta potential after conjugating the DNPs with antibody may be due to the masking of the anionic surface charge by the antibody coating. The entrapment efficiency was lower in ADNPs compared with nonconjugated NPs, probably because of leaching of drug during the antibody conjugation process. The resulting lower entrapment efficiency in ADNPs as compared with DOX-loaded NPs consistent with the literature.33,34 Release of DOX from PLGA NPs occurred via two mechanisms: burst release and diffusion-controlled release. Burst release from the NPs was due to the release of DOX that was adsorbed onto the surface of PLGA NPs during formulation. After this phase, release was primarily due to diffusion from the core of the PLGA NPs and by hydrolysis or degradation of the PLGA. Similar results have been reported by Misra and Sahoo,25 who observed a total 63% release from DOX-loaded NPs over a period of 60 days. Tewes et al.35 also reported a very slow release of DOX from DOX NPs prepared using the single-emulsion method (1.5% in 24 hours). Such slow release might be due to the delayed degradation of the PLGA, because Stevanović et al.36 reported a degradation time of ~60 days for PLGA polymer with 50:50 lactide co-glycolide ratio. In addition, this slow release could be due to the low aqueous solubility of DOX and low diffusivity of the drug within the polymer matrix.37
The cellular uptake results are consistent with existing literature,7,38 in that free DOX is delivered into the cells mainly through diffusion,25 whereas the major delivery mechanism of PLGA NPs is endocytosis.39 We observed improved cellular uptake compared with free DOX in cancer cell lines over-expressing P-gp due to endocytosis. However, in P-gp–negative cancer cell lines, DOX-loaded NPs (without antibody attachment) did not have an obvious advantage in delivery compared to free DOX probably because diffusion is not affected. The anti-HER-2-conjugated DOX-loaded NPs were found to enhance DOX uptake in SKOV-3 cancer cells compared with their nonconjugated counterparts. Similar HER-2 conjugation results have been reported for anti-HER-2 conjugated PLGA-montmorillonite NPs encapsulating paclitaxel,33 and for anti-HER-2 conjugated magnetic PLGA NPs encapsulating DOX.23 Thus, the higher uptake that is evident from our cellular uptake experiments and our fluorescence images with conjugated NPs could be justified by the presence of the antibody, which is complementary to the overexpressed receptors on the cancer cell surface. The conjugated NPs are believed to be taken up into the tumor cells by means of receptor-mediated endocytosis.40
In the subcellular localization experiments, our fluorescence images are consistent with existing literature that describes rapid intercalation of DOX with nuclear DNA in living cells,41 as well as decreased nuclear localization with the presence of P-gp efflux pump mechanisms in MDR cancer cells.
We observed cytotoxicity results consistent with the cellular uptake results at 10 μM extracellular DOX concentration. The cytotoxicity experiment results were similar among all the drug formulations for 0.1 μM and 1 μM extracellular DOX concentration when tested in MES-SA. However, we did not observe any obvious improvement in the 24-hour cytotoxic effect for either DNPs or ADNPs compared to free DOX in Dx5 and SKOV-3 with 0.1 μM or 1 μM extracellular DOX concentration. According to the literature, PLGA NPs are probably entrapped in endosomes or lysosomes, so DOX would be released at this location.42,43 Once released, the weakly basic anthracycline molecules will tend to remain in these cellular compartments because of their acidity.42,43 The endosomes and lysosomes in drug-resistant cancer cells are more acidic, whereas their cytosol and nucleus are more alkaline compared with drug-sensitive cells. Changes in compartmental pH are an important mechanism in cell resistance phenomena, because cells can slow drug release from endosomes and lysosomes by modulating pH gradients between intracellular compartments.44-46 Therefore, it may be easier for DOX to escape from endosomes or lysosomes and be delivered from the cytosol into the nucleus in MES-SA than in Dx5 and SKOV-3 after they are released from NPs. However, a cell has only a limited amount of endosomes or lysosomes, and it is possible that the recycling rate of endosomes cannot keep up with the demand when the accumulation of DOX inside the cells is high. This may explain the higher cytotoxicity effect of DNPs or ADNPs compared with free DOX in Dx5 or SKOV-3, which was only observable at high extracellular DOX concentrations. For MES-SA, on the other hand, all the DOX formulations have comparable cytotoxicity for all concentrations tested.
Both p53 mutations and P-gp overexpression are involved in cellular resistance to chemotherapy, and developing a drug to overcome them would be an important aid in the design of customized targeted therapy. Dx5 and SKOV-3 cancer cells represent examples of cell lines that exhibit these two different drug resistance mechanisms. Our results indicate that decorating the NP surface with antibodies will not affect cellular uptake or cytotoxicity in drug-sensitive or P-gp–overexpressing cancer cells if these cell types do not express the corresponding receptors. NP surface decoration with antibodies added a positive charge to the otherwise negatively charged NPs. Because tumor cell membranes exhibit negative charge, some groups suggest that incorporation of a positive moiety would enhance the interaction of the NPs with the tumor cell membrane by electrostatic interaction, and improve the uptake of NPs.47 However, there was not a significant increase in cellular uptake in HER-2–negative MES-SA and Dx5, indicating that the main factor in determining cellular uptake is still specific receptor-mediated endocytosis, rather than a generic electrostatic interaction with the membrane. Moreover, we did not observe significantly higher cytotoxicity in SKOV-3 from ADNPs treatment in comparison to free DOX and DNPs, even though we obtained much higher cellular uptake from ADNPs. The reason is probably related to the genetic deficiency or mutation of p53 in ovarian cancer cells that leads to DOX chemoresistance. It seems that increasing the accumulation of DOX inside SKOV-3 cancer cells does not result in an optimized therapeutic effect. It is possible that adding another therapy modality, such as hyperthermia, could be an adjuvant to improve the effect of DOX chemotherapy. Targeted and nontargeted PLGA NPs also provide the possibility to encapsulate additional agents along with the anticancer drug. For instance, in a previous study we observed a significant synergistic cytotoxic effect on SKOV-3 cells when combining the photothermal agent indocyanine green (ICG) and DOX treatment.28,48 We have obtained preliminary in vitro results when encapsulating DOX and ICG together in PLGA NPs (ICG-DOX-PLGANPs).49 Our future studies will focus on decorating antibodies onto ICG-DOX-PLGANPs and measuring the response of SKOV-3 and Dx5 during hyperthermia and chemotherapy, including the exploration of possible synergistic effects between the two therapeutic strategies.
This study has shown improved cellular uptake of DOX in drug-resistant human ovarian cancer and human uterine cancer cell lines. By encapsulating DOX into PLGA NPs we were able to successfully overcome the MDR effect in Dx5 human uterine cancer cells, obtaining increased cellular uptake and enhanced nuclear retention. Additionally, these DOX-loaded PLGA NPs were conjugated with HER-2 antibodies, which can be recognized by receptors overexpressed on the surface of SKOV-3 cancer cells. This approach resulted in a significant increase in cellular uptake of DOX by SKOV-3 cells. However, this did not translate into significant increases in cytotoxicity. We observed an increase in cytotoxicity in SKOV-3 cells at high extracellular DOX concentration, but the differences did not reach statistical significance. We predict that DOX antibody-conjugated PLGA NPs will result in higher delivery efficacy, and have promising potential for in vivo clinical applications.
Supplementary Material
Acknowledgments
Sources of support for research: This work was conducted using the facilities of the Biomedical Engineering Department at Florida International University, and partially funded by Florida Department of Health grant no. 08-BB-11, the Biomedical Engineering Young Inventor Award from the Wallace H. Coulter Foundation to R.M., the Florida International University Dissertation Year Fellowship to Y.T., and support from the National Institutes of Health/National Institute of General Medical Sciences grant R25 GM061347 to A.F.F.
Footnotes
Appendix A. Supplementary data Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.nano.2010.11.004.
References
- 1.Vermorken JB. The role of anthracyclines in second-line therapy of ovarian cancer. Int J Gynecol Cancer. 2003;13(Suppl 2):178–84. doi: 10.1111/j.1525-1438.2003.13364.x. [DOI] [PubMed] [Google Scholar]
- 2.Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–27. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- 3.Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med. 1998;339:900–5. doi: 10.1056/NEJM199809243391307. [DOI] [PubMed] [Google Scholar]
- 4.Brigger I, Dubernet C, Couvreur P. Nanoparticles in cancer therapy and diagnosis. Adv Drug Deliv Rev. 2002;54:631–51. doi: 10.1016/s0169-409x(02)00044-3. [DOI] [PubMed] [Google Scholar]
- 5.Yuan F, Dellian M, Fukumura D, Leunig M, Berk DA, Torchilin VP, et al. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res. 1995;55:3752–6. [PubMed] [Google Scholar]
- 6.Hobbs SK, Monsky WL, Yuan F, Roberts WG, Griffith L, Torchilin VP, et al. Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci USA. 1998;95:4607–12. doi: 10.1073/pnas.95.8.4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Panyam J, Labhasetwar V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv Drug Deliv Rev. 2003;55:329–47. doi: 10.1016/s0169-409x(02)00228-4. [DOI] [PubMed] [Google Scholar]
- 8.Sapra P, Allen TM. Ligand-targeted liposomal anticancer drugs. Prog Lipid Res. 2003;42:439–62. doi: 10.1016/s0163-7827(03)00032-8. [DOI] [PubMed] [Google Scholar]
- 9.Vasir JK, Reddy MK, Labhasetwar V. Nanosystems in drug targeting: opportunities and challenges. Curr Nanosci. 2005;1:47–64. [Google Scholar]
- 10.Adams GP, Weiner LM. Monoclonal antibody therapy of cancer. Nat Biotechnol. 2005;23:1147–57. doi: 10.1038/nbt1137. [DOI] [PubMed] [Google Scholar]
- 11.Garnett MC. Targeted drug conjugates: principles and progress. Adv Drug Deliv Rev. 2001;53:171–216. doi: 10.1016/s0169-409x(01)00227-7. [DOI] [PubMed] [Google Scholar]
- 12.Funaro A, Horenstein AL, Santoro P, Cinti C, Gregorini A, Malavasi F. Monoclonal antibodies and therapy of human cancers. Biotechnol Adv. 2000;18:385–401. doi: 10.1016/s0734-9750(00)00043-4. [DOI] [PubMed] [Google Scholar]
- 13.Nobs L, Buchegger F, Gurny R, Allémann E. Current methods for attaching targeting ligands to liposomes and nanoparticles. J Pharm Sci. 2004;93:1980–92. doi: 10.1002/jps.20098. [DOI] [PubMed] [Google Scholar]
- 14.Jaracz S, Chen J, Kuznetsova LV, Ojima I. Recent advances in tumor-targeting anticancer drug conjugates. Bioorg Med Chem. 2005;13:5043–54. doi: 10.1016/j.bmc.2005.04.084. [DOI] [PubMed] [Google Scholar]
- 15.Dinauer N, Balthasar S, Weber C, Kreuter J, Langer K, Briesen H. Selective targeting of antibody-conjugated nanoparticles to leukemic cells and primary T-lymphocytes. Biomaterials. 2005;26:5898–906. doi: 10.1016/j.biomaterials.2005.02.038. [DOI] [PubMed] [Google Scholar]
- 16.Astete CE, Sabliov CM. Synthesis and characterization of PLGA nanoparticles. J Biomater Sci Polym Ed. 2006;17:247–89. doi: 10.1163/156856206775997322. [DOI] [PubMed] [Google Scholar]
- 17.Danhier F, Lecouturier N, Vroman B, Jerome C, Marchand-Brynaert J, Feron O, et al. Paclitaxel-loaded PEGylated PLGA-based nanoparticles: in vitro and in vivo evaluation. J Control Release. 2009;133:11–7. doi: 10.1016/j.jconrel.2008.09.086. [DOI] [PubMed] [Google Scholar]
- 18.Yoo HS, Oh JE, Lee KH, Park TG. Biodegradable nanoparticles containing doxorubicin-PLGA conjugate for sustained release. Pharm Res. 1999;16:1114–8. doi: 10.1023/a:1018908421434. [DOI] [PubMed] [Google Scholar]
- 19.Yoo HS, Park TG. Biodegradable polymeric micelles composed of doxorubicin conjugated PLGA-PEG block copolymer. J Control Release. 2001;70:63–70. doi: 10.1016/s0168-3659(00)00340-0. [DOI] [PubMed] [Google Scholar]
- 20.Lee ES, Na K, Bae YH. Doxorubicin loaded pH-sensitive polymeric micelles for reversal of resistant MCF-7 tumor. J Control Release. 2005;103:405–18. doi: 10.1016/j.jconrel.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 21.Liu JR, Opipari AW, Tan L, Jiang Y, Zhang Y, Tang H, et al. Dysfunctional apoptosome activation in ovarian cancer: implications for chemoresistance. Cancer Res. 2002;62:924–31. [PubMed] [Google Scholar]
- 22.Wolf BB, Schuler M, Li W, Eggers-Sedlet B, Lee W, Tailor P, et al. Defective cytochrome c-dependent caspase activation in ovarian cancer cell lines due to diminished or absent apoptotic protease activating factor-1 activity. J Biol Chem. 2001;276:34244–51. doi: 10.1074/jbc.M011778200. [DOI] [PubMed] [Google Scholar]
- 23.Yang J, Lee CH, Park J, Seo S, Lim EK, Song YJ, et al. Antibody conjugated magnetic PLGA nanoparticles for diagnosis and treatment of breast cancer. J Mater Chem. 2007;17:2695–9. [Google Scholar]
- 24.Manchanda R, Fernandez-Fernandez A, Nagesetti A, McGoron AJ. Preparation and characterization of a polymeric (PLGA) nanoparticulate drug delivery system with simultaneous incorporation of chemotherapeutic and thermo-optical agents. Colloids Surf B Biointerfaces. 2010;75:260–7. doi: 10.1016/j.colsurfb.2009.08.043. [DOI] [PubMed] [Google Scholar]
- 25.Misra R, Sahoo SK. Intracellular trafficking of nuclear localization signal conjugated nanoparticles for cancer therapy. Eur J Pharm Sci. 2010;39:152–63. doi: 10.1016/j.ejps.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 26.Mo Y, Lim LY. Preparation and in vitro anticancer activity of wheat germ agglutinin (WGA)-conjugated PLGA nanoparticles loaded with paclitaxel and isopropyl myristate. J Control Release. 2005;107:30–42. doi: 10.1016/j.jconrel.2004.06.024. [DOI] [PubMed] [Google Scholar]
- 27.Monks A, Scudiero D, Skehan P, Shoemaker R, Paull K, Vistica D, et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor-cell lines. J Natl Cancer Inst. 1991;83:757–66. doi: 10.1093/jnci/83.11.757. [DOI] [PubMed] [Google Scholar]
- 28.Tang Y, McGoron AJ. Combined effects of laser-ICG photothermotherapy and doxorubicin chemotherapy on ovarian cancer cells. J Photochem Photobiol B. 2009;97:138–44. doi: 10.1016/j.jphotobiol.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 29.Sahoo SK, Panyam J, Prabha S, Labhasetwar V. Residual polyvinyl alcohol associated with poly (d,l-lactide-co-glycolide) nanoparticles affects their physical properties and cellular uptake. J Control Release. 2002;82:105–14. doi: 10.1016/s0168-3659(02)00127-x. [DOI] [PubMed] [Google Scholar]
- 30.Redhead HM, Davis SS, Illum L. Drug delivery in poly(lactide-co-glycolide) nanoparticles surface modified with poloxamer 407 and poloxamine 908: in vitro characterisation and in vivo evaluation. J Control Release. 2001;70:353–63. doi: 10.1016/s0168-3659(00)00367-9. [DOI] [PubMed] [Google Scholar]
- 31.Barnes TJ, Prestidge CA. PEO-PPO-PEO block copolymers at the emulsion droplet-water interface. Langmuir. 2000;16:4116–21. [Google Scholar]
- 32.Stolnik S, Dunn SE, Garnett MC, Davies MC, Coombes AG, Taylor DC, et al. Surface modification of poly(lactide-co-glycolide) nanospheres by biodegradable poly(lactide)-poly(ethylene glycol) copolymers. Pharm Res. 1994;11:1800–8. doi: 10.1023/a:1018931820564. [DOI] [PubMed] [Google Scholar]
- 33.Sun B, Ranganathan B, Feng SS. Multifunctional poly(d,l-lactide-co-glycolide)/montmorillonite (PLGA/MMT) nanoparticles decorated by trastuzumab for targeted chemotherapy of breast cancer. Biomaterials. 2008;29:475–86. doi: 10.1016/j.biomaterials.2007.09.038. [DOI] [PubMed] [Google Scholar]
- 34.Chittasupho C, Xie SX, Baoum A, Yakovleva T, Siahaan TJ, Berkland CJ. ICAM-1 targeting of doxorubicin-loaded PLGA nanoparticles to lung epithelial cells. Eur J Pharm Sci. 2009;37:141–50. doi: 10.1016/j.ejps.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tewes F, Munnier E, Antoon B, Ngaboni-Okassa L, Cohen-Jonathan S, Marchais H, et al. Comparative study of doxorubicin-loaded poly (lactide-co-glycolide) nanoparticles prepared by single and double emulsion methods. Eur J Pharm Biopharm. 2007;66:488–92. doi: 10.1016/j.ejpb.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 36.Stevanović M, Maksin T, Petković J, Filipič M, Uskoković D. An innovative, quick and convenient labeling method for the investigation of pharmacological behavior and the metabolism of poly(d,l-lactide-co-glycolide) nanospheres. Nanotechnology. 2009;20:335102. doi: 10.1088/0957-4484/20/33/335102. [DOI] [PubMed] [Google Scholar]
- 37.Arifin DY, Lee LY, Wang CH. Mathematical modeling and simulation of drug release from microspheres: implications to drug delivery systems. Adv Drug Deliv Rev. 2006;58:1274–325. doi: 10.1016/j.addr.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 38.Panyam J, Labhasetwar V. Sustained cytoplasmic delivery of drugs with intracellular receptors using biodegradable nanoparticles. Mol Pharm. 2004;1:77–84. doi: 10.1021/mp034002c. [DOI] [PubMed] [Google Scholar]
- 39.Qaddoumi MG, Gukasyan HJ, Davda J, Labhasetwar V, Kim KJ, Lee VH. Clathrin and caveolin-1 expression in primary pigmented rabbit conjunctival epithelial cells: role in PLGA nanoparticle endocytosis. Mol Vis. 2003;9:559–68. [PubMed] [Google Scholar]
- 40.Zuhorn IS, Kalicharan R, Hoekstra D. Lipoplex-mediated transfection of mammalian cells occurs through the cholesterol-dependent clathrin-mediated pathway of endocytosis. J Biol Chem. 2002;277:18021–8. doi: 10.1074/jbc.M111257200. [DOI] [PubMed] [Google Scholar]
- 41.Belloc F, Lacombe F, Dumain P, Lopez F, Bernard P, Boisseau MR, et al. Intercalation of anthracyclines into living cell DNA analyzed by flow cytometry. Cytometry. 1992;13:880–5. doi: 10.1002/cyto.990130811. [DOI] [PubMed] [Google Scholar]
- 42.Cartiera MS, Johnson KM, Rajendran V, Caplan MJ, Saltzman WM. The uptake and intracellular fate of PLGA nanoparticles in epithelial cells. Biomaterials. 2009;30:2790–8. doi: 10.1016/j.biomaterials.2009.01.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Panyam J, Zhou WZ, Prabha S, Sahoo SK, Labhasetwar V. Rapid endolysosomal escape of poly(d,l-lactide-co-glycolide) nanoparticles: implications for drug and gene delivery. FASEB J. 2002;16:1217–26. doi: 10.1096/fj.02-0088com. [DOI] [PubMed] [Google Scholar]
- 44.Miraglia E, Viarisio D, Riganti C, Costamagna C, Ghigo D, Bosia A. Na+/H+ exchanger activity is increased in doxorubicin-resistant human colon cancer cells and its modulation modifies the sensitivity of the cells to doxorubicin. Int J Cancer. 2005;115:924–9. doi: 10.1002/ijc.20959. [DOI] [PubMed] [Google Scholar]
- 45.Gong Y, Duvvuri M, Krise JP. Separate roles for the Golgi apparatus and lysosomes in the sequestration of drugs in the multidrug-resistant human leukemic cell line HL-60. J Biol Chem. 2003;278:50234–9. doi: 10.1074/jbc.M306606200. [DOI] [PubMed] [Google Scholar]
- 46.Duvvuri M, Krise JP. Intracellular drug sequestration events associated with the emergence of multidrug resistance: a mechanistic review. Front Biosci. 2005;10:1499–509. doi: 10.2741/1634. [DOI] [PubMed] [Google Scholar]
- 47.Yang R, Shim WS, Cui FD, Cheng G, Han X, Jin QR, et al. Enhanced electrostatic interaction between chitosan-modified PLGA nanoparticle and tumor. Int J Pharm. 2009;371:142–7. doi: 10.1016/j.ijpharm.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 48.Tang Y, Lei T, Manchanda R, Nagesetti A, Fernandez-Fernandez A, Srinivasan S, et al. Simultaneous delivery of chemotherapeutic and thermal-optical agents to cancer cells by a polymeric (PLGA) nanocarrier: an in vitro study. Pharm Res. 2010;27:2242–53. doi: 10.1007/s11095-010-0231-6. [DOI] [PubMed] [Google Scholar]
- 49.Manchanda R, Lei T, Tang Y, Fernandez-Fernandez A, McGoron AJ. Cellular uptake and cytotoxicity of a novel ICG-DOX-PLGA dual agent polymer nanoparticle delivery system. IFMBE Proc. 2010;32:228–31. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.