

Abstract
We have sequenced and annotated two genomic regions located in the Giemsa negative band q22 of human chromosome 7. The first region defined by the erythropoietin (EPO) locus is 228 kb in length and contains 13 genes. Whereas 3 genes (GNB2, EPO, PCOLCE) were known previously on the mRNA level, we have been able to identify 10 novel genes using a newly developed automatic annotation tool RUMMAGE-DP, which comprises >26 different programs mainly for exon prediction, homology searches, and compositional and repeat analysis. For precise annotation we have also resequenced ESTs identified to the region and assembled them to build large cDNAs. In addition, we have investigated the differential splicing of genes. Using these tools we annotated 4 of the 10 genes as a zonadhesin, a transferrin homolog, a nucleoporin-like gene, and an actin gene. Two genes showed weak similarity to an insulin-like receptor and a neuronal protein with a leucine-rich amino-terminal domain. Four predicted genes (CDS1–CDS4) CDS that have been confirmed on the mRNA level showed no similarity to known proteins and a potential function could not be assigned. The second region in 7q22 defined by the CUTL1 (CCAAT displacement protein and its splice variant) locus is 416 kb in length and contains three known genes, including PMSL12, APS, CUTL1, and a novel gene (CDS5). The CUTL1 locus, consisting of two splice variants (CDP and CASP), occupies >300 kb. Based on the G,C profile an isochore switch can be defined between the CUTL1 gene and the APS and PMSL12 genes.
[Clones 37G3, 164c7, and 235f8 are deposited in GenBank under accession no. AF053356; clone 123e15, accession no. AF024533; 186d2, accession no. AF024534; 46f6, accession no. AF006752; 50h2, accession no. AF047825; and 76h2, accession no. AF030453]
Human chromosome 7 accounts for ∼5% of the human genome and contains >4000 genes and ∼170 Mb of DNA. The Giemsa negative band 7q22 is ∼20 Mb in length and represents one of the most gene-dense bands in the genome.
Two intervals surrounding the erythropoietin (EPO) and CCAAT displacement protein (CUTL1) genes within 7q22 have been the focus of many cytogenetic and molecular studies because of the correlation of this region with breakpoints observed in acute myeloid leukemias and myelodysplastic syndromes (Fischer et al. 1997), as well as leiomyoma (Ishwad et al. 1997; Zeng et al. 1997). These regions of chromosome 7 have been particularly difficult to analyze, as they are not represented in a single contiguous yeast artificial chromosome (YAC) contig in any existing map. Moreover, the immediate region surrounding EPO is not represented in YAC libraries, which has inhibited gene identification studies in search of additional biologically important proteins.
In the context of a comprehensive analysis of 7q22 we initiated genomic sequencing and annotation of two regions within this chromosomal band: a 228-kb contig around the locus for the EPO and a 416-kb contig around the locus for CUTL1. The two regions analyzed are separated from each other by 1–2 Mb of DNA (Fig. 1). Therefore, their analysis should show whether bands with a uniform Giemsa stain are also uniform in their structural features. Here we present the exon/intron organization of 17 genes found in the two regions, assign possible functions for four new genes, and describe alternative splicing for two genes. Our study shows that genomic sequencing in combination with high-quality annotation using automatic and manual tools is a very effective method to discover the complete coding potential of large genomic regions. In addition, we describe a number of structural features in these regions including a possible isochore switch in the CUTL1 contig. The genes identified in this study will provide an important source for future biological studies of this chromosomal region.
Figure 1.
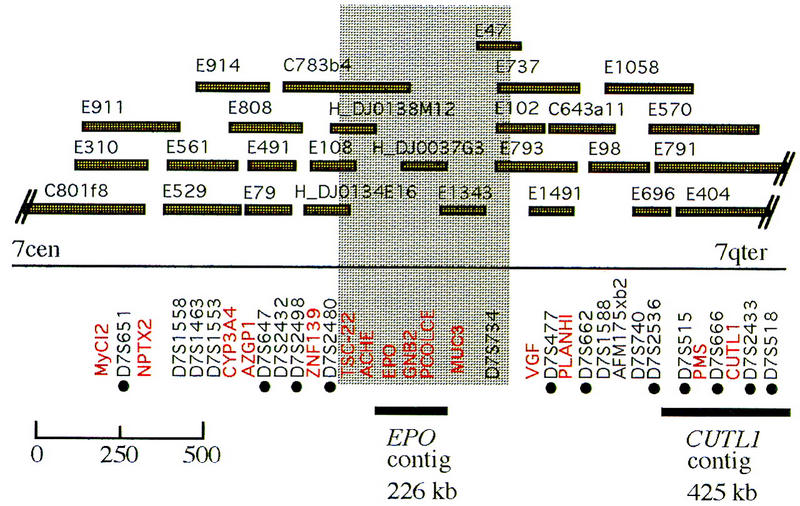
Location of the EPO and CUTL1 contigs in the 7q22 region. The position of representative genetic markers [(•) genes (in red) and STSs (unmarked)] with respect to genomic clones in the region are shown. The clones preceded by C represent CEPH–Généthon mega-YACs. Clones preceded by an E are from the HSC7E chromosome 7-specific YAC library. Three PAC clones are also shown (preceded by H_DJ). Information on additional markers represented on these genomic clones is available on the World Wide Web at http://www.genet.sickkids.on.ca/chromosome7/. The shaded area represents an interval within the contig where the orientation of the markers is still not confirmed.
RESULTS
Sequence-Ready Map
For DNA sequencing we chose two bacterial clone contigs positioned within defined regions in 7q22 (Fig. 1). We have named the 228- and 416-kb segments surrounding EPO and CUTL1 the EPO contig and the CUTL1 contig, respectively. Based on a sequence-ready map for the two contigs (Takahara et al. 1996; Zeng et al. 1997) we sequenced four PAC and four cosmid clones with minimal overlap (Fig. 2A,B). The two contigs span 650 kb of DNA in 7q22 and are separated by ∼1–2 Mb of DNA (Fig. 1). The CUTL1 region still contains a clone gap between cosmid 186d2 and PAC50h2 that could not be filled in by cosmid or PAC clones despite screening various large-insert bacterial clone libraries. Subsequent analysis showed that this cloning gap is located within a single intron of at least 40 kb of the CUTL1 locus.
Figure 2.

Map of the sequenced contigs. Sequenced clones are drawn as open rectangles containing the clone names. Arrows indicate the genes and their transcriptional directions. The genes are (1) ZAN, (2) EPO, (3) CDS1, (4) CDS2, (5) GNB2, (6) ACTL6, (7) TFR2, (8) CDS3, (9) PCOLCE, (10) CDS4, (11) LRN, (12) IRS3L, (13) HRBL, (14) CDS5, (15) PMSL12, (16) APS, and (17) CUTL1. The GC content is drawn below the gene arrows with a step of 1000 and a sliding window of 100. (A) EPO contig. The single sequencing gap is indicated by a star. (B) CUTL1 contig. The cloning gap is not drawn to scale.
Genomic DNA Sequence
Using the shotgun method we have sequenced 644 kb of genomic DNA in 7q22 spread over two regions, the EPO contig of 228 kb and the CUTL1 contig of 416 kb (Fig. 2A,B). In the EPO contig one sequencing gap remains that could not be closed despite numerous attempts with various sequencing chemistries and a PCR approach using several primer sets. This may be due to its repetitive nature, that is, a SVA repeat (Zhu et al. 1992) is flanking this gap. Using the overlap between individual bacterial clones we determined the accuracy of the final sequence to be >99.99%.
Automated First-Pass Annotation by RUMMAGE-DP
The sequence was first analyzed using a software package RUMMAGE-DP developed in our laboratory for automated first-pass annotation (R. Schattevoy, J. Weber, B. Drescher, G. Glöckner, and A. Rosenthal, in prep.). RUMMAGE-DP contains 26 different programs including tools for predicting repetitive and compositional sequence elements (REPEATMASKER, CENSOR), several exon prediction engines (GENESCAN, XGRAIL, MZEF, XPOUND, FEXHB), homology search programs (BLAST, DPS), as well as tools for verifying predicted exons using expressed sequences (EXONSAMPLER). RUMMAGE-DP is based on a distributed processing mode and is run on a farm of UNIX workstations. RUMMAGE-DP is available via a server at the IMB in Jena (http://genome.imb-jena.de). The RUMMAGE-DP results are converted into three different formats: HTML, ACeDB, and GenBank. Automated first-pass annotation of the 228-kb EPO sequence by RUMMAGE-DP is graphically displayed in Figure 3. Strand-specific information is compiled in the uppermost part for the forward strand and in the lower part for the reverse strand, respectively. This 228-kb contig is extremely gene rich, as indicated by the large number of exon clusters shown in red. Known genes and genes similar to known homologs in other species (EPO, GNB2, PCOLCE, and ZAN) are easily detected by the presence of BLASTX hits against the GenPept database. BLAST hits of individual exons showing similarity to the same mRNA or EST are connected by green lines to facilitate the detection of entire genes. The light green and yellow blocks show the local GC content as well as CpG islands (Fig. 3, middle). The inverted repeat regions are also shown here. The tandem repeat structures are depicted on the first line followed by the plus strand-specific Alu and non-Alu repeats.
Figure 3.

Graphic output of RUMMAGE results of the EPO contig. The vertical line represents the sequencing gap in the EPO contig. Rectangles define the regions in which matches were found. Corresponding matches are connected by green lines. Repeat structures are derived from Repeatmasker, Censor; exon predictions from GENESCAN, GRAIL2, FEXHB, MZEF, XPOUND; promotor prediction from ProScan; motif matches from DPS; poly(A) signals from Pol II; protein motifs from PROSITE; BLAST matches from BLASTS of various databases, Exon sampler. For references, see Methods. The graphic and tabular output of the automated first-pass annotation of both the EPO and the CUTL1 contig with RUMMAGE-DP is viewable via our Home page at http://genome.imb-jena.de/.
GC Content
Despite their location in the same Giemsa band the GC content of the two regions is very different. Most of the sequence in the EPO contig shows an average GC content of 53.7%. Some stretches with a lower GC content contain fewer exons but more repetitive elements. All genes identified in the EPO contig are associated with CpG islands, which is in good correlation with previous findings (Cross and Bird 1995). In contrast, the average GC content of the CUTL1 contig is 49%. However, between the APS gene and the 3′ end of the CUTL1 locus the GC content approaches 60% (Fig. 2).
Distribution of Repetitive Elements
Table 1 summarizes the content and distribution of genome wide repeats for both sequenced regions. To find these repeats the default settings of the programs used were applied. In the gene-dense EPO contig, as well as in the first part of the CUTL1 region (CUTL1-1) containing the genes CDS5, PMSL12, and APS, a similar overall repeat content of 38.3% and 37.3% was found. The major difference between these two regions is the amount of LTRs. The second part of the CUTL1 locus (CUTL1-2) contains twice and three times as many LINE repeats compared with the EPO contig and the CUTL1-1 region, respectively. Hence, CUTL1-2 has an overall repeat content of 44.8%. There are three clusters of LINE repeats in PAC clone 50H2 belonging to the CUTL1-2 region. A fourth LINE cluster was found in PAC 37G3.
Table 1.
Repeats Found in the Three Contigs
| EPO | CUTLI-1 | CUTLI-2 | ||||
|---|---|---|---|---|---|---|
| no. | % | no. | % | no. | % | |
| SINE | 304 | 34.13 | 268 | 32.35 | 232 | 32.5 |
| Alu | 295 | 33.77 | 249 | 31.48 | 215 | 31.41 |
| Mir | 9 | 0.35 | 19 | 0.87 | 17 | 1.09 |
| LINE | 32 | 3.08 | 18 | 2.04 | 35 | 7.3 |
| LINE1 | 16 | 1.87 | 8 | 0.83 | 22 | 6.07 |
| LINE2 | 16 | 1.21 | 10 | 1.21 | 13 | 1.23 |
| LTR | 4 | 0.38 | 16 | 2.1 | 15 | 1.99 |
| MaLRs | 4 | 0.38 | 3 | 0.6 | 6 | 1.27 |
| Retrov. | 11 | 0.8 | 7 | 0.12 | ||
| MER4_group | 2 | 0.6 | 1 | 0.24 | ||
| DNA | 8 | 0.61 | 6 | 0.4 | 19 | 3.05 |
| MER1 type | 6 | 0.40 | 3 | 0.22 | 10 | 1.20 |
| MER2 type | 1 | 0.13 | 2 | 0.16 | 8 | 1.82 |
| Mariners | 1 | 0.03 | 1 | 0.03 | ||
| 1 | 0.08 | |||||
| UNCL | 1 | 0.13 | 5 | 0.42 | 0 | 0 |
| Small RNA | 3 | 0.09 | 1 | 0.01 | ||
| Total | 38.33 | 37.35 | 44.84 | |||
Exon Prediction
RUMMAGE-DP comprises five exon prediction programs that are based on different search algorithms and predicts a total of 302 exon positions within the 228-kb contig (Table 3, below; compiled exon predictions in Fig. 3). Many of these exons are only predicted by one or two of these programs. Our detailed analysis showed that these can be estimated as false positives. However, if an exon is predicted by three or more programs it is probably a true exon and was considered as a part of the real gene structure (Table 2). To support this general finding we used the known genes in both contigs (EPO, GNB2, PCOLCE, APS, CUTL1) as an internal control. In the EPO contig and the exon-dense regions of the CUTL1 contig, all confirmed exons of the known genes were correctly predicted by at least three programs within RUMMAGE-DP. However, most programs failed to predict the correct exons at the 5′ end of the CUTL1 locus and around the AT-rich PMSL12 gene. In these two regions gene structures can only be defined using EST or cDNA data.
Table 3.
Genes Found in the Analyzed Regions
| Gene | Description | Accession/ homolog | No. of exons on contig | Minimum length (bp) of mRNA | Minimum length (bp) on genome | ORF length (aa) | mRNA covered by ESTs (%) | Detection method/comment |
|---|---|---|---|---|---|---|---|---|
| A. EPO contig | ||||||||
| ZAN | zonadhesin | U83191 partial human mRNA | 34 | 7233 | >48,500 | 2177 | 0 | alignment to S. scrofa zan U40024 |
| EPO | erythropoietin | M11329 human mRNA | 6 | 783 | 2,700 | 193 | 10 | genomic structure and mRNA sequence already known |
| CDS1 | unknown | EST aa 158469 | 2 | 461 | 980 | 168 | 100 | Alignment to aa 158469 |
| CDS2 | unknown | U90567G. gallus | 19 | 3065 | 7,250 | 817 | 30 | one EST, exon prediction programs |
| GNB2 | G-nucleotidebinding factor | M16514 human mRNA | 10 | 1438 | 2,950 | 340 | 100 | mRNA sequence already known |
| ACT16 | actin-like protein | D32140C. merolae | 13 | 1541 | 10,100 | 475 | 77 | overlapping ESTs, cDNA sequencing |
| TFR2 | transferrin receptor | X01060-related human mRNA | 18 | 2531 | 20,500 | 786 | 95 | overlapping ESTs, cDNA sequencing, 60% homology to X01060 |
| CDS3A | unknown | C34D10 C. elegans | 6 | 1034 | 3,200 | 182–235 | 100 | overlapping ESTs, cDNA sequencing; only homology to C. elegans ORF; splicing variants lead to different N amino termini |
| CDS3B | 6 | 1013 | 2,800 | 100 | ||||
| CDS3C | 4 | 842 | 2,330 | 100 | ||||
| POLCE | procollagen C-proteinase enhancer | L33799 mRNA | 9 | 1480 | 5,800 | 449 | 100 | mRNA sequence already known |
| CDS4 | unknown | EST aa 251566 | 8 | 1530 | 10,750 | 318 | 25 | gene prediction programs |
| LRN | leucine-rich neuronal protein | X79682Felis catus | 19 | 2407 | 13,000 | 612 | 70 | overlapping ESTs; gene prediction programs |
| IRS3L | insulin receptor substrate 3-like protein | U93880Rattus norvegicus | 5 | 1228 | 3,600 | 256 | 0 | gene prodiction programs |
| HRBL | nucleoporin-like protein | D14689-related human mRNA | 9 | 1321 | 17,000 | 327 | 85 | overlapping ESTs; gene prediction programs |
| B. CUTL1 contig | ||||||||
| CDS5 | unknown | EST T11673 | 7 | 1499 | 7,400 | 235 | 80 | overlapping ESTs; prolin rich protein |
| PMSL12 | mismatch repair gene | U14658-related human mRNA | 6 | 1454 | 18,300 | 219 | ? | Alignment with pms2 |
| APS | adaptor protein | AB000520 human mRNA | 9 | 2111 | 36,500 | 632 | 30 | mRNA sequence already known |
| CUTL1 (CDP) | human displacement protein | M74099 human mRNA | 21 | 5376 | >285,000 | 1505 | 15 | mRNA sequence already known |
| (CASP) | alternatively spliced CDP | L12579 human mRNA | 22 | 2855 | >320,000 | 678 | 60 | mRNA sequence already known |
Table 2.
Success of Exon Predictions for Selected Known and Unknown Genes
| Exon | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EPO known | |||||||||||||
| MZEF | − | − | + | + | + | + | |||||||
| GRAIL | − | + | + | + | + | − | |||||||
| GENESCAN | − | + | + | + | + | + | |||||||
| XPOUND | − | + | + | + | + | + | |||||||
| FEXHB | − | − | + | − | − | + | |||||||
| GNB2 known | |||||||||||||
| MZEF | − | + | + | + | + | + | − | + | + | − | |||
| GRAIL | + | + | + | + | + | + | − | + | + | − | |||
| GENESCAN | + | + | + | + | + | + | + | + | + | − | |||
| XPOUND | + | + | + | + | + | + | + | − | + | − | |||
| FEXHB | + | + | + | − | + | − | − | − | − | − | |||
| ActL6 unknown | |||||||||||||
| MZEF | − | − | + | + | + | + | + | + | + | − | + | + | + |
| GRAIL | + | + | + | + | + | + | + | + | + | + | + | + | + |
| GENESCAN | − | + | + | + | + | + | + | + | + | + | + | + | + |
| XPOUND | + | + | + | + | + | + | + | − | + | + | + | + | + |
| FEXHB | − | + | + | + | + | − | − | − | − | − | − | − | − |
| PCOLCE known | |||||||||||||
| MZEF | − | + | + | + | + | + | + | + | − | ||||
| GRAIL | + | + | − | + | + | + | + | + | + | ||||
| GENESCAN | + | + | + | + | + | + | + | + | + | ||||
| XPOUND | + | + | + | + | + | + | + | − | − | ||||
| FEXHB | − | + | − | + | − | − | + | + | + | ||||
| CDS4 unknown | |||||||||||||
| MZEF | + | − | + | − | + | + | + | − | + | − | |||
| GRAIL | + | + | + | − | + | + | + | − | − | − | |||
| GENESCAN | + | + | + | + | + | + | + | + | − | − | |||
| XPOUND | + | + | − | − | + | + | + | − | − | − | |||
| FEXHB | − | − | + | + | + | − | − | − | − | − | |||
EXON SAMPLER is a special analysis tool within RUMMAGE-DP that has been developed in our laboratory (J. Weber, B. Drescher, R. Schattevoy, and A. Rosenthal, unpubl.). It compiles matching ESTs and the positions of coding sequences of related genes for the construction of complete gene structures.
For gene prediction best results are obtained if exons suggested by three or more exon prediction engines and summarized in the Compile Exon results of RUMMAGE-DP are combined with the exons defined by EXON SAMPLER. However, a serious analysis problem for EXON SAMPLER are the many genomic sequences contaminating the EST database that may lead to the construction of artificial or wrong mRNAs. To minimize this problem we used only those ESTs containing at least one intron.
Identification of Transcription Units
Table 3 summarizes the main features of 17 genes in the two genomic regions of 7q22 analyzed. Five genes (CDS1–CDS5) did not show similarity to any known protein or mRNA; hence, no function could be assigned. Seven unknown genes (CDS1, CDS3, CDS5, ACTL6, TFR2, LRN, and HRBL) showed nearly complete EST coverage. For detailed analyses selected EST clones were resequenced to obtain the complete coding information for these genes. This expressed sequence information was then used to confirm predicted exons and to obtain exon/intron structures for genes CDS2, ACTL6, and CDS3. Two genes (CDS4, IRS3LL) did not show any similarity to closely related genes or ESTs and their existence is based only on the prediction of exon clusters by RUMMAGE-DP. Nevertheless, gene models were built that remain speculative. However, the CDS genes spanning a large portion of these predicted genes may be used as a hint for their functionality.
Zonadhesin
The zonadhesin (ZAN) gene encodes a sperm membrane protein that binds to the extracellular matrix of the egg in a species-specific manner. In human, only the 3′ portion of the mRNA was known, whereas in Sus scrofa the complete mRNA was described previously (Hardy and Garbers 1995). The human gene was recently localized to 7q22 (Gao et al. 1997). Alignment of the pig ZAN mRNA to the human genomic sequence showed a similarity of ∼61% and allowed the identification of 33 coding exons of the human ZAN gene spanning a chromosomal region of >48 kb.
EPO
The glycoprotein hormone EPO regulates the level of oxygen in the blood by modulating the number of circulating erythrocytes (Cowling and Dexter 1992). Both the mRNA and the gene structure have been known for a long time (Lin et al. 1985).
CDS1
CDS1, a small gene of unknown function is defined by several matching ESTs, most of which cover only one of the two predicted exons. Database entries define CDS1 as a single exon gene. It shows no striking similarity to any known gene. Our studies show that two ESTs are spliced, thus confirming the two exons predicted from the genomic sequence.
CDS2
CDS2 describes a gene of unknown function spanning a genomic region of∼5 kb. A cluster of 19 exons has been predicted within this interval. Similarity searches revealed only one single EST clone matching the 3′ end of the gene. The gene product derived by translation from the spliced exons shows a good similarity score of 62% in a segment of 261 amino acids to a glutamine-rich protein from Gallus gallus (U90567), as well as to a yeast protein (GenBank accession no. M90654) that is assumed to suppress the MYO2 gene, which is essential for the vectorial transport of vesicles (Schaaff-Gerstenschlager et al. 1993).
Guanine nucleotide binding factor 2
A variety of genes has been identified that specify the synthesis of the components of guanine nucleotide-binding proteins (G proteins). Guanine nucleotide binding factor 2 (GNB2) encodes a β subunit of G proteins. Its complete mRNA sequence and localization to chromosome 7 have been described previously (Fong et al. 1987; Blatt et al. 1988). Alignment of GNB2 mRNA with the genomic sequence revealed the complete exon/intron structure. As a gene transcribed at a high level it is also completely covered by matching ESTs.
Actin (ACTL6)-like
More than 75% of the predicted gene is covered by overlapping ESTs. Exon prediction and resequencing of selected ESTs revealed 13 exons spanning a region of 10 kb. A similarity of 52% with a Cyanidioschizon merolae actin (D32140) suggests that the Actin (ACT)-like protein is a member of the actin family of proteins. Interestingly, multiple alignments show that the ACT-like protein does not share the high degree of conservation known among actins from vertebrates, including mammals and possesses a smaller number of actin-typical domains (data not shown). Tree analysis using the neighbor joining method shows that the ACT-like protein is more closely related to actins from lower organisms like Nagleria fowleri (M90311) than to actins from vertebrates and mammals (Fig. 4). Thus, the ACT-like protein may represents the first example of a new subclass of actin-related proteins. Several lines of evidence suggest that there are many actin pseudogenes spread over the whole genome. One possible actin pseudogene, ACTBP5, has been located in 7q22–7ter (Ng et al. 1985). But because the act-like gene described here is almost completely covered with ESTs and has an open reading frame (ORF) it is unlikely that it is identical with ACTBP5.
Figure 4.

Phylogenetic tree of act genes. Sequences were aligned using CLUSTALW. The phylogenetic tree was constructed using PHYLIPP. The random seed value was 75 with 15× to jumble.
Transferrin
Transferrin receptors are involved in the cellular transport of iron (Richardson and Ponka 1997). In human, only transferrin receptor 1 (TRR1) has been described (Enns and Sussmann 1981). It is a transmembrane glycoprotein with a molecular mass of 180 kD. In trypanosomes, several variants of this protein exist to avoid interference by antireceptor antibodies (Borst 1991). TFR2, which shows a 60% similarity to TRR1, is the second transferrin receptor found in human. TFR2 may bind other ligands than transferrin; therefore, its role in iron metabolism may be different.
CDS3
CDS3, which shows only similarity to a putative gene in Caenorhabditis elegans (Wilson et al. 1994), is a novel human gene with unknown function. Five overlapping human ESTs have been found that, after resequencing, were used to establish the exon/intron organization of this gene. Detailed analysis of all human ESTs showed that this gene exists in at least three splice variants (Fig. 5). All three splice variants allowed the translation of an ORF comprising the same carboxyl terminus with two transmembrane domains. Based on PSort (http://psort.nibb.ac.jp) we hypothesize that the three splice variants of CDS3 may be used in different compartments of the cell: variant a in the endoplasmic reticulum and variants b and c in the plasma membrane.
Figure 5.

The splice variants of CDS3. Rectangles connected by lines indicate the different gene structures of the splice variants. The minimal EST coverage of the splice variants is drawn below the gene structures. The gene location on the EPO contig, together with the transcription direction, is given above the line representing the sequence.
Procollagen C-proteinase enhancer
Procollagen C-proteinase enhancer (PCOLCE) is a specific glycoprotein in the connective tissue that is likely to regulate the processing of procollagen in vivo (Kessler et al. 1990). Alignment of the known PCOLCE mRNA (Takahara et al. 1994) with the genomic sequence revealed nine exons spread over 6 kb.
CDS4
For CDS4, a cluster of eight exons has been predicted in a genomic region of 11 kb. The last two exons at the 3′ end have been confirmed by the existence of a single matching EST. Translation of all predicted exons yields a long coding sequence that shows no similarity to any protein in the databases.
Leucine-rich neuronal protein
A combination of exon prediction and resequencing of overlapping EST was used to establish the exon/intron organisation of the leucine-rich neuraonl (LRN) gene. It has 19 exons spanning a region of 13 kb. The predicted LRN protein contains leucine-rich repeats (LRR) at its amino terminus (amino acid positions 40–262) known to be involved in ligand binding (Kobe and Deisenhofer 1995). These repeats are followed by a region (amino acids 462–538) that shows a similarity of 60% to a partially characterized gene from Felis catus (GenBank accession no. X79682) encoding a neuronal protein and the carboxyl terminus (amino acids 588–604), which may be a membrane anchor. The identified structural elements suggest that LRN resembles a receptor.
Insulin receptor substrate-like protein
For the insulin receptor substrate 3-like (IRS3L) protein, a cluster of five exons have been predicted within 3 kb of genomic DNA by several programs. No EST or mRNA matches have been found to confirm the putative gene structure. A BLAST search of the translated putative protein revealed a similarity of 62% in the carboxy-terminal part of the predicted protein to insulin receptor substrate proteins which, however, is restricted to a short stretch between amino acids 168–246 of the rat insulin receptor substrate protein (Lavan et al. 1997). The lack of expressed sequences suggests that IRS3L may be transcribed at a very low level or alternatively represents a pseudogene.
HIV-1 Rev binding like protein
The HIV-1 Rev-binding-like protein (HRBL) that encodes a nucleoporin-like protein which may be involved in nuclear transport of viral RNAs (Bogerd et al. 1995; Fritz et al. 1995). The HRB protein is 200 amino acids shorter than the related nucleoporin. Although at the amino terminus the HRBL protein is very similar to nucleoporin the similarity between both proteins declines in the carboxy-terminal part. The gene structure of the HRBL protein described here is incomplete. The promoter region and the first 48 amion acids are not contained on our sequence.
CDS5
A cluster of seven exons in 7.4 kb has been predicted to represent CDS5. Although 80% of this novel gene is matched by ESTs no significant similarity to known genes or proteins has been found. The translated protein of unknown function contains many prolines and glutamic acids and is predicted to reside in the nucleus according to the results of PSort.
Postmeiotic segregation
The postmeiotic segregation gene PMSL12 belongs to a large family of genes localized on human chromosome 7, which is involved in mismatch repair. One important member of this family is the human PMS2 mismatch repair gene that has been mapped previously to 7q22 and shown to be causative in hereditary nonpolyposis colon cancer (Peltomaki and de la Chapelle 1997). Seventeen other PMS-related genes have been mapped to various positions of chromosome 7 (Nicolaides et al. 1995; Osborne et al. 1997). The exon prediction programs within RUMMAGE-DP failed to reveal the gene structure of the PMSL12 gene. Although ESTs could be identified that match the PMSL12 locus with ∼90%, they possibly originated from different PMS-related genes and were therefore of limited use for assigning the correct splice sites. The structure of the PMSL12 gene comprising 6 exons within 18 kb was predicted by alignment with PMS2 (GenBank accession no. U38964).
Adaptor protein with PH and SH2 domain
The adaptor protein with PH and SH2 domain (APS) gene consists several domains including a pleckstrin homology (PH) domain, a Src homology 2 (SH2) domain, and a tyrosine phosphorylation site. Several lines of evidence suggest that APS may link immune receptors to signalling pathways involved in tyrosine phosphorylation (Yokouchi et al. 1997). The APS mRNA was known previously. We describe here its genomic structure and map position.
Cut-like homeobox
The CUTL1 (cut-like homeobox) locus has been mapped previously to 7q22 (Scherer et al. 1993b) and was shown to encode a transcriptional repressor that down-modulates the expression of c-MYC (Dufort and Nepveu 1994). In uterine leiomyomas, deletion of 7q22 or reduced expression of CUTL1 has often been observed. It has been suggested that this locus may be involved in the etiology of these tumors (Zeng et al. 1997). The CUTL1 locus spans a genomic region of >300 kb and consists of 31 exons that have been deducted by alignment with the known mRNA. The 5′ exons are separated by very large introns, with the largest ∼85 kb in size. CUTL1 exists in two alternative splice variants: CDP and CASP. CDP has 21 exons including the homeobox domains and the cut repeats. CASP lacks these domains and comprises 22 exons. CASP therefore may have lost its ability to bind DNA. The genomic structure of the CUTL1 locus comprising splice variants CDP and CASP differs from the corresponding Cux/Cdp/mCasp locus in mouse. It has been shown that the 3′ exons of CASP are interposed between cut repeats 2 and 3 of the murine Cux/Cdp gene (Lievens et al. 1997). In the human CUTL1 locus the 3′ exons of CASP are attached to the CDP splice variant.
DISCUSSION
Human genes are not uniformly distributed along the chromosomes. The chromosomal band 7q22 is extremely gene rich and, like Xq28, represents a prime target region for large-scale sequencing and analysis. We have sequenced and carefully annotated 650 kb of genomic DNA in 7q22.
As sequencing of human DNA in the scale of hundreds of kilobases has become more routine the analysis and annotation of these large blocks of genomic DNA is still a very tedious manual procedure. To make full use of human genomic sequence through comprehensive annotation we have developed a new automated annotation tool RUMMAGE-DP for automated first-pass analysis. We used it extensively to annotate the 650 kb of genomic sequence from the EPO and the CUTL1 region in 7q22.
The key problem in analysis and annotation is gene finding by exon prediction and homology searches or a combination of both followed by the construction of complete exon/intron structures of confirmed human genes. Using RUMMAGE-DP, we were able to predict 17 genes within 650 kb of genomic sequence. Both the EPO and CUTL1 contigs contain very short intergenic sequences. This suggests that we may have found all human genes in this interval, although we cannot exclude that an additional gene resides in the cloning gap of the CUTL1 locus.
Gene finding and establishing complete genomic structures for human genes on the bases of their complete mRNAs should be highly reliable. Although gene finding can be done automatically and in a few hours, establishing the exact genomic structure of a human gene requires the execution of various manual procedures like EST resequencing, careful alignments, and evaluations of homology searches. Three resources can be used for verifying predicted genes on the mRNA level: human ESTs, ESTs or mRNAs from other species, and ESTs or mRNAs from gene families. If no significant EST or mRNA matches can be found gene structures have to be based entirely on predicted exons. There are several problems connected with all of these methods. First, according to our findings, EST databases are contaminated with ESTs that seem to be of genomic origin. Therefore, only those ESTs that are spliced and span at least two predicted exons were used for the reconstruction of mRNAs. Second, in general the coverage of genes with ESTs is not uniform. This may be due to different expression profiles of individual genes in particular tissues and the methods used for generating these EST databases. In cases where overlapping ESTs cover all or most of the predicted exons of a single gene (Table 3, genes CDS1, ACTL6, TFR2, CDS3, CDS5, LRN, and HRBL) the gene structure should be correct. Figure 6 shows the overall coverage of the gene-rich EPO contig with ESTs. GNB2 and PCOLCE show the highest EST coverage, followed by CDS1 and the ACT-like protein. In contrast, ZAN, EPO, CDS2, and CDS4 are not or only poorly covered by ESTs. All other genes of the EPO contig show modest EST coverage. We assume that this EST map may reflect the transcription level of the genes quite accurately. In general, genes that are transcribed at low levels, or transcribed only, for example, in embryonic tissues at certain times are not or only poorly represented in the EST database. These genes can be detected more easily by genomic sequencing.
Figure 6.

EST coverage of the total EPO contig. All matches of ESTs to the EPO contig in either strand are shown as lines. Matches of the same EST are represented by lines covering the whole region within the matches. Regions of the genes found are drawn above the EST matches.
As discussed, another possibility to reveal the genomic and mRNA structure of a gene is to make use of genes from other organisms or families that are highly similar (Table 3; ZAN and PMSL12). In these cases, we were able to detect many coding exons easily, but we cannot exclude that some exons with no similarity to the mRNA from other species are omitted.
When no EST or mRNA matches could be found we used only the predicted exons to construct gene models. It is well known that single exon prediction programs often fail to predict certain exons (especially 5′- and 3′-untranslated exons, very small exons, or exons followed or preceded by very large introns) and often detect false positives (Lopez at al. 1994; Burge and Karlin 1997). The ability to predict correct exons is often correlated with the GC content of a region that also reflects gene density. To discriminate between correct and false positives exons more precisely, five exon prediction programs (XGRAIL, XPOUND, MZEF, FEXHB, and GENESCAN) were used. In our experience, exons predicted by more than two of these programs are most likely true exons. This was confirmed by comparison of predicted exons with true exons, which were confirmed on the EST/mRNA level. CDS2 and CDS4 are examples of this situation, as they are scarcely covered by ESTs, but exons have been predicted by several programs. The putative gene models of CDS2 and CDS4 need to be confirmed. Another problem is the prediction of exons in AT-rich isochores of the chromosome (Fig. 2). No exon of the PMSL12 gene could be predicted by more than one exon prediction program. The same problem occurred in the 5′ portion of the CUTL1 locus, where a similar AT content was observed. Thus, in AT-rich isochores the detection of genes may depend more on the existence of human ESTs and on the knowledge of related genes (ESTs or mRNAs) in other species.
Detailed GC content analysis showed that the EPO contig with its 13 genes has an average GC content of 53% and represents an H3 isochore. Although some local regions show a GC content well over 60%, other sections have a GC content below 50%. Concerning the GC content the CUTL1 contig can be divided into two blocks. Most of the CUTL1 gene has an average GC content of 48% and can be defined as an H2 isochore. The region including the PMSL12, APS, and the very 3′ end of the CUTL1 locus shows a GC content of almost 60% and represents a H3 isochore. This interesting finding of a possible isochore switch within a region of 450 kb is supported by statistical analysis, showing that long genes are scarce in GC-rich isochores (Duret et al. 1995). Our data suggest that 7q22 is heterogeneous concerning gene density and GC content and may be composed of different isochores.
METHODS
DNA Sequencing
The PAC and cosmid clone DNAs were isolated using a standard alkaline lysis method (Birnboim and Doly 1979). The DNA was purified on a CsCl radient (Radloff et al. 1967). The closed circle band was sonicated, size fractionated, and ligated into M13 vector after filling the protruding ends by T4 plymerase treatment (NEB) (Craxton 1993). The M13 templates were prepared by the Triton method (Mardis 1994). In the shotgun phase of a sequencing project all templates were sequenced by dye-terminator chemistries (Perkin Elmer). Data were collected using ABI 377 automated sequencers and assembled with GAP4 (Staden 1996). Most gaps were closed by performing long runs using dye primer chemistry for sequencing of the M13 templates. With custom-made primers, M13 templates and PCR products derived from the cosmid or PAC clone were sequenced. PCR bands were purified using the Genomed Gel Extraction Kit. With Big dye chemistry (ABI) eight remaining gaps on PAC 37G3 were closed by sequencing with custom-made primers on the PAC clone. cDNA clones of matching ESTs were received from the ResourceCenter/Primary Database (Berlin). The sequences of the cDNA clones were directly determined using universal and custom made primers.
Resequencing of EST Cones
Clones IMAGp998A21255, IMAGp998C17736, IMAGp998B16677, IMAGp998G011743, IMAGp998G23194, IMAGp998G13270, IMAGp998G15286, IMAGp998J181536, IMAGp998J161008, IMAGp998K211672, IMAGp998K231889, IMAGp998K031714, IMAGp998K13435, IMAGp998N101745, IMAGp998N201717, IMAGp998N23584, IMAGp998N17972, IMAGp998P10406, IMAGp998P171429, IMAGp998P091672, IMAGp998O17311, IMAGp998L021744, and IMAGp998P221167 were used for the confirmation of cDNA structures.
Clone Names
In other studies clones PAC37g3, 164c7, 235f8, PAC76h2, PAC123e15, 186d2, PAC59h2, and 46f6 are also called H DJ0037G03, cos164c7, cos235f8, H DJ0076H02, H DJ0123E15, cos 186d2, H DJ0050H02, and cos46f6, respectively.
Computer Analysis
Repetitive sequences were tagged and removed for subsequent analyses using Censor (Jurka et al. 1996) and RepeatMasker (A.F.A. Smit, and P. Green, http://ftp.genome.washington.edu/RM/RepeatMasker.html) with default settings. Tandem and inverted repeat regions were determined using the algorithms of the ACeDB package (J. Thierry-Mieg and R. Durbin, pers. comm.). The Wisconsin Sequence Analysis Package (Genetics Computer Group, Inc.) and the algorithm of Huang (1994) were used to determine the GC content and distribution. Programs XPOUND (Thomas and Skolnick 1994), XGRAIL (Xu et al. 1994), GENESCAN (Burge and Karlin 1997), FEXHB (Solovyew and Salamov 1996), and MZEF (Zhang 1997) were used with default settings for exon predictions.
Homology searches against various databases were performed using BLAST (Altschul et al. 1990). BlastN of predicted exons was performed against the human, EST, and emnew subdivision of the EMBL database. BlastN of the whole sequence was done against the EST, genembl, and the codseq database. Codseq an inhouse built database (J. Weber, unpubl.) comprises all nonredundant CDS entries of the EMBL database. BlastX against the translated EMBL database (genpept) was done using the translated genomic sequence. The exon sampler integrated predicted exons information with EST databases (J. Weber and B. Drescher, pers. comm.). DPS was used for the comparison of DNA sequences with protein databases (Huang 1996). Promotor predictions were done with Pol II (Prestridge 1995). PROSITE was used for the analysis of patterns in predicted exons (Bairoch 1993). PSORT (http://psort.nibb.ac.jp) was used for the prediction of protein localizations.
Acknowledgments
We thank S. Förste and S. Landmann for expert technical assistance. G.G., R.S., and J.W. were funded by a grant from the German BMBF, Projektträger BEO Förderungsnummer 0311108.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL arosenth@imb-jena.de; FAX 49-3641-656255.
REFERENCES
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Bairoch A. The PROSITE dictionary of sites and patterns in proteins, its current status. Nucleic Acids Res. 1993;21:3097–3103. doi: 10.1093/nar/21.13.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnboim HC, Doly J. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 1979;7:1513–1523. doi: 10.1093/nar/7.6.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blatt C, Eversole-Cire P, Cohn VH, Zollman S, Fournier RE, Mohandas L T, Nesbitt M, Lugo T, Jones DT, Reed RR. Chromosomal localization of genes encoding guanine nucleotide-binding protein subunits in mouse and human. Proc Natl Acad Sci. 1988;85:7642–7646. doi: 10.1073/pnas.85.20.7642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogerd HP, Fridell RA, Madore S, Cullen BR. Identification of a novel cellular cofactor for the Rev/Rex class of retroviral regulatory proteins. Cell. 1995;82:485–494. doi: 10.1016/0092-8674(95)90437-9. [DOI] [PubMed] [Google Scholar]
- Borst P. Transferrin receptor, antigenic variation and the prospect of a trypanosome vaccine. Trends Genet. 1991;7:307–309. doi: 10.1016/0168-9525(91)90406-g. [DOI] [PubMed] [Google Scholar]
- Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- Craxton M. Cosmid sequencing. Methods Mol Biol. 1993;23:149–167. doi: 10.1385/0-89603-248-5:149. [DOI] [PubMed] [Google Scholar]
- Cross SH, Bird AP. CpG islands and genes. Curr Opin Genet Dev. 1995;5:309–314. doi: 10.1016/0959-437x(95)80044-1. [DOI] [PubMed] [Google Scholar]
- Cowling GJ, Dexter TM. Erythropoietin and myeloid colony stimulating factors. Trends Biotechnol. 1992;10:349–357. doi: 10.1016/0167-7799(92)90267-y. [DOI] [PubMed] [Google Scholar]
- Dufort D, Nepveu A. The human cut homeodomain protein represses transcription from the c-myc promoter. Mol Cell Biol. 1994;14:4251–4257. doi: 10.1128/mcb.14.6.4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, Mouchiroud D, Gautier C. Statistical analysis of vertebrate sequences reveals that long genes are scarce in GC-rich isochores. J Mol Evol. 1995;40:308–317. doi: 10.1007/BF00163235. [DOI] [PubMed] [Google Scholar]
- Enns CA, Sussmann HH. Similiarities between the transferrin receptor proteins on human reticulocytes and human placentae. J Biol Chem. 1981;256:12620–12623. [PubMed] [Google Scholar]
- Fischer K, Frohling S, Scherer SW, McAllister Brown J, Scholl C, Stilgenbauer S, Tsui LC, Lichter P, Dohner H. Molecular cytogenetic delineation of deletions and translocations involving chromosome band 7q22 in myeloid leukemias. Blood. 1997;89:2036–2041. [PubMed] [Google Scholar]
- Fong HK, Amatruda TT, Birren BW, Simon MI. Distinct forms of the beta subunit of GTP-binding regulatory proteins identified by molecular cloning. Proc Natl Acad Sci. 1987;84:3792–3796. doi: 10.1073/pnas.84.11.3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz CC, Zapp ML, Green MR. A human nucleoporin-like protein that specifically interacts with HIV Rev. Nature. 1995;376:530–533. doi: 10.1038/376530a0. [DOI] [PubMed] [Google Scholar]
- Gao Z, Harumi T, Garbers DL. Chromosome localization of the mouse zonadhesin gene and the human zonadhesin gene (ZAN) Genomics. 1997;41:119–122. doi: 10.1006/geno.1997.4620. [DOI] [PubMed] [Google Scholar]
- Hardy DM, Garbers DL. A sperm membrane protein that binds in a species-specific manner to the egg extracellular matrix is homologous to von Willebrand factor. J Biol Chem. 1995;270:26025–26028. doi: 10.1074/jbc.270.44.26025. [DOI] [PubMed] [Google Scholar]
- Huang X. An algorithm for identifying regions of a DNA sequence that satisfy a content requirement. Comput Appl Biosci. 1994;10:219–225. doi: 10.1093/bioinformatics/10.3.219. [DOI] [PubMed] [Google Scholar]
- ————— Fast comparison of a DNA sequence with a protein sequence database. Microb Comp Genomics. 1996;1:281–291. doi: 10.1089/mcg.1996.1.281. [DOI] [PubMed] [Google Scholar]
- Ishwad CS, Ferrell RE, Hanley K, Davare J, Meloni AM, Sandberg AA, Surti U. Two discrete regions of deletion at 7q in uterine leiomyomas. Genes Chromo Cancer. 1997;19:156–160. doi: 10.1002/(sici)1098-2264(199707)19:3<156::aid-gcc4>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Jurka J, Klonowski P, Dagman V, Pelton P. CENSOR—A program for identification and elimination of repetitive elements from DNA sequences. Comput Chem. 1996;20:119–121. doi: 10.1016/s0097-8485(96)80013-1. [DOI] [PubMed] [Google Scholar]
- Kessler E, Mould AP, Hulmes DJ. Procollagen type I C-proteinase enhancer is a naturally occurring connective tissue glycoprotein. Biochem Biophys Res Commun. 1990;173:81–86. doi: 10.1016/s0006-291x(05)81024-1. [DOI] [PubMed] [Google Scholar]
- Kobe B, Deisenhofer J. A structural basis of the interactions between leucin-rich repeats and protein ligands. Nature. 1995;374:183–186. doi: 10.1038/374183a0. [DOI] [PubMed] [Google Scholar]
- Lavan BE, Lane WS, Lienhard GE. The 60-kDa phosphotyrosine protein in insulin-treated adipocytes is a new member of the insulin receptor substrate family. J Biol Chem. 1997;272:11439–11443. doi: 10.1074/jbc.272.17.11439. [DOI] [PubMed] [Google Scholar]
- Lievens PM, Tufarelli C, Donady JJ, Stagg A, Neufeld EJ. CASP, a novel, highly conserved alternative-splicing product of the CDP/cut/cux gene, lacks cut-repeat and homeo DNA-binding domains, and interacts with full-length CDP in vitro. Gene. 1997;197:73–81. doi: 10.1016/s0378-1119(97)00243-6. [DOI] [PubMed] [Google Scholar]
- Lin F-K, Suggs S, Lin C-H, Browne JK, Smalling R, Egrie JC, Chen K, Fox GM, Martin F, Stabinsky Z, et al. Cloning and expression of the human erythropoietin gene. Proc Natl Acad Sci. 1985;82:7580–7584. doi: 10.1073/pnas.82.22.7580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez R, Larsen F, Prydz H. Evaluation of the exon predictions of the GRAIL software. Genomics. 1994;24:133–136. doi: 10.1006/geno.1994.1590. [DOI] [PubMed] [Google Scholar]
- Mardis ER. High-throughput detergent extraction of M13 subclones for fluorescent DNA sequencing. Nucleic Acids Res. 1994;22:2173–2175. doi: 10.1093/nar/22.11.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SY, Gunning P, Eddy R, Ponte P, Leavitt J, Shows T, Kedes L. Evolution of the functional human beta-actin gene and its multi-pseudogene family: Conservation of noncoding regions and chromosomal dispersion of pseudogenes. Mol Cell Biol. 1985;5:2720–2732. doi: 10.1128/mcb.5.10.2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolaides NC, Carter KC, Shell BK, Papadopoulos N, Vogelstein B, Kinzler KW. Genomic organization of the human PMS2 gene family. Genomics. 1995;30:195–206. doi: 10.1006/geno.1995.9885. [DOI] [PubMed] [Google Scholar]
- Osborne LR, Herbrick JA, Greavette T, Heng HHQ, Tsui LC, Scherer SW. PMS2-Related genes flank the rearrangement breakpoints associated with williams syndrome and other diseases on human chromosome 7. Genomics. 1997;45:402–406. doi: 10.1006/geno.1997.4923. [DOI] [PubMed] [Google Scholar]
- Peltomaki P, de la Chapelle A. Mutations predisposing to hereditary nonpolyposis colorectal cancer. Adv Cancer Res. 1997;71:93–119. doi: 10.1016/s0065-230x(08)60097-4. [DOI] [PubMed] [Google Scholar]
- Prestridge DS. PolII Promoter Prediction v2.0. J Mol Biol. 1995;249:923–932. doi: 10.1006/jmbi.1995.0349. [DOI] [PubMed] [Google Scholar]
- Radloff R, Bauer W, Vinograd J. A dye-buoyant-density method for the detection and isolation of closed circular duplex DNA: The closed circular DNA in HeLa cells. Proc Natl Acad Sci. 1967;57:1514–1521. doi: 10.1073/pnas.57.5.1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson DR, Ponka P. The molecular mechanisms of the metabolism and transport of iron in normal and neoplastic cells. Biochim Biophys Acta. 1997;1331:1–40. doi: 10.1016/s0304-4157(96)00014-7. [DOI] [PubMed] [Google Scholar]
- Schaaff-Gerstenschlager I, Baur A, Boles E, Zimmermann FK. Sequence and function analysis of a 4.3 kb fragment of Saccharomyces cerevisiae chromosome II including three open reading frames. Yeast. 1993;9:915–921. doi: 10.1002/yea.320090811. [DOI] [PubMed] [Google Scholar]
- Scherer SW, Rommens JM, Soder S, Wong E, Plavsic N, Tompkins BJ, Beattie A, Kim J, Tsuia LC. Refined localization and yeast artificial chromosome (YAC) contig-mapping of genes and DNA segments in the 7q21-q32 region. Hum Mol Genet. 1993a;2:751–760. doi: 10.1093/hmg/2.6.751. [DOI] [PubMed] [Google Scholar]
- Scherer SW, Neufeld EJ, Lievens PM, Orkin SH, Kim J, Tsui LC. Regional localization of the CCAAT displacement protein gene (CUTL1) to 7q22 by analysis of somatic cell hybrids. Genomics. 1993b;15:695–696. doi: 10.1006/geno.1993.1130. [DOI] [PubMed] [Google Scholar]
- Staden R. The Staden sequence analysis package. Mol Biotechnol. 1996;5:223–241. doi: 10.1007/BF02900361. [DOI] [PubMed] [Google Scholar]
- Takahara K, Kessler E, Biniaminov L, Brusel M, Eddy RL, Jani-Sait S, Shows TB, Greenspan DS. Type I procollagen COOH-terminal proteinase enhancer protein: Identification, primary structure, and chromosomal localization of the cognate human gene (PCOLCE) J Biol Chem. 1994;269:26280–26285. [PubMed] [Google Scholar]
- Takahara K, Osborne L, Elliott EW, Tsui LC, Scherer SW, Greenspan DS. Fine mapping of the human and mouse genes for the type I procollagen COOH-terminal proteinase enhancer protein. Genomics. 1996;31:253–256. doi: 10.1006/geno.1996.0043. [DOI] [PubMed] [Google Scholar]
- Thomas A, Skolnick MH. A probabilistic model for detecting coding regions in DNA sequences. IMA J Math Appl Med Biol. 1994;11:149–160. doi: 10.1093/imammb/11.3.149. [DOI] [PubMed] [Google Scholar]
- Wilson R, Ainscough R, Anderson K, Baynes C, Berks M, Bonfield J, Burton J, Connell M, Copsey T, Cooper J, et al. 2.2 Mb of contiguous nucleotide sequence from chromosome III of C. elegans. Nature. 1994;368:32–38. doi: 10.1038/368032a0. [DOI] [PubMed] [Google Scholar]
- Xu Y, Mural R, Shah M, Uberbacher E. Recognizing exons in genomic sequence using GRAIL II. Genet Eng. 1994;16:241–253. [PubMed] [Google Scholar]
- Yokouchi M, Suzuki R, Masuhara M, Komiya S, Inoue A, Yoshimura A. Cloning and characterization of APS, an adaptor molecule containing PH and SH2 domains that is tyrosine phosphorylated upon B-cell receptor stimulation. Oncogene. 1997;15:7–15. doi: 10.1038/sj.onc.1201163. [DOI] [PubMed] [Google Scholar]
- Zeng WR, Scherer SW, Koutsilieris M, Huizenga JJ, Filteau F, Tsui LC, Nepveu A. Loss of heterozygosity and reduced expression of the CUTL1 gene in uterine leiomyomas. Oncogene. 1997;14:2355–2365. doi: 10.1038/sj.onc.1201076. [DOI] [PubMed] [Google Scholar]
- Zhang MQ. Identification of protein coding regions in the human genome by quadratic discriminant analysis. Proc Natl Acad Sci. 1997;94:565–568. doi: 10.1073/pnas.94.2.565. [DOI] [PMC free article] [PubMed] [Google Scholar]