

Abstract
Several genes in an interval of human and mouse chromosome 1 are associated with a predisposition for systemic lupus erythematosus. Congenic mouse strains that contain a 129-derived genomic segment, which is embedded in the B6 genome, develop lupus because of epistatic interactions between the 129-derived and B6 genes, e.g. in B6.129chr1b mice. If a gene that is located on chromosome 1 is altered through homologous recombination in 129-derived embryonic stem cells (ES cells) and if the resultant knockout mouse is backcrossed with B6, interpretation of the phenotype of the mutant mouse may be affected by epistatic interactions between the 129 and B6 genomes. Here, we report that knockout mice of two adjacent chromosome 1 genes, Slamf1−/− and Slamf2−/−, which were generated with the same 129-derived ES cell line, develop features of lupus, if backcrossed on to the B6 genetic background. By contrast, Slamf1−/− [BALB/c.129] and Slamf2−/− [BALB/c.129] do not develop disease. Surprisingly, Slamf1−/− [B6.129] mice develop both auto-antibodies and glomerulonephritis between 3 and 6 months of age, while disease fully develops in Slamf1−/− [B6.129] mice after 9–14 months. Functional analyses of CD4+ T cells reveals that Slamf2−/− T cells are resistant to tolerance induction in vivo. We conclude that the Slamf2−/− mutation may have a unique influence on T-cell tolerance and lupus.
Keywords: CD48, congenic, lupus, SLAM, SLE
Introduction
Systemic lupus erythematosus (SLE) is a multisystem autoimmune disease, marked by a range of auto-antibodies with a long prodromal phase of auto-antibody development and epitope spreading. This pre-diagnosis phase [positive antinuclear antibody (ANA) and musculoskeletal discomfort] is often marked by elevated serum BLyS/BAFF and MIF levels, which implicates B-cell activation and myeloid (macrophage and dendritic cells) stimulation. The major hallmark of SLE is the production of auto-antibodies to self-determinants and these auto-antibodies are predominantly directed against intracellular and nuclear antigens. Even if initial events are more focused on the B-cell arm, overt clinical disease involves a ‘network’ of immunological cells (T, B, dendritic cells and macrophage) and the repertoire of mechanisms for an inflammatory response. A comprehensive genetic dissection of the immunoregulatory pathways that lead to the SLE in humans and mice is therefore necessary.
Genome-wide linkage scans in SLE families have identified several lupus susceptibility loci (1). Evidence for one or more lupus susceptibility loci on human 1q23 comes from multiple genome-wide linkage scans in humans, which has been replicated (2–8). In mice, genome-wide linkage studies have implicated the syntenic region to human 1q23 in three different models of spontaneous lupus: the (NZB × NZW)F2 intercross, the NZM/Aeg2410 New Zealand mice and the BXSB mice (9–11). The phenotype of these mice is very similar to that in SLE patients, with the production of auto-antibodies as well as multiorgan involvement, including severe nephritis. In congenic mice derived from crossing the NZM2410 mouse strain with B6 mice, the locus on chromosome 1, i.e. Sle1, by itself was sufficient to generate a strong, spontaneous humoral ANA response, reacting primarily with H2A/H2B/DNA subnucleosomes. Sle1 also led to an expanded pool of histone-reactive T cells. Sle1 is thought to be a major player in orchestrating selective loss of B-cell and T-cell tolerance to chromatin. Fine mapping of the Sle1 locus determined that three loci within this congenic interval, termed Sle1a, Sle1b and Sle1c, could independently cause a loss of tolerance to chromatin, a necessary step for full disease induction (12). More recently, the Sle1b region has been defined as an ∼0.9 Mb segment (0.4 cM) that includes seven polymorphic signaling lymphocytic activation molecule family (Slamf) cell surface receptor genes (13). Slamf members regulate T cell, macrophage, dendritic cell, neutrophil and platelet functions, as well as humoral immune responses. Thus, Slamf members are ideal candidates for controlling SLE relevant cellular and signal transduction pathways.
Recent studies suggest that the two alternative splice forms of the Slamf receptor Ly108 (CD352 / Slamf6), each of which is found in one of the major haplotypes, could be key contributors to role of Sle1b in tolerance (13, 14). Here, we investigate the roles of two other Slamf members, Slamf1 (CD150 / SLAM) and Slamf2 (CD48) in tolerance to chromatin and susceptibility to lupus. Both Slamf1 and Slamf2 have IgV-like and IgC-like extracellular domains, but Slamf1 is a type I transmembrane glycoprotein, while Slamf2 has a glycosylphosphatidylinositol membrane anchor. Slamf1 is a self-ligand, whereas Slamf2 interacts with Slamf4 (CD244) and CD2. In addition, Slamf1 is one of the two known receptors for measles virus and Slamf2 is a receptor for the lectin FimH present on pili of certain enterobacteriaceae. Slamf1 is expressed on the surface of activated and memory T cells as well as on activated B cells, dendritic cells, macrophages and platelets, while Slamf2 is expressed on T cells, B cells, dendritic cells, macrophages, NK cells and eosinophils. Slamf1 plays a key role in controlling T-cell and macrophage functions. Slamf2 regulates T-cell activation and differentiation (15).
Here, we use Slamf1−/− and Slamf2−/− mice, which were generated with the same 129-derived ES cell line and then backcrossed on to the B6 or BALB/c genetic background to compare the roles of Slamf1 and Slamf2 in the development of lupus. While we detect auto-antibodies and glomerulonephritis in both Slamf1−/− [B6.129] and Slamf2−/− [B6.129] mice, disease develops at a much younger age in Slamf2−/− [B6.129] mice. Slamf1−/− and Slamf2−/− [BALB/c.129] mice do not manifest any sign of lupus. Functional analyses of CD4+ T cells from the mutant mice reveal that Slamf2−/− T cells are resistant to tolerance induction in vivo. The differences in development of lupus in Slamf2−/− [B6.129] and Slamf1−/− [B6.129] mice are discussed in the context of lupus pathogenesis in other chromosome 1 congenic mouse strains.
Methods
Mice
Wild-type B6 and B6.MRL-Faslpr/J mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). 129/SvEvTac (129) and NZM2410 mice were purchased from Taconic. Slamf2−/− mice (16) were generated with J1 129 embryonic stem cells (ES cells) and backcrossed onto the C57BL/6 (B6) background for 12 generations to generate the Slamf2−/− [B6.129] strain and onto the BALB/c background for 10 generations to generate the Slamf2−/− [BALB/c.129] strain. Slamf1−/− mice (17) also were generated with J1 129 ES cells and backcrossed for nine generations onto the C57BL/6 background to generate the Slamf1−/− [B6.129] strain and onto the BALB/c background to generate the Slamf1−/− [BALB/c.129] strain. B6.Sle1b mice (12) were generously provided by Dr Laurence Morel (University of Florida).
DO11.10 TCR transgenic mice were intercrossed with Slamf2−/− [BALB/c.129] mice to generate DO11 Slamf2−/− [BALB/c.129] mice. All mice were maintained in a pathogen-free facility and used according to institutional and National Institutes of Health guidelines. Harvard Medical School and Beth Israel Deaconess Medical Center are accredited by the American Association of Accreditation of Laboratory Animal Care.
Cell isolation, antibodies and staining
Single-cell suspensions of spleen and lymph node were prepared by mechanical dissociation. Following RBC lysis with ACK buffer (Gibco, Carlsbad, CA, USA), cells were washed and stained with the following antibodies after blocking non-specific binding with CD16/32: anti-CD3 (17A2; 145-2C11), anti-CD4 (GK1.5), anti-CD8α (53-6.7), anti-CD19 (ID3), anti-B220 (RA3-6B2), anti-CD25 (PC61), anti-CD44 (IM7), anti-CD62L (MEL-14), anti-CD69(H1.2F3), anti-CD80(16-10A1), anti-CD86(GL-1) (BD Biosciences, San Jose, CA, USA and Biolegend, San Diego, CA, USA). Data were acquired with FACScalibur or LSRII cytometer (BD Pharmingen, San Jose, CA, USA) and analyzed using FlowJo software (Treestar, San Carlos, CA, USA).
Anti-single stranded DNA (anti-ssDNA), anti-double stranded DNA (anti-dsDNA), anti-chromatin and anti-nucleosome antibody analyses
ELISA assays were performed to quantitate levels of anti-ssDNA, anti-dsDNA, anti-chromatin and anti-nucleosome antibodies in sera of mice. For dsDNA ELISA, salmon sperm DNA or mung bean nuclease (New England Biolabs, Ipswich, MA, USA)-treated dsDNA (Sigma-Aldrich, Saint Louis, Mo, USA) (3 μg ml−1) was coated overnight at 4°C on Immunolon plates (Dynatech, Alexandria, VA, USA). Test or control (ANA-positive NZM2410 sera) was serially diluted and applied for 2 h. After extensive washing, plates were incubated with alkaline phosphatase-conjugated anti-mouse IgG and developed with alkaline phosphate substrate. For anti-nucleosome (anti-histone/DNA complex) ELISAs, met-BSA precoated Immunolon plates were coated overnight with dsDNA and then with total histone solutions (Sigma-Aldrich). Alternatively, commercial human nucleosome-coated plates (Orgentec, Mainz, Germany) were used where indicated. Sera were serially diluted and incubated on plates for 2 hours. Plates were washed extensively and then incubated with HRP-conjugated anti-mouse IgG and developed with substrate. Antibody titers are expressed as ELISA units (EU) comparing optical density (OD) values of test samples with a standard curve prepared using serial dilutions of ANA-positive NZM2410 or MRL.lpr sera.
Antinuclear antibodies
To detect ANA, sera were applied to slides with permeabilized Hep-2 cells (Antibodies Incorporated, Davis, CA, USA) for 45 min at room temperature. After washing, Alexa 427-conjugated anti-mouse IgG antibody (Molecular Probes, Eugene, OR, USA) was applied for 30 min at room temperature followed by extensive washing. Nuclei were visualized by fluorescence microscopy.
Histopathology and immunohistochemisty
Kidneys were fixed in 10% phosphate-buffered formalin (PBS), dehydrated and embedded in paraffin. Five-micron paraffin tissue sections were stained with hematoxylin and eosin (H and E) or periodic acid-Schiff (PAS) for microscopic analysis. All slides were examined by at least one pathologist in a blinded fashion. H and E-stained sections of kidney were evaluated for the presence of glomerular inflammation and PAS preparations were examined for better assessment of glomerular basement membranes, mesangial areas and the presence of large aggregated immune complexes. Kidneys were scored by a board-certified pathologist with subspeciality expertise in kidney pathology using the International Society of Nephrology/Renal Pathology Society (ISN/RPS) classification of lupus nephritis (18) for general classification into kidneys with active (‘proliferative’) glomerulonephritis (classes III and IV) and inactive (‘non-proliferative’) glomerulonephritis (classes I and II). The NIH lupus criteria (19) were also determined for each kidney in order to semiquantitatively assess the degree of lupus-like inflammatory activity (activity index) and chronic parenchymal damage (chronicity index). To evaluate germinal center formation, snap frozen spleens in OCT media (Sakura Finetek, Torrance, CA, USA) were cryosectioned and stained with PE-conjugated anti-CD45R/B220 (eBioscience, San Diego, CA, USA), PE- or FITC-conjugated anti-CD4 (Pharmingen) and FITC-labeled Peanut agglutinin (PNA–FITC) (Vector Laboratories, Burlingame, CA, USA).
Antibody responses and basal Ig detection
Mice were injected intra-peritoneally with 100 μg of alum-precipitated nitrophenol (NP)–keyhole limpet haemocyanin (KLH) (Biosearch Technologies, Novato, CA, USA). Imject® alum was purchased from Pierce (Woburn, MA, USA). Mice were boosted after 14–21 days with an intra-peritoneal (i.p.) injection of 50 μg of NP–KLH in PBS and sacrificed 7 days later. Sera were collected and were tested for NP-specific antibodies by ELISA.
Basal serum Igs were determined by ELISA using isotype-specific goat antibodies to mouse Ig. Trinitrophenol (TNP)–Ficoll and TNP–LPS were used to assess responses to T-cell-independent antigens.
In vitro cell death assays
To measure the level of activation-induced cell death (AICD), 2 × 106 DO11+ or DO11+ Slamf2−/− [BALB/c.129] T cells were activated in vitro for 4 days with 1 μg ml−1 of ovalbumin peptide 323-339 (OVA323-339) and 2 × 106 mitomycin C-treated antigen-presenting cells (APCs). On day 4, the cells were collected and cultured overnight in media containing 50 U ml−1 of IL-2 (BD Biosciences). To measure AICD, T cells were cultured for 24 h in the absence or presence of plate-bound anti-CD3 (0–1.0 μg ml−1) plus 50 U ml−1 of IL-2. Cells were incubated with anti-CD4-FITC, fixed in 70% ethanol solution stained with propidium iodide. The percentage of apoptotic cells was evaluated from the subdiploid population of a cell cycle analysis plot.
Adoptive transfers, immunizations and restimulation assays
For T-cell transfers, DO11+ or DO11+ Slamf2−/− [BALB/c.129] spleen and lymph node cells were collected and cell suspensions pooled and washed through a 70-μM filter. Cells were washed with RPMI-1640 plating media containing 10% fetal bovine serum, 2 mM glutamine, 0.01 M Hepes buffer and 6 × 10−5 M 2-mercaptoethanol. CD4 T cells were purified by positive selection using CD4 (L3T4) Dynal beads. Prior to adoptive transfer, an aliquot of purified CD4 T cells was stained for flow cytometry analysis with CD4-PE and KJ1-26-FITC, the anti-idiotypic antibody that binds to the DO11 TCR (BD Biosciences). KJ1-26+ CD4+ cells (3 × 106) were adoptively transferred intravenously into non-irradiated syngeneic wild-type recipients. At day 1 after cell transfer, mice were either untreated (naive), immunized subcutaneously with 50 μg of OVA323–339 peptide emulsified in incomplete Freund’s adjuvant (primed group) or tolerized with 300 μg of OVA323–339 peptide in PBS intravenously (tolerized group). Four to seven days later, cells were harvested from inguinal lymph nodes and restimulated with OVA323–339 in vitro. Proliferative responses were measured by culturing 1 × 104 KJ1-26+ T cells with 2 × 105 mitomycin C-treated CD4 and CD8 depleted APCs with 0–1 μg ml−1 peptide for 72 h as described previously (20). To measure proliferation, cells were pulsed with 1 μCi of 3H-thymidine on day 3 for 6 h and harvested for liquid scintillation counting. Data reflect the mean of triplicates from each peptide concentration. Supernatants were harvested from 24-well plates in which 1 × 105 antigen-specific T cells and 2 × 106 APCs were stimulated with 0 or 1 μg ml−1 OVA323–339 peptide. Supernatants were collected on days 1, 2 and 3, and cytokine levels were determined by ELISA.
Genotypic analysis
Genomic DNA from Slamf2−/− [B6.129] mice was analyzed with polymorphic genetic markers. PCR was performed using 10 markers on chromosome 1 and at least one marker per other chromosomes. Primer sequences were obtained from Mouse Genome Informatics database (http://www.informatics.jax.org/)
Results
Slamf1−/− [B6.129] mice but not Slamf1−/− [BALB/c.129] develop auto-antibodies
To begin to evaluate humoral immune responses in Slamf1−/− [B6.129] and Slamf1−/− [BALB/c.129] mice, which had been generated using homologous recombination in 129-derived ES cells and subsequently backcrossed with B6 or BALB/c mice for nine generations, we first compared basal Ig levels in the sera of both strains. No significant differences were observed in IgM, IgG subclass or IgA levels between mutant mice and their wild-type (wt) littermates at 3 months of age (Supplementary Figure S1A is available at International Immunology Online). Surprisingly, sera of 1-year-old Slamf1−/− [B6.129] mice contained high levels of ANAs, as judged by ELISA (Figure 1A) and immunofluorescence staining of Hep-2 cells (Figure 1B). However, no auto-antibodies were found in Slamf1−/− [BALB/c.129] mice (Figure 1A). Anti-nucleosome IgG production developed gradually over time (Figure 1C), 100% of Slamf1−/− [B6.129] mice had anti-nucleosome antibodies over 15 months of age without a significant difference between male and female mice (Supplementary Figure S2 is available at International Immunology Online). In spite the presence of high titer auto-antibodies, total IgG and IgM levels were lower in the sera of aged Slamf1−/− [B6.129] mice than their age-matched wt counterparts (Supplementary Figure S1B is available at International Immunology Online) indicating specific autoimmune response. The notion that Slamf1−/− [B6.129] mice developed features of autoimmunity was supported by development of splenomegaly (Figure 1D) and increased levels of activated CD4+ T cells in 10- to 13-month-old mice (Figure 1E).
Fig. 1.

SLE-like humoral autoimmunity in Slamf1−/− [B6.129] mice. (A) Anti-nucleosome auto-antibodies in aged Slamf1−/− [B6.129] mice but not in Slamf1−/− [BALB/c.129] mice. Anti-nucleosome auto-antibody titers in the serum of 10- to 13-month-old females were determined by ELISA, as described in Methods. B6 n = 38; Slamf1−/− [B6.129] n = 16; Slamf1−/− [BALB/c.129] n = 5; BALB/c n = 9. (B) Antinuclear auto-antibodies in Slamf1−/− [B6.129] mice. Permeabilized Hep-2 cells were incubated with sera from 1-year-old female Slamf1−/− [B6.129] (n = 7) and B6 (n = 7) mice. After washes, bound IgG was detected with anti-mouse IgG-Alexa488 (see Methods). Demonstrated staining pattern is representative for the Slamf1−/− [B6.129] group. (C) Progressive humoral autoimmunity in Slamf1−/− [B6.129]. Penetrance of anti-nucleosome IgG in aged female Slamf1−/− [B6.129] mice by age groups (±1 month). A serum was judged positive if auto-antibody titer was above mean titer of the B6 group plus 2 SD. (D) Mild splenomegaly in aged Slamf1−/− [B6.129] mice. (E) Increased percentage of effector/memory CD4+ T cells in Slamf1−/−[B6.129] mice. Determined by flow cytometry as described in Methods. Ten- to 13-month-old female B6 (n = 25) and Slamf1−/− [B6.129] (n = 10) mice were used for both panels.
Upon further analyses, we found that the sera of aged Slamf1−/− [B6.129] mice only contained antibodies directed against nucleosomes but not ssDNA, dsDNA or histones (Figure 2). These auto-antibodies were similar to those found in the B6.Sle1b mice (Figure 2) (21), while serum of the NZM2410 mice, from which B6.Sle1b had been derived, contained relatively more anti-ssDNA and dsDNA than anti-nucleosome antibodies (Figure 2). Similarly, auto-antibodies in B6.MRL-FASlpr/J mice had a wider spectrum of reactivity with chromatin components.
Fig. 2.

Nucleosome-specific auto-antibodies in Slamf1−/− [B6.129] mice. Auto-antibody titers were determined by ELISA as described in Methods. Bars represent mean titer of auto-antibodies measured from B6 n =17; Slamf1−/− [B6.129] n = 15; Sle1b n = 6; NZM2410 n = 6; B6.MRL-Faslpr/J n = 11. *P < 0.05, **P < 0.01, *** P < 0.001.
These findings suggest that mice which harbor the Slamf1−/− mutation in a 129-derived segment (as documented by Slamf haplotype analyses; Supplementary Figure S3 is available at International Immunology Online) in B6 background develop a lupus-like syndrome that is likely to be dependent upon epistatic interactions with B6 genes.
Normal antibody responses in immunized Slamf1−/− [BALB/c.129] mice
Slamf1 has been shown to be a co-regulator of antigen-driven T-cell responses (15, 22). TCR-induced IL-4 secretion but not IL-2 production of Slamf1−/− CD4+ cells is down-regulated (17). To assess whether the Slamf1−/− mutation leads to an increase in all humoral immune responses, T-cell-independent and T-dependent responses to NP–KLH were determined in the non-autoimmune prone Slamf1−/− [BALB/c.129] mice. T-independent responses were identical those in wt mice, as judged by TNP-specific IgM and IgG responses induced by either TNP–Ficoll or TNP–LPS injections (Fig. 3A and B). While T-dependent IgM responses to immunization with NP–KLH were normal, we have seen a slight defect in NP-specific IgG production compared with wt mice (Fig. 3C). Germinal center formation was similar in mutant mice and their wt littermates (Supplementary Figure S4 is available at International Immunology Online). We conclude that the Slamf1−/− mutation per se does not dramatically increase general humoral responses.
Fig. 3.
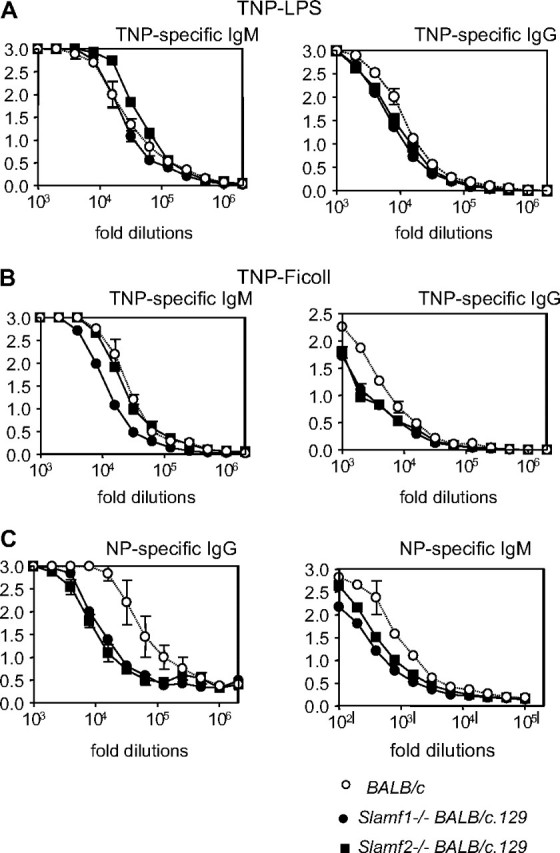
Hapten-specific antibody responses in Slamf1−/− [BALB/c.129] and Slamf2−/− [BALB/c.129] mice. (A) Type I T-independent B-cell responses in Slamf1−/− [BALB/c.129] and Slamf2−/− [BALB/c.129] mice. BALB/c, Slamf1−/− [BALB/c.129] and Slamf2−/− [BALB/c.129] mice (n = 4) received one i.p. injection of 10 μg of TNP–LPS. TNP-specific IgM (left panel) and IgG (right panel) titers were determined at day 6 by ELISA after serial dilutions of the serum (y-axis = OD 405). Results are representative of two independent experiments. (B) Type II T-independent B-cell responses in Slamf1−/− [BALB/c.129] and Slamf2−/− [BALB/c.129] mice. BALB/c, Slamf1−/− [BALB/c.129] and Slamf2−/− [BALB/c.129] mice (n = 4) received one i.p. injection of 30 μg of TNP-Ficoll. TNP-specific IgM (left panel) and IgG (right panel) titers were determined at day 6 by ELISA after serial dilutions of the serum (y-axis = OD 405). Results are representative of two independent experiments. (C) Antibody responses to T–D antigens in Slamf1−/− [BALB/c.129] and Slamf2−/− [BALB/c.129] mice. NP-specific antibody production in the sera of BALB/c, Slamf1−/− [BALB/c.129] and Slamf2−/− [BALB/c.129] mice (n = 4) was determined 7 days after i.p. immunization with NP–KLH antigen in normal saline of mice previously immunized with alum-precipitated NP–KLH. NP-specific IgG and IgM antibody titers were determined by ELISA after serial serum dilutions (y-axis = OD 405; OD at 405 nm). NP-specific IgM antibodies were determined 10 days after primary immunization.
Increased frequency of glomerulonephritis in Slamf1−/−[B6.129] mice
Because the presence of anti-nucleosome IgG in the sera of aged Slamf1−/− [B6.129] mice, we set out to examine whether they also develop glomerulonephritis. Histological evaluation of kidneys from 1-year-old Slamf1−/− [B6.129] revealed active proliferative glomerulonephritis (ISN/RPS class III or IV) in 7 of 11 mice as compared with 1 of 13 control B6 mice (Fig. 4B), manifesting as hypercellular enlarged glomeruli with accumulation of leukocytes in the glomerular capillaries (Fig. 4A). Immune complexes were frequently identified as PAS-positive aggregates within glomerular capillaries (‘microthrombi’) or as deposits conforming to the contours of the glomerular basement membranes (‘wire loops’).
Fig. 4.

Increased frequency of glomerulonephritis in Slamf1−/−[B6.129] mice. Kidney sections from 10- to 13-month-old female B6 and Slamf1−/− [B6.129] mice were processed and stained with hematoxylin and eosin as described in Methods. (A) Example of lesions observed in aged Slamf1−/− [B6.129] mice. Images were taken at ×400 magnification. Glomerulonephritis manifesting as hypercellular enlarged glomeruli with accumulation of leukocytes in the glomerular capillaries. (B) Quantitative evaluation of renal histology. ISN/RPS scores were determined as described in Methods. Scores III–IV (above dashed line) indicate active proliferative glomerulonephritis. B6 n = 13; Slamf1−/−[B6.129] n = 11; Sle1b n = 8.
Auto-antibody responses in Slamf2−/− [B6.129] mice
To begin to characterize humoral immune responses in Slamf2−/− [B6.129] mice, we first examined basal serum Ig levels in these mice over time. Basal serum Ig levels were comparable in young wt and Slamf2−/− [B6.129] mice (6- to 8-week old) (Supplementary Figure S1 is available at International Immunology Online). However, at 6 months of age, there was a significant increase in total IgG1, IgG2b and IgG3 subclasses in Slamf2−/− [B6.129] mice compared with wt controls (data not shown). There was also an increase seen in total IgM at 6 months of age in Slamf2−/−[B6.129] mice compared with wt controls, but this did not reach statistical significance.
We next investigated whether Slamf2−/− [B6.129] mice developed antibodies to DNA and chromatin components. Anti-nucleosome IgG antibodies were elevated in 3-month-old Slamf2−/− [B6.129] mice and rose to higher levels in 6-month-old Slamf2−/− [B6.129] mice (Fig. 5A). At 3 and 6 months of age, Slamf2−/− [B6.129] female mice had elevated levels of IgG anti-dsDNA antibodies compared with wt controls (Supplementary Figure S5A is available at International Immunology Online) but an increase in IgM anti-dsDNA in Slamf2−/− [B6.129] mice was seen only at 6 months (Supplementary Figure S5B is available at International Immunology Online).
Fig. 5.
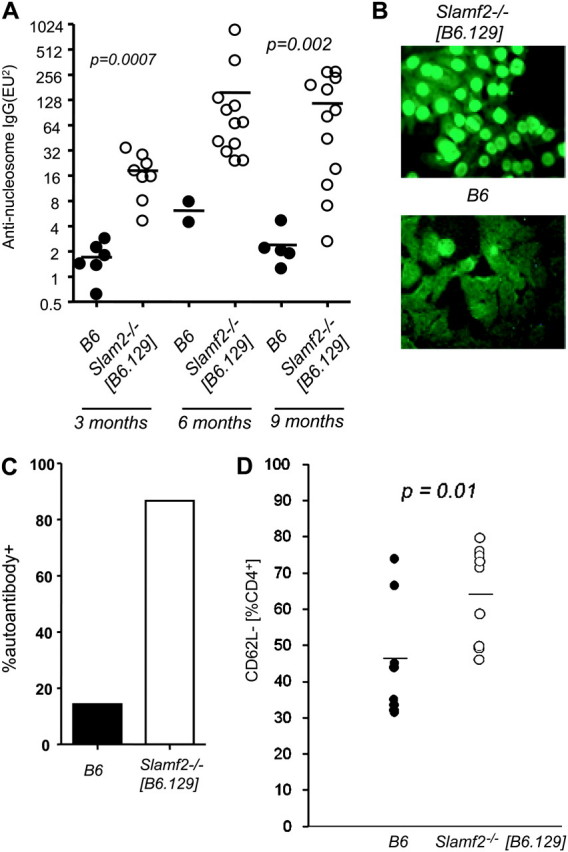
Slamf2−/− [B6.129] mice have elevated levels of anti-nucleosome IgG antibodies and activated T cells. (A) Anti-nucleosome auto-antibodies in aged Slamf2−/− [B6.129] mice. Anti-nucleosome auto-antibody titers in the serum of 3-, 6- and 9-month-old females were determined by ELISA, as described in Methods. Three-month-old B6 n = 5; 3-month-old Slamf2−/− [B6.129] n = 8; 6-month-old B6 n = 2; 6-month-old Slamf2−/− [B6.129] n = 12; 9-month-old B6 n = 5; 9-month-old Slamf2−/− [B6.129] n = 12; (B) Antinuclear auto-antibodies in Slamf2−/− [B6.129] mice. Sera from 6-month-old B6 and Slamf2−/− [B6.129] mice were incubated with Hep-2 cells at a dilution of 1/100. ANAs were detected with FITC-labeled anti-mouse IgG secondary antibody. Magnification was ×40. B6 n = 7; Slamf2−/− [B6.129] n = 15; (C) Penetrance of antinuclear auto-antibodies. Percentage of Slamf2−/− [B6.129] mice positive for ANAs. (D) Increased percentage of activated CD4+ T cells in Slamf2−/−. Splenocytes from B6 or Slamf2−/− [B6.129] at 6 months were stained with anti-CD4 and anti-CD62L and analyzed by flow cytometry.Plots show the percentage of CD62L− cells within the CD4+ gate. B6 n = 8; Slamf2−/− [B6.129] n = 9.
The sera of 13 of 15 Slamf2−/− [B6.129] female mice were positive for antinuclear staining at 6 months of age (Figs 5B and 5C), whereas one of seven sera from 6-month-old wt female mice had ANAs. Similarly, four of four sera from Slamf2−/− [B6.129] male mice were positive for antinuclear staining at 9 months of age, while sera from none of three wt male mice had ANAs. Thus, both female and male Slamf2−/− [B6.129] mice develop ANAs. Both Slamf2−/− [BALB/c.129] and BALB/c mice were negative for ANAs at 6 months of age. As shown in Fig. 3, T-independent humoral responses were comparable in Slamf2−/− [B6.129] and wt mice, while there was a reduction in NP-specific T-dependent response, similarly to the Slamf1−/− [B6.129] mice.
Taken together, these findings suggest that there is an influence of background genes on the production of auto-antibodies. We also conclude that the lupus manifestations in Slamf2−/− [B6.129] mice occur at earlier age and are more severe than those in Slamf1−/− [B6.129] mice as they appear with more rapid kinetics.
An activated phenotype in lymphocytes isolated from ageing Slamf2−/− [B6.129] mice
Slamf2−/− mice develop normally and are healthy at 6–8 weeks of age. Lymphoid development in Slamf2−/− mice appears grossly normal, as judged from the total number of lymphocytes in thymus, spleen and lymph node. However, there was a modest increase in CD4+ thymocytes and peripheral CD4+ T cells in Slamf2−/− mice (16). At 6–8 weeks of age, Slamf2−/−[B6.129] mice had a modestly increased percentage of CD4+ T cells in the spleen, but these cells had a naive phenotype. In contrast, at 6 months of age, CD4+ T cells from Slamf2−/− [B6.129] mice exhibited an activated phenotype with an up-regulation of CD69 and down-regulation of CD62L (Fig 5D and Supplementary Table I is available at International Immunology Online). There also was a down-regulation of CD62L on Slamf2−/− CD8+ T cells. There was no difference in CD80 or CD86 expression on Slamf2−/− [B6.129] and wt B cells (Supplementary Table I is available at International Immunology Online). Taken together, the results of these analyses are comparable with the outcomes of similar studies in Slamf1−/− female mice but with earlier onset of CD4+ T-cell activation.
Slamf2−/− [B6.129] mice develop glomerulonephritis
Histopathological analyses of kidneys from Slamf2−/− [B6.129] female mice revealed active glomerulonephritis, marked by hypercellular glomeruli with leukocyte infiltration in glomerular capillaries and expansion of mesangial areas in two-thirds of these mice by 6 months of age. None of the wt B6 mice showed evidence of active glomerulonephritis at these times (Fig 6A and B).
Fig. 6.
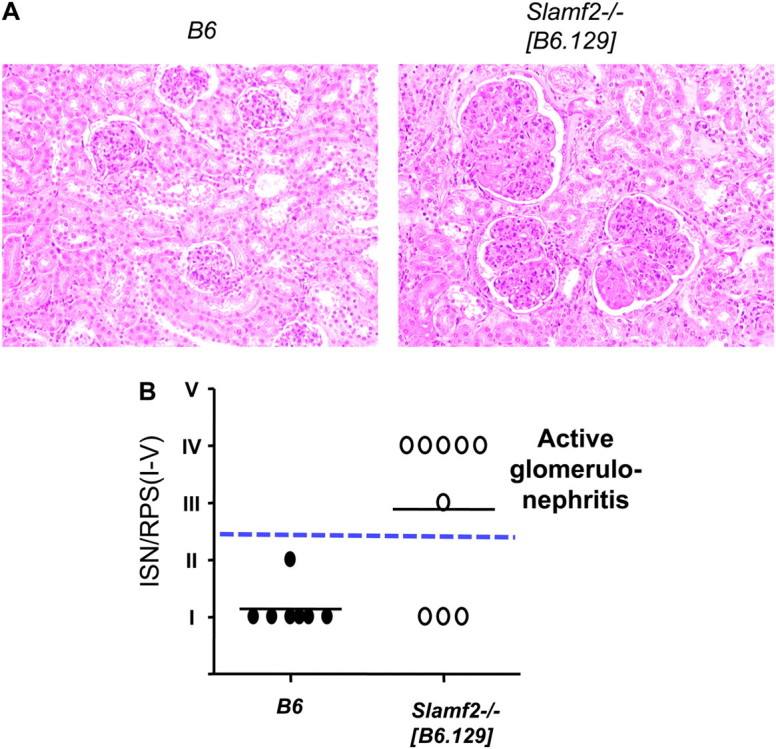
Glomerulonephritis in the kidneys of Slamf2−/− [B6.129] mice. (A) Example of lesions observed in aged Slamf2−/− [B6.129] mice. Images were taken at ×400 magnification. (B) Quantitative evaluation of renal histology. Kidney sections from 6- to 9-month-old female and male B6 and Slamf2−/− [B6.129] mice were stained with hematoxylin and eosin or PAS. ISN/RPS scores were determined as described in Methods. Scores III–IV (above dashed line) indicate active proliferative glomerulonephritis. B6 n = 7; Slamf2−/−[B6.129] n = 9.
Slamf2−/− [B6.129] mice exhibit defects in peripheral tolerance
The phenotype of Slamf2−/− [B6.129] led us to question whether Slamf2 plays a role in T-cell tolerance. Peripheral T-cell tolerance is achieved by several mechanisms: ignorance (failure to recognize antigens), anergy, deletion and suppression. To evaluate whether Slamf2 is involved in regulating any of the mechanism of peripheral T-cell tolerance, we used DO11.10 TCR transgenic Slamf2−/− mice, generated by breeding DO11.10 TCR transgenic mice (OVA specific) with Slamf2−/− [BALB/c.129] mice, as tools. Slamf2−/− DO11.10 mice have normal thymic development. Using Slamf2−/− mice on the BALB/c background circumvented the potentially confounding issues of autoimmune disease seen in Slamf2−/−[B6.129] mice on interpretation of our results.
AICD, a form of apoptosis is important in the deletion of self-reactive T cells. AICD is dependent on IL-2 production and the ligation of Fas with its ligand (23, 24). Support for ACID as a mechanism of deletion of self-reactive T cells is seen by the SLE-like phenotype of MRL/lpr or MRL/gld mice in which there is a mutation in the Fas or Fas ligand gene, respectively. To determine if there is a defect in AICD in the absence of Slamf2, we utilized DO11.10 transgenic CD4+ T cells deficient in Slamf2. DO11.10 or Slamf2−/− DO11.10 splenocytes were activated with 1μg/ml OVA323–339 peptide for 4 days. Viable cells were recovered and rested overnight with IL-2. CD4+ T cells were then activated with anti-CD3. Cells were stained with KJ-126, the clonotypic antibody against the DO11.10 TCR, fixed with 70% ethanol and stained with propidium iodide. The percentage of apoptotic cells was evaluated from the subdiploid population of a cell cycle analysis plot. As seen in Fig. 7, there was no difference in AICD between Slamf2−/− and wt CD4+ T cells. These results indicate that Slamf2 does not have an obligatory role in AICD in previously activated T cells.
Fig. 7.

No defects in AICD in Slamf2−/− mice. DO11.10 or Slamf2−/− DO11.10 splenocytes were activated with 1 μg ml−1 OVA323–339 peptide for 4 days. Viable cells were recovered by Ficoll purification and rested overnight with IL-2. CD4+ T cells were activated with anti-CD3 (0–1.0 μg ml−1) for 24h. Cells were fixed with ethanol and stained with KJ126, the clonotypic antibody against the DO11.10 TCR and propidium iodide to evaluate the apoptotic cells. The data are shown as the % apoptotic cells of the KJ-126 population and are representative of three similar experiments.
We also used naive DO11.10 TCR T cells lacking Slamf2−/− to investigate the requirement for Slamf2 for peripheral T-cell tolerance in vivo, using the well-established DO11 adoptive transfer model (25). For these studies, we adoptively transferred wt or Slamf2−/− DO11.10+ T cells into wt BALB/c or Slamf2−/− [BALB/c.129] recipients, respectively, and compared responses to an immunogenic (OVA peptide in incomplete Freund's adjuvant) versus a tolerogenic (OVA peptide in PBS) stimulus. In vivo responses of T cells from wt and Slamf2−/− mice were evaluated by harvesting lymph nodes from the recipients and assessing T-cell proliferation and cytokine production following restimulation with antigen in vitro. Primed DO11+ wt and Slamf2−/− DO11+T cells exhibited similarly strong proliferative responses and secrete comparably high amounts of IL-2 and IFN-γ. In contrast, when tolerance-inducing conditions were applied, wt and Slamf2−/− DO11.10 T cells responded differently. Whereas DO11+ wt T cells that encountered a tolerogenic stimulus in vivo proliferated poorly and produced little cytokines, DO11+ Slamf2−/− T cells proliferated strongly and secreted high amounts of IL-2 and IFN-γ upon re-stimulation in vitro, following an in vivo tolerogenic stimulus (high-dose aqueous peptide) (Fig. 8). Taken together, these results demonstrate that Slamf2 has an essential role in peripheral T-cell tolerance.
Fig. 8.

Tolerance cannot be induced in absence of Slamf2. DO11.10 wt CD4+ T cells were adoptively transferred into wt recipients and Slamf2−/− DO11.10 CD4+ T cells were adoptively transferred into Slamf2−/− recipients. Adoptively transferred recipients were given OVA323–339 peptide plus incomplete Freund’s adjuvant (for priming) or OVA323–339 peptide in PBS (for tolerizing). Three days later, T-cell responses were evaluated by re-stimulation of lymph node cells with OVA peptide in vitro. Proliferation was measured by 3H-thymidine incorporation (A and B), IL-2 (C and D) and IFN-γ (E and F) by ELISA. The data are representative of three similar experiments.
Discussion
Recent genetic and functional studies have pointed to roles for SLAM family members in controlling susceptibility to murine SLE. The highly polymorphic nature of SLAM family receptors, together with their potential to control innate and adaptive immunity, have made them attractive candidate genes in the mouse chromosome 1 lupus congenic models. While recent work has identified Slamf6 as a major contributor to the role of Sle1b in tolerance (13, 14), the functional diversity and overlapping signaling of SLAM family members suggest that their roles in self-tolerance may not be confined to only one Slamf gene. Here, we have utilized Slamf1−/− [B6.129] and Slamf2−/− [B6.129] congenic mouse strains to evaluate the roles of Slamf1 and Slamf2 in the development of lupus. We find that both Slamf1−/− [B6.129] and Slamf2−/− [B6.129] congenic mice develop features of lupus. However, while both strains develop auto-antibodies to nuclear antigens, Slamf2−/− [B6.129] mice develop glomerulonephritis at a much earlier age and with a more severe outcome than Slamf1−/− [B6.129] mice.
The loss of tolerance to nuclear antigens, i.e. increased ANA titers and ANAs, in Slamf1−/− [B6.129] mice is remarkably similar to that of B6.Sle1b (12) and B6.129chr1b (26) mice. Furthermore, Slamf1−/− [B6.129] mice, like B6.129chr1b and B6.Sle1b mice, had only a modest renal pathology. All three mouse strains also developed splenomegaly and spontaneous CD4 T-cell activation by 1 year of age.
The phenotype of Slamf2−/− [B6.129] mice was similar to the phenotype of Slamf1−/− [B6.129] and B6.129chr1b or B6.Sle1b mice with respect to the development of significantly increased titers of ANA, increased ANAs, splenomegaly and CD4 T-cell activation at 1 year of age. Slamf2−/−[B6.129] mice, however, already exhibited auto-antibodies at 3 months of age and severe proliferative glomerulonephritis as early as 6 months of age. This contrast between B6.129chr1b and Slamf2−/− [B6.129] is remarkable because the congenic boundaries largely overlap (Supplementary Figure S6 is available at International Immunology Online).
To begin to understand why Slamf2−/− [B6.129] mice develop severe spontaneous autoimmunity, we investigated whether Slamf2 regulates peripheral T-cell tolerance. We used Slamf2−/− [BALB/c.129] mice to analyze whether Slamf2 has an essential role in regulating AICD or T-cell anergy because Slamf2−/− [BALB/c.129] mice do not develop any evidence of spontaneous T-cell activation. This approach provides a means to study Slamf2 function while avoiding potential confounding issues related to spontaneous autoimmune disease seen in Slamf2−/− [B6.129] mice. We first investigated AICD since MRL/lpr and MRL/gld mice developed a lupus-like phenotype and have defects in AICD but found no defect in ACID in Slamf2−/− [BALB/c.129] mice. Using the D011.10 adoptive transfer model (25), we found that Slamf2−/− DO11.10 T cells were resistant to tolerance induction following adoptive transfer into Slamf2−/− recipients in vivo. These findings demonstrate that Slamf2 has critical role in peripheral T-cell tolerance and suggest that a defect in peripheral T-cell tolerance may be key to the development of SLE-like phenotype in Slamf2−/− [B6.129] mice. Further studies are needed to individually assess the function of Slamf2 on T cells and APCs in peripheral T-cell tolerance, as well as the roles of Slamf2:Slamf4 versus Slamf2:CD2 interactions in peripheral T-cell tolerance.
In summary, our studies of Slamf1−/− [B6.129] and Slamf2−/− [B6.129] congenic mice have revealed roles for both Slamf1 and Slamf2 in self-tolerance and suggest that both these SLAM family members may contribute to the role of Sle1b in tolerance and the development of features of lupus in chromosome 1 congenic mouse strains. The shared and distinct phenotypes of Slamf1−/− [B6.129] and Slamf2−/− [B6.129] congenic mice suggest that these SLAM family members may have overlapping and unique roles in the pathogenesis of SLE and give impetus to further studies on Slamf1 and Slamf2 in B-and T-cell tolerance.
Supplementary data
Supplementary data are available at International Immunology Online.
Funding
National Institutes of Health (AI-065687 to C.T., A.H.S. and Y.E.L.; T32 HL007627 to Brigham and Women's Hospital for V.K.V; K08AR057861 to D.B.).
Supplementary Material
Acknowledgments
We are grateful to Aimee Julien for technical assistance. The authors have no conflicting financial interest.
References
- 1.Graham RR, Hom G, Ortmann W, Behrens TW. Review of recent genome-wide association scans in lupus. J. Intern. Med. 2009;265:680. doi: 10.1111/j.1365-2796.2009.02096.x. [DOI] [PubMed] [Google Scholar]
- 2.Moser KL, Neas BR, Salmon JE, et al. Genome scan of human systemic lupus erythematosus: evidence for linkage on chromosome 1q in African-American pedigrees. Proc. Natl Acad. Sci. USA. 1998;95:14869. doi: 10.1073/pnas.95.25.14869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsao BP, Cantor RM, Kalunian KC, et al. Evidence for linkage of a candidate chromosome 1 region to human systemic lupus erythematosus. J. Clin. Invest. 1997;99:725. doi: 10.1172/JCI119217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsao BP, Cantor RM, Grossman JM, et al. Linkage and interaction of loci on 1q23 and 16q12 may contribute to susceptibility to systemic lupus erythematosus. Arthritis Rheum. 2002;46:2928. doi: 10.1002/art.10590. [DOI] [PubMed] [Google Scholar]
- 5.Cantor RM, Yuan J, Napier S, et al. Systemic lupus erythematosus genome scan: support for linkage at 1q23, 2q33, 16q12-13, and 17q21-23 and novel evidence at 3p24, 10q23-24, 13q32, and 18q22-23. Arthritis Rheum. 2004;50:3203. doi: 10.1002/art.20511. [DOI] [PubMed] [Google Scholar]
- 6.Johanneson B, Lima G, von Salome J, Alarcon-Segovia D, Alarcon-Riquelme ME. A major susceptibility locus for systemic lupus erythemathosus maps to chromosome 1q31. Am. J. Hum. Genet. 2002;71:1060. doi: 10.1086/344289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shai R, Quismorio FP, Jr., Li L, et al. Genome-wide screen for systemic lupus erythematosus susceptibility genes in multiplex families. Hum. Mol. Genet. 1999;8:639. doi: 10.1093/hmg/8.4.639. [DOI] [PubMed] [Google Scholar]
- 8.Wakeland EK, Liu K, Graham RR, Behrens TW. Delineating the genetic basis of systemic lupus erythematosus. Immunity. 2001;15:397. doi: 10.1016/s1074-7613(01)00201-1. [DOI] [PubMed] [Google Scholar]
- 9.Rozzo S, Vyse T, Drake C, Kotzin B. Effect of genetic background on the contribution of New Zealand Black loci to autoimmune lupusánephritis. Proc. Natl Acad. Sci. USA. 1996;93:15164. doi: 10.1073/pnas.93.26.15164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kono DH, Burlingame RW, Owens DG, et al. Lupus susceptibility loci in New Zealand mice. Proc. Natl Acad. Sci. USA. 1994;91:10168. doi: 10.1073/pnas.91.21.10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hogarth MB, Slingsby JH, Allen PJ, et al. Multiple lupus susceptibility loci map to chromosome 1 in BXSB mice. J. Immunol. 1998;161:2753. [PubMed] [Google Scholar]
- 12.Morel L, Blenman KR, Croker BP, Wakeland EK. The major murine systemic lupus erythematosus susceptibility locus, Sle1, is a cluster of functionally related genes. Proc. Natl Acad. Sci. USA. 2001;98:1787. doi: 10.1073/pnas.031336098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wandstrat AE, Nguyen C, Limaye N, et al. Association of extensive polymorphisms in the SLAM/CD2 gene cluster with murine lupus. Immunity. 2004;21:769. doi: 10.1016/j.immuni.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 14.Kumar KR, Li L, Yan M, et al. Regulation of B cell tolerance by the lupus susceptibility gene Ly108. Science. 2006;312:1665. doi: 10.1126/science.1125893. [DOI] [PubMed] [Google Scholar]
- 15.Calpe S, Wang N, Romero X, et al. The SLAM and SAP gene families control innate and adaptive immune responses. Adv. Immunol. 2008;97:177. doi: 10.1016/S0065-2776(08)00004-7. [DOI] [PubMed] [Google Scholar]
- 16.Gonzalez-Cabrero J. CD48-deficient mice have a pronounced defect in CD4+ T cell activation. Proc. Natl Acad. Sci. USA. 1999;96:1019. doi: 10.1073/pnas.96.3.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang N, Satoskar A, Faubion W, et al. The cell surface receptor SLAM controls T cell and macrophage functions. J. Exp. Med. 2004;199:1255. doi: 10.1084/jem.20031835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weening JJ, D'Agati VD, Schwartz MM, et al. The classification of glomerulonephritis in systemic lupus erythematosus revisited. Kidney Int. 2004;65:521. doi: 10.1111/j.1523-1755.2004.00443.x. [DOI] [PubMed] [Google Scholar]
- 19.Austin HA, III, Muenz LR, Joyce KM, et al. Prognostic factors in lupus nephritis. Contribution of renal histologic data. Am. J. Med. 1983;75:382. doi: 10.1016/0002-9343(83)90338-8. [DOI] [PubMed] [Google Scholar]
- 20.Greenwald RJ, Boussiotis VA, Lorsbach RB, Abbas AK, Sharpe AH. CTLA-4 regulates induction of anergy in vivo. Immunity. 2001;14:145. doi: 10.1016/s1074-7613(01)00097-8. [DOI] [PubMed] [Google Scholar]
- 21.Mohan C, Alas E, Morel L, Yang P, Wakeland EK. Genetic dissection of SLE pathogenesis. Sle1 on murine chromosome 1 leads to a selective loss of tolerance to H2A/H2B/DNA subnucleosomes. J. Clin. Invest. 1998;101:1362. doi: 10.1172/JCI728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cocks BG, Chang CC, Carballido J, Yssel H, de Vries JE, Aversa G. A novel receptor involved in T-cell activation. Nature. 1995;376:260. doi: 10.1038/376260a0. [DOI] [PubMed] [Google Scholar]
- 23.Alderson MR, Tough TW, Davis-Smith T, et al. Fas ligand mediates activation-induced cell death in human T lymphocytes. J. Exp. Med. 1995;181:71. doi: 10.1084/jem.181.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ju ST, Matsui K, Ozdemirli M. Molecular and cellular mechanisms regulating T and B cell apoptosis through Fas/FasL interaction. Int. Rev. Immunol. 1999;18:485. doi: 10.3109/08830189909088495. [DOI] [PubMed] [Google Scholar]
- 25.Kearney ER, Pape KA, Loh DY, Jenkins MK. Visualization of peptide-specific T cell immunity and peripheral tolerance induction in vivo. Immunity. 1994;1:327. doi: 10.1016/1074-7613(94)90084-1. [DOI] [PubMed] [Google Scholar]
- 26.Carlucci F, Cortes-Hernandez J, Fossati-Jimack L, et al. Genetic dissection of spontaneous autoimmunity driven by 129-derived chromosome 1 Loci when expressed on C57BL/6 mice. J. Immunol. 2007;178:2352. doi: 10.4049/jimmunol.178.4.2352. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.