

Abstract
The SET oncoprotein participates in cancer progression by affecting multiple cellular processes, inhibiting the tumor suppressor PP2A and inhibiting the metastasis suppressor nm23-H1. Based upon these multiple activities, we hypothesized that targeted inhibition of SET would have multiple discrete and measurable effects on cancer cells. Here, the effects of inhibiting SET oncoprotein function on intracellular signaling and proliferation of human cancer cell lines was investigated. We observed the effects of COG112, a novel SET interacting peptide, on PP2A activity, Akt signaling, nm23-H1 activity, and cellular migration/invasion in human U87 glioblastoma and MDA-MB-231 breast adenocarcinoma cancer cell lines. We found that COG112 interacted with SET protein and inhibited the association between SET and PP2A-c or nm23-H1. The interaction between COG112 and SET caused PP2A phosphatase, and nm23-H1 exonuclease activities, to increase. COG112-mediated increases in PP2A activity resulted in the inhibition of Akt signaling and cellular proliferation. Additionally, COG112 inhibited SET association with Rac1 leading to decreased cellular migration and invasion. COG112 treatment releases the SET-mediated inhibition of the tumor suppressor PP2A, as well as the metastasis suppressor nm23-H1. These results establish SET as a novel molecular target, and that the inhibition of SET may have beneficial effects in cancer chemotherapy.
Keywords: SET/I2PP2A, PP2A, Akt, nm23-H1, Rac1
Introduction
The protein encoded by the oncogene SET (TAF-1/I2PP2A) has multiple cellular functions including control of cell cycle (Canela et al., 2003; Carujo et al., 2006), gene transcription (Compagnone et al., 2000), apoptosis (Madeira et al., 2005), cell migration (ten Klooster et al., 2007) and epigenetic regulation (Seo et al., 2001). SET was discovered as the SET-CAN fusion gene associated with myeloid leukemogenesis (Adachi et al., 1994; von Lindern et al., 1992). SET is highly expressed in Wilms’ tumors (Carlson et al., 1998) and may contribute to liver carcinogenesis (Fukukawa et al., 2000). Furthermore, SET expression is high in rapidly dividing cells, but low in quiescent or contact-inhibited cells, indicating a potential selectivity for SET expression levels and cell growth potential (Carlson et al., 1998). Taken together, SET plays an important role in facilitating cellular growth and proliferation, and interacts with pathways that promote tumorigenesis and metastasis.
SET contributes to tumorigenesis, in part, by forming an inhibitory protein complex with protein phosphatase 2A (PP2A) (Li et al., 1995; Li et al., 1996). The tumor suppressor PP2A is a major cellular phosphatase that negatively regulates multiple pro-growth/pro-survival signaling pathways associated with cancer progression such as Akt, β-catenin and c-Myc (Arnold and Sears, 2008; Gotz et al., 2000; Resjö et al., 2002). These pathways are often dysregulated in cancers and SET expression further exacerbates the effect of uncontrolled signaling by inhibiting the endogenous regulators of these pathways. Interestingly, expression of the SET-CAN fusion gene in mice correlates with overexpression of β-catenin in stomach mucosa and indicates a role in cellular proliferation for this oncogene (Ozbek et al., 2007). As PP2A is the endogenous regulator of multiple pathways associated with cancer progression, the pharmacological activation of PP2A is a desirable goal for cancer chemoprevention and chemotherapy (Eichhorn et al., 2009; Westermarck and Hahn, 2008). Although commonly repressed in cancer cells, PP2A activity can be pharmacologically manipulated (Feschenko et al., 2002; Guichard et al., 2006; Neviani et al., 2007; Perrotti and Neviani, 2008; Switzer et al., 2009).
SET also forms an inhibitory complex with nm23-H1 (Fan et al., 2003), a metastasis suppressor whose expression level is inversely related to the metastatic potential of a cancer cell (Rosengard et al., 1989; Steeg et al., 1988). Nm23-H1 is a multi-tasking protein with histidine kinase, nucleoside-diphosphate kinase and 3’-5’ exonuclease activities (Freije et al., 1998). The DNA exonuclease activity of nm23-H1 is central to its metastasis suppressor function (Zhang et al., 2010) which SET modulates by forming a complex that sequesters nm23-H1 into the cytosol thereby preventing its exonuclease activity and promoting metastasis (Fan et al., 2003). Upon attack by cytotoxic lymphocytes (CTL), SET is cleaved by Granzyme A releasing nm23-H1 to translocate into the nucleus and exert its exonuclease activity (Fan et al., 2003). Therefore, SET plays a key role in numerous pathways that lead to more aggressive phenotypes and provides a unique opportunity to target multiple pathways involved in tumor progression.
Due to the diverse roles that SET plays in multiple pathways leading to cancer progression and metastasis, the pharmacological targeting of the SET oncoprotein could provide a novel approach to anti-cancer therapy. Recently COG112, a novel peptide based on a short fragment of apolipoprotein-E, was reported to bind SET expressed in immune cells that led to an increase in PP2A activity (Christensen et al.). We report here that COG112 targets the oncoprotein functions of SET in human cancer cells and results in the dissociation of SET from PP2A, nm23-H1 and Rac1. COG112 treatment also resulted in increased PP2A activity that corresponded to decreased Akt signaling and c-Myc stability. Furthermore, Nm23-H1 translocated to the nucleus under conditions where cellular migration and invasion was inhibited by this novel peptide. In contrast to cancer cells, COG112 shows limited toxicity and good bioavailability in whole animals, primary cells in culture and various cell lines, making it an attractive candidate for anticancer therapy in humans (Christensen et al.; Li et al., 2006). The data presented here indicate that pharmacological targeting of the oncoprotein SET results in multiple beneficial effects in the potential treatment of human cancer.
Results
COG112 Binds to SET Protein
COG112 was recently reported to interact with SET protein in immune cells (Christensen et al.). To confirm that COG112 interacted with SET from MDA-MB-231 human breast adenocarcinoma cells, either biotin or biotin-labeled COG112 was incubated with whole cell lysates and proteins bound to COG112 peptide were pulled down using streptavidin-conjugated agarose beads. Immunoprecipitated proteins were Western blotted and probed with anti-SET antibodies to show a predominant 39 KDa SET protein band that was not present in biotin-only controls (Figure 1A) suggesting a specific interaction between COG112 and the SET protein.
Figure 1. COG112 Interacts with SET and Increases PP2A Activity.

(A) COG112 interacts with SET. MDA-MB-231 whole cell lysate was incubated with biotin or biotin-COG112. Proteins pulled-down with streptavidin-agarose beads were immunobloted with anti-SET/I2PP2A. (B) COG112 inhibits SET:PP2A-c association. MDA-MB-231 cells were incubated with COG112 for 2 hr prior to EGF. SET and PP2A-c protein complexes were immunoprecipitated and analyzed for SET and PP2A-c co-precipitation. COG112 inhibited SET:PP2A-c complex formation in a concentration-dependent manner. (Inset: SET:PP2A-c association in serum starved cells treated with or without EGF) (C) COG112 increases PP2A activity. PP2A-c was immunoprecipitated from COG112 treated U87 cells and phosphatase activity measured using a synthetic phospho-peptide. Data represent mean values (± SEM) of released phosphate from three independent experiments. Significance is relative to EGF controls.
COG112 Disrupts PP2A-c and SET Association
To determine if COG112 can specifically inhibit PP2A-c from forming a complex with SET, serum-starved MDA-MB-231 cells were treated with COG112 for 2 hours prior to EGF stimulation. Co-immunoprecipitation studies show that COG112 caused a concentration-dependent decrease in SET bound to PP2A-c in EGF-stimulated cells (Figure 1B), indicating that COG112 reduced the interaction of SET with PP2A-c.
COG112 Increases PP2A Phosphatase Activity
To determine if COG112 altered PP2A activity, MDA-MB-231 and U87 cells were treated as above and phosphatase activity was measured from immunoprecipitated PP2A catalytic subunit (PP2A-c) using a specific phospho-peptide substrate. The activity of PP2A-c from untreated U87 cells had significantly higher phosphatase activity compared to EGF stimulated cells (Figure 1C). EGF-stimulated cells pretreated with COG112 (≥ 10 nM) had significantly higher phosphatase activity than EGF-stimulated cells (Figure 1C). Although COG112 pretreatment significantly increased PP2A phosphatase activity, it did not completely restore the activity to those measured in serum-starved untreated control cells.
COG112 Inhibits Akt Signaling via PP2A Activation
Akt is a critical target in cancer therapy (LoPiccolo et al., 2007) and is negatively regulated by PP2A (Resjö et al., 2002). We investigated the effects of COG112 on Akt signaling in response to EGF stimulation of MDA-MB-231 and U87 cells. Cells were treated with COG112 in the presence or absence of okadaic acid (OA), an inhibitor of PP2A, for 2 hours prior to stimulation with EGF. Akt activation, as measured by phospho-Akt (phospho-serine-473) levels, was concentration dependently inhibited by COG112 in the absence of OA, but had no effect on Akt activation in the presence of OA (Figure 2A). Analysis of three independent experiments shows that 100 and 1000 nM COG112 significantly decreased Akt activation compared to COG112 and OA treatment (Figure 2B). Furthermore, COG112 did not alter proximal EGF signaling as EGFR and PDK1 activation were not affected by treatments as shown by the lack of changes in phospho-EGFR (tyr 1045) and phospho-PDK1 (ser 241) levels (Figure 2C&D), indicating that the peptide acts downstream of Akt activation, consistent with PP2A activation.
Figure 2. COG112 Inhibits Akt Activation via PP2A.
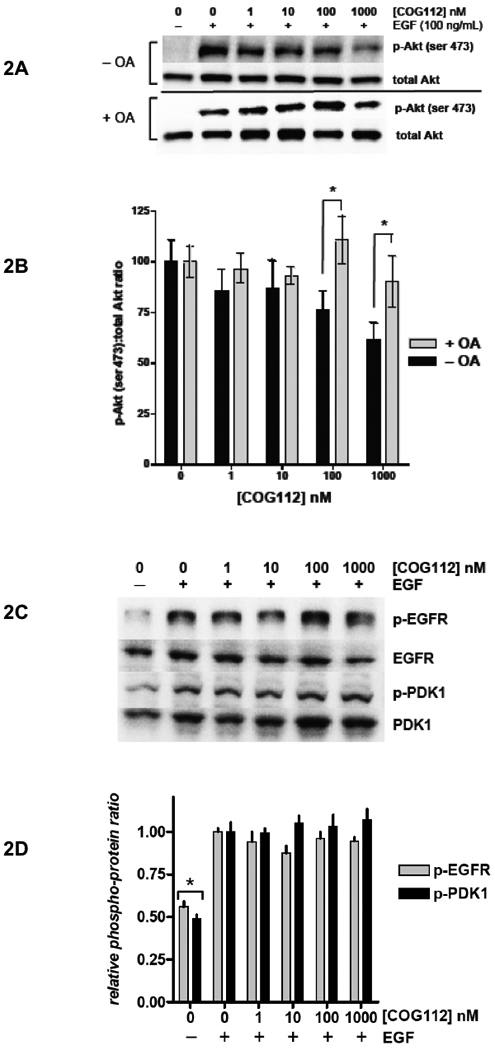
Serum-starved MDA-MB-231 cells were exposed to COG112 for 2 h and stimulated with EGF. (A) Western blot analysis of Akt activation in response to COG112 and EGF in the presence or absence of OA. (B) COG112 inhibits Akt activation in a concentration- and OA- dependent manner. Densitometry analysis of Akt (ser-473) phosphorylation versus COG112 is shown ± OA. Data represent mean ± SEM and significance was determined by paired t test. (C) Representative western blot and (D) densitometry analysis of EGFR and PDK1 activation in response to COG112 and EGF. Data represent mean values (± SEM) of the relative densitometry of phospho-protein:total protein from three independent experiments. Significance is relative to EGF controls.
Akt exerts its signal to proliferate by phosphorylating protein substrates such as mTOR and GSK-3β. To examine the effects of COG112 on Akt signaling partners in U87 cells, mTOR activation and GSK-3β inhibition were measured by western blot analysis. EGF stimulation caused a marked increase in mTOR serine-2448 phosphorylation in both cell lines tested (Figure 3A). However, COG112 pre-incubation decreased mTOR activation with a threshold concentration > 10 nM. In OA treated cells, mTOR phosphorylation is not decreased with respect to COG112 concentration. Similarly, GSK-3β serine-9 phosphorylation was increased upon EGF stimulation, which was decreased in COG112 treated cells (Figure 3B). OA blocked the decrease in GSK-3β phosphorylation due to COG112.
Figure 3. PP2A Mediates COG112 Inhibition of Cancer Cell Proliferation.


Serum-starved U87 cells were exposed to COG112 for 2 h and stimulated with EGF. Representative western blots of Akt substrates (A) mTOR (phosphoserine 2448), (B) GSK-3b (phosphoserine 9) show that COG112 inhibits Akt signaling but not in the presence of OA. (C) c-Myc protein levels are similarly down-regulated by COG112 in an OA-sensitive manner. (D) Nuclear protein from COG112 treated U87 cells were assayed for c-Myc binding to its consensus sequence DNA using an ELISA based kit. Data represent mean values (± SD) of relative c-Myc-DNA levels from three independent experiments. Significance is relative to untreated controls. (E) The effect of COG112 on Detroit 551 fibroblasts, MDA-MB-231 and U87 cell proliferation in serum-containing media. Data represent mean values (± SEM) and significance is relative to FBS positive control.
c-Myc is also a substrate for, and is negatively regulated by, PP2A activity (Arnold and Sears, 2008; Arnold and Sears, 2006). To determine the activity of PP2A on this endogenous protein substrate in response to COG112, MDA-MB-231 cells were treated as above. Total c-Myc protein levels were measured by western blot analysis and normalized to actin (Figure 3C). COG112 caused a concentration dependent decrease in c-Myc protein levels in the absence, but not in the presence of OA, indicating that the decrease of c-Myc protein levels by COG112 is mediated by PP2A. To further assess the ability of COG112 to inhibit c-Myc transcriptional activity, nuclear fractions were isolated from COG112 treated cells and the ability of c-Myc to bind its consensus DNA sequence was measured by an ELISA-based kit. COG112 caused a concentration-dependent decrease in the amount of c-Myc able to bind DNA (Figure 3D).
Because COG112 inhibited Akt activation and its downstream signaling, we examined the effects of COG112 on cellular proliferation. Adherent MDA-MB-231, U87 and Detroit 551 human fibroblast cells were grown for 24 hours in the presence of COG112 and cellular proliferation was measured by MTT reduction. Serum stimulation approximately doubled the number of cells compared to serum-starved control cells (Figure 3E). In MDA-MB-231 and U87 cells, COG112 caused a significant decrease in proliferation at 100 and 1000 nM. However COG112 did not affect the human fibroblast cell line indicating that this potential agent is innocuous to non-cancerous cells.
COG112 Dissociates nm23-H1 from SET
To determine if COG112 can displace nm23-H1 from its inhibitory complex with SET, MDA-MB-231 cells were treated with COG112 for 3 hours. Western blot of input lysate show equal amounts of both SET and nm23-H1 in treated cells (Figure 4A). Co-immunoprecipitation studies show that SET is bound to nm23-H1 in untreated control cells, however COG112 caused a significant decrease in nm23-H1 bound with SET (Figure 4A&B). COG112 treatment for 3 hours also resulted in the increased nuclear localization of nm23-H1 as determined by western blot analysis of fractionated cells (Figure 4C). To observe the localization of nm23-H1 with respect to time of COG112 administration, MDA-MB-231 cells were treated with 1 µM COG112 for various times. Western blotting of fractionated cells shows a marked increase in nuclear nm23-H1 after 3 and 18 hours of COG112 treatment. To examine the effect of COG112 on nm23-H1 exonuclease activity, MDA-MB-231 cells were incubated with COG112 and nuclear fractions were isolated. The exonuclease activity of the nuclear extract was assessed by S1 nuclease-hypersensitive site (SHS) oligonucleotide digestion (Ma et al., 2002). COG112 caused a marked increase in exonuclease activity (Figure 4E), corresponding to increased nm23-H1 nuclear translocation.
Figure 4. COG112 Increases nm23-H1 Metastasis Suppressor Activity.
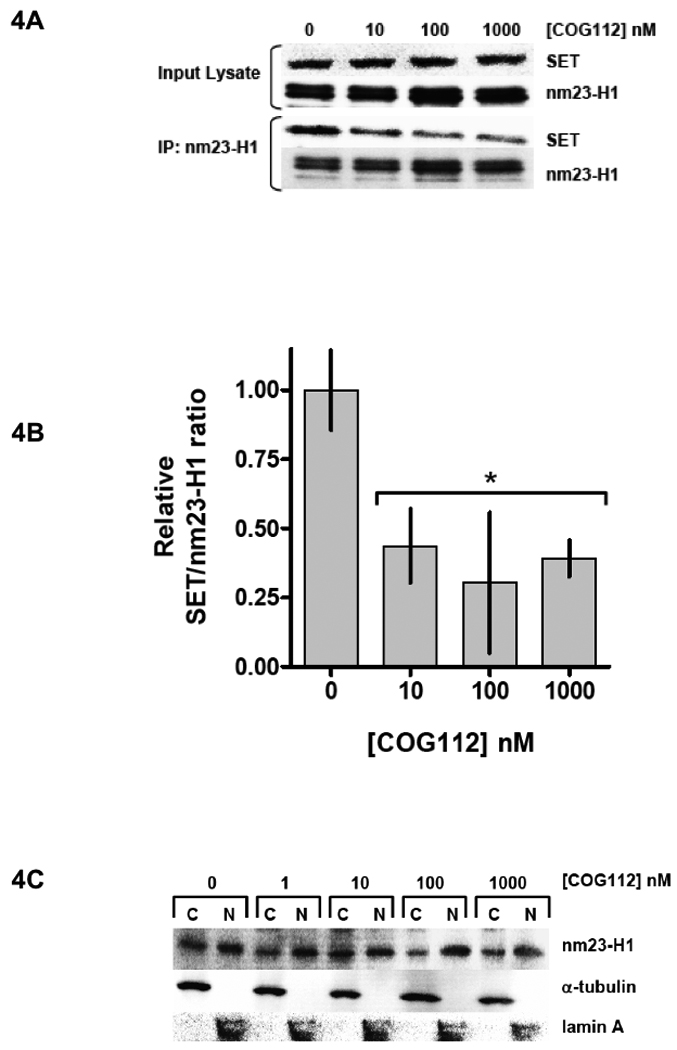

Treating MDA-MB-231 cells with COG112 for 3 hr caused SET to dissociate from nm23-H1. (A) Representative co-immunoprecipitation experiment showing reduced SET bound to nm23-H1 in COG112 treated cells. (B) Densitometry analysis of the SET:nm23-H1 complex in response to COG112. Data represent mean values (± SD) of relative SET:nm23-H1 densitometry from three independent experiments. Significance is relative to untreated controls. (B) Western blot of nuclear extract showing that COG112 caused the nuclear accumulation of nm23-H1. (C) Fractionated MDA-MB-231 cells were incubated with COG112 for 3 hr. COG112 resulted in the nuclear accumulation of nm23-H1. (D) MDA-MB-231 cells were treated with COG112 (1 µM) for indicated times and nm23-H1 levels were determined from fractionated cells. (E) COG112 increased the nuclear exonuclease activity of MDA-MB-231 cells. SHS oligonucleotides were incubated with nuclear protein from treated cells; DNA cleavage products are shown from a representative experiment.
COG112 Inhibits SET Association with Rac1, Cell Migration and Invasion
SET association is necessary for Rac1 mediated cell migration (ten Klooster et al., 2007). To determine if COG112 can inhibit SET/Rac1 association, serum-starved MDA-MB-231 cells were incubated with COG112 for 3 hours and then stimulated with EGF for 30 minutes. Co-immunoprecipitation experiments show that Rac1 complexes to SET in EGF-stimulated cells, but not in serum-starved cells (Figure 5A). Pre-treating cells with COG112 prior to EGF stimulation caused a concentration-dependent decrease in Rac1 association with SET. To measure the effect of COG112 on Rac1-GTPase activity, cells were treated as above and active Rac1 was pulled-down with a GST-Pak1 p21-binding domain fusion protein. Western blot shows that COG112 caused a concentration-dependent decrease in active Rac1-GTPase consistent with decreased SET/Rac1 binding (Figure 5B). The effect of COG112 on cell migration and invasion was assessed in MDA-MB-231 cells. Using 5% FBS as a chemotractant, MDA-MB-231 cells were incubated with COG112 and allowed to migrate or invade for 24 hours. COG112 treatment (≥ 10 nM) significantly decreased cell migration compared to vehicle-treated control cells (Figure 5C). At 1 µM COG112, migration was almost completely inhibited. Furthermore, cell invasion through matrigel correlated with the cell migration data to show that treatment with ≥ 10 nM COG112 significantly decreased invasion compared to vehicle-treated control cells and invasion was almost completely inhibited at 1 µM COG112 (Figure 5D).
Figure 5. COG112 Inhibits SET/Rac1 Association and Cell Migration.

(A) Co-imunoprecipitation experiments show that COG112 pretreatment inhibits SET/Rac1 association in EGF-stimulated MDA-MB-231 cells. (B) Active Rac1-GTPase was pulled-down using GST-PAK1-p21 protein binding domain fusion protein with glutathione-conjugated beads from COG112 pre-treated MDA-MB-231 cells. COG112 caused a concentration-dependent decrease in activated Rac1-GTPase in response to EGF. MDA-MB-231 cells were incubated with COG112 and allowed to (C) migrate or (D) invade using 5% FBS as a chemotractant for 18 hours. Migrating/invading cells were fixed, stained and counted. Data represent mean values (± SD) of cell counts from three independent experiments.
Discussion
This study demonstrates that targeting SET, an oncoprotein with numerous cellular functions, has multiple effects in human cancer cell lines. COG112, a chimeric peptide consisting of a short antennapedia sequence fused to the receptor recognition sequence of ApoE, was recently shown to increase PP2A activity in murine microglia cells by interacting with SET (Christensen et al.). Here we demonstrate that COG112 directly interacts with SET protein from MDA-MB-231 breast cancer cells, confirming that the peptide interacts with SET regardless of cell type. Furthermore, whole cell experiments show that the interaction between COG112 and SET inhibited the formation of the SET:PP2A-c complex in EGF-stimulated cells (Figure 1B). COG112 mediated inhibition of SET also correlated with increased cellular PP2A activity levels. COG112 (≥ 10 nM) pretreatment resulted in a significant increase in PP2A phosphatase activity compared to EGF-treated controls (Figure 1C), but did not completely restore PP2A activity levels to those found in controls. These results suggest that PP2A is negatively regulated by additional factors beyond SET in EGF-stimulated cancer cells. One of these factors may be Jak2 as EGFR activation induces Jak2 signaling (Olayioye et al., 1999), which has recently been shown to increase SET expression (Samanta et al., 2009). Furthermore Jak2 inhibition caused an increase in PP2A activity, suggesting a role for Jak2 in SET/PP2A regulation (Samanta et al., 2009). Other protein inhibitors of PP2A have been described such as CIP2A (Junttila et al., 2007) and I1PP2A (Li et al., 1995), and chemical factors such as reactive oxygen species, may also contribute to the inhibition or negative regulation of PP2A activity (Kim et al., 2003; Sommer et al., 2002).
PP2A has tumor suppressor functions and therefore its activity in cancer cells is commonly decreased by multiple mechanisms (Westermarck and Hahn, 2008). An attractive approach to cancer therapy is to pharmacologically increase PP2A activity since PP2A negatively regulates many signaling pathways associated with cancer progression and metastasis such as p38, Akt, β-catenin and c-Myc. Although PP2A is involved in many processes and recognizes a variety of substrates, PP2A is a highly regulated enzyme complex that is comprised of three subunits: a catalytic subunit, a scaffolding subunit and a regulatory subunit (Eichhorn et al., 2009). PP2A specific activity, substrate affinity and cellular localization are determined by the holoenzyme’s subunit composition. While releasing PP2A-c from its inhibitory complex with SET does not affect the formation of subsequent PP2A complexes, it does represent a strategy resulting in an overall increase in PP2A activity. Thus we cannot conclude that specific PP2A-heterotrimeric complexes are promoted by COG112, but rather that the levels of active PP2A heterotrimers, which are free of associated SET are increased by COG112.
COG112 resulted in decreased Akt, mTOR and GSK-3β phosphorylation in response to EGF. Akt and its downstream substrates mTOR and GSK-3β are regulated by PP2A (Westermarck and Hahn, 2008). However in the presence of OA, COG112 did not affect Akt, mTOR or GSK-3β activation indicating that PP2A mediated the inhibition of Akt signaling. Additionally c-Myc is regulated by PP2A activity (Arnold and Sears, 2006) and COG112 caused c-Myc protein and DNA binding to decrease in an OA-sensitive manner. Additionally these data suggest that PP2A activation results in decreased cancer cell proliferation, which is consistent with previous studies on pharmacological activation of PP2A in leukemias (Neviani et al., 2007; Perrotti and Neviani, 2006; Perrotti and Neviani, 2008). These results strongly support a mechanism where COG112 increases PP2A activity and that this increased phosphatase activity decreased Akt signaling and c-Myc protein levels.
The SET oncoprotein functions to inhibit the metastasis suppressor protein nm23-H1 by sequestering it in the cytosol (Fan et al., 2003). We show that COG112 treatment resulted in the decreased association of nm23-H1 with its inhibitor SET (Figure 4A&B), together with a concomitant increase in nuclear accumulation of nm23-H1 (Figures 4C&D). Nm23-H1 is a multi-functional protein, however its DNA 3’-5’ exonuclease activity is important to its metastasis suppressor function (Zhang et al., 2010). Furthermore, we show that COG112 increased the nuclear exonuclease activity of nm23-H1 (Figure 4E). SET is a substrate for Granzyme A (GzmA) and upon cytotoxic T-lymphocyte (CTL) attack, GzmA cleaves SET, which then allows nm23-H1 to exert its exonuclease activity (Fan et al., 2003). Our results indicate that COG112 may serve as an anti-metastasis agent for primary tumors through a mechanism where nm23-H1 exonuclease function is increased when COG112:SET complexes are increased. Cancer cells escape the immune response predominantly via immunosuppression (Stewart and Abrams, 2008), so an agent such as COG112 that can mimic CTL attack in the absence of a natural immune assault may have beneficial clinical impact. Nm23-H1 expression is pharmacologically increased by medroxyprogesterone (MPA) (Ouatas et al., 2003) that reduces metastases in vivo (Palmieri et al., 2005) and we hypothesize that COG112 may increase the efficacy of MPA even further by inhibiting the naturally occurring SET-inhibitor of nm23-H1 metastasis suppressor activity.
SET also contributes to tumor metastasis by binding to Rac1 during cell migration (ten Klooster et al., 2007). Cellular migration is necessary for cancer metastasis, as individual cancer cells must actively move to distal sites. COG112 inhibited the formation of Rac1 complexes with SET as well as active Rac1-GTPase in EGF-stimulated MDA-MB-231 cells. Preventing the Rac1:SET complex resulted in the inhibition of migration and invasion in COG112 treated cells. In addition to affecting migration by inhibiting the Rac1:SET complex and Rac1 activation, COG112 may also inhibit migration by other mechanisms. For example the activation of Rac1 initiates signaling via PAK1 and p38 MAPK (Zhang et al., 1995) and PP2A is known to suppress p38 and PAK1 activation (Sundaresan and Farndale, 2002; Westphal et al., 1999). Akt signaling is also known to contribute to migration (Qiao et al., 2008) and this is another mechanism by which COG112 may stimulate PP2A activity to inhibit migration. Also, nm23-H1 reduces Rac1 activity by interacting with the Rac1-specific nucleotide exchange factor Tiam1 (Otsuki et al., 2001). The potential for COG112 to simultaneously affect Rac1 function via PP2A and nm23-H1 activation highlights the multi-pathway approach of SET inhibition.
The remarkable success in developing agents that target specific pathways has advanced our understanding of the molecular mechanisms that cause cancer. Even with this success, the clinical reality is that cancer persists, and even recurs within the same patient, due to tumor heterogeneity, which leads to an attenuated response to an individual anti-cancer therapy. Thus it is necessary to develop agents that do not target just one pathway, but instead, concurrently target several cancer pathways without toxicity toward healthy tissues (Zhu et al., 2008). COG112 showed selective proliferation inhibition for aggressive cancer cells but did not affect normal fibroblasts (Figure 3E), suggesting that COG112 may fulfill these ideal requirements. The multi-functional action of COG112 exerted through the inhibition of SET indicates that SET may be a versatile molecular target due to its involvement with numerous tumor and metastatic promoting functions (Figure 6). Furthermore, the tumor suppressor PP2A is emerging as a target with inhibitory activity toward multiple cellular signaling pathways critical to proliferation, metastasis and angiogenesis in cancer maturation (Eichhorn et al., 2009). We propose that targeting the oncoprotein SET, an inhibitor of PP2A and other anticancer proteins, is a novel strategy for simultaneously affecting multiple pathways that are implicated in cancer progression and metastasis.
Figure 6. Targeting SET Oncoprotein Function has Multiple Effects.

An overview scheme of COG112 inhibition of SET functions. (A) SET functions to inhibit both PP2A and nm23-H1. SET is also required for Rac1-mediated cell migration. (B) In the presence of COG112, SET is unable to form protein complexes with PP2A-c, nm23-H1 and Rac1. We conclude that COG112 inhibits multiple oncoprotein functions of SET in human cancer cell lines.
We conclude that a novel ApoE-based peptide, COG112, inhibits the ability of SET to associate with PP2A-c, which correlates with increased PP2A activity in EGF-stimulated cancer cells (Figure 6). Furthermore, COG112 inhibited Akt and c-Myc activity by a mechanism including increasing PP2A activity. We also conclude that COG112 releases SET from nm23-H1 resulting in increased metastasis suppressor function of nm23-H1. Finally we conclude that COG112 inhibits cancer cell migration and invasion by inhibiting the binding of SET to Rac1. These conclusions point to the need for PP2A activators and/or Akt inhibitors, and to the desire to pharmacologically activate the tumor suppressor nm23-H1, as a combinatorial approach to control cancer. These results demonstrate that targeting SET oncoprotein functions is a novel approach to cancer therapy that achieves these goals.
Materials and Methods
Cell culture and reagents
U87-MG (U87) human glioblastoma, MDA-MB-231 human breast adenocarcinoma cells and Detroit 551 human skin fibroblasts (ATCC, VA) were cultured in DMEM, RPMI and MEM respectively (Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum (FBS) (Atlanta Biologicals, GA), 100 U/ml penicillin and 100 µg/ml streptomycin (Sigma-Aldrich, MO) and passaged two to three times per week. Human recombinant EGF was purchased from R&D Systems (Minneapolis, MN). Okadaic acid was purchased from Millipore (Billerica, MA). Pan Akt, p-Akt (serine 473), p-EGFR (tyrosine 992), EGFR, p-mTOR (serine 2448), total mTOR, p-GSK-3β, total GSK-3β, c-Myc, p-PDK1 (ser-2421), PDK1, lamin A, anti-mouse and anti-rabbit HRP antibodies were purchased from Cell Signaling (Danvers, MA), Rac1 antibody was from BD Biosciences (San Jose, CA) and SET/I2PP2A, PP2A-c, nm23-H1, α-tubulin and actin antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA).
Peptide Synthesis
COG112 is a chimeric peptide containing the antennapedia protein transduction domain (RQIKIWFQNRRMKWKKC) followed by COG133 (LRVRLASHLRKLRKRLL) (Laskowitz et al., 2001). The resulting sequence of COG112 (acetyl-RQIKIWFQNRRMKWKKCLRVRLASHLRKLRKRLL-amide) (Li et al., 2006) was synthesized using standard Fmoc chemistries and purified by NeoMPS, Inc. (San Diego, CA). Biotin was added at the amino-terminus during synthesis to produce biotin-COG112 (Polypeptide Systems, San Diego, CA). Lyophilized peptides were resuspended in sterile PBS and stock solutions were stored at −20°C.
Immunoprecipitation Western Blot Identification of SET:COG112 Complexes
MDA-MB-231 cells were grown to log phase and lysates prepared in NP40 buffer (100mM Tris, 0.2% NP40, 150 mM NaCl, pH 7.4). Protein concentrations of the lysate were adjusted to 1mg of total protein per ml of solution. Streptavidin agarose beads (1 mL) were washed with 10 mL of NP40 buffer and 0.1 mL of beads added to 2 ml of total protein extract (2 mg). After incubation for 1 hr at 4°C, beads were collected by centrifugation and the cleared lysate was removed and split into 2 tubes. Biotin-COG112 (10 µg) was added to one tube and biotin (1 µg) added to the other as a negative control. After incubation at 2 hrs at 4°C, 50 µl of washed beads were added to the lysate/peptide mixtures and incubated for an additional 2 hrs. Beads were collected by centrifugation, washed 5× with chilled NP40 buffer and centrifugation cycles. 15 µl of 4× LDS sample buffer (Invitrogen) was added to the beads and then heated to 90°C for 10 min, and briefly centrifuged. Supernatant proteins were analyzed by Western blot as detailed below.
Western blot analysis
Serum starved cells were treated for 2 hours with COG112 and stimulated with EGF for 5 minutes at 37°C. The cells were placed on ice and the media was aspirated and the cells were washed with cold PBS. Cells were harvested in cold 2X RIPA lysis buffer supplemented with 1 mM Na2VO4, 1 mM NaF, Protease Inhibitor Cocktail Set I (EMD Chemicals, NJ) and 0.2 mM PMSF (Sigma-Aldrich), and stored at −80°C. The whole cell lysates were cleared and the protein content was determined by BCA assay (Thermo Scientific, IL). Nuclear protein extracts were isolated using commercial kit (Active Motif, CA). Western blotting was performed as previously described (Switzer et al., 2009). PVDF membranes were imaged using ECF Western blotting kit (GE Healthcare, NJ) and recorded on a FluoroChem™ SP imaging system using AlphaEase® FC software (Alpha Innotech, CA). Images were further analyzed using ImageQuant software (Molecular Dynamics, CA).
Co-Immunoprecipitation Experiments and Active Rac1-GTPase Pull-down
Serum-starved MDA-MB-231 or U87 cells were incubated with COG112 for 2 hours prior to EGF stimulation for 30 minutes. Cells were lysed as above and Rac1 protein complexes were immunoprecipitated using Dynabeads® Protein G conjugated with either anti-Rac1 or anti-SET. Isolated protein complexes were washed with PBS (3×) and analyzed for both Rac1 and SET by western blot as described above. Active Rac1-GTPase was measured using a commercially available kit as directed (Active Rac1 Pull-Down and Detection Kit, Thermo Scientific).
Phosphatase Activity Assay
Serum starved MDA-MB-231 or U87 cells were treated with COG112 for 2 hours and then stimulated with EGF (100 ng/ml) for 5 minutes. Cells were lysed with NP40 buffer supplemented with 1 mM Na2VO4, 1 mM NaF, Protease Inhibitor Cocktail Set I (EMD Chemicals, NJ) and 0.2 mM PMSF (Sigma-Aldrich), and stored at −80°C. PP2A phosphatase activity was measured as previously described (Switzer et al., 2009). Briefly immunopreciptated PP2A-c is incubated with a PP2A-specific phospho-peptide substrate Arg-Arg-Ala-pThr-Val-Ala (BIOMOL) for 30 minutes at 30°C. The amount of phosphate released is determined using the Malachite green assay (Echelon Bioscience, Salt Lake City, USA).
c-Myc Activation assay
c-Myc activation was examined using the TransAM c-Myc ELISA (Active Motif). Briefly, nuclear protein extracts were allowed to bind to a c-Myc consensus sequence. Wells were washed 3 times and c-Myc antibody was added to each well for 1 hour. Wells were washed 3 times and incubated for 1 hour with HRP-conjugated antibody. Wells were washed 4 times before developing solution (100 µl) was added to each well and incubated for 10 minutes. Stop solution was added and absorbance measured at 450 nm.
Proliferation assays
Cells were seeded in media containing 10 % FBS and incubated for 6 hours to allow the cells adhere. The cells are washed (3X) with serum free media and serum-starved overnight. The cells were then incubated 24 hours in serum-replete media containing the indicated concentration of COG112. Proliferation was determined by the MTT assay and data represent 6 replicate experiments.
Exonuclease Assay
Nm23-H1 exonuclease activity was assessed as previously described with modification (Ma et al., 2002). Subconfluent MDA-MB-231 or U87 cells were treated with COG112 for 6 hr in serum-containing media and nuclear extracts were isolated as above. Single-stranded 5’-Alexa Fluor® 488-labeled SHS oligonucleotides were incubated with 5 µg of nuclear protein in Tris-buffered saline supplemented with 2 mM MgCl2 for 2 hr at room temperature. DNA cleavage products were separated by electrophoresis through 2% polyacrylamide gels and visualized by UV-fluorescent imaging using a FluoroChem™ SP system from Alpha Innotech. The SHS antisense sequence used was 3'-AGAGAGGTGGAGGGGGGGTGGGGAGGTGTGTAG-5'.
Cell Migration and Invasion Assays
MDA-MB-231 cells (5,000 cells in 0.5 mL serum-free RPMI with indicated concentration of COG112) were placed into either BD multiwell insert systems (for migration assays) or BD Matrigel Invasion Chambers (for invasion assays) and 0.75 mL of RPMI + 5% FBS was added to the bottom chamber. Cells were incubated for 24 hr at 37°C; non-migrating/invading cells were removed by aspiration and membranes were scrubbed with wet cotton swabs. Migrating/invading cells were then fixed in cold methanol for 10 min at −20°C, rinsed with water, stained with crystal violet, rinsed again and dried. The cells were then manually counted under 40X magnification to determine the number of migrating or invading cells.
Statistical Analysis
Data represented are mean of at least three independent experiments ± SEM. Statistical comparisons were performed by t test or one-way ANOVA with Dunnett’s post-test analysis using GraphPad Prism software (La Jolla, CA). Significance was indicated by p<0.05.
Footnotes
Conflict of interest
Michael P. Vitek and Dale J. Christensen are stockholders in Cognosci, Inc., which has patent rights on a reagent used in this study. The remaining authors declare no potential conflict of interest.
References
- Adachi Y, Pavlakis GN, Copeland TD. Identification and characterization of SET, a nuclear phosphoprotein encoded by the translocation break point in acute undifferentiated leukemia. J Biol Chem. 1994;269:2258–2262. [PubMed] [Google Scholar]
- Arnold H, Sears R. A tumor suppressor role for PP2A-B56α through negative regulation of c-Myc and other key oncoproteins. Cancer and Metastasis Reviews. 2008;27:147–158. doi: 10.1007/s10555-008-9128-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold HK, Sears RC. Protein Phosphatase 2A Regulatory Subunit B56{alpha} Associates with c-Myc and Negatively Regulates c-Myc Accumulation. Mol. Cell. Biol. 2006;26:2832–2844. doi: 10.1128/MCB.26.7.2832-2844.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canela N, Rodriguez-Vilarrupla A, Estanyol JM, Diaz C, Pujol MJ, Agell N, et al. The SET protein regulates G2/M transition by modulating cyclin B-cyclin-dependent kinase 1 activity. J Biol Chem. 2003;278:1158–1164. doi: 10.1074/jbc.M207497200. [DOI] [PubMed] [Google Scholar]
- Carlson SG, Eng E, Kim EG, Perlman EJ, Copeland TD, Ballermann BJ. Expression of SET, an inhibitor of protein phosphatase 2A, in renal development and Wilms' tumor. J Am Soc Nephrol. 1998;9:1873–1880. doi: 10.1681/ASN.V9101873. [DOI] [PubMed] [Google Scholar]
- Carujo S, Estanyol JM, Ejarque A, Agell N, Bachs O, Pujol MJ. Glyceraldehyde 3-phosphate dehydrogenase is a SET-binding protein and regulates cyclin B-cdk1 activity. Oncogene. 2006;25:4033–4042. doi: 10.1038/sj.onc.1209433. [DOI] [PubMed] [Google Scholar]
- Christensen D, Ohkubo N, Oddo J, Van Kanegan MJ, Neil J, Li F, et al. Apolipoprotein-E and Peptide Mimetics Modulate Inflammation by Binding the SET Protein and Activating Protein Phosphatase 2A. Journal of Immunology. doi: 10.4049/jimmunol.1002847. in press. [DOI] [PubMed] [Google Scholar]
- Compagnone NA, Zhang P, Vigne J-L, Mellon SH. Novel Role for the Nuclear Phosphoprotein SET in Transcriptional Activation of P450c17 and Initiation of Neurosteroidogenesis. Mol Endocrinol. 2000;14:875–888. doi: 10.1210/mend.14.6.0469. [DOI] [PubMed] [Google Scholar]
- Eichhorn PJA, Creyghton MP, Bernards R. Protein phosphatase 2A regulatory subunits and cancer. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer. 2009;1795:1–15. doi: 10.1016/j.bbcan.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Fan Z, Beresford PJ, Oh DY, Zhang D, Lieberman J. Tumor suppressor NM23-H1 is a granzyme A-activated DNase during CTL-mediated apoptosis, and the nucleosome assembly protein SET is its inhibitor. Cell. 2003;112:659–672. doi: 10.1016/s0092-8674(03)00150-8. [DOI] [PubMed] [Google Scholar]
- Feschenko MS, Stevenson E, Nairn AC, Sweadner KJ. A Novel cAMP-Stimulated Pathway in Protein Phosphatase 2A Activation. J Pharmacol Exp Ther. 2002;302:111–118. doi: 10.1124/jpet.302.1.111. [DOI] [PubMed] [Google Scholar]
- Freije JM, MacDonald NJ, Steeg PS. Nm23 and tumour metastasis: basic and translational advances. Biochem Soc Symp. 1998;63:261–271. [PubMed] [Google Scholar]
- Fukukawa C, Shima H, Tanuma N, Ogawa K, Kikuchi K. Up-regulation of I-2(PP2A)/SET gene expression in rat primary hepatomas and regenerating livers. Cancer Lett. 2000;161:89–95. doi: 10.1016/s0304-3835(00)00598-x. [DOI] [PubMed] [Google Scholar]
- Gotz J, Probst A, Mistl C, Nitsch RM, Ehler E. Distinct role of protein phosphatase 2A subunit Calpha in the regulation of E-cadherin and beta-catenin during development. Mech Dev. 2000;93:83–93. doi: 10.1016/s0925-4773(00)00267-7. [DOI] [PubMed] [Google Scholar]
- Guichard C, Pedruzzi E, Fay M, Marie J-C, Braut-Boucher F, Daniel F, et al. Dihydroxyphenylethanol induces apoptosis by activating serine/threonine protein phosphatase PP2A and promotes the endoplasmic reticulum stress response in human colon carcinoma cells. Carcinogenesis. 2006;27:1812–1827. doi: 10.1093/carcin/bgl009. [DOI] [PubMed] [Google Scholar]
- Junttila MR, Puustinen P, Niemelä M, Ahola R, Arnold H, Böttzauw T, et al. CIP2A Inhibits PP2A in Human Malignancies. Cell. 2007;130:51–62. doi: 10.1016/j.cell.2007.04.044. [DOI] [PubMed] [Google Scholar]
- Kim HS, Song MC, Kwak IH, Park TJ, Lim IK. Constitutive induction of p-Erk1/2 accompanied by reduced activities of protein phosphatases 1 and 2A and MKP3 due to reactive oxygen species during cellular senescence. J Biol Chem. 2003;278:37497–37510. doi: 10.1074/jbc.M211739200. [DOI] [PubMed] [Google Scholar]
- Laskowitz DT, Thekdi AD, Thekdi SD, Han SK, Myers JK, Pizzo SV, et al. Downregulation of microglial activation by apolipoprotein E and apoE-mimetic peptides. Exp Neurol. 2001;167:74–85. doi: 10.1006/exnr.2001.7541. [DOI] [PubMed] [Google Scholar]
- Li F-Q, Sempowski GD, McKenna SE, Laskowitz DT, Colton CA, Vitek MP. Apolipoprotein E-Derived Peptides Ameliorate Clinical Disability and Inflammatory Infiltrates into the Spinal Cord in a Murine Model of Multiple Sclerosis. J Pharmacol Exp Ther. 2006;318:956–965. doi: 10.1124/jpet.106.103671. [DOI] [PubMed] [Google Scholar]
- Li M, Guo H, Damuni Z. Purification and Characterization of Two Potent Heat-Stable Protein Inhibitors of Protein Phosphatase 2A from Bovine Kidney. Biochemistry. 1995;34:1988–1996. doi: 10.1021/bi00006a020. [DOI] [PubMed] [Google Scholar]
- Li M, Makkinje A, Damuni Z. The Myeloid Leukemia-associated Protein SET Is a Potent Inhibitor of Protein Phosphatase 2A. J. Biol. Chem. 1996;271:11059–11062. doi: 10.1074/jbc.271.19.11059. [DOI] [PubMed] [Google Scholar]
- LoPiccolo J, Granville CA, Gills JJ, Dennis PA. Targeting Akt in cancer therapy. Anticancer Drugs. 2007;18:861–874. doi: 10.1097/CAD.0b013e3280cc2c6f. [DOI] [PubMed] [Google Scholar]
- Ma D, Xing Z, Liu B, Pedigo NG, Zimmer SG, Bai Z, et al. NM23-H1 and NM23-H2 Repress Transcriptional Activities of Nuclease-hypersensitive Elements in the Platelet-derived Growth Factor-A Promoter. Journal of Biological Chemistry. 2002;277:1560–1567. doi: 10.1074/jbc.M108359200. [DOI] [PubMed] [Google Scholar]
- Madeira A, Pommet JM, Prochiantz A, Allinquant B. SET protein (TAF1beta, I2PP2A) is involved in neuronal apoptosis induced by an amyloid precursor protein cytoplasmic subdomain. FASEB J. 2005;19:1905–1907. doi: 10.1096/fj.05-3839fje. [DOI] [PubMed] [Google Scholar]
- Neviani P, Santhanam R, Oaks JJ, Eiring AM, Notari M, Blaser BW, et al. FTY720, a new alternative for treating blast crisis chronic myelogenous leukemia and Philadelphia chromosome-positive acute lymphocytic leukemia. Journal of Clinical Investigation. 2007;117:2408–2421. doi: 10.1172/JCI31095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olayioye MA, Beuvink I, Horsch K, Daly JM, Hynes NE. ErbB Receptor-induced Activation of Stat Transcription Factors Is Mediated by Src Tyrosine Kinases. Journal of Biological Chemistry. 1999;274:17209–17218. doi: 10.1074/jbc.274.24.17209. [DOI] [PubMed] [Google Scholar]
- Otsuki Y, Tanaka M, Yoshii S, Kawazoe N, Nakaya K, Sugimura H. Tumor metastasis suppressor nm23H1 regulates Rac1 GTPase by interaction with Tiam1. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:4385–4390. doi: 10.1073/pnas.071411598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouatas T, Halverson D, Steeg PS. Dexamethasone and medroxyprogesterone acetate elevate Nm23-H1 metastasis suppressor gene expression in metastatic human breast carcinoma cells: new uses for old compounds. Clin Cancer Res. 2003;9:3763–3772. [PubMed] [Google Scholar]
- Ozbek U, Kandilci A, van Baal S, Bonten J, Boyd K, Franken P, et al. SET-CAN, the Product of the t(9;9) in Acute Undifferentiated Leukemia, Causes Expansion of Early Hematopoietic Progenitors and Hyperproliferation of Stomach Mucosa in Transgenic Mice. Am J Pathol. 2007;171:654–666. doi: 10.2353/ajpath.2007.060934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri D, Halverson DO, Ouatas T, Horak CE, Salerno M, Johnson J, et al. Medroxyprogesterone acetate elevation of Nm23-H1 metastasis suppressor expression in hormone receptor-negative breast cancer. J Natl Cancer Inst. 2005;97:632–642. doi: 10.1093/jnci/dji111. [DOI] [PubMed] [Google Scholar]
- Perrotti D, Neviani P. ReSETting PP2A tumour suppressor activity in blast crisis and imatinib-resistant chronic myelogenous leukaemia. Br J Cancer. 2006;95:775–781. doi: 10.1038/sj.bjc.6603317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrotti D, Neviani P. Protein phosphatase 2A (PP2A), a drugable tumor suppressor in Ph1(+) leukemias. Cancer and Metastasis Reviews. 2008;27:159–168. doi: 10.1007/s10555-008-9119-x. [DOI] [PubMed] [Google Scholar]
- Qiao M, Sheng S, Pardee AB. Metastasis and AKT activation. Cell Cycle. 2008;7:2991–2996. doi: 10.4161/cc.7.19.6784. [DOI] [PubMed] [Google Scholar]
- Resjö S, Göransson O, Härndahl L, Zolnierowicz S, Manganiello V, Degerman E. Protein phosphatase 2A is the main phosphatase involved in the regulation of protein kinase B in rat adipocytes. Cellular Signalling. 2002;14:231–238. doi: 10.1016/s0898-6568(01)00238-8. [DOI] [PubMed] [Google Scholar]
- Rosengard AM, Krutzsch HC, Shearn A, Biggs JR, Barker E, Margulies IM, et al. Reduced Nm23/Awd protein in tumour metastasis and aberrant Drosophila development. Nature. 1989;342:177–180. doi: 10.1038/342177a0. [DOI] [PubMed] [Google Scholar]
- Samanta AK, Chakraborty SN, Wang Y, Kantarjian H, Sun X, Hood J, et al. Jak2 inhibition deactivates Lyn kinase through the SET-PP2A-SHP1 pathway, causing apoptosis in drug-resistant cells from chronic myelogenous leukemia patients. Oncogene. 2009;28:1669–1681. doi: 10.1038/onc.2009.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo SB, McNamara P, Heo S, Turner A, Lane WS, Chakravarti D. Regulation of histone acetylation and transcription by INHAT, a human cellular complex containing the set oncoprotein. Cell. 2001;104:119–130. doi: 10.1016/s0092-8674(01)00196-9. [DOI] [PubMed] [Google Scholar]
- Sommer D, Coleman S, Swanson SA, Stemmer PM. Differential susceptibilities of serine/threonine phosphatases to oxidative and nitrosative stress. Arch Biochem Biophys. 2002;404:271–278. doi: 10.1016/s0003-9861(02)00242-4. [DOI] [PubMed] [Google Scholar]
- Steeg PS, Bevilacqua G, Pozzatti R, Liotta LA, Sobel ME. Altered expression of NM23, a gene associated with low tumor metastatic potential, during adenovirus 2 Ela inhibition of experimental metastasis. Cancer Res. 1988;48:6550–6554. [PubMed] [Google Scholar]
- Stewart TJ, Abrams SI. How tumours escape mass destruction. Oncogene. 2008;27:5894–5903. doi: 10.1038/onc.2008.268. [DOI] [PubMed] [Google Scholar]
- Sundaresan P, Farndale RW. P38 mitogen-activated protein kinase dephosphorylation is regulated by protein phosphatase 2A in human platelets activated by collagen. FEBS Lett. 2002;528:139–144. doi: 10.1016/s0014-5793(02)03277-5. [DOI] [PubMed] [Google Scholar]
- Switzer CH, Ridnour LA, Cheng RYS, Sparatore A, Del Soldato P, Moody TW, et al. Dithiolethione compounds inhibit Akt signaling in human breast and lung cancer cells by increasing PP2A activity. Oncogene. 2009;28:3837–3846. doi: 10.1038/onc.2009.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Klooster JP, Leeuwen Iv, Scheres N, Anthony EC, Hordijk PL. Rac1-induced cell migration requires membrane recruitment of the nuclear oncogene SET. EMBO J. 2007;26:336–345. doi: 10.1038/sj.emboj.7601518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Lindern M, van Baal S, Wiegant J, Raap A, Hagemeijer A, Grosveld G. Can, a putative oncogene associated with myeloid leukemogenesis, may be activated by fusion of its 3' half to different genes: characterization of the set gene. Mol Cell Biol. 1992;12:3346–3355. doi: 10.1128/mcb.12.8.3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermarck J, Hahn WC. Multiple pathways regulated by the tumor suppressor PP2A in transformation. Trends Mol Med. 2008;14:152–160. doi: 10.1016/j.molmed.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Westphal RS, Coffee RL, Jr, Marotta A, Pelech SL, Wadzinski BE. Identification of kinase-phosphatase signaling modules composed of p70 S6 kinase-protein phosphatase 2A (PP2A) and p21-activated kinase-PP2A. J Biol Chem. 1999;274:687–692. doi: 10.1074/jbc.274.2.687. [DOI] [PubMed] [Google Scholar]
- Zhang Q, McCorkle JR, Novak M, Yang M, Kaetzel DM. Metastasis suppressor function of NM23-H1 requires its 3′-5′ exonuclease activity. International Journal of Cancer. 2010;128:40–50. doi: 10.1002/ijc.25307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Han J, Sells MA, Chernoff J, Knaus UG, Ulevitch RJ, et al. Rho family GTPases regulate p38 mitogen-activated protein kinase through the downstream mediator Pak1. J Biol Chem. 1995;270:23934–23936. doi: 10.1074/jbc.270.41.23934. [DOI] [PubMed] [Google Scholar]
- Zhu F, Zheng CJ, Han LY, Xie B, Jia J, Liu X, et al. Trends in the exploration of anticancer targets and strategies in enhancing the efficacy of drug targeting. Curr Mol Pharmacol. 2008;1:213–232. doi: 10.2174/1874467210801030213. [DOI] [PubMed] [Google Scholar]