

Abstract
The efficacy of dimethyl sulfoxide (DMSO) as a supercharging reagent for protein ions formed by electrospray ionization from aqueous solution and the mechanism for supercharging were investigated. Addition of small amounts of DMSO to aqueous solutions containing hen egg white lysozyme or equine myoglobin results in a lowering of charge, whereas a significant increase in charge occurs at higher concentrations. Results from both near-UV circular dichroism spectroscopy and solution-phase hydrogen/deuterium exchange mass spectrometry indicate that DMSO causes a compaction of the native structure of these proteins at low concentration, but significant unfolding occurs at ~63% and ~43% DMSO for lysozyme and myoglobin, respectively. The DMSO concentrations required to denature these two proteins in bulk solution are ~3–5 times higher than the concentrations required for the onset of supercharging, consistent with a significantly increased concentration of this high boiling point supercharging reagent in the ESI droplet as preferential evaporation of water occurs. DMSO is slightly more basic than m-nitrobenzyl alcohol and sulfolane, two other supercharging reagents, based on calculated proton affinity and gas-phase basicity values both at the B3LYP and MP2 levels of theory, and all three of these supercharging reagents are significantly more basic than water. These results provide additional evidence that the origin of supercharging from aqueous solution is the result of chemical and/or thermal denaturation that occurs in the ESI droplet as the concentration of these supercharging reagents increases, and that proton transfer reactivity does not play a significant role in the charge enhancement observed.
Key words: Electrospray ionization, Supercharging, DMSO, Protein conformation, Charge state distribution
Introduction
Electrospray ionization (ESI) of proteins generally results in a characteristic distribution of charge states that can be readily mass analyzed on virtually any type of mass spectrometer owing to the lowering of the m/z that occurs as a result of multiple charging for large molecules. A major factor in the extent of charging is the conformation of a protein in solution. Narrow charge state distributions at lower charge are generally formed from solutions in which a protein has a compact native or native-like structure, whereas significantly higher charge states and broader charge state distributions are typically formed from solutions in which the protein is denatured [1–4]. Multiple distinct charge state distributions corresponding to different conformations of the same protein in solution are often observed, and information about the populations of solution-phase conformers has been obtained by modeling these distributions [4]. A number of factors, in addition to protein conformation, can affect charging in ESI, including solvent and analyte basicity [5–7], solvent surface tension [8–10], and instrument parameters [11–14].
Addition of small quantities of “supercharging” reagents, such as m-nitrobenzyl alcohol (m-NBA), to either aqueous “native” solutions [15–21] or “denaturing” solutions [8–10, 22–27] can cause an increase in the average and maximum charge states of peptides, proteins, or protein complexes compared with the same solutions without the reagents. Increasing ion charge is often desirable because the performance of most mass analyzers improves at lower m/z and both the dissociation efficiency and the information obtained from many tandem mass spectrometry methods, such as electron capture or transfer dissociation, can increase significantly with charge [28–31].
Because many factors affect charging in ESI, attributing the increased charging observed with these supercharging reagents to just a single predominant factor can be challenging. Supercharging reagents generally have relatively low vapor pressures compared with other components in the initial solutions, resulting in a significant increase in their relative concentration in the ESI droplet as solvent evaporation occurs [32]. For example, the boiling point of m-NBA is 177 °C at 3 Torr [33], from which a value of ~405 °C at 760 Torr can be estimated [34]. The composition of ESI droplets that initially contain low concentrations of the supercharging reagent will rapidly become enhanced in the supercharging reagent as the more volatile components preferentially evaporate. This will change a number of physical parameters, including the droplet surface tension [8–10], which can either be increased or decreased by the supercharging reagent depending on the composition of the initial droplet. In the absence of any significant analyte conformational changes, the charging observed with these supercharging reagents has been related to effects of surface tension [8–10]. The surface tension of m-NBA (50 ± 5 mN/m [9]) is higher than many organic solvents, e.g., methanol (22.1 mN/m at 25 °C [33]), but lower than water (72.0 mN/m at 25 °C [33]). Addition of m-NBA to methanol solutions containing the amine-functionalized dendrimer, poly(propyl)eneimine (DAB-16), which cannot undergo significant conformational changes [35], resulted in an increase in charging, whereas addition of m-NBA to aqueous solutions containing this same molecule resulted in a decrease in charging, consistent with the changes in surface tension of the ESI droplet as evaporation occurs [10].
We have proposed that the primary origin of supercharging proteins and protein complexes from purely aqueous solution is conformational changes that can occur as a result of chemical and/or thermal denaturation, and that the decrease in droplet surface tension is a mitigating factor in the extent of supercharging [19–21]. Sulfolane, a supercharging reagent shown to be effective at increasing charge for proteins and protein complexes formed from aqueous solutions in which the structures are native [16], destabilizes the native structure of myoglobin by ~1.5 kcal/mol/M at 25 °C, making it ~30% as effective as guanidinium chloride for unfolding this protein [20]. The higher charge state ions produced from aqueous solutions with sulfolane and m-NBA have higher collision cross sections measured using ion mobility, consistent with more unfolded structures [20, 21]. At the typically low initial concentrations of reagent required for supercharging, no significant change to the native structure was observed using circular dichroism spectroscopy (CD) [20] or hydrogen/deuterium exchange (HDX) MS [21]. However, as the ESI droplet becomes enriched with the reagent due to its relatively high boiling point, the concentration threshold for chemical and/or thermal denaturation can be reached and increased charging as a result of protein unfolding can occur. This mechanism has been controversial [16–18], in part, because many other factors, including proton transfer reactivity [5–7], also affect charging. Loo and coworkers proposed that the relative proton transfer reactivity of the supercharging reagent compared with that of the protein may be a significant factor in supercharging from aqueous solutions [16].
Dimethyl sulfoxide (DMSO) added to denaturing solutions containing high concentrations of organic solvent can result in supercharging of peptides and proteins, an effect that can be attributed largely to surface tension [10, 27]. Here, the efficacy of DMSO for supercharging hen egg white lysozyme and equine myoglobin from aqueous solutions is investigated. Both near-UV CD and solution-phase HDX-MS are used to determine what conformational changes occur in these proteins in the initial ESI solutions as a function of DMSO concentration. These and other results indicate that protein conformational changes are the major factor in supercharging with DMSO from these solutions, and that proton transfer reactivity of DMSO or other supercharging reagents cannot explain the enhanced charging observed with supercharging from aqueous solutions.
Experimental
Mass spectra were acquired using either a LTQ-mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) or a 9.4 T Fourier-transform ion cyclotron resonance mass spectrometer (FT-ICR) described elsewhere [36]. Ions were formed using nanoelectrospray emitters prepared by pulling borosilicate capillaries (1.0 mm o.d./0.78 mm i.d.; Sutter Instruments, Novato, CA, USA) to a tip i.d. of ~1 μm with a Flaming/Brown micropipette puller (Model P-87; Sutter Instruments). A platinum wire (0.127 mm diameter, Sigma, St. Louis, MO, USA) was inserted into the nanoelectrospray emitter in contact with the solution, and ESI was initiated and maintained by applying 0.8–1.5 kV to the wire relative to instrument ground. Hen egg white lysozyme and horse heart myoglobin were purchased from Sigma and were used without further purification. Solutions were prepared with extra dry DMSO (Acros Organics, Waltham, MA, USA) as vol/vol fractions. Estimates of DMSO enrichment during ESI were calculated by converting the vol/vol percents of the reagent to mole fractions using the known densities and molecular weights of DMSO and water.
Hydrogen/Deuterium Exchange
Deuterated lysozyme and myoglobin were prepared for HDX studies by dissolving the solid proteins to 500 μM in D2O (Sigma) and heating these solutions for three hours at 50 °C. The deuterated protein solutions were held at 4 °C prior to use. The deuterium incorporation of lysozyme and myoglobin was measured at ~99% and ~80%, respectively, using a 9.4 T FT-ICR mass spectrometer where isotopic resolution is readily achieved. The deuterated proteins were diluted 50-fold into 200 mM ammonium acetate (pH 7.4) solutions containing various concentrations of DMSO, mixed, and the resulting HDX monitored continuously using a LTQ mass spectrometer for 10 min. Mass spectra acquired between 3.5 and 4.5 min of exchange were scan-averaged and the peak intensities of the scan-averaged mass spectra were used to calculate the relative extents of HDX for the analytes. The temperature inside the nanoelectrospray source was 44 °C.
Circular Dichroism
Circular dichroism (CD) spectra were acquired on a Jasco model 810 spectropolarimeter (JASCO, Inc., Easton, MD, USA). Twenty μM protein solutions were prepared in 200 mM ammonium acetate (pH 7.4) solution containing various concentrations of DMSO and held at 44 °C for 5 min in a 1.0 cm quartz cuvette with constant mixing using a Teflon stir bar prior to wavelength scan analysis in the range 260 to 300 nm.
Computations
Initial structures for neutral and protonated water, sulfolane, and m-nitrobenzyl alcohol were generated using Maestro 9.1 (Schrödinger, Inc., Portland, OR, U.S.A.). Energy-minimized geometries calculated at the B3LYP/6-31 G* level of theory were further optimized at the B3LYP/6-311++G(2d,2p) or MP2(full)/6-311++G(2d,2p) level of theory using Q-Chem 3.2, and vibrational frequencies were computed. The resulting electronic energies and unscaled vibrational frequencies were used to obtain 298 K enthalpies and Gibbs free energies. Proton affinities and gas-phase basicities (−ΔH° and –ΔG° for protonation of the neutral molecule, respectively) for each molecule were calculated using the neutral and protonated structures of lowest energy at 298 K.
Results and Discussion
Supercharging with DMSO
Lysozyme
Nanoelectrospray mass spectra of hen egg white lysozyme obtained from purified water with 0%–99% (vol/vol) DMSO were obtained, and mass spectra with 0%, 2%, and 50% DMSO are shown in Figure 1a–c, respectively. Without DMSO, the charge state distribution is narrow and the average charge, calculated as the abundance-weighted average of the observed charge states, is 7.7+ (Figure 1a). Both the narrow distribution and low charge states are consistent with a native or native-like conformation of the protein in solution (Figure 1a). With 2% DMSO, the average charge decreases to 6.6+, but a similarly narrow distribution of charge states is observed (Figure 1b). In contrast, the charge state distribution obtained from a solution containing 50% DMSO (Figure 1c) is slightly broader with a much higher average charge of 8.8+. The average charge as a function of DMSO concentration over the entire range measured is shown in Figure 1d. Small amounts of DMSO, up to 10%, result in narrow charge state distributions with lower average charge. More charging and broader charge state distributions are observed with higher DMSO concentrations. The maximum average charge (9.3+) occurs with 80% DMSO, and the average charge with a DMSO concentration of 99% is nearly one charge state lower (8.5+).
Figure 1.
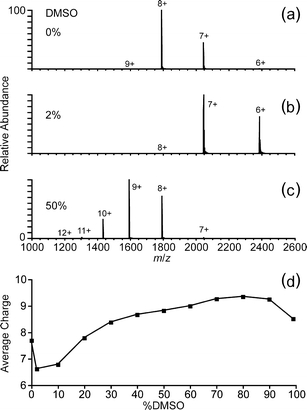
Nanoelectrospray mass spectra of hen egg white lysozyme (10 μM) in unbuffered water solutions containing (a) 0%, (b) 2%, and (c) 50% DMSO; (d) the average charge, calculated as the abundance-weighted average of the observed charge states, as a function of DMSO concentration
Equine Myoglobin
Similar trends in charging with DMSO concentration were obtained for equine myoglobin, although there are subtle differences. Nanoelectrospray mass spectra of equine myoglobin aqueous solutions containing up to 50% (vol/vol) DMSO were acquired, and these data are shown in Figure 2. Higher concentrations of DMSO resulted in nearly complete loss of signal for myoglobin ions, consistent with previous observations of aggregation of this protein with ~66%–92% DMSO in D2O [37]. The mass spectrum of myoglobin without DMSO (Figure 2a) is dominated by a narrow distribution of holomyoglobin ions with an average charge of 8.2+, with a low abundance of apomyoglobin ions with an average charge of 7.7+ also observed. The charge state distributions for both holo- and apomyoglobin are relatively narrow, and these data are consistent with predominantly native or native-like structures for both forms of this protein in solution. Although there are differences, the structure of apomyoglobin in aqueous solution is similar to that of holomyoglobin [38]. With 2% DMSO, the average charge for holo- and apomyoglobin decreases to 7.5+ and 6.5+, respectively (Figure 2b), but there is no change in the range of charge states produced. The maximum average charge states for both holo- (10.1+) and apomyoglobin (9.7+) were obtained from the solution containing 30% DMSO (Figure 2c). In contrast to the spectra with 0% and 2% DMSO, the charge state distributions in the spectrum with 30% DMSO are very broad and shifted to significantly higher charge. In addition, the abundance of apomyoglobin ions is significantly greater than that of holomyoglobin.
Figure 2.
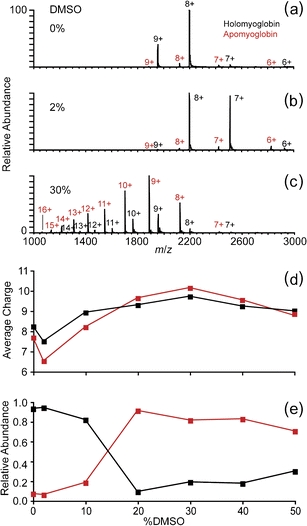
Nanoelectrospray mass spectra of equine myoglobin (10 μM) in unbuffered water solutions containing (a) 0%, (b) 2%, and (c) 30% DMSO; (d) the average charge, calculated as the abundance-weighted average of the observed charge states, and (e) the relative abundances of holo- versus apomyoglobin as a function of DMSO concentration
The average charge of holo- (black lines) and apomyoglobin (red lines) as a function of DMSO concentration are shown in Figure 2d, and the trends shown in these data are similar to that obtained for lysozyme. The relative abundances of the holo- and apomyoglobin ions as a function of DMSO concentration are shown in Figure 2e. A nearly complete inversion of the abundances of these ions occurs between 10% and 20% DMSO. These data, along with the broader and higher charge state distributions with higher DMSO concentration are consistent with a loss of native structure of myoglobin and concomitant loss of heme at the higher DMSO concentrations used.
Conformations of Lysozyme and Myoglobin in Aqueous Solutions Containing DMSO
In order to understand how DMSO affects protein charging from aqueous solution, it is important to know how it affects the conformation(s) of the proteins in the initial solution prior to ESI. Both near-UV CD spectroscopy and solution-phase HDX experiments were performed, and these results are compared with results of supercharging experiments performed with identical solution conditions. Changes in near-UV CD spectra reflect changes in the environment around aromatic residues [39]. Secondary structure analysis by far-UV CD was not done owing to interference by DMSO. Extents of HDX can reflect solution-phase accessibility of backbone amide hydrogen atoms from which information about relative protein conformations can be inferred. These data for both lysozyme and for myoglobin are described below.
Near-UV CD Spectroscopy
CD spectra at wavelengths between 260 and 300 nm were acquired as a function of DMSO concentration (20 μM lysozyme in 200 mM ammonium acetate; pH = 7.4; T = 44 °C) with 0%–90% DMSO (Figure 3a, inset). The maximum signal intensity in this wavelength range without DMSO occurs at 289 nm. A plot of the signal intensity at this wavelength as a function of DMSO concentration is shown in Figure 3a. The signal increases from 0% to 50% DMSO, consistent with a previous study of lysozyme in unbuffered water and DMSO at ambient temperature, which showed that compaction of the tertiary structure of this protein occurs with up to a DMSO mole fraction of 0.6 (~86% vol/vol) [40]). As was observed previously [40], there is a subtle blue shift of the wavelength where the maximum intensity is observed with increasing DMSO concentration up until the point at which CD signal in this region is essentially lost (Figure 3a, inset). At 60% DMSO, there is a small decrease in signal followed by a dramatic loss of signal in the solutions with ≥70% DMSO, indicating significant loss of protein higher-order structure [40]. Protein denaturation under these experimental conditions occurs at a slightly lower DMSO concentration than observed previously [40]. Torreggiani et al. showed that the denaturation peak temperature of lysozyme decreases with increasing DMSO concentration between 0% and 50% [41]. The lower concentration of DMSO required for unfolding in our experiments at higher temperature (>60% at 44 °C) is consistent with the slightly higher concentration required (>~86%) at ambient temperature [40]. These data indicate that lysozyme initially becomes more compact in solution with up to 50% DMSO, but then unfolds when the concentration of DMSO is between ~50%–70%.
Figure 3.
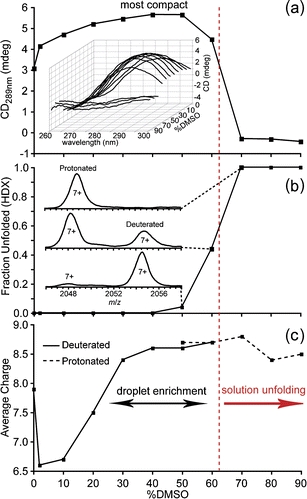
Changes in tertiary structure of hen egg white lysozyme (20 μM) in 200 mM ammonium acetate (pH 7.4) solutions as a function of DMSO concentration measured by (a) circular dichroism at 289 nm (wavelength scans inset) and (b) solution hydrogen/deuterium exchange mass spectrometry (HDX-MS); (c) the average charge of ions in these HDX-MS experiments calculated as the abundance-weighted average of all the charge states in the distributions
Similar experiments were performed for myoglobin, and these data are shown in Figure 4a. Significant precipitation occurred with ≥60% DMSO in these buffered solutions, so 50% was the maximum DMSO concentration used. The maximum signal intensity without DMSO is at 269 nm (Figure 4a inset), and signal intensity at this wavelength is plotted as a function of DMSO concentration in Figure 4a. Similar to the result for lysozyme, the CD signal increases with increasing DMSO concentration up to 30%, suggesting that some compaction of the myoglobin structure occurs within this concentration range. The signal decreases slightly at 40% DMSO, and complete loss of signal occurs with 50% DMSO. As was the case for lysozyme, the curve maximum shifts to shorter wavelengths with increasing DMSO concentration (waterfall plot inset) up to the point at which CD is lost. In contrast to lysozyme, there is a local maximum in the curves at ~294 nm that decreases continuously from 0 to 50% DMSO. This may be a result of partial unfolding of a region of the myoglobin molecule even at low DMSO concentrations. These results indicate that significant myoglobin unfolding occurs in these solutions with ~40–50% DMSO.
Figure 4.
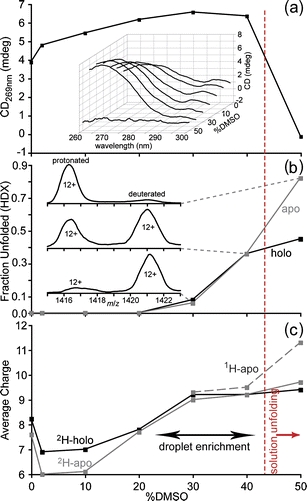
Changes in tertiary structure of equine myoglobin (20 μM) in 200 mM ammonium acetate (pH 7.4) solutions as a function of DMSO concentration measured by (a) circular dichroism at 269 nm (wavelength scans inset) and (b) solution hydrogen/deuterium exchange mass spectrometry (HDX-MS); (c) the average charge of the ions in HDX-MS experiments calculated as the abundance-weighted average of all the charge states in the distributions
Solution-phase H/D Exchange
To further probe the changes in protein higher-order structure that occur with increasing DMSO concentration, fully deuterated lysozyme (500 μM in D2O) was diluted 1:50 into 200 mM ammonium acetate (pH 7.4) solutions containing 0%–90% (vol/vol) DMSO. Mass spectra were acquired continuously for 10 min, during which time HDX occurred. Two isotopic distributions of the molecular ions owing to two distinct levels of HDX were observed, indicating the presence of two forms of the protein. The conformer that undergoes less exchange corresponds to a native or native-like structure, whereas the higher exchanging form has sampled unfolded conformations, resulting in more complete exchange. The presence of two discrete forms (or two families of indistinguishable folded and unfolded structures) of the protein is consistent with two-state unfolding in the EX1 regime [42], and we refer to these two forms as compact and unfolded. Spectra acquired from 3.5 to 4.5 min of exchange were scan-averaged, and the relative abundances of the compact and unfolded forms of the protein were calculated from the sum of the relative abundances of the distinct isotopic distributions for each molecular ion corresponding to less or more HDX, respectively. The fraction of the population that is unfolded as a function of DMSO concentration is shown in Figure 3b, which indicates no significant change in the tertiary structure of lysozyme occurs between 0% and 40% DMSO. A small fraction of unfolded conformers is observed with 50% DMSO (partial mass spectra of the 7+ showing both distributions inset). With 60% DMSO, the fraction of unfolded conformers increases to ~0.44. The corresponding partial mass spectrum of the 7+ charge state in the inset shows a larger population of unfolded conformers, whereas the fraction unfolded value is calculated from all of the observed charge states (12+ through 6+). For all of the solutions with ≥ 70% DMSO, no peaks corresponding to compact conformations are observed for any charge state.
These HDX results indicate that lysozyme unfolds between 50% and 70% DMSO in these buffered solutions, consistent with the concentration range where unfolding is observed by CD (Figure 3a). Intersection of the interpolated CD and HDX data (when overlaid) occurs at 62% DMSO and is indicated by the dashed red line in Figure 3. The HDX results show that there is a ~17% decrease in the average hydrogen incorporation for the compact conformations of lysozyme with increasing DMSO concentrations between 2% and 60%, consistent with compaction of the native structure demonstrated by Voets et al. using CD, small-angle neutron scattering, Rayleigh scattering, and Fourier transform-infrared spectroscopy [40]. Even for a 60% DMSO solution, the molarity of water (~22 M) is high enough that the intrinsic rate of hydrogen/deuterium exchange should be fast enough to exchange the majority of solvent-exposed backbone amides after 4 min, so that the average deuterium incorporation should still reflect global protection. Rapid intrinsic exchange is indicated by the two discrete exchange populations observed throughout the entire range of DMSO concentrations, although a subtle decrease in the extent of exchange at the higher DMSO concentrations occurs (Figure 3b).
Similar HDX-MS experiments were performed with myoglobin. Deuterated myoglobin (~80% 2H incorporation) was diluted 1:50 into 200 mM ammonium acetate (pH 7.4) solutions containing 0%–50% (vol/vol) DMSO. Mass spectra were acquired continuously for 10 min, and the relative abundances of the compact and unfolded forms of the protein after 4 min of exchange were calculated as described above for lysozyme. The relative population of the unfolded form as a function of DMSO concentration is shown in Figure 4b and indicates no significant change in the tertiary structure of myoglobin occurs in solutions with 0%–30% DMSO. Inset in Figure 4b are partial mass spectra of the 12+ charge state of apomyoglobin from the solutions containing 30%, 40%, and 50% DMSO, again showing discrete populations of exchange indicative of two-state unfolding in the EX1 regime [42]. The fraction of unfolded conformers in this concentration range (average of all charge states) increases from 8% to 44% for holomyoglobin and from 6% to 82% for apomyoglobin. Both the CD and HDX-MS show that protein unfolding occurs between 30% and 50% DMSO concentration in these solutions, with the intersection of these interpolated data (when overlaid) equal to ~43% DMSO (vertical red line in Figure 4).
In contrast to lysozyme, a much smaller decrease in the average hydrogen content (~8% for holo- and apomyoglobin) was observed for the folded forms of these proteins when the DMSO concentration was increased from 2% to 30%. This could be the result of less global compaction for this protein, or it may be due to competing effects of global compaction and some unfolding in an isolated region of the protein, as suggested by the CD spectroscopy. A high spatial resolution analysis of the HDX kinetics could potentially distinguish these two possibilities, but such an analysis is beyond the scope of the current work.
Conformational Changes and Supercharging
Lysozyme
The average charge of the compact and unfolded conformers of lysozyme from the HDX experiments as a function of DMSO concentration is shown in Figure 3c. The trends in charging with DMSO concentration from these buffered solutions (200 mM ammonium acetate) are similar to those obtained from unbuffered solutions (Figure 1d), indicating that the ammonium acetate has only a minor effect on supercharging. There is a similar onset of supercharging at around 20% DMSO from both solutions, but the maximum charge state is slightly lower for the buffered solution (8.8+ versus 9.3+), perhaps due to increased stabilization of the folded forms of the protein in the higher ionic strength solutions.
The data in Figure 1d and 3c show that onset of supercharging occurs at a significantly lower DMSO concentration than is required to unfold lysozyme in the initial solution prior to ESI. Furthermore, no significant change in charging occurs within the range of DMSO concentration where the change from a folded to an unfolded protein occurs in the initial solutions. Because DMSO has a higher boiling point than water (bp of DMSO is 189 °C at 760 Torr) [33], preferential evaporation of water in the ESI droplet will occur resulting in an enrichment in DMSO in the later stages of the droplet lifetime. Because DMSO denatures proteins at high concentration, unfolding of the protein in the DMSO-enriched droplet can occur, and this unfolding can result in significant enhancement in charge at higher DMSO concentrations.
DMSO does not form an azeotrope with water [43], so the extent to which droplets have to become enriched in DMSO in order for unfolding to occur can be estimated from these data. The unfolding indicated by the trend in charging occurs at ~20% DMSO, whereas this onset in the initial solutions occurs at ~63% indicating that the droplet becomes enriched in DMSO by about 5× (mole fraction). These results are consistent with the decrease in charging observed at low concentration (2% and 10% DMSO), which indicate that compaction of the native structure occurs in the initial solutions containing up to 50% DMSO. These values are rough estimates because other factors influence charging as well, including relative gas-phase basicities and droplet surface tension. For example, the droplet surface tension will be lowered as the DMSO concentration is enriched, resulting in less charging than predicted without taking this effect into account.
Myoglobin
The average charge of myoglobin ions from the HDX experiments as a function of DMSO concentration is shown in Figure 4c. The compact and unfolded conformers of apomyoglobin are distinguished in the figure with either a solid or dashed line, respectively, whereas the average charge of the compact and unfolded (present in the 30%–50% DMSO solutions only) conformers of holomyoglobin have the same average charge, so the lines overlap and are indistinguishable in the figure. The onset of supercharging occurs between 10% and 20% DMSO (Figure 4c) compared with between 2% and 10% DMSO (Figure 2d) in the unbuffered solutions. The transition from holo- to apomyoglobin occurs at ~40% DMSO in the buffered solutions compared with ~15% DMSO in the unbuffered water solutions (Figure 2e). Both of these results are consistent with greater stabilization at higher ionic strength for this protein. Considerably more DMSO is required to unfold myoglobin in the initial buffered solution (red vertical line in Figure 4) compared with that required to induce supercharging. From these results, we can estimate enrichment of DMSO in the ESI droplet by ~3× (mole fraction). This is comparable with the ~5× enrichment deduced from the lysozyme data and does not take into account any kinetic effects that could limit conformational changes that can occur in a rapidly evaporating droplet.
Supercharging of myoglobin from aqueous solution also occurs with both m-NBA and sulfolane [20]. It is of interest to note that the extent of supercharging, on a per mole basis, appears to relate well with the ordering of the boiling points of these reagents [20]: m-NBA (177 °C at 3 Torr) > sulfolane (287.6 °C at 760 Torr) > DMSO (189 °C at 760 Torr) [33]. More enrichment of the higher boiling point supercharging reagents will occur, but other factors, such as the relative denaturing strengths of the three reagents, droplet surface tension, relative gas-phase basicities, etc., will also affect the extent of supercharging observed.
Proton Transfer Reactivity of Supercharging Reagents
Gas-phase Basicity of DMSO
The maximum charge states of multiply protonated ions formed by ESI can be limited by several factors, including the relative proton transfer reactivity between the multiply protonated ion and neutral molecules in the solvent or with the solvent itself [6, 7, 44]. Solvents with high basicity can reduce the extent of charging [7] and the maximum charge states of protein and peptide ions formed from denaturing solutions has been related to solvent gas-phase basicity [6, 44, 45], indicating that proton transfer reactivity can have a significant affect on the maximum charge states produced by ESI. Loo and coworkers have suggested that proton transfer reactivity can play a significant role in supercharging from aqueous solutions [16]. However, the data shown here appear to rule out this mechanism for supercharging observed with DMSO. In the absence of any conformation changes, it is difficult to rationalize how a single supercharging reagent could reduce protein charge at low concentration, and then increase charge at high concentration based on proton transfer reactivity. DMSO has a gas-phase basicity (GB) of 853 kJ/mol, which is significantly higher than that of water (660 kJ/mol) [46]. The highest charge state of lysozyme formed from water is 9+, and the apparent GB of this charge state formed from solutions where the structure is native-like is between 933 and 958 kJ/mol depending on the conformer formed [47]. The apparent GB values of lysozyme ions with lower charge states or this same charge state of more unfolded ions are even higher [47]. Thus, proton transfer from the protein to either DMSO or to water should not occur spontaneously. Conversely, proton transfer from protonated water or protonated DMSO to the protein is exothermic based on the apparent GB of the protein, but proton transfer from protonated water is more exothermic so more charging should take place without DMSO present in the solution if this mechanism played a role.
Although water dimers and higher order clusters are more basic than water itself, and the basicities are influenced by solvent and the local environment of the molecules, the disparate trends in charging of these proteins at low and high DMSO concentration cannot be explained by proton transfer reactivity. The slight reduction in supercharging of lysozyme at 99% versus 90% DMSO may occur as a result of proton transfer owing to the much higher concentration of DMSO compared with protein and the potential for any energetic collisions that may occur in the ESI interface to drive endothermic proton transfer reactions to some extent. Protonated DMSO clusters were observed at high DMSO concentration. This may be due to competitive ionization, but the higher basicity of such clusters may cause some limited proton transfer from the proteins to occur at the highest DMSO concentrations used. Alternatively, the slight reduction in charging at 99% in the unbuffered solution may be related to the “partially collapsed” conformations observed by Voets et al. at DMSO mole fraction > 0.9 [40].
Calculated Proton Affinities and Gas-phase Basicities of Sulfolane and m-NBA
Both sulfolane and m-NBA are commonly used supercharging reagents and have the advantage that even greater extents of supercharging can be typically achieved with much lower initial solution concentrations. The proton affinities (PA) and GB values for these two supercharging reagents have not been previously reported, so these values were determined using computational chemistry. The lowest-energy structures of protonated sulfolane and m-NBA are shown in Figure 5. The energetically most favorable protonation site is the sulfonyl oxygen atom of sulfolane, and one of the nitro oxygen atoms for m-NBA. From the computed 298 K enthalpies of these structures as well as those for H2O and H3O+, PA and GB values were computed at the B3LYP/6-311++G(2d,2p) and MP2(full)/6-311++G(2d,2p) levels of theory, and these values are given in Table 1. B3LYP results in slightly higher values than MP2. Although there are some uncertainties in calculated PA and GB values, those obtained here for water are only slightly below their experimentally measured values (691 and 660 kJ/mol, respectively). These results indicate that GB values of m-NBA and sulfolane are similar and that these molecules are less basic (lower GB) than DMSO by roughly 50–75 kJ/mol, but more basic than water by roughly 125–150 kJ/mol. Thus, all three of these supercharging reagents are significantly more basic than water. These results indicate that proton transfer reactivity or protection of protonation sites on the protein by any of these three supercharging reagents is not a significant factor in the origin of supercharging from purely aqueous solutions.
Figure 5.
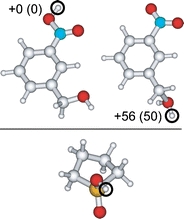
Low-energy calculated structures for protonated m-NBA (top) and sulfolane (bottom), with protonation sites circled. Relative Gibbs free energies (kJ/mol at 298 K) indicated for protonated m-NBA using B3LYP and (full MP2) with the 6-311++G(2d,2p) basis set
Table 1.
Proton Affinities (PA) and Gas-phase Basicities (GB). Values for Water and DMSO from Reference [46] and Values for Sulfolane and m-NBA Calculated Using B3LYP and (MP2 full) with the 6-311++G(2d,2p) Basis Set
| PA (kJ/mol) | GB (kJ/mol) | |
|---|---|---|
| Water | 691 | 660 |
| DMSO | 884 | 853 |
| Sulfolane | 830 (817) | 801 (787) |
| m-NBA | 834 (811) | 800 (779) |
Conclusions
DMSO is an effective supercharging reagent for protein ions formed from aqueous solutions, although considerably higher concentrations are required to obtain significant charge increases compared with either m-NBA or sulfolane. A decrease in the average charge of both lysozyme and myoglobin occurs at low concentrations of DMSO in the initial solutions, consistent with a compaction of the native structure as indicated by both near UV-CD spectroscopy and solution HDX-MS. Protein unfolding occurs at high DMSO concentrations, where extensive charge enhancement is also observed. The concentrations required for solution-phase unfolding of myoglobin and lysozyme are ~3–5× higher (mole fraction) than is required for the onset of supercharging. This is consistent with the ESI droplets becoming enriched with the reagent during solvent evaporation due to its much lower vapor pressure compared with water. As the reagent concentration increases in the droplet, chemical and/or thermal denaturation occurs, and the unfolded protein molecules can carry away more charge. These supercharging reagents are much more basic than water, indicating that the proton-transfer reactivities of these reagents are not the cause of charge increases, but as with surface tension effects, may limit the extent of supercharging observed at high reagent concentrations.
Acknowledgments
The authors thank Professor Bryan Krantz of the University of California, Berkeley for use of the spectropolarimeter and the National Science Foundation (grant CHE-1012833 and CHE-0840505) and the National Institutes of Health (training grant T32GM08295 for H.J.S) for generous financial support.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Chowdhury SK, Katta V, Chait BT. Probing Conformational Changes in Proteins by Mass Spectrometry. J. Am. Chem. Soc. 1990;112:9012–9013. doi: 10.1021/ja00180a074. [DOI] [Google Scholar]
- 2.Konermann L, Douglas DJ. Equilibrium Unfolding of Proteins Monitored by Electrospray Ionization Mass Spectrometry: Distinguishing Two-State from Multi-State Transitions. Rapid Commun. Mass Spectrom. 1998;12:435–442. doi: 10.1002/(SICI)1097-0231(19980430)12:8<435::AID-RCM181>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 3.Loo JA, Loo RRO, Udseth HR, Edmonds CG, Smith RD. Solvent-Induced Conformational Changes of Polypeptides Probed by Electrospray-Ionization Mass-Spectrometry. Rapid Commun. Mass Spectrom. 1991;5:101–105. doi: 10.1002/rcm.1290050303. [DOI] [PubMed] [Google Scholar]
- 4.Dobo A, Kaltashov IA. Detection of Multiple Protein Conformational Ensembles in Solution Via Deconvolution of Charge-State Distributions in ESI MS. Anal. Chem. 2001;73:4763–4773. doi: 10.1021/ac010713f. [DOI] [PubMed] [Google Scholar]
- 5.Loo RRO, Smith RD. Proton-Transfer Reactions of Multiply-Charged Peptide and Protein Cations and Anions. J. Mass Spectrom. 1995;30:339–347. doi: 10.1002/jms.1190300217. [DOI] [Google Scholar]
- 6.Williams ER. Proton Transfer Reactivity of Large Multiply Charged Ions. J. Mass Spectrom. 1996;31:831–842. doi: 10.1002/(SICI)1096-9888(199608)31:8<831::AID-JMS392>3.0.CO;2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iavarone AT, Jurchen JC, Williams ER. Effects of Solvent on the Maximum Charge State and Charge State Distribution of Protein Ions Produced by Electrospray Ionization. J. Am. Soc. Mass Spectrom. 2000;11:976–985. doi: 10.1016/S1044-0305(00)00169-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iavarone AT, Jurchen JC, Williams ER. Supercharged Protein and Peptide Ions Formed by Electrospray Ionization. Anal. Chem. 2001;73:1455–1460. doi: 10.1021/ac001251t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iavarone AT, Williams ER. Supercharging in Electrospray Ionization: Effects on Signal and Charge. Int. J. Mass Spectrom. 2002;219:63–72. doi: 10.1016/S1387-3806(02)00587-0. [DOI] [Google Scholar]
- 10.Iavarone AT, Williams ER. Mechanism of Charging and Supercharging Molecules in Electrospray Ionization. J. Am. Chem. Soc. 2003;125:2319–2327. doi: 10.1021/ja021202t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thomson BA. Declustering and Fragmentation of Protein Ions from an Electrospray Ion Source. J. Am. Soc. Mass Spectrom. 1997;8:1053–1058. doi: 10.1016/S1044-0305(97)00129-3. [DOI] [Google Scholar]
- 12.Benkestock K, Sundqvist G, Edlund PO, Roeraade J. Influence of Droplet Size, Capillary-Cone Distance and Selected Instrumental Parameters for the Analysis of Noncovalent Protein–Ligand Complexes by Nano-Electrospray Ionization Mass Spectrometry. J. Mass Spectrom. 2004;39:1059–1067. doi: 10.1002/jms.685. [DOI] [PubMed] [Google Scholar]
- 13.Yang PX, Cooks RG, Ouyang Z, Hawkridge AM, Muddiman DC. Gentle Protein Ionization Assisted by High-Velocity Gas Flow. Anal. Chem. 2005;77:6174–6183. doi: 10.1021/ac050711l. [DOI] [PubMed] [Google Scholar]
- 14.Page JS, Kelly RT, Tang K, Smith RD. Ionization and Transmission Efficiency in an Electrospray Ionization-Mass Spectrometry Interface. J. Am. Soc. Mass Spectrom. 2007;18:1582–1590. doi: 10.1016/j.jasms.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 15.Lomeli SH, Yin S, Loo RRO, Loo JA. Increasing Charge While Preserving Noncovalent Protein Complexes for ESI-MS. J. Am. Soc. Mass Spectrom. 2009;20:593–596. doi: 10.1016/j.jasms.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lomeli SH, Peng IX, Yin S, Loo RRO, Loo JA. New Reagents for Increasing ESI Multiple Charging of Proteins and Protein Complexes. J. Am. Soc. Mass Spectrom. 2010;21:127–131. doi: 10.1016/j.jasms.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yin, S., Loo, J.A.: Top-Down Mass Spectrometry of Supercharged Native Protein-Ligand Complexes. Int. J. Mass Spectrom.300, 118–122 (2011) [DOI] [PMC free article] [PubMed]
- 18.Hogan CJ, Loo RRO, Loo JA, de la Mora JF. Ion Mobility-Mass Spectrometry of Phosphorylase B Ions Generated with Supercharging Reagents but in Charge-Reducing Buffer. Phys. Chem. Chem. Phys. 2010;12:13476–13483. doi: 10.1039/c0cp01208d. [DOI] [PubMed] [Google Scholar]
- 19.Sterling HJ, Williams ER. Origin of Supercharging in Electrospray Ionization of Noncovalent Complexes from Aqueous Solution. J. Am. Soc. Mass Spectrom. 2009;20:1933–1943. doi: 10.1016/j.jasms.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sterling HJ, Daly MP, Feld GK, Thoren KL, Kintzer AF, Krantz BA, Williams ER. Effects of Supercharging Reagents on Noncovalent Complex Structure in Electrospray Ionization from Aqueous Solutions. J. Am. Soc. Mass Spectrom. 2010;21:1762–1774. doi: 10.1016/j.jasms.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sterling HJ, Williams ER. Real-Time Hydrogen/Deuterium Exchange Kinetics Via Supercharged Electrospray Ionization Tandem Mass Spectrometry. Anal. Chem. 2010;82:9050–9057. doi: 10.1021/ac101957x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iavarone AT, Williams ER. Collisionally Activated Dissociation of Supercharged Proteins Formed by Electrospray Ionization. Anal. Chem. 2003;75:4525–4533. doi: 10.1021/ac034144i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sze SK, Ge Y, Oh H, McLafferty FW. Top-Down Mass Spectrometry of a 29-kDa Protein for Characterization of Any Posttranslational Modification to within One Residue. Proc. Natl. Acad. Sci. U.S.A. 2002;99:1774–1779. doi: 10.1073/pnas.251691898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davies NW, Wiese MD, Browne SGA. Characterization of Major Peptides in 'Jack Jumper' Ant Venom by Mass Spectrometry. Toxicon. 2004;43:173–183. doi: 10.1016/j.toxicon.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 25.Kjeldsen F, Giessing AMB, Ingrell CR, Jensen ON. Peptide Sequencing and Characterization of Post-Translational Modifications by Enhanced Ion-Charging and Liquid Chromatography Electron-Transfer Dissociation Tandem Mass Spectrometry. Anal. Chem. 2007;79:9243–9252. doi: 10.1021/ac701700g. [DOI] [PubMed] [Google Scholar]
- 26.Madsen JA, Brodbelt JS. Comparison of Infrared Multiphoton Dissociation and Collision-Induced Dissociation of Supercharged Peptides in Ion Traps. J. Am. Soc. Mass Spectrom. 2009;20:349–358. doi: 10.1016/j.jasms.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 27.Valeja SG, Tipton JD, Emmett MR, Marshall AG. New Reagents for Enhanced Liquid Chromatographic Separation and Charging of Intact Protein Ions for Electrospray Ionization Mass Spectrometry. Anal. Chem. 2010;82:7515–7519. doi: 10.1021/ac1016858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jockusch RA, Schnier PD, Price WD, Strittmatter EF, Demirev PA, Williams ER. Effects of Charge State on Fragmentation Pathways, Dynamics, and Activation Energies of Ubiquitin Ions Measured by Blackbody Infrared Radiative Dissociation. Anal. Chem. 1997;69:1119–1126. doi: 10.1021/ac960804q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reid GE, Wu J, Chrisman PA, Wells JM, McLuckey SA. Charge-State-Dependent Sequence Analysis of Protonated Ubiquitin Ions Via Ion Trap Tandem Mass Spectrometry. Anal. Chem. 2001;73:3274–3281. doi: 10.1021/ac0101095. [DOI] [PubMed] [Google Scholar]
- 30.Breuker K, Oh HB, Horn DM, Cerda BA, McLafferty FW. Detailed Unfolding and Folding of Gaseous Ubiquitin Ions Characterized by Electron Capture Dissociation. J. Am. Chem. Soc. 2002;124:6407–6420. doi: 10.1021/ja012267j. [DOI] [PubMed] [Google Scholar]
- 31.Iavarone AT, Paech K, Williams ER. Effects of Charge State and Cationizing Agent on the Electron Capture Dissociation of a Peptide. Anal. Chem. 2004;76:2231–2238. doi: 10.1021/ac035431p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grimm RL, Beauchamp JL. Evaporation and Discharge Dynamics of Highly Charged Multicomponent Droplets Generated by Electrospray Ionization. J. Phys. Chem. A. 2010;114:1411–1419. doi: 10.1021/jp907162w. [DOI] [PubMed] [Google Scholar]
- 33.CRC Handbook of Chemistry and Physics. In [Online] 89th ed.; D. R. Lide, Ed. 2008–2009.
- 34.Goodman JM, Kirby PD, Haustedt LO. Some Calculations for Organic Chemists: Boiling Point Variation, Boltzmann Factors and the Eyring Equation. Tetrahedron Lett. 2000;41:9879–9882. doi: 10.1016/S0040-4039(00)01754-8. [DOI] [Google Scholar]
- 35.Scherrenberg R, Coussens B, van Vliet P, Edouard G, Brackman J, de Brabander E, Mortensen K. The Molecular Characteristics of Poly(Propyleneimine) Dendrimers as Studied with Small-Angle Neutron Scattering, Viscosimetry, and Molecular Dynamics. Macromolecules. 1998;31:456–461. doi: 10.1021/ma9618181. [DOI] [Google Scholar]
- 36.Jurchen JC, Williams ER. Origin of Asymmetric Charge Partitioning in the Dissociation of Gas-Phase Protein Homodimers. J. Am. Chem. Soc. 2003;125:2817–2826. doi: 10.1021/ja0211508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jackson M, Mantsch HH. Beware of Proteins in DMSO. Biochim. Biophys. Acta. 1991;1078:231–235. doi: 10.1016/0167-4838(91)90563-F. [DOI] [PubMed] [Google Scholar]
- 38.Fontana A, Zambonin M. deLaureto, P. P.; DeFilippis, V.; Clementi, A.; Scaramella, E. Probing the Conformational State of Apomyoglobin by Limited Proteolysis. J. Mol. Biol. 1997;266:223–230. doi: 10.1006/jmbi.1996.0787. [DOI] [PubMed] [Google Scholar]
- 39.Kahn PC. The Interpretation of Near-Ultraviolet Circular Dichroism. Methods Enzymol. 1979;61:339–378. doi: 10.1016/0076-6879(79)61018-2. [DOI] [PubMed] [Google Scholar]
- 40.Voets IK, Cruz WA, Moitzi C, Lindner P, Areas EPG, Schurtenberger P. DMSO-Induced Denaturation of Hen Egg White Lysozyme. J. Phys. Chem. B. 2010;114:11875–11883. doi: 10.1021/jp103515b. [DOI] [PubMed] [Google Scholar]
- 41.Torreggiani A, Di Foggia M, Manco I, De Maio A, Markarian SA, Bonora S. Effect of Sulfoxides on the Thermal Denaturation of Hen Lysozyme: A Calorimetric and Raman Study. J. Mol. Struct. 2008;891:115–122. doi: 10.1016/j.molstruc.2008.03.007. [DOI] [Google Scholar]
- 42.Kaltashov IA. Probing Protein Dynamics and Function under Native and Mildly Denaturing Conditions with Hydrogen Exchange and Mass Spectrometry. Int. J. Mass Spectrom. 2005;240:249–259. doi: 10.1016/j.ijms.2004.09.021. [DOI] [Google Scholar]
- 43.Gmehling J, Mollman C. Synthesis of Distillation Processes Using Thermodynamic Models and the Dortmund Data Bank. Ind. Eng. Chem. Res. 1998;37:3112–3123. doi: 10.1021/ie970782d. [DOI] [Google Scholar]
- 44.Schnier PD, Gross DS, Williams ER. On the Maximum Charge-State and Proton-Transfer Reactivity of Peptide and Protein Ions Formed by Electrospray-Ionization. J. Am. Soc. Mass Spectrom. 1995;6:1086–1097. doi: 10.1016/1044-0305(95)00532-3. [DOI] [PubMed] [Google Scholar]
- 45.Schnier PD, Price WD, Williams ER. Modeling the Maximum Charge State of Arginine-Containing Peptide Ions Formed by Electrospray Ionization. J. Am. Soc. Mass Spectrom. 1996;7:972–976. doi: 10.1016/1044-0305(96)00071-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hunter EPL, Lias SG. Evaluated Gas Phase Basicities and Proton Affinities of Molecules: An Update. J. Phys. Chem. Ref. Data. 1998;27:413–656. doi: 10.1063/1.556018. [DOI] [Google Scholar]
- 47.Gross DS, Schnier PD, Rodriguez-Cruz SE, Fagerquist CK, Williams ER. Conformations and Folding of Lysozyme Ions In Vacuo. Proc. Natl. Acad. Sci. U S.A. 1996;93:3143–3148. doi: 10.1073/pnas.93.7.3143. [DOI] [PMC free article] [PubMed] [Google Scholar]