

Abstract
An early substantial loss of basal forebrain cholinergic neurons (BFCN) is a constant feature of Alzheimer's disease and is associated with deficits in spatial learning and memory. The ability to selectively control the differentiation of human embryonic stem cells (hESCs) into BFCN would be a significant step toward a cell replacement therapy. We demonstrate here a method for the derivation of a predominantly pure population of BFCN from hESC cells using diffusible ligands present in the forebrain at developmentally relevant time periods. Overexpression of two relevant human transcription factors in hESC-derived neural progenitors also generates BFCN. These neurons express only those markers characteristic of BFCN, generate action potentials, and form functional cholinergic synapses in murine hippocampal slice cultures. siRNA-mediated knockdown of the transcription factors blocks BFCN generation by the diffusible ligands, clearly demonstrating the factors both necessary and sufficient for the controlled derivation of this neuronal population. The ability to selectively control the differentiation of hESCs into BFCN is a significant step both for understanding mechanisms regulating BFCN lineage commitment and for the development of both cell transplant-mediated therapeutic interventions for Alzheimer's disease and high-throughput screening for agents that promote BFCN survival.
Keywords: Alzheimer's disease, Cholinergic fibers, Cell differentiation, Growth differentiation factor 2, Embryonic stem cell, Neural stem cell
Introduction
The basal forebrain cholinergic system is the predominant source of cortical cholinergic input. Alzheimer's disease-related tauopathies arise earliest in cholinergic neurons of the basal forebrain and loss of these neurons parallels cognitive decline [1]. Human lesion [2–4] and magnetic resonance imaging [5] studies have demonstrated the role of basal forebrain cholinergic neurons (BFCN) in memory function. Studies in monkeys, rats, and mice have further demonstrated the role of these cells in memory function [6–9], hippocampal neurogenesis [10–12], and functional plasticity of the cortex [13–17]. The group of BFCN arising from the median ganglionic eminence (MGE) is the largest and best characterized [18, 19]. Several proteins are characteristic of BFCN in vivo and in vitro. Choline acetyltransferase (ChAT), which catalyzes the formation of acetylcholine (ACh), is expressed by cholinergic neurons of both the basal forebrain and the motor system. In the cortex, the nerve growth factor receptor, TrkA, is expressed by BFCN from development through adulthood [20], and is necessary for nerve growth factor (NGF)–mediated survival of these neurons [21]. Another neurotrophin receptor (p75NTR, p75), is also expressed by more than 95% of ChAT-positive cells in the basal forebrain [22]. BFCN should also express acetylcholinesterase (AChE) and calbindin, but not nicotinamide adenine dinucleotide phosphate diaphorase, somatostatin, or HB9, which are specific to cortical, amygdalar, and motor cholinergic neuronal subpopulations.
Bone morphogenetic protein-9 (BMP9) is transiently expressed in vivo in the septum during the period of BFCN development and treatment of mouse septal cultures with BMP9 increases expression of cholinergic markers. Injection of BMP9 into E14 and E16 mouse ventricles increases levels of ACh detectable in the forebrain [23], and BMP9 induces the transcriptome of BFCN in cultured murine septal progenitors [24]. The effects of BMP9 on cholinergic neurogenesis are both spatially and temporally limited, having minimal effects outside of the E14-16 septum [23]. BFCN are also neurotrophin responsive, but TrkA, p75, and NGF [25] are not required for BFCN lineage commitment [26, 27].
Several transcription factors have been implicated in the differentiation of BFCN. Lhx8, a LIM-family homeodomain transcription factor, is expressed in the developing MGE [28], and Lhx8-expressing cells become cholinergic neurons [29, 30]. Lhx8-positive cells which become BFCN also express Gbx1 [31]. Murine Lhx8 knockout lines have reductions in BFCN with minimal effects on other neuronal subtypes [32–34], and Lhx8 is a pivotal factor for cholinergic differentiation of murine embryonic stem cells [35]. Nkx2.1, necessary for encoding the regional identity of the MGE, is expressed during development in all cells derived from this region [36]. Nkx2.1 knockout animals lack BFCN [37] and all other populations generated in or migrating through the MGE.
Transplantation of rodent fetal cholinergic neurons into the cortex of BFCN-lesioned adult primates restores memory function [38]. Transplantation of murine septal precursors, porcine cholinergic precursors, or the human embryonic septal/diagonal band region into physiologically relevant cortical areas of rodents resulted in the stable engraftment of the introduced cells, some of which differentiated into cholinergic neurons [39–42]. Although limited by the inability to generate highly purified BFCNs from precursor cells, these studies demonstrate the potential for transplanted exogenous BFCNs to ameliorate memory deficits through stable engraftment in adult cortex. Additionally, the use of human embryonic stem cell (hESC)-derived cells to screen novel drugs has been established [43], indicating that populations of purified human BFCN have the potential for use in discovery of drugs able to act specifically on this neuronal population.
Materials and Methods
Generation and Programming of Neural Progenitors with BMP9 Treatment
Using retinoic acid, we generated neural progenitors from hESC using modifications of an existing protocol [44] (for a schematic diagram of the protocol and media transitions, please refer to Supporting Information Fig. 12). Cells from the H7 hESC line were grown in adherent culture on Matrigel in hESC media conditioned for 24 hours on a feeder layer of 56,000 γ-irradiated mouse embryonic fibroblasts per centimeter square and supplemented to 4 ng ml−1 basic fibroblast growth factor (bFGF). All growth factors used were human recombinant isoforms from R&D Systems (Minneapolis, MN, www.rndsystems.com). Prior to splitting, all colonies were assayed for morphology and all imperfect colonies were manually removed (Supporting Information Fig. 13). Two days after splitting, cells were treated with 10 μM retinoic acid (RA) in conditioned media for 7 days. Fresh RA aliquots were used every week, and RA stock was made at 6 mM in 100% EtOH. RA-treated hESCs were dissociated for 4 minutes with 5 ml accutase/10-cm dish at 37°C followed by enzyme inactivation with media and gentle scraping with a cell scraper (Corning). After pelleting at 150g for 7 minutes, the small clumps were resuspended and grown in nonadherent flasks for 4 days in 25 ml hESC media without either conditioning, RA, or bFGF to begin neurosphere formation; half of the media volume was changed after 48 hours and the flasks were agitated twice a day to minimize sphere aggregation. These nascent neurosphere cultures were then allowed to gravity settle for 10 minutes, and then were moved by pipetting them into nonadherent 10-cm dishes for expansion in neurosphere media for 4 days with half the volume replaced after 48 hours. Plates were kept on a slow rotary shaker (in the 37°C incubator) to minimize neurosphere aggregation. To separate the neurospheres from debris and into equal numbers for sonic hedgehog (SSH)/fibroblast growth factor 8 (FGF8) and control pretreatment conditions, individual neurospheres were moved using a pipettor under a sterile microscope to the same media supplemented with 100 ng ml−1 FGF8 and 200 ng ml−1 SHH for 72 hours with half the volume replaced after 48 hours. Neurospheres were dissociated in 500 μl accutase at 37°C for 10 minutes, with gentle agitation at 5 and 10 minutes, pelleted, then treated with trypsin inhibitor (Invitrogen, Carlsbad, CA, www.invitrogen.com) at 37°C for a further 10 minutes. Cells were rinsed in Hank's balanced salt solution (HBSS; Invitrogen) and titrated gently through a p200 tip until all neurospheres had dissociated. Cells were plated on poly-D-lysine (PDL)–laminin (either 10-cm dishes or 12-mm glass coverslips, both BD Biosciences, San Jose, CA, www.bdbiosciences.com/home.jsp) in neuron media 1 for 5 days. For the first 24 hours, media was supplemented with 100 ng ml−1 SHH, 100 ng ml−1 FGF8, and 10 ng ml−1 BMP9. For the next 48 hours, media was supplemented with only BMP9, after which the cells were never again exposed to BMP9. Cells grew without additional mitogens for a subsequent 48 hours when they were moved to neuron media 2, which has been shown optimal for the growth of murine BFCN [45], from D5 to D16-19. From D5 to D10, media was supplemented with 2.66 μM arabinosylcytosine (AraC) to eliminate the growth of bFGF-responsive cells arising from fragments of undissociated neurospheres.
Generation of BFCN Through Nucleofection
Neurospheres were similarly pretreated for 72 hours with 100 ng ml−1 FGF8 and 200 ng ml−1 SHH for 72 hours. For nucleofections, we used 4 μg of DNA (a plasmid we cloned, which encodes both Lhx8 expressed from the cytomegalovirus promoter and Gbx1 expressed from the elongation factor 1-alpha promoter with a third promoter driving a constitutively active CAG-driven enhanced-green fluorescent protein (eGFP) [Supporting Information Fig. 14 online]) in hESC Nucleofection Solution-1 (Lonza, Basel, Switzerland, www.lonza.com) with program C-30 on an Amaxa nucleofector. For nucleofection, the neurospheres were again dissociated in 500 μl accutase at 37°C for 10 minutes, with gentle agitation at 5 and 10 minutes, pelleted, then treated with trypsin inhibitor at 37°C for a further 10 minutes. Cells were rinsed in HBSS and titrated gently through a p200 tip until all neurospheres had dissociated. The HBSS was completely aspirated to ensure that only nucleofection solution was present during nucleofection. Immediately following nucleofection the cuvette was gently rinsed with neuron media, which had been kept separately aliquoted at 37°C to ensure the cells were immediately warm following nucleofection; cells were subsequently kept for 10 minutes in a 37°C water bath. Following this warming step, cells were plated into 100 ng ml−1 SHH and 100 ng ml−1 FGF8 for 24 hours, but in the absence of BMP9. Control neurons were nucleofected with the same vector without the Lhx8 and Gbx1 sequences. After 48 hours of culture, the neurons were detached with a combination of 5 ml accutase and 5 ml 0.05% trypsin (Invitrogen) on the rotary shaker, pelleted, rinsed in HBSS, resuspended in neuron media 1, strained through a 100-μm filter (BD Biosciences) to remove debris, and kept on ice until they were FACS-purified at low pressure using a Beckman Coulter MoFlo after gating to exclude remaining debris and dead cells. Following FACS, the cells were rewarmed in a 37°C water bath for 10 minutes before being replated on PDL-laminin glass coverslips. Neurons were grown in the same media for the same periods as the BMP9 cells above (without BMP9 treatment) but without the AraC treatment as all of the FACS-purified cells differentiated following transcription factor expression.
For media composition and methods related to confirming the identity of the BFCN generated, please refer to the Supporting Information methods section
Results
Generation of BFCN Using Diffusible Ligands
We initially developed methods to differentiate hESCs into BFCN through treatment with diffusible ligands expressed in the developing murine MGE. hESC-derived neural progenitors (hNSCs) were first generated using retinoic acid and published protocols [44]. As FGF8 and SHH are necessary for patterning the developing neural tube and specification of the primordial forebrain [46], and together induce the transcriptome of the MGE while inhibiting lateral ganglionic eminence–specific factors [47], pretreatment with SHH and FGF8 was used to differentiate hNSCs toward a forebrain progenitor fate. Immunostaining for FORSE1, an immunohistochemical marker of forebrain progenitors [48], increases greatly after FGF8/SHH pretreatment (Supporting Information Fig. 4). FGF8/SHH preprogrammed hNSCs were then dissociated and treated transiently with BMP9. Quantitative real-time polymerase chain reaction (qRT-PCR) analysis of these neurons at DIV16 (day in vitro) (Fig. 1G) shows a large and significant increase in the expression of markers for the BFCN lineage, including ChAT, p75, TrkA, and AChE, whereas markers for other populations of cholinergic neurons, such as somatostatin and nitric oxide synthase, are expressed at levels below control neuronal cultures (data not shown). TuJ1 levels remain unchanged, indicating that BMP9/FGF8/SHH treatment alters lineage selection within progenitors already committed to a neural fate (data not shown). Immunostaining at DIV19 for ChAT or p75 shows a vast increase in numbers of marker+/microtubule-associated protein 2 (Map2)+ cells (immunocytochemistry Fig. 1A, 1C; counts Fig. 4A), with 85.59% ± 1.31% of cells becoming ChAT-positive neurons with a projection neuron morphology, whereas 9.24% ± 1.19% become ChAT-negative cells with an interneuron morphology (n = 5 replicate cultures; 2,582 cells). The absence of HB9 (HLXB9) immunopositivity demonstrates that these cholinergic neurons are not motor neurons (Supporting Information Fig. 1A, 1B). Control neurospheres, derived in parallel but neither pretreated with FGF8/SHH nor treated with BMP9, yielded 0.89% ± 0.24% cholinergic neurons (n = 4 cultures; 4,700 cells) (Fig. 1B, 1D), and expressed markers consistent with glutamatergic neurons (Supporting Information Fig. 2). Neither control nor BMP9-treated cultures contained cells immunopositive for markers of an astroglial (GFAP) or oligodendroglial (MBP) lineage (Supporting Information Fig. 3C) at DIV16. BMP9-derived neuronal axon growth and RNA expression were NGF-responsive, indicating their expression of TrkA (data not shown), which was confirmed through immunohistochemistry (Supporting Information Fig. 1C). Immunohistochemistry for calbindin (Supporting Information Fig. 1D) confirms that these cells express all known markers specific to the BFCN lineage. Direct treatment of hESC (instead of the FGF8/SHH restricted hNSC) with BMP9 did not generate neurons; without SHH/FGF8 pretreatment neurospheres fail to become BFCN after BMP9 treatment (data not shown). These data indicate that a temporally precise regime of neural restriction, followed by pretreatment with forebrain-specifying factors, followed by exposure to a factor expressed in the MGE during BFCN generation is able to generate a population of neurons significantly enhanced for markers of the BFCN lineage. Experiments also suggest that this protocol is also able to generate cholinergic neurons from the H1 hESC line (data not shown) in addition to the H7 hESC used for all other data presented. Only hESC with a passage number less than 35 were used in these studies to limit variability. These data demonstrate that BMP9 treatment of human forebrain progenitors is able to drive their differentiation into human BFCN.
Figure 1.
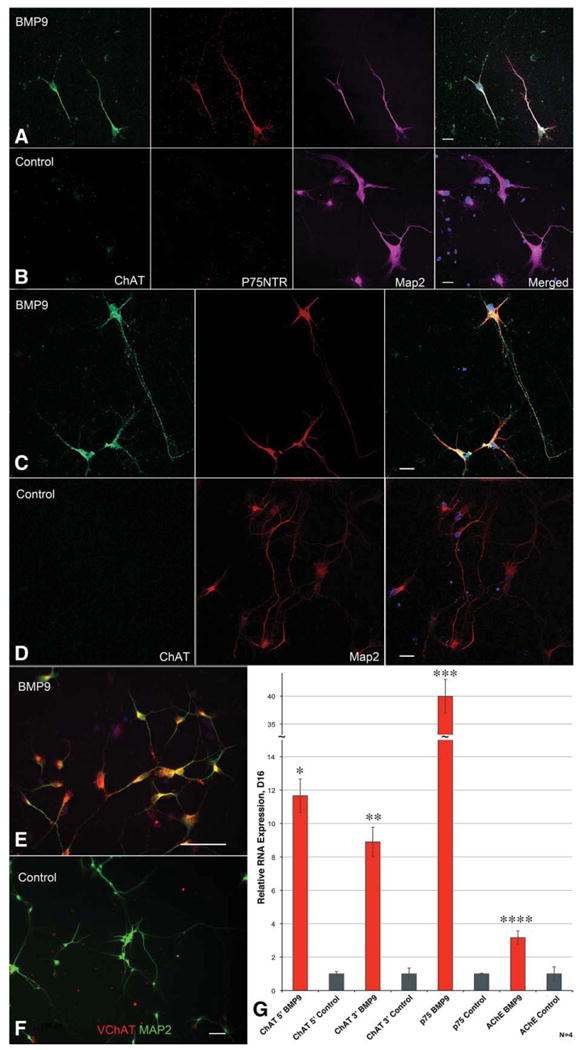
Generation of basal forebrain cholinergic neurons (BFCN) through sequential growth factor treatments. (A, B): Confocal microscopy demonstrates that cells generated through BMP9 treatment of fibroblast growth factor 8/sonic hedgehog pretreated neural progenitors express ChAT, p75, and Map2 and have a projection neuron morphology, whereas control neurons are only Map2-positive. (C, D): Confocal microscopy of equivalent cells stained only for ChAT and Map2 show the same BMP9 response and long ChAT+, Map2-axons. Scale bar = 20 μM. (E, F): BMP9-mediated ChAT immunopositivity is associated with expression of the VChaT. Lower magnification analysis of fields of neurons stained for VChAT. Scale bar = 100 μM. (G): qRT-PCR analysis shows 12- to 40-fold increases of RNA levels for markers characteristic of the BFCN. Bars are standard error, n = 4. All increases were significant by ANOVA (*, p < .0001; **, p = .0014; ***, p < .0001; ****, p < .0001). Data are from four replicate experiments; error bars show SEM. Data in (A–F) are from five replicate experiments, data in (G) is from four replicate experiments; error bars show SEM. Abbreviations: AChE, acetylcholinesterase; BMP9, bone morphogenetic protein-9; ChAT, choline acetyltransferase; Map2, microtubule-associated protein 2; VChAT, vesicular acetylcholine transporter.
Figure 4.
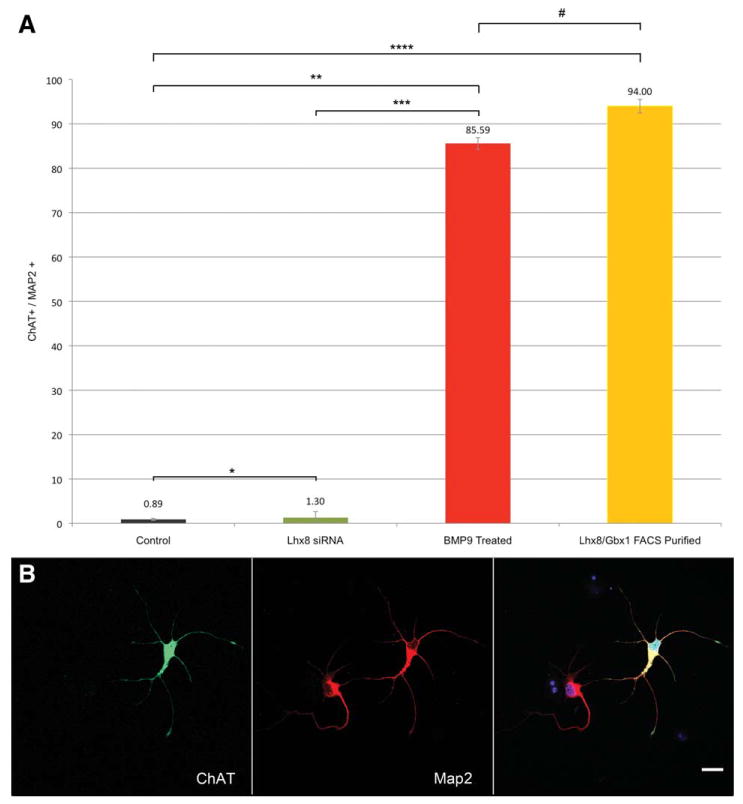
Quantification of neuronal differentiation into BFCN. (A): A high percentage of neurons from the FACS-purified (94% ± 1.53%) or BMP9-treated (85.59% ± 1.31%) populations are ChAT immunopositive, whereas control (0.89% ± 0.24%) and Lhx8 siRNA-treated (1.26% ± 1.33%) populations fail to express ChAT. All populations are significantly different by Mann-Whitney U test (*, p = .035; **, p < .001; ***, p = .001; ****, p = .006) except BMP9 versus nucleofected (#, p = .066). n = 4,700 control, 2,565 siRNA, 2,582 BMP9, or 1,718 nucleofected cells from four (control, BMP9 and nucleofected) or three (siRNA) replicate cultures. Error bars show SEM. (B): Representative ChAT immunohistochemistry demonstrates the clear distinction of ChAT immunopositivity between positive and negative cells. Scale bar = 20 μM. Abbreviations: BMP9, bone morphogenetic protein-9; ChAT, choline acetyltransferase; FACS, fluorescence-activated cell sorting; Map2, microtubule-associated protein 2.
Generation of BFCN Through Controlled Gene Expression
We then asked whether BFCN could also be generated through controlled expression of specific human genes. Based on murine embryology, the transcription factors Lhx8 and Gbx1 were chosen for further study. As no complete human Gbx1 sequence has been published, the full-length human sequence, with an open reading frame encoding 439 amino acids (Supporting Information Fig. S5) was derived through rapid amplification of cDNA ends experiments using a human 13-week fetal brain mRNA library. Involvement of these genes as downstream effectors of the BMP9 signaling cascade was confirmed through treating either control or FGF8/SHH-pretreated neurospheres with BMP9 and analyzing the transcription factor responses (Supporting Information Fig. 6). Lhx8 transcripts increased 72-fold at 6 hours following BMP9 treatment and remained elevated for 12 hours before decreasing. Gbx1 had a delayed 4.62-fold increase at 48 hours after treatment, whereas related factors with high-sequence homology (Lhx-6) or related to other neuronal populations (Islet1) were unaffected by BMP9 treatment (data not shown). These Lhx8 induction data were confirmed in dissociated progenitors as they differentiated into BFCN; BMP9 treatment resulted in Lhx8 transcript expression 2.59 times higher than equivalent FGF8/Shh-pretreated progenitors in the absence of BMP9 at 48 hours (data not shown). qRT-PCR (data not shown) and immunohistochemistry of FGF8/SHH pretreated neurospheres 24 hours after dissociation demonstrated that these neurons are derived from an Nkx2.1-expressing progenitor population: confocal analysis shows that over 85% of nuclei have robust expression of the Nkx2.1 transcription factor protein, as expected for forebrain progenitor cells (Supporting Information Fig. 7A, 7B). FoxG1, another transcription factor marker of forebrain-specific progenitors, should be highly expressed in forebrain progenitor cells and translocates from the nucleus to the cytoplasm as the cells differentiate [49]. As expected, the FGF8/SHH-pretreated progenitors express high levels of FoxG1 at both the protein and RNA levels. Staining 24 hours after dissociation shows strong FoxG1 expression clustered directly outside of the nucleus of a vast majority of cells, as expected of cells differentiating into BFCN (Supporting Information Fig. 7C, 7D), whereas 48 hours after dissociation the cells continue to express FoxG1 RNA at a level 13.45 times higher than cells from control neurospheres (Supporting Information Fig. 7E).
As Lhx8 and Gbx1 are specifically upregulated by BMP9 treatment of hNSCs, a technique was developed for nucleofection-based overexpression of these factors with a constitutively active eGFP in FGF8/SHH-pretreated hNSCs, allowing for FACS-based purification of overexpressing cells. FGF8/SHH-treated forebrain progenitors were nucleofected and grown without BMP9. For RNA experiments, populations of cells were used without FACS-purification. Although only ∼40% of cells were successfully nucleofected in each group (Supporting Information Fig. 8), this was sufficient to significantly increase expression of markers for the BFCN lineage (Fig. 2); these data were confirmed with FACS-purified neurons. Cells were FACS-purified 48 hours after nucleofection and then cultured for 19 days. Immunostaining for ChAT showed that 94% ± 1.53% of cells became cholinergic neurons with long projecting axons, whereas the remainder became neurons without ChAT expression or with an interneuron morphology (n = 3 cultures, 1,718 cells). Confocal analysis confirmed that the ChAT immunopositivity was entirely contained within the neuronal cytoplasm (Supporting Information Fig. 9A–9C and Fig. 4B). Dual immunostaining for ChAT/p75 demonstrated that these cells are positive for both markers, whereas control neurons are 18.67% ± 0.88% p75 positive. As the high magnification of these confocal micrographs allows only a few neurons to be visualized in each image, representative light microscopy was used to demonstrate the high density of the neuronal cultures prior to staining at 19 days (Supporting Information Fig. 13D). These RNA and immunohistochemical data show that the FACS-purified transiently Lhx8/Gbx1-overexpressing cells from SHH/FGF8 pretreated neural progenitors, after culture for 19 days, are a highly purified population of human neurons expressing only those markers characteristic of the BFCN. These experiments demonstrate that overexpression of the transcription factors Lhx8 and Gbx1 is able to generate neurons seemingly identical to the population generated by BMP9 treatment, suggesting that BMP9 treatment and transcription factor expression work through a similar mechanism.
Figure 2.
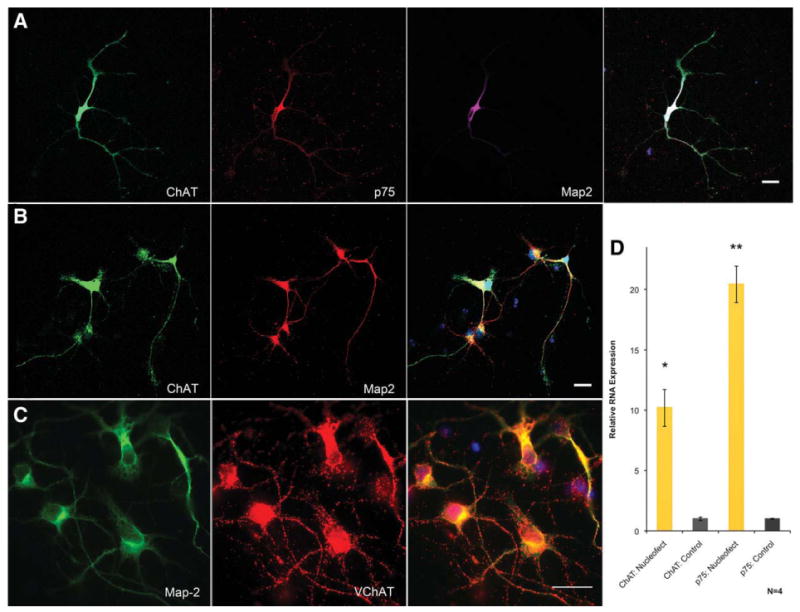
Generation of basal forebrain cholinergic neurons through transcription factor overexpression. (A): Confocal analysis of FACS-purified neurons shows strong expression of ChAT, p75, and Map2. (B): Confocal microscopy shows FACS-purified neurons immunostained only for ChAT and Map2. (C): FACS-purified neurons shown at higher power contain large numbers of vesicles positive for VChAT. Scale bars = 20 μM. (D): qRT-PCR analysis shows large increases in ChAT and p75 RNA transcript levels. Bars show SEM, n = 4. Both increases were shown to be significant by ANOVA (*, p = .0002; **, p = .0031). Abbreviations: ChAT, choline acetyltransferase; Map2, microtubule-associated protein 2; VChAT, vesicular acetylcholine transporter.
BMP9 and Lhx8/Gbx1 Overexpression Function Through One Pathway
Although these experiments demonstrate that the transcription factors Lhx8 and Gbx1 respond to BMP9 signaling after FGF8/SHH pretreatment and differentiate progenitors toward a BFCN phenotype, it remained unproven that the transcription factors mediate BMP9 signaling along a common pathway. To determine this, an siRNA-mediated knockdown of Lhx8 was performed on FGF8/SHH pretreated neural progenitors simultaneously with the BMP9 treatment paradigm. siRNA treatment blocked the BMP9-mediated upregulation of Lhx8 levels, reducing Lhx8 levels detectable by qRT-PCR to below basal expression levels (Fig. 3A). qRT-PCR analysis at D16 after Lhx8 knockdown demonstrates greatly reduced BFCN marker expression compared with equivalent BMP9-treated cells without siRNA (Fig. 3B), with expression levels not significantly different than untreated control cells nucleofected with control siRNA; this demonstrates that the knockdown is specific to the Lhx8 siRNA used and is not an artifact of the nucleofection procedure. Immunohistochemistry showed an equivalent reduction to 1.26% ± 1.33% of Map2-positive neurons expressing ChAT (immunocytochemistry Fig. 3C, counts Fig. 4A) (n = 3 cultures, 2,565 cells). That the Lhx8-specific knockdown blocks the effects of BMP9 further supports the conclusion that the ChAT+, p75+, Map2+ cells generated by both the small molecule and transcription factor treatments are BFCN as this is the only neuronal population specifically lost after null mutation of Lhx8 [32–34]. The ability of Lhx8 knockdown to block the effects of BMP9 treatment indicates that it is a necessary downstream effector of the BMP9-mediated signaling cascade in FGF8/SHH-pretreated neural progenitors, and demonstrates that the BMP9 treatment and the Lhx8/Gbx1 overexpression studies function through the same pathway.
Figure 3.
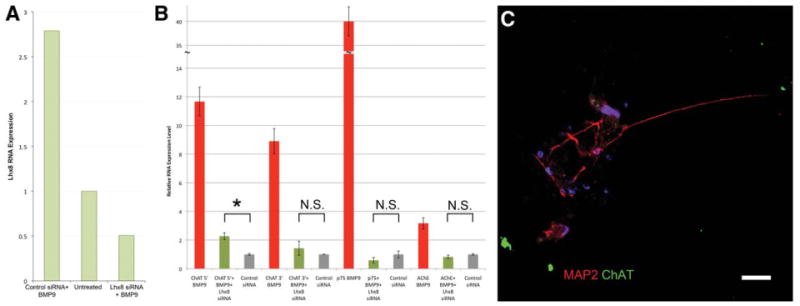
Lhx8 siRNA blocks BMP9 effects on basal forebrain cholinergic neurons (BFCN) differentiation. (A): qRT-PCR analysis demonstrates that Lhx8 siRNA nucleofection blocks the BMP9-mediated increase in Lhx8 levels, causing a reduction in Lhx8 transcript to levels below basal expression when compared with scrambled siRNA nucleofection after BMP9 treatment of dissociated and plated neural progenitors. (B): qRT-PCR analysis indicates that Lhx8 siRNA inhibits the BMP9-mediated BFCN differentiation of human neural progenitors, with only a twofold but still significant increase in levels of ChAT mRNA (*, p = .0225) after the siRNA treatment. (C): Neurons generated from Lhx8 siRNA-expressing neural progenitors fail to become ChAT immunopositive. Scale bar = 20 μM. Data in (B, C) are from three replicate experiments; error bars show SEM. Abbreviations: BMP9, bone morphogenetic protein-9; ChAT, choline acetyltransferase; Map2, microtubule-associated protein 2; N.S., not significant.
ChAT Staining Correlates with Enzymatically Functional ChAT
The functionality of the ChAT detected by immunohistochemistry was confirmed through direct detection of ACh. The cytoplasm of neurons from both derivation methods had markedly increased ACh levels: 5.11 ± 0.66 ng ACh per microgram of protein for BMP9 treatment and 6.94 ± 3.06 ng Ach per microgram of protein from nucleofections versus 0.83 ± 0.02 ng ACh per microgram of protein for control neurons (n = 3 replicate cultures per condition); as expected, Lhx8 siRNA expression reduced ACh levels toward basal levels: 1.79 ± 0.16 ng ACh per microgram of protein. These data are similar to or higher than the ACh concentration within cultured primary rat septal cholinergic neurons [50], confirming that neuronal ChAT immunopositivity correlates with physiologically relevant ChAT enzymatic activity.
Neurons Engraft in Mouse Ex Vivo Slice Cultures
Although neurons generated through Lhx8/Gbx1 nucleofection almost uniformly express markers characteristic of BFCN and produce ACh, it remained to be determined whether these cells would be functional after transplantation. To address this, a FACS-purified population was virally labeled with eGFP and allowed to engraft in cultured entorhinal-hippocampal murine cortical slices (i.e., ex vivo slice cultures) for 7–19 days. The cells migrated and extended long axonal projections deep into the cortical slices (Fig. 5A). Immunohistochemistry for synapsin1, a marker specific for the presynaptic terminal, showed a large number of presynaptic regions directly abutting neurite outgrowths from the engrafted cells (Fig. 5B), indicating, as confirmed electrophysiologically below, that murine neurons form functional synapses with these neurons. Confocal analysis also confirmed the presence of presynaptic regions within the engrafted cells (Fig. 5C; orthogonal view confirming synapsin1 inclusion Supporting Information Fig. 9D), suggesting that the BFCN are initiating synaptic transmission with murine cells. Immunohistochemistry for additional markers of mature presynaptic terminals shows the inclusion of puncta of synaptotagmin1 (the Ca2+ sensor necessary for vesicle release), synaptophysin1 (a protein intrinsic to presynaptic vesicles), bassoon (a presynaptic scaffolding molecule necessary for vesicle cycling), and SV2A (a synaptic vesicle protein involved in maintaining the presynaptic vesicle pool) (Supporting Information Fig. 10A–10D), indicating that these mature neurons express proteins performing diverse functions specific to the presynaptic area. The ability of these neurons to receive synaptic transmission was indicated by their expression of densin180, a factor specific to the postsynaptic density (Supporting Information Fig. 10E). To further characterize the presynaptic densities, we treated live cultures with fluorescently conjugated α-bungarotoxin, a highly selective and permanent α7-nicotinic cholinergic receptor antagonist. Axons from the engrafted BFCN terminated on α-bungarotoxin-positive processes (Fig. 5D), confirming that these neurons are capable of generating cholinergic synapses. These studies demonstrate that the engrafted human BFCN are able to stably engraft in the region of the murine hippocampus and form structures consistent with functional cholinergic synapses.
Figure 5.
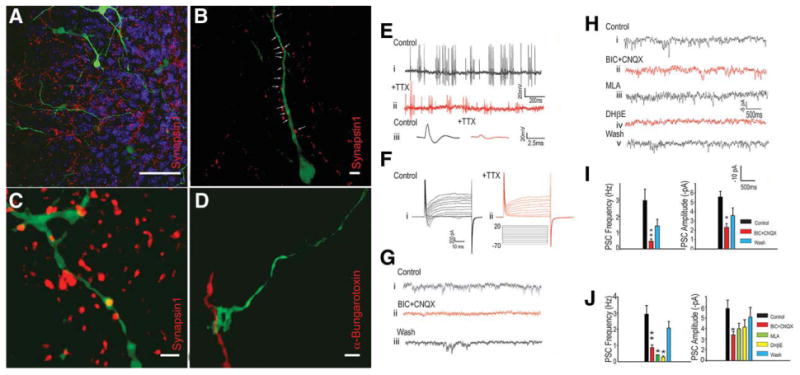
Immunohistochemical and electrophysiological evidence for and characterization of functional synaptic transmission after engraftment of FACS-purified neurons into murine ex vivo hippocampal slice cultures. (A): FACS-purified neuronal populations stably engraft in mouse hippocampal ex vivo slice cultures and project long networks of axons. All green fluorescence in Figure 5 is the enhanced-green fluorescent protein (eGFP) expression from the adenovirally labeled FACS-purified Lhx8/Gbx1 transiently overexpressing neurons. Scale bar = 50 μM. (B): Murine presynaptic terminals ((synapsin1, red channel) line the axons of the engrafted cells, giving immunohistochemical verification of the electrophysiologically detected synaptic inputs to these cells. Scale bar = 5 μM. (C): Transcription factor-generated basal forebrain cholinergic neurons contain presynaptic terminals (synapsin1, red channel) within their axons, indicating that they are generating synapses with other neurons. Please refer to Supporting Information Figure 8 for orthogonal view confirming inclusion of synapsin1 immunopositivity within the engrafted neuron. Scale bar = 2 μM. (D): α-Bungarotoxin labeling indicates that eGFP+ axons from FACS-purified neurons terminate on regions with postsynaptic α7 nicotinic acetylcholine receptors, strongly indicating the presence of cholinergic neurotransmission. Scale bar = 2 μM. (E): (i) Spontaneous action potentials recorded from an EGFP-expressing neuron. This cell was held at −60 mV in current clamp mode (n = 3). Second trace (ii) illustrates the gradual decrease in action potentials amplitude after addition of TTX (500 nM). Lower traces (iii) illustrate action potentials before and after TTX addition (from i and ii). (F): Voltage-dependent Na currents recorded under voltage clamp conditions from an eGFP-positive cell (i), illustrating block of the inward current by TTX (ii) (n = 4). (G): Spontaneous γ-aminobutyric acid-ergic PSCs were detected in eGFP-expressing cells (i). Representative traces of PSCs recorded under voltage clamp conditions. Using high KCl in the pipette, at −70 mV eGFP cells displayed numerous PSCs. The frequency of PSCs was greatly reduced after application of BIC (100 mM) and CNQX (10 mM) (*, p < .01; n = 7) (ii). PSCs reappeared after 10 minutes washing (iii). We observed that all PSCs were blocked by BIC when this question was specifically examined. Nevertheless, we always also included CNQX as well to block any 2-amino-3-(5-methyl-3-oxo-1,2-oxazol-4-yl)propanoic acid–mediated currents were they to occur. (H): (i) PSCs recorded from an eGFP-negative cell in close proximity to an eGFP-expressing cell. PSCs were recorded under whole-cell voltage clamp (−70 mV) conditions. (ii) These PSCs were partially blocked by BIC (100 mM) and CNQX (10 mM). (iii, iv) The frequencies of PSCs were further blocked by the nicotinic antagonists MLA (10 nM) and DHβE (1 mM). (v) PSCs reappeared following washout of these drugs. (I): In eGFP-expressing cells the frequency (**, p < .01) and amplitude (*, p < .05) of PSCs were significantly blocked by BIC (100 mM) plus CNQX (10 μM, n = 5). (J): In non-eGFP-expressing cells closely juxtaposed to eGFP-expressing cells MLA and DHβE produced a significant decrease in PSC frequency, (n = 7). Abbreviations: BIC, bicuculline; CNQX, 6-cyano-7-nitroquinoxaline-2,3-dione; DHβE, dihydro-β-erythroidine; MLA, methyllycaconitine; PSC, postsynaptic current; TTX, tetrodotoxin.
Electrophysiological Function After Engraftment in Murine Slice Cultures
To demonstrate that the neurons we generated are functional, we performed electrophysiological recordings from FACS-purified eGFP-expressing cells engrafted into hippocampal slice cultures. eGFP-expressing cells fired numerous spontaneous tetrodotoxin (TTX)–sensitive action potentials (Fig. 5E). Consistent with these observations voltage clamp recordings from these cells demonstrated TTX-sensitive voltage-dependent Na currents (Fig. 5F). The presence of these currents and associated action potentials expressed by eGFP-positive cells is clearly consistent with their neuronal phenotype. Furthermore, we observed that engrafted eGFP-positive cells exhibited numerous postsynaptic currents (PSCs) under voltage clamp conditions (Fig. 5G, 5H). These currents were blocked by the γ-aminobutyric acid A (GABA-A) receptor antagonist bicuculline and had a measured reversal potential consistent with their identity as GABA-A receptor currents (Supporting Information Fig. 11A, 11B). No PSCs recorded from eGFP expressing engrafted neurons were sensitive to nicotinic receptor blocking drugs including methyllycaconitine (MLA), a blocker of α7-nicotinic receptors, or dihydro-β-erythroidine (DHβE), a blocker of α4β2-nicotinic receptors. In contrast, when we recorded from neurons in close proximity to these eGFP-expressing neurons, we consistently observed that a significant percentage of small PSCs were blocked by MLA or DHβE, indicating the presence of nicotinic cholinergic synapses on these cells (Fig. 5I, 5J). Measurements of the reversal potentials for these PSCs were also consistent with their nicotinic identity (Supporting Information Fig. 11C, 11D). On the other hand, when we recorded from neurons distal to eGFP-expressing cells, we never observed cholinergic PSCs (not shown, n = 5). Thus, neurons generated through Lhx8/Gbx1 overexpression are electrically excitable, being capable of generating sodium currents and action potentials; they receive synaptic inputs, which are mediated by GABA-A receptors, and they are capable of quantal release of ACh at nicotinic synapses formed with other neurons; these properties establish these cells as bona fide cholinergic neurons.
Discussion
These experiments demonstrate the controlled generation of functional human BFCN from pluripotent stem cells. Treatment of hNSCs with human orthologs of ligands known to be present in the MGE during murine embryogenesis generated a relatively pure (85%) population of these neurons. Even higher yields and purity of these cells (94%) could be achieved through transient overexpression of the transcription factors Lhx8 and Gbx1 in hNSC. These neurons express all relevant markers of the BFCN lineage at both the RNA and protein levels, produce ACh in vitro and do not express markers of other cholinergic lineages. Further, they generate electrophysiologically functional cholinergic synapses and fire spontaneous and induced TTX-sensitive action potentials when engrafted into murine hippocampal slice cultures. Although the neurons generated though these experiments express all the markers known to be associated with the BFCN lineage while expressing no markers of other neuronal lineages, there are a number of highly related cortical cholinergic neuronal populations, and thus the precise subtype of cholinergic neuron generated cannot be rigorously demonstrated. Previous studies demonstrating the ability of transplanted BFCN to partially restore memory function following BFCN lesions [38–40] suggest a therapeutic role for these cells in Alzheimer's disease. Because ACh produced by BFCN functions as a hippocampal neuromodulator [51], it seems unnecessary for transplanted neurons to replace the exact synapses lost in Alzheimer's disease to enhance memory function. Additionally, the ability to derive these cells with high efficiency will allow the direct study of mechanisms regulating the survival and function of this critical population of human neurons, and also allows for the screening of drugs with actions specific to these neurons [43].
As is standard for nucleofecting hNSC, approximately 30% of cells die during the combined dissociation/nucleofection protocol; at DIV16–19, ∼100% of these plated progenitors become neurons in the conditions used for these experiments, and 94% of the successfully nucleofected surviving cells become BFCN. Thus, the overall efficiency considering all phases of the protocol is ∼65% (i.e., each 100 successfully nucleofected hESC-derived neurosphere cells generate 65 BFCNs) (Supporting Information Table 1). Neurosphere generation amplifies cell number so the yield expressed per each starting hESC is higher. The high efficiency of our protocol is one of its most important features. It is possible that other cell types transiently exist in the early neuronal cultures but are selected against through the media conditions used: due to the fragility of the newly plated neurons and the lack of marker expression or morphology early after cell adhesion, there is no way to accurately discern the transient presence of dying cells of other lineages from the natural cell death of the early neurons following routine plating. We only used hESC below passage number 35 for these studies to limit variability as responses of high-passage cells to epigenetic signals may change [52, 53].
Mirroring the narrow spatial and temporal window of cortical BMP9 response during embryogenesis, almost no BFCN were generated using these neural progenitors either 24 hours before or after the times used here or with other departures from this protocol. Complete dissociation of neurospheres is critical as undissociated fragments generate bFGF-responsive non-neuronal cells able to overgrow the cultures; similarly, cell–cell signaling in progenitors plated at too high a density blocks their proper differentiation into BFCN. Accordingly, we found no protocol able to generate BFCN using undissociated neurospheres. It remains unclear if Lhx8 and Gbx1 activity are the final mediators of signaling toward the BFCN lineage or if they are upstream of other genes along a common pathway. The differing responses of transcripts for these factors after BMP9 treatment suggests independent regulation, as does the prevalence of Gbx1 expression in presumptive BFCN in Lhx8 null mutants [38]. In the nucleofection experiments, both Gbx1 and Lhx8 were necessary for the induction of the cholinergic fate. Interestingly, nucleofection of each factor alone reproducibly generated a specific and different morphology of neuron but never BFCN, whereas individual overexpression of either Lhx8 or Gbx1 caused a small upregulation of the other factor. The BMP9-derived BFCN population contains some cells not committed to neuronal lineages but all cells generated from the FACS-purified nucleofections differentiated into neurons, suggesting that this population is suitable for transplantation; if necessary FACS-purification based on p75 expression [45] could remove the 6% non-BFCN neurons.
Conclusion
Together, our findings demonstrate that BMP9 signaling in hESC-derived forebrain precursor cells induces expression of Lhx8 and Gbx1, which are both necessary and sufficient to drive the differentiation of the cells into functional BFCN. We define here the pathways necessary for generation of functional human BFCN from hESC cells and demonstrate derivation of a predominantly pure population using either diffusible ligands present in forebrain at developmentally relevant time periods or overexpression of two relevant human transcription factors. siRNA-mediated knockdown of the transcription factors blocks BFCN generation by the diffusible ligands indicating a common pathway. These neurons express only those markers characteristic of BFCN at both the protein and RNA levels, stably engraft in murine ex vivo hippocampal slices, generate electrophysiologically functional cholinergic synapses expressing multiple mature synaptic markers, and have both spontaneous and evoked action potentials.
The data presented here describe all factors both necessary and sufficient for the controlled generation of this highly important neuronal population. The ability to selectively control the differentiation of human embryonic stem cells (hESCs) into BFCN is a significant step both for understanding mechanisms regulating BFCN lineage commitment and for the development of both cell-transplant-mediated therapeutic interventions for Alzheimer's disease and high-throughput screening for agents that promote BFCN survival.
Supplementary Material
Acknowledgments
We thank R. Awatramani for assistance with growth factor selection for forebrain lineage restriction; E. McMillan for neuron media one formulation and hNSC nucleofection program recommendation; M. Mesulam for BFCN marker advice; J. Nelson and P. Mehl for Northwestern University Flow Cytometry Core Facility; and V. Sahni for eGFP adenovirus purification. This work was supported by NIH grants NS 20013 and NS 20778 and by a grant from the Brinson Foundation.
Footnotes
Author contributions: C.B.: conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing; L.L.: provision of study material; B.B.: collection and assembly of data; A.B.: collection of data; R.M.: conception and design; J.K.: conception and design, financial support, data analysis and interpretation, final approval of manuscript.
Disclosure of Potential Conflicts of Interest: The authors indicate no potential conflicts of interest.
References
- 1.Mesulam M, Shaw P, Mash D, et al. Cholinergic nucleus basalis tauopathy emerges early in the aging-MCI-AD continuum. Ann Neurol. 2004;55:815–828. doi: 10.1002/ana.20100. [DOI] [PubMed] [Google Scholar]
- 2.Abe K, Inokawa M, Kashiwagi A, et al. Amnesia after a discrete basal forebrain lesion. J Neurol Neurosurg Psychiatry. 1998;65:126–130. doi: 10.1136/jnnp.65.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Salmond CH, Chatfield DA, Menon DK, et al. Cognitive sequelae of head injury: Involvement of basal forebrain and associated structures. Brain. 2005;128(part 1):189–200. doi: 10.1093/brain/awh352. [DOI] [PubMed] [Google Scholar]
- 4.Benke T, Koylu B, Delazer M, et al. Cholinergic treatment of amnesia following basal forebrain lesion due to aneurysm rupture—An open-label pilot study. Eur J Neurol. 2005;12:791–796. doi: 10.1111/j.1468-1331.2005.01063.x. [DOI] [PubMed] [Google Scholar]
- 5.Fujii T, Okuda J, Tsukiura T, et al. The role of the basal forebrain in episodic memory retrieval: a positron emission tomography study. Neuroimage. 2002;15:501–508. doi: 10.1006/nimg.2001.0995. [DOI] [PubMed] [Google Scholar]
- 6.Easton A, Ridley RM, Baker HF, et al. Unilateral lesions of the cholinergic basal forebrain and fornix in one hemisphere and inferior temporal cortex in the opposite hemisphere produce severe learning impairments in rhesus monkeys. Cereb Cortex. 2002;12:729–736. doi: 10.1093/cercor/12.7.729. [DOI] [PubMed] [Google Scholar]
- 7.Book AA, Wiley RG, Schweitzer JB. Specificity of 192 IgG-saporin for NGF receptor-positive cholinergic basal forebrain neurons in the rat. Brain Res. 1992;590:350–355. doi: 10.1016/0006-8993(92)91121-t. [DOI] [PubMed] [Google Scholar]
- 8.Pizzo DP, Thal LJ, Winkler J. Mnemonic deficits in animals depend upon the degree of cholinergic deficit and task complexity. Exp Neurol. 2002;177:292–305. doi: 10.1006/exnr.2002.7993. [DOI] [PubMed] [Google Scholar]
- 9.Moreau PH, Cosquer B, Jeltsch H, et al. Neuroanatomical and behavioral effects of a novel version of the cholinergic immunotoxin mu p75-saporin in mice. Hippocampus. 2008;18:610–622. doi: 10.1002/hipo.20422. [DOI] [PubMed] [Google Scholar]
- 10.Mohapel P, Leanza G, Kokaia M, et al. Forebrain acetylcholine regulates adult hippocampal neurogenesis and learning. Neurobiol Aging. 2005;26:939–946. doi: 10.1016/j.neurobiolaging.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 11.Kaneko N, Okano H, Sawamoto K. Role of the cholinergic system in regulating survival of newborn neurons in the adult mouse dentate gyrus and olfactory bulb. Genes Cells. 2006;11:1145–1159. doi: 10.1111/j.1365-2443.2006.01010.x. [DOI] [PubMed] [Google Scholar]
- 12.Cooper-Kuhn CM, Winkler J, Kuhn HG. Decreased neurogenesis after cholinergic forebrain lesion in the adult rat. J Neurosci Res. 2004;77:155–165. doi: 10.1002/jnr.20116. [DOI] [PubMed] [Google Scholar]
- 13.Conner JM, Culberson A, Packowski C, et al. Lesions of the Basal forebrain cholinergic system impair task acquisition and abolish cortical plasticity associated with motor skill learning. Neuron. 2003;38:819–829. doi: 10.1016/s0896-6273(03)00288-5. [DOI] [PubMed] [Google Scholar]
- 14.Conner JM, Chiba AA, Tuszynski MH. The basal forebrain cholinergic system is essential for cortical plasticity and functional recovery following brain injury. Neuron. 2005;46:173–179. doi: 10.1016/j.neuron.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 15.Kuczewski N, Aztiria E, Leanza G, et al. Selective cholinergic immunolesioning affects synaptic plasticity in developing visual cortex. Eur J Neurosci. 2005;21:1807–1814. doi: 10.1111/j.1460-9568.2005.04014.x. [DOI] [PubMed] [Google Scholar]
- 16.Ma X, Suga N. Augmentation of plasticity of the central auditory system by the basal forebrain and/or somatosensory cortex. J Neurophysiol. 2003;89:90–103. doi: 10.1152/jn.00968.2001. [DOI] [PubMed] [Google Scholar]
- 17.Thiel CM, Friston KJ, Dolan RJ. Cholinergic modulation of experience-dependent plasticity in human auditory cortex. Neuron. 2002;35:567–574. doi: 10.1016/s0896-6273(02)00801-2. [DOI] [PubMed] [Google Scholar]
- 18.Sweeney JE, Hohmann CF, Oster-Granite ML, et al. Neurogenesis of the basal forebrain in euploid and trisomy 16 mice: An animal model for developmental disorders in Down syndrome. Neuroscience. 1989;31:413–425. doi: 10.1016/0306-4522(89)90384-9. [DOI] [PubMed] [Google Scholar]
- 19.Schambra UB, Sulik KK, Petrusz P, et al. Ontogeny of cholinergic neurons in the mouse forebrain. J Comp Neurol. 1989;288:101–122. doi: 10.1002/cne.902880109. [DOI] [PubMed] [Google Scholar]
- 20.Li Y, Holtzman DM, Kromer LF, et al. Regulation of TrkA and ChAT expression in developing rat basal forebrain: Evidence that both exogenous and endogenous NGF regulate differentiation of cholinergic neurons. J Neurosci. 1995;15:2888–2905. doi: 10.1523/JNEUROSCI.15-04-02888.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sofroniew MV, Galletly NP, Isacson O, et al. Survival of adult basal forebrain cholinergic neurons after loss of target neurons. Science. 1990;247:338–342. doi: 10.1126/science.1688664. [DOI] [PubMed] [Google Scholar]
- 22.Mufson EJ, Ginsberg SD, Ikonomovic MD, et al. Human cholinergic basal forebrain: Chemoanatomy and neurologic dysfunction. J Chem Neuroanat. 2003;26:233–242. doi: 10.1016/s0891-0618(03)00068-1. [DOI] [PubMed] [Google Scholar]
- 23.Lopez-Coviella I, Berse B, Krauss R, et al. Induction and maintenance of the neuronal cholinergic phenotype in the central nervous system by BMP-9. Science. 2000;289:313–316. doi: 10.1126/science.289.5477.313. [DOI] [PubMed] [Google Scholar]
- 24.Lopez-Coviella I, Follettie MT, Mellott TJ, et al. Bone morphogenetic protein 9 induces the transcriptome of basal forebrain cholinergic neurons. Proc Natl Acad Sci USA. 2005;102:6984–6989. doi: 10.1073/pnas.0502097102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crowley C, Spencer SD, Nishimura MC, et al. Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergic neurons. Cell. 1994;76:1001–1011. doi: 10.1016/0092-8674(94)90378-6. [DOI] [PubMed] [Google Scholar]
- 26.Van der Zee CE, Hagg T. p75NGFR mediates death of cholinergic neurons during postnatal development of the neostriatum in mice. J Chem Neuroanat. 1998;14:129–140. doi: 10.1016/s0891-0618(98)00002-7. [DOI] [PubMed] [Google Scholar]
- 27.Lee KF, Li E, Huber LJ, et al. Targeted mutation of the gene encoding the low affinity NGF receptor p75 leads to deficits in the peripheral sensory nervous system. Cell. 1992;69:737–749. doi: 10.1016/0092-8674(92)90286-l. [DOI] [PubMed] [Google Scholar]
- 28.Matsumoto K, Tanaka T, Furuyama T, et al. L3, a novel murine LIM-homeodomain transcription factor expressed in the ventral telencephalon and themesenchyme surrounding the oral cavity. Neurosci Lett. 1996;204:113–116. doi: 10.1016/0304-3940(96)12341-7. [DOI] [PubMed] [Google Scholar]
- 29.Asbreuk CH, van Schaick HS, Cox JJ, et al. The homeobox genes Lhx7 and Gbx1 are expressed in the basal forebrain cholinergic system. Neuroscience. 2002;109:287–298. doi: 10.1016/s0306-4522(01)00466-3. [DOI] [PubMed] [Google Scholar]
- 30.Nobrega-Pereira S, Kessaris N, Du T, et al. Postmitotic Nkx2-1 controls the migration of telencephalic interneurons by direct repression of guidance receptors. Neuron. 2008;59:733–745. doi: 10.1016/j.neuron.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao Y, Guo YJ, Tomac AC, et al. Isolated cleft palate in mice with a targeted mutation of the LIM homeobox gene lhx8. Proc Natl Acad Sci USA. 1999;96:15002–15006. doi: 10.1073/pnas.96.26.15002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao Y, Marin O, Hermesz E, et al. The LIM-homeobox gene Lhx8 is required for thedevelopment of many cholinergic neurons in the mouse forebrain. Proc Natl Acad Sci USA. 2003;100:9005–9010. doi: 10.1073/pnas.1537759100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mori T, Yuxing Z, Takaki H, et al. The LIM homeobox gene, L3/Lhx8, is necessary for proper development of basal forebrain cholinergic neurons. Eur J Neurosci. 2004;19:3129–3141. doi: 10.1111/j.0953-816X.2004.03415.x. [DOI] [PubMed] [Google Scholar]
- 34.Fragkouli A, Hearn C, Errington M, et al. Loss of forebrain cholinergic neurons and impairment in spatial learning and memory in LHX7-deficient mice. Eur J Neurosci. 2005;21:2923–2938. doi: 10.1111/j.1460-9568.2005.04141.x. [DOI] [PubMed] [Google Scholar]
- 35.Manabe T, Tatsumi K, Inoue M, et al. L3/Lhx8 is a pivotal factor for cholinergic differentiation of murine embryonic stem cells. Cell Death Differ. 2007;14:1080–1085. doi: 10.1038/sj.cdd.4402106. [DOI] [PubMed] [Google Scholar]
- 36.Price M, Lazzaro D, Pohl T, et al. Regional expression of the homeobox gene Nkx-2.2 in the developing mammalian forebrain. Neuron. 1992;8:241–255. doi: 10.1016/0896-6273(92)90291-k. [DOI] [PubMed] [Google Scholar]
- 37.Sussel L, Marin O, Kimura S, et al. Loss of Nkx2.1 homeobox gene function results in a ventral to dorsal molecular respecification within the basal telencephalon: Evidence for a transformation of the pallidum into the striatum. Development. 1999;126:3359–3370. doi: 10.1242/dev.126.15.3359. [DOI] [PubMed] [Google Scholar]
- 38.Ridley RM, Baker JA, Baker HF, et al. Restoration of cognitive abilities by cholinergic grafts in cortex of monkeys with lesions of the basal nucleus of Meynert. Neuroscience. 1994;63:653–666. doi: 10.1016/0306-4522(94)90512-6. [DOI] [PubMed] [Google Scholar]
- 39.Nilsson OG, Brundin P, Widner H, et al. Human fetal basal forebrain neurons grafted to the denervated rat hippocampus produce an organotypic cholinergic reinnervation pattern. Brain Res. 1988;456:193–198. doi: 10.1016/0006-8993(88)90363-0. [DOI] [PubMed] [Google Scholar]
- 40.Cassel JC, Gaurivaud M, Lazarus C, et al. Grafts of fetal septal cells after cholinergic immunotoxic denervation of the hippocampus: A functional dissociation between dorsal and ventral implantation sites. Neuroscience. 2002;113:871–882. doi: 10.1016/s0306-4522(02)00226-9. [DOI] [PubMed] [Google Scholar]
- 41.LeBlanc CJ, Deacon TW, Whatley BR, et al. Morris water maze analysis of 192-IgG-saporin-lesioned rats and porcine cholinergic transplants to the hippocampus. Cell Transplant. 1999;8:131–142. doi: 10.1177/096368979900800105. [DOI] [PubMed] [Google Scholar]
- 42.Deacon T, Whatley B, LeBlanc C, et al. Pig fetal septal neurons implanted into the hippocampus of aged or cholinergic deafferented rats grow axons and form cross-species synapses in appropriate target regions. Cell Transplant. 1999;8:111–129. doi: 10.1177/096368979900800104. [DOI] [PubMed] [Google Scholar]
- 43.Jensen J, Hyllner J, Bjorquist P. Human embryonic stem cell technologies and drug discovery. J Cell Physiol. 2009;219:513–519. doi: 10.1002/jcp.21732. [DOI] [PubMed] [Google Scholar]
- 44.Lee JP, Jeyakumar M, Gonzalez R, et al. Stem cells act through multiple mechanisms to benefit mice with neurodegenerative metabolic disease. Nat Med. 2007;13:439–447. doi: 10.1038/nm1548. [DOI] [PubMed] [Google Scholar]
- 45.Schnitzler AC, Lopez-Coviella I, Blusztajn JK. Purification and culture of nerve growth factor receptor (p75)-expressing basal forebrain cholinergic neurons. Nat Protoc. 2008;3:34–40. doi: 10.1038/nprot.2007.477. [DOI] [PubMed] [Google Scholar]
- 46.Rhinn M, Picker A, Brand M. Global and local mechanisms of forebrain and midbrain patterning. Curr Opin Neurobiol. 2006;16:5–12. doi: 10.1016/j.conb.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 47.Tucker ES, Segall S, Gopalakrishna D, et al. Molecular specification and patterning of progenitor cells in the lateral and medial ganglionic eminences. J Neurosci. 2008;28:9504–9518. doi: 10.1523/JNEUROSCI.2341-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Allendoerfer KL, Magnani JL, Patterson PH. FORSE-1, an antibody that labels regionally restricted subpopulations of progenitor cells in the embryonic central nervous system, recognizes the Le(x) carbohydrate on a proteoglycan and two glycolipid antigens. Mol Cell Neurosci. 1995;6:381–395. doi: 10.1006/mcne.1995.1029. [DOI] [PubMed] [Google Scholar]
- 49.Regad T, Roth M, Bredenkamp N, et al. The neural progenitor-specifying activity of FoxG1 is antagonistically regulated by CKI and FGF. Nat Cell Biol. 2007;9:531–540. doi: 10.1038/ncb1573. [DOI] [PubMed] [Google Scholar]
- 50.Takei N, Tsukui H, Hatanaka H. Nerve growth factor increases the intracellular content of acetylcholine in cultured septal neurons from developing rats. J Neurochem. 1988;51:1118–1125. doi: 10.1111/j.1471-4159.1988.tb03076.x. [DOI] [PubMed] [Google Scholar]
- 51.Disterhoft JF, Oh MM. Pharmacological and molecular enhancement of learning in aging and Alzheimer's disease. J Physiol Paris. 2006;99:180–192. doi: 10.1016/j.jphysparis.2005.12.079. [DOI] [PubMed] [Google Scholar]
- 52.Tanasijevic B, Dai B, Ezashi T, et al. Progressive accumulation of epigenetic heterogeneity during human ES cell culture. Epigenetics. 2009;4:330–338. doi: 10.4161/epi.4.5.9275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maitra A, Arking DE, Shivapurkar N, et al. Genomic alterations in cultured human embryonic stem cells. Nat Genet. 2007;37:1099–1103. doi: 10.1038/ng1631. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.