

Abstract
A number of studies have suggested that Basques might be a relic of Mesolithic Europeans who escaped much of the homogenization brought about by the Neolithic expansion. In an attempt to add new insights into this hypothesis, MS205 minisatellite diversity has been investigated by Minisatellite Variant Repeat (MVR) analysis in a sample of >100 autochthonous individuals from the Basque Country, along with 24 Castilian (N. Spain) and 23 individuals from the United Kingdom. These populations were examined in the context of the available world database for MS205 alleles. To deduce the similarities among populations, we have applied a phylogenetic approach that takes into account similarity between alleles. The variability of these populations seems to be a subset of the greater and presumably older African diversity, as has been suggested previously for non-Africans. Within non-Africans, Basques seem to cluster with other Northern European populations; however, some apparently Basque-specific alleles can be dated back to post-Aurignacian times, supporting the continuity of some lineages of this population since the Upper Paleolithic period.
The most distinctive characteristic of Basques is their language. Basque (or Euskara) not only lacks a common Indoeuropean root with the majority of European languages but is a linguistic isolate with no known living European relatives. Although linguistic differentiation cannot always be linked to an underlying genetic distinctiveness (Cavalli-Sforza et al. 1994; Sajantila and Pääbo 1995; Sajantila et al. 1995), a number of genetic studies on classical markers (Mourant 1947; Aguirre et al. 1991; Bertranpetit and Cavalli-Sforza 1991; Calafell and Bertranpetit 1994; Manzano et al. 1996a,b) have argued in favor of a parallel evolution of language and genes in the case of Basques. Similarly, they led to suggestions that Basques might be a relic of Mesolithic Europeans (Cavalli-Sforza et al. 1994), who escaped the homogenization brought about by the Neolithic demic expansion from the Near East (Ammerman and Cavalli-Sforza 1984). Recently, others have challenged this hypothesis and claim a colonization of the Basque region ∼5000 BP by a small Neolithic population from the North Caucasus region (Calderon et al. 1998).
Polymorphisms at the DNA level are much more abundant and informative than gene products and allow phylogenetic analysis. In an attempt to provide new insights into the origin of Basques, Bertranpetit et al. (1995) focused on the hypervariable segment I of the control region of mitochondrial DNA (mtDNA); the Basque distinctiveness was not confirmed by genetic distances when compared to other world populations but, instead, seemed to support the lack of geographical clustering of mtDNA variants in Europeans. This has been explained as a function of the high mutation rates of mtDNA, at least at some mutational hot spots, which tend to homogenize summary distances between populations (Cavalli-Sforza and Minch 1997; Richards et al. 1997). However, Richards et al. (1996) demonstrated by comparison of mtDNA sequences that the only consistently different European population in their study was the Basques and proposed an alternative explanation for the Basque distinctiveness, based not on Basques being the only pre-Neolithic relic in Europe but on a period of long isolation and genetic drift.
However, mtDNA is still a single effectively nonrecombining molecule, and additional forces, such as selective hitchhiking and/or background selection, may influence its distribution. Selection may have also had a role in the distribution of the variability observed in classical markers, especially in the HLA system, for which not only strong balancing selection has been proposed to explain its variability but also recombination or convergence (Hedrick 1994; Hickson and Cann 1997; and references therein). Nevertheless, principal component analyses of HLA class I and II loci still demonstrate some distinctive features of the Basques (Comas et al. 1998).
Repetitive DNA sequences such as microsatellites have become an increasingly popular alternative in evolutionary analysis and have also been applied to the study of Basque diversity (Iriondo et al. 1997; Garcia et al. 1998). However, size constraint may limit the range of microsatellite alleles (Garza et al. 1995), with recurrent mutation causing an eventual loss of their phylogenetic information (Nauta and Weising 1996; Feldman et al. 1997). Additionally, different microsatellite loci are likely to show different size constraints and different mutation rates.
In this regard, the analysis of loci with lower effective population size, in which drift tends to dominate over mutation, such as those on the Y chromosome (effective size one-fourth of nuclear loci), showed the Basques as an extreme of the European frequency distribution because of their low haplotype-diversity value (Scozzari et al. 1997; but see Perez-Lezaun et al. 1997). Other Y-chromosome polymorphisms support the “outlier” behavior of Basques (Lucotte and Hazout 1996; Semino et al. 1996).
Here, we examine Basque diversity in the context of other world populations already studied, using another class of variable number of tandem repeat (VNTR) loci, minisatellites (Armour et al. 1998). The autosomal minisatellite MS205 not only shows huge allelic diversity with a heterozygosity value close to 100% (Armour et al. 1993, 1996) but other additional useful properties: Its size distribution, so far restricted to between 1 and 5 kb, allows Minisatellite Variant Repeat analysis by PCR (MVR–PCR) (Jeffreys et al. 1991). Thus, minisatellite alleles are not only differentiated by length but also by the internal arrangements of variant repeat units. Furthermore, a considerable body of data is available on its mutational processes, shown by pedigree and small-pool PCR (SP–PCR) analysis of MS205 mutants (Jeffreys et al. 1994; May et al. 1996). Mutation events (average mutation rate 0.4% per gamete; Royle et al. 1992), are highly restricted to the 3′ end of the minisatellite, whereas the 5′ end remains more stable. This polarity allows some of the deeper evolutionary history of the alleles to be retained, whereas differences confined to the unstable 3′ end tell us about more recent relationships between alleles.
RESULTS
A total of MS205 alleles from 218 Basque (Northern Spain), 48 Castilian (Northern Spain), and 46 individuals from the United Kingdom were MVR analyzed in this work for their A/T variant repeat interspersion pattern. Analyses were performed on this data set, along with that described in Armour et al. (1996).
From a total sample size of 321 individuals representing the global world population, 393 different alleles could be observed, many of them specific to each population sample (287 alleles were singletons). In Basques, the most represented sample, 146 different alleles were detected from a total of 118 individuals [including the 9 individuals analyzed in Armour et al. (1996)]. Of those, 25 alleles are shared with some of the other samples of European origin (the Northern European sample from the CEPH panel, UK individuals, Finns, and Castilians), and 34 when considering all of the populations worldwide. Therefore, most of the shared Basque variability is of European origin. The unshared alleles were mainly singletons (88 of the 112 unshared alleles).
Basques, Castilians, and UK individuals show high levels of heterozygosity (>0.99), as do the majority of populations described (Armour et al. 1996). Although Africans show a slightly lower heterozygosity value, it is not significant (one-tail P value = 0.11), as expected for loci with high mutation rates (Relethford 1997), confirming that European ascertainment bias is eliminated at highly polymorphic loci (Rogers and Jorde 1996).
No significant departure from panmictic Hardy–Weinberg expectations was observed for the Basques (χ2 = 0.856, P = 0.635), thus supporting the idea of lack of significant Basque heterogeneity. However, for this kind of highly polymorphic system the effect of substructuring on heterozygosity deficiency is minimized (Jin and Chakraborty 1995).
Allele t10.7 (5′-ttttttttttattatatattatttttttttttattatatattatta-3′), termed CE1 in Armour et al. (1996), with high frequencies in Saami (0.21) and Japanese (0.3) and a very high frequency in Surui (0.75), has also been found in the Basque sample, but as a singleton. However, two other very similar alleles (t10.8, 5′-ttttttttttattatatattatttttttttttattatatattattaa-3′ and t10.9, 5′-ttttttttttattatatattatttttttttttattatatattattaaa-3′) are present in this sample with high frequencies: 0.04 and 0.03 (nine and eight counts) respectively, the highest individual allele frequencies found in the Basque sample. Only the Japanese and Saami samples, along with the Castilian sample, share t10.8 with the Basques, and only the Saami sample share t10.9 with the Basques. In general, the t10 group (to which t10.7, t10.8, and t10.9 belong) is very homogeneous. Only 33 different t10 alleles, very similar in internal structure, have been observed in comparison to the 81 found for the t8 group, the 155 for t11, or the 87 observed for t12, and most of its lineages share alleles that can be related by a single apparent mutational event. This may be a reflection of a lower mutation rate for this group; in this regard, at least for t10.7, a 10-fold reduction in mutation rate has been observed in analysis of sperm DNA (May et al. 1996). Another slowly mutating allele (May et al. 1996), belonging to the t12 group (5′-ttttttttttttattatatattattttttaa-3′) is also found at relatively high frequency in European samples (four times in 236 Basque alleles, three in the 106 alleles of the Northern European sample, three in the 46 UK alleles, and once in the Castilian sample of 48 alleles). A similar allele (5′-ttttttttttttattatatattatttttttaaa-3′) is found five times in Basques only. Differential allele mutation rates may therefore have favored drift to higher frequency for those alleles with the lowest mutation rate. This is especially conspicuous in the Surui population, in which t10.7 accounts for 75% of alleles. The extent to which varying average mutation rates at this minisatellite may explain different evolutionary rates for populations with different allele compositions remains to be studied.
The parameter θ = 4Neμ has been estimated as θH = 135.9 ± 23.6. This allows us to estimate the long-term mean effective population size. Thus, for the total Basque population, using an average mutation rate of 0.4%, an approximate value of 8500 is obtained, in fair agreement with the conventional Figure of 10,000 (Harpending et al. 1998). An estimate (θk) of the same parameter θ from the number of observed alleles can be used instead to obtain the expected number of rare alleles (in this case, arbitrarily, those found only once) under an infinite allele model (IAM) and compare this to the observed value. For the Basque population, there is a nonsignificant excess of observed rare alleles (100 observed vs. 96.4 ± 9.8 expected, P(n ⩾ 100) = 0.37; θk = 162.3 ± 23.1), suggesting a fit to the model.
To represent the similarities between populations graphically, the allele frequency distributions were used as an initial approach by means of correspondence analysis (Fig. 1). The first two factors explained only ∼17% of the total inertia. Axis 1 showed differentiation of the African populations, whereas axis 2, apart from displaying the outlier behavior of Melanesians, shows Basques forming a homogeneous group with the rest of the European populations.
Figure 1.

Representation of the first two principal factors. (Bas) Basques; (Cas) Castilians; (NE) Northern Europeans from the CEPH panel; (Fin) Finns; (Jap) Japanese; (Ken) Kenyans; (Mel) Melanesians; (Mok) Moksha; (Saa) Saami; (Sur) Surui; (UK) British; (Waf) West Africans; (Zbw) Zimbabweans. Statistical significance for this representation can be obtained by working out the absolute contribution of each population to the definition of the axes. Thus, the percentage values indicate the contribution of the African populations to the first axis; the contribution of the rest of the populations refer to the second axis.
It can be argued that this approach disregards any information on phylogenetic relationships between alleles. We have therefore applied a procedure using values intended to reflect the average phylogenetic dissimilarity of alleles within and between populations. The permutation test of genetic differentiation between populations (Table 1) shows in principle the same population relationships. Although we need to bear in mind that the small sample size for some of the populations analyzed may reduce the power of the test employed (Hudson et al. 1992) and that more extensive bootstrapping would help reducing the variance associated with the significance values, in the particular case of the Basques no significant differences with other European populations are found.
Table 1.
Test for Genetic Differentiation between Populations
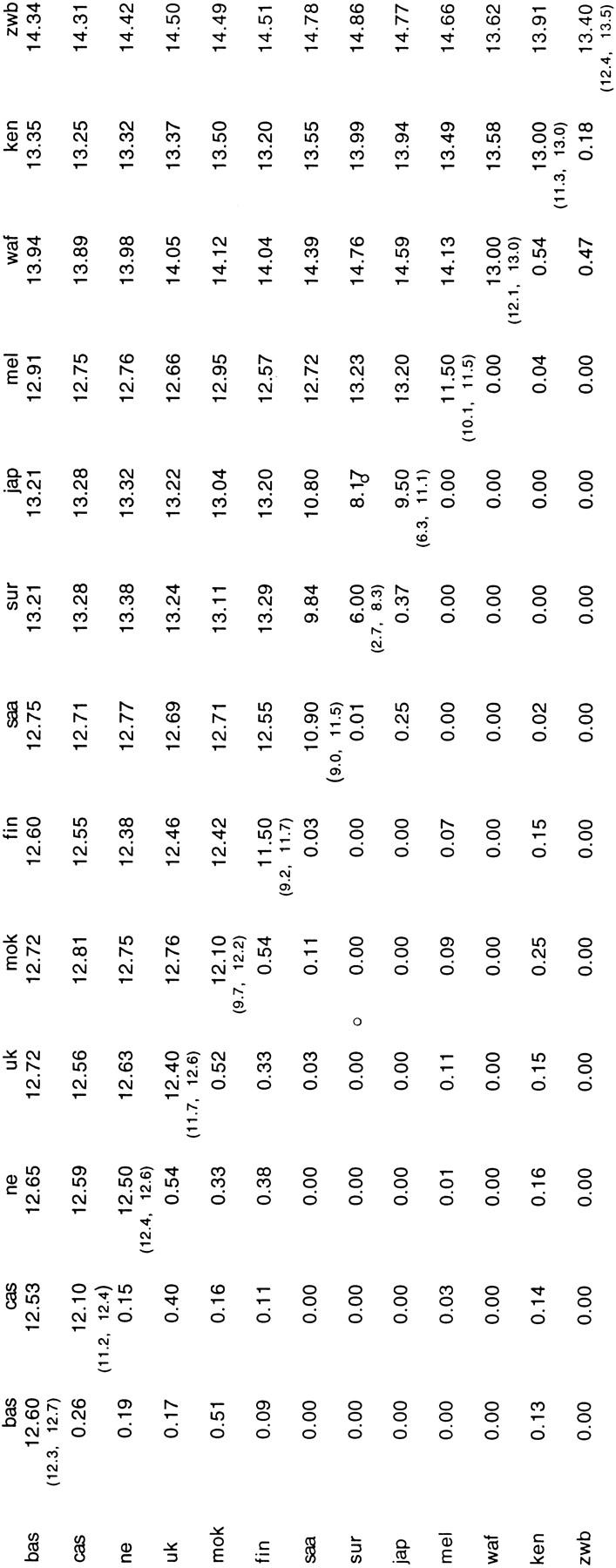 |
Upper matrix: Averaged interpopulational dissimlarity values. Lower matrix: Permutation significance value for the differentiation test. Diagonal: Intrapopulational averaged diversity values and 5%–95% confidence limits obtained by bootstrapping 100 times over alleles. Abbreviations as in Fig. 1.
However, the neighbor-joining tree in Figure 2a shows that although Basques lie within the European cluster, their long branch indicates a divergent behavior. The same pattern is observed under a Fitch algorithm (data not shown). This divergence is more conspicuous in the consensus tree of the bootstrapped data sets, which supports a shift for the Basques from within the European populations to a position between Europeans and Africans. These apparently discordant results can be reconciled as Basques being a divergent European population that has retained a greater proportion of more ancestral alleles.
Figure 2.

Neighbor-joining tree using the similarity values between alleles. Abbreviations as in Fig. 1. (a) Tree obtained from the original distance matrix; (b) Consensus tree of the 100 bootstrapped data sets. Bootstrap values are indicated as percentages.
In this sense, estimates of allele age may provide a clue to the minimum age of a population. The advantage of MS205 in this regard is that its high mutation rate and its fit to IAM increase the chances for the generation of a number of population-specific alleles (or groups of alleles). If it is possible to estimate their age, we can infer a minimum estimate of the age of the population to which they belong. However, reconstructing the phylogeny of all groups of alleles is not straightforward, as some mutational events may produce, by chance, similar-by-state alleles (the chances of producing an allele identical to previously existing ones by recurring mutation are more remote, especially if alleles with an unusual internal structure are considered). Thus, estimation of allele ages has been restricted to independent single alleles rather than lineages until additional information is gathered.
For the Basques, allele t14.1 (5′-ttttttttttttttatattattttatta-3′) is the best example of a population-specific allele; it has only been found in Basques (four copies have been observed in 236 Basque alleles), and no other allele belonging to the t14 group has been detected in any world population.
We have based our analysis in a present day population size for the (autochthonous) Basques, of 0.5 million individuals. Calderon et al. (1998) argue that the present-day population in the Basque Country would be ∼3 million people and that not more than one-fourth of the population settled in the Basque region can be considered autochthonous Basque for genetic studies. Therefore, half a million seems to be a conservative estimate. Calderon et al. (1998) also suggest that the estimated population density figure for Paleolithic times was on the order of 0.1 inhabitants/km2, ∼2000 people. Assuming an exponential increase in population size, we obtain (using No = 2,000, Nt = 500,000, t = 40,000 years), a value for the exponential growth rate r of 0.00014 per year (i.e., 0.0028 per generation). To be conservative in the estimate of allele ages, we have used r = 0.005 as estimated for the general Western European populations (Slatkin and Ranala 1997), which gives younger estimates of allele ages.
Thus, according to Slatkin and Ranala (1997), the maximum likelihood estimate of the age of allele t14.1, for instance, would be 19,441 years, with confidence limits of 11,260–31,700 years. Other examples shown in Table 2 yield similarly pre-Neolithic estimates for the allele ages.
Table 2.
Estimated Ages for Some Basque-Specific Alleles
| Alleles | No. of copies each | Age (years) | Confidence interval |
|---|---|---|---|
| t8.39, t8.56, t10.23, t11.43, t11.61, t11.76, t11.115, t11.129, t12.33, t12.44, t12.65 , t12.77, t12.92, t17.6 | 2 | 15,079 | (3,720–28,240) |
| t8.51, t11.37, t11.143, t12.39, t12.72 | 3 | 17,805 | (8,860–30,360) |
| t12.57, t14.01 | 4 | 19,441 | (11,260–31,700) |
| t8.63, t12.21 | 5 | 20,554 | (12,900–32,760) |
| t8.45 | 6 | 21,442 | (14,100–33,580) |
It can be argued that sample sizes analyzed so far are responsible for the nondetection of at least some of the alleged Basque-specific alleles in other world populations. Although this can hold true for some of them, many alleles observed twice have only been detected in a specific Basque subpopulation. On the other hand, one should expect alleles with a high frequency to be observed at least once in some other world population sample if they represent common ancestral alleles.
Calculated allele ages are likely to represent minimum ages, as we cannot ascertain if other closely related alleles, population specific or not, are derived from the allele under consideration or from other alleles observed in other populations. If the latter holds true, our inferences are not affected; if the former, these alleles would be part of the same lineage, and in discarding them we are not taking into account their frequency, thus producing younger estimates of the population age.
DISCUSSION
Although the Upper Paleolithic period offers a number of archaeological sites in the Basque Country, all of them in caves, the anthropological reconstruction of the local ethnogenic processes suffers from a scarcity and fragmentation of human paleontological remains (de la Rua 1995 and references therein). In this scenario, the analysis of genetic polymorphisms in the extant Basque (and world) population stands as a useful and complementary approach to unveil our links to the past.
As regards our more distant past, the comparison of the classical heterozygosity values to the intrapopulational averaged diversity values (Table 1) shows clearly that although most of the non-African populations have acquired high levels of diversity, this is mainly associated with mutations occurring in the hypervariable 3′ end that have generated a huge number of closely related alleles. This in principle would agree with the accumulation of diversity after an expansion event, in which genetic traits associated with low mutation rates show less postexpansion diversity compared to traits with higher mutation rates (Relethford 1997). Africans, on the other hand, despite showing similar heterozygosity values to Europeans, have a higher intrapopulational averaged allele dissimilarity (Table 1), reflecting a higher degree of slower-evolving, older diversity. This is consistent with African populations evolving for longer times and/or with greater long-term effective population sizes.
This study points to a European affiliation for the Basques. Nonetheless, the observation that a significant proportion of apparently Basque-specific alleles can be dated back to post-Aurignacian indicates a continuity of this population from prehistoric times several thousand years after the arrival of modern Homo sapiens in Europe, to the present day.
Archeological data suggest that the expansion of typically Aurignacian technology [attributed specifically to modern H. sapiens and most likely originating in the Middle East ∼100 thousand years ago (kya)] into Europe, and the dispersal of the associated populations, could be linked to an East–West cline. However, strong evidence indicates that Aurignacian was already present in Northwestern and Northeastern Spain at least by 40 kya, earlier than in Southwestern France (∼35 kya); here, it coexisted for several thousand years with the typical Neanderthal-associated Chatelperronian culture, which penetrated for a short distance into the Pyrenees and adjacent Northern Spain (Mellars 1992). Similarly, late Mousterian (Neanderthal) also coexists in some sites in Southern Spain, like the Zafarraya site, where it seems to have persisted until well after the Last Pleniglacial. Solutrean and Magdalenian industries appear to arrive at the Cantabrian fringe (North Spanish Coast) as already developed cultures from Southwestern France, whereupon they seem to evolve locally as an adaptation to this new environment (Cerdeño and Vega 1995). In addition, Neolithic culture in the Basque Country seems to be a late and partial process (Cava 1990).
Thus, preservation of these Upper Paleolithic alleles by a certain degree of isolation rather than by drift seems a more plausible explanation as, for the inferred long-term effective population size (close to 10,000), mutation appears to be the major driving force for the evolution of this minisatellite (Armour et al. 1996).
Although caution must be taken about the population specificity of the alleles from which ages have been estimated, these data suggest that Basques may at least have retained some of the ancestral European Paleolithic diversity, therefore supporting the previous hypothesis of Basques predating the Neolithic expansion from the Near East (Cavalli-Sforza et al. 1994). Thus, assuming that Neanderthals left no significant contribution from their gene pool (Krings et al. 1997), a southward Upper Paleolithic expansion from an area in Northern Spain–Southwestern France containing the Basque Country [it is well known from toponymy that Basque was once a language widely spoken over Southwestern France (Cavalli-Sforza et al. 1994)] would in this context correlate with the first principal component of gene frequencies for the Iberian Peninsula (Calafell and Bertranpetit 1994). The dense concentration of cave art within this region (i.e., Lascaux in Southwestern France, Altamira in Northern Spain; between 20000 and 15000 BP) has been related to the high density and concentration of human populations within this area, probably to the particular economic and ecologic opportunities (Mellars 1998). However, more data on similar loci and more extensive samples from a greater number of populations, especially surrounding the Basque area, are necessary to add support to this hypothesis. Whether or not other European populations conceal specific alleles that can be traced back to a pre-Neolithic period can be approached similarly.
The story involving MS205 is just one of the many that shape the genomes of the populations. To untangle them, future work will encompass the analysis of the 5′-flanking sequences of the minisatellite, not only to get a better resolution of the phylogenetic relationship between MS205 alleles but to make full genealogical use of this nuclear sequence information.
METHODS
Samples
Blood samples (10 ml) were obtained by venipuncture with the informed consent of volunteers. DNA was extracted from frozen blood samples using Nucleon (Scotlab) or standard phenol–chloroform techniques. A total of 312 MVR alleles were obtained from 109 autochthonous individuals from the Basque Country, Northern Spain, covering the provinces of Alava (Araba), Vizcaya (Bizkaia), Guipuzcoa (Gipuzkoa), and Navarra (Nafarroa); 24 Castilian individuals from the northern area of the province of Burgos (Northern Spain), and 23 individuals from the UK. These alleles were analyzed, along with a previously described world survey including African (Kenyans, West Africans, and Zimbabweans), Finns, Saami, Moksha (Finno-Ugric speakers from the Urals), Japanese, Surui (Amazonians), and Melanesian populations (Armour et al. 1996).
MVR Analysis of MS205
Internal structures of MS205 variant repeats were analyzed as described (Armour et al. 1993), except that primer 205TAG-T was used (Armour et al. 1996). In short, 50 ng of DNA was amplified in a first round of PCR with primers 205A and 205B for 16 cycles; amplification products were run on 0.8% agarose gels and identified by hybridization of the corresponding Southern blots to a 32P-labeled MS205 probe. A second independent reaction of amplification for 25 cycles allowed the direct visualization of MS205 alleles on an ethidium bromide-stained agarose gel under UV light. Positive identification of PCR-amplified minisatellite alleles was accomplished by size comparison with the corresponding hybridization autoradiograph. Bands were subsequently cut out and DNA was extracted in 1ml of water by freezing and thawing repeatedly.
Four PCR reactions per allele were performed, using the flanking primer 205A in combination with TAG-RA or TAG-RT reverse repeat-specific internal primers, or flanking primer 205B and TAG-A or TAG-T forward primers. The four sets of PCR reactions allowed us to obtain the full unambiguous MVR alleles after hybridization to probe MS205, especially in the cases of long (4–5 kb) MS205 alleles. In the case of alleles of very close or identical length (but different internal structures), the use of primers specific for flanking substitutional polymorphisms allowed us to amplify one allele or the other.
Allele Nomenclature
Alleles were designated according to the length of the run of t-type repeat units at the beginning of the 5′ end, followed by an arbitrary number to identify the different internal structures of the alleles within the group. Thus, allele t10.7 represents allele number 7 of the group of alleles with 10t-type repeat units at the beginning of the 5′ end.
Data Analysis
An unbiased estimate (θH) of θ(4 Ne μ) was obtained by iteration from the expected heterozygosity under the infinite allele model, following the procedure of Chakraborty and Daiger (1991). The expected number of rare alleles, and the significance of the difference from the observed value was also calculated as in Chakraborty and Daiger (1991) using θk, the iteration estimator of θ based on the expected number of alleles.
Multivariate Factor Correspondence analyses of allele frequencies in populations were performed using the ADE-4 package (Thioulouse et al. 1997).
To gain a deeper insight into the phylogenetic relationships among populations, customized computer programs were developed. One of them compares all pairs of alleles in the global sample and establishes a distance value for each comparison. In this procedure, data on the rate and pattern of mutational events at MS205, revealed by analysis of sperm mutants by SP–PCR (Jeffreys et al. 1994; May et al. 1996) were considered. The polarity of the mutation (with a greater penalty going to those mutations inferred to have taken place at the 5′ end compared to those at the 3′ end), the length of the region involved, and the kind of mutation involved are factors in the program. Thus, relationships among alleles differing by small insertion/deletion events, terminal and subterminal duplications of small blocks of repeats, and terminal and subterminal short gene-conversion events are included, as well as comparisons of the numbers of t-repeat types in the first run of ts from the 5′ end.
Based on Nei’s minimum distance (1987) and the works of Hudson et al. (1992) and Shriver et al. (1995), the averaged intrapopulational diversity, a value representing the average dissimilarity between pairs of alleles for each population, was estimated as
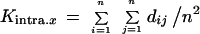 |
where n is the total number of alleles in the sample and dij the calculated (as indicated above) distance value for each corresponding allele pair. This figure represents the comparison of a population to itself and matches the classical heterozygosity
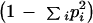 |
when a binary code (0 for identical, 1 for different alleles) is used as the distance value between alleles, disregarding any phylogenetic information. Confidence intervals were estimated by bootstrapping 100 times over alleles.
Subsequently, an averaged interpopulation diversity value between populations x and y,
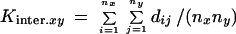 |
was calculated by comparing all alleles in population x to all alleles in population y, where nxand ny are the corresponding sample sizes (numbers of alleles). This dissimilarity value was used as a measure of genetic distance as
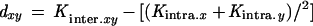 |
An estimate of the sampling distribution of this parameter dxy was obtained by lumping both populations under comparison together and generating 100 pairs of random partitions of the total, each time of the same size as the two original samples, by shuffling alleles 20,000 times. The number of times (in the 100 simulations) the simulated parameter was equal to or greater than the observed one was used as the significance value of a test for differentiation between populations.
The genetic distance among populations defined above was used to generate neighbor-joining trees by means of PHYLIP 3.572 (Felsenstein 1985) NEIGHBOR program; the bootstrap consensus tree was obtained using CONSENSE. Trees were drawn using TreeView (Page 1996).
Programs for pair-wise comparison have been written in Future BASIC II (STAZ Software, Inc.) for PowerPC Macintosh and are available on request.
To estimate the age of specific alleles, we have applied, the maximum-likelihood approach based on the number of copies of the allele in a sample from the population proposed by Slatkin and Ranala (1997). The estimator
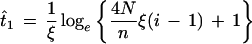 |
provides an estimate of the age of an allele with i copies in a sample of n alleles from a present-day population of size N, where ξ = s + r, s being the selection coefficient for heterozygotes and r the exponential growth-rate parameter. The value s is assumed to be 0 (neutrality), and a value r of 0.005 per generation (Slatkin and Ranala 1997) was used to represent an exponentially growing population. This value is intended to represent an upper limit for the growth rate of the Basque population to obtain conservative estimates of the allele ages. The confidence intervals of the estimate were obtained by determining the support interval of the likelihood, the support function being defined as the natural logarithm of the likelihood function (Edwards 1972).
Acknowledgments
We express our gratitude, first, to all of the volunteers that kindly donated their blood for this work. We are indebted also to the schools, institutions, and people that facilitated the sampling. We thank M. Slatkin for his help with allele-age estimates, and J.F.Y. Brookfield, R. Badge, L. Williams, E. Rogers, S. Miles. A. Davison, C. Wade, and B. Lafay for fruitful discussions about the manuscript. This work was funded by The Wellcome Trust (grant 047696/Z/96).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL pdzsaa@granby.nott.ac.uk; FAX 115-9709906.
REFERENCES
- Aguirre A, Vicario A, Mazon LI, Estomba A, Martinez de Pancorbo M, Arrieta-Pico V, Perez-Elortondo F, Lostao CM. Are the Basques a single and unique population? Am J Hum Genet. 1991;49:450–458. [PMC free article] [PubMed] [Google Scholar]
- Ammerman AJ, Cavalli-Sforza LL. Neolithic transition and population genetics in Europe. Princeton, NJ: Princeton University Press; 1984. [Google Scholar]
- Armour JAL, Harris PC, Jeffreys AJ. Allelic diversity at minisatellite MS205 (D16S309): Evidence for polarized variability. Hum Mol Genet. 1993;2:1137–1145. doi: 10.1093/hmg/2.8.1137. [DOI] [PubMed] [Google Scholar]
- Armour JAL, Anttinen T, May C, Vega EE, Sajantila A, Kidd JR, Kidd KK, Bertranpetit J, Pääbo S, Jeffreys AJ. Minisatellite diversity supports a recent African origin for modern humans. Nat Genet. 1996;13:154–160. doi: 10.1038/ng0696-154. [DOI] [PubMed] [Google Scholar]
- Armour JAL, Alonso S, Miles S, Williams LJ, Badge RM. Minisatellites and mutation processes in tandemly repetitive DNA. In: Goldstein DB, Schlötterer C, editors. Microsatellites: Evolution and applications. Oxford, UK: Oxford University Press; 1998. [Google Scholar]
- Bertranpetit J, Cavalli-Sforza LL. A genetic reconstruction of the history of the Iberian Peninsula. Ann Hum Genet. 1991;55:51–67. doi: 10.1111/j.1469-1809.1991.tb00398.x. [DOI] [PubMed] [Google Scholar]
- Bertranpetit J, Sala J, Calafell F, Underhill PA, Moral P, Comas D. Human mitochondrial DNA variation and the origin of Basques. Ann Hum Genet. 1995;59:63–81. doi: 10.1111/j.1469-1809.1995.tb01606.x. [DOI] [PubMed] [Google Scholar]
- Calafell F, Bertranpetit J. Principal component analysis of gene frequencies and the origins of Basques. Am J Phys Anthropol. 1994;93:201–215. doi: 10.1002/ajpa.1330930205. [DOI] [PubMed] [Google Scholar]
- Calderon R, Vidales C, Peña JA, Perez-Miranda A, Dogoujon JM. Immunoglobulin allotypes (GM and KM) in Basques from Spain: Approach to the origin of the Basque population. Hum Biol. 1998;70:667–698. [PubMed] [Google Scholar]
- Cava A. El Neolitico en el Pais Vasco. Munibe Antropol Arkeol. 1990;42:97–106. [Google Scholar]
- Cavalli-Sforza LL, Minch E. Paleolithic and Neolithic lineages in the European mitochondrial gene pool. Am J Hum Genet. 1997;61:247–250. doi: 10.1016/S0002-9297(07)64303-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli-Sforza LL, Menozzi P, Piazza A. The history and geography of human genes. Princeton, NJ: Princeton University Press; 1994. [Google Scholar]
- Cerdeño ML, Vega G. La España de Altamira. Prehistoria en la Peninsula Iberica. Madrid, Spain: Temas de Hoy S.A.; 1995. [Google Scholar]
- Chakraborty R, Daiger SP. Polymorphisms at VNTR loci suggest homogeneity of the white population of Utah. Hum Biol. 1991;63:571–587. [PubMed] [Google Scholar]
- Comas D, Mateu E, Calafell F, Perez-Lezaun A, Bosch E, Martinez-Arias R, Bertranpetit J. HLA class I and class II DNA typing and the origin of Basques. Tissue Antigens. 1998;51:30–40. doi: 10.1111/j.1399-0039.1998.tb02944.x. [DOI] [PubMed] [Google Scholar]
- de la Rua C. La historia del poblamiento del Pais Vasco desde una perspectiva antropologica. In: Bertranpetit J, Vives E, editors. El passat dels Pirineus des d’una perspectiva multidisciplinaria. Muntanyes I Poblacio. Andorra la Vella, Spain: Centre de Trobada de les Cultures Pirinenques; 1995. pp. 301–316. [Google Scholar]
- Edwards AWF. Likelihood. Cambridge, UK: Cambridge University Press; 1972. [Google Scholar]
- Feldman MW, Bergman A, Pollock DD, Golstein DB. Microsatellite genetic distances with range constraint: analytical description and problems of estimation. Genetics. 1997;145:207–216. doi: 10.1093/genetics/145.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP: Phylogeny inference package version 3.2. Cladistics. 1985;5:164–166. [Google Scholar]
- Garcia O, Martin P, Budowle B, Uriarte J, Albarran C, Alonso A. Basque Country autochtonous population data on 7 short tandem repeat loci. Int J Leg Med. 1998;111:162–164. doi: 10.1007/s004140050140. [DOI] [PubMed] [Google Scholar]
- Garza JC, Slatkin M, Freimer NB. Microsatellite allele frequencies in humans and chimpanzees with implications for constraints on allele size. Mol Biol Evol. 1995;12:594–603. doi: 10.1093/oxfordjournals.molbev.a040239. [DOI] [PubMed] [Google Scholar]
- Harpending HC, Batzer MA, Gurven M, Jorde LB, Rogers A, Sherry ST. Genetic traces of ancient demography. Proc Natl Acad Sci. 1998;95:1961–1967. doi: 10.1073/pnas.95.4.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick PW. Evolutionary genetics of the major histocompatibility complex. Am Nat. 1994;143:945–964. [Google Scholar]
- Hickson RE, Cann RL. MHC allelic diversity and modern human origins. J Mol Evol. 1997;45:589–598. doi: 10.1007/pl00006263. [DOI] [PubMed] [Google Scholar]
- Hudson RR, Boos DD, Kaplan NL. A statistical test for detecting geographic subdivision. Mol Biol Evol. 1992;9:138–151. doi: 10.1093/oxfordjournals.molbev.a040703. [DOI] [PubMed] [Google Scholar]
- Iriondo M, Barbero C, Izagirre N, Manzano C. Data on six short tandem repeat polymorphisms in an autochtonous Basque population. Hum Hered. 1997;47:131–137. doi: 10.1159/000154401. [DOI] [PubMed] [Google Scholar]
- Jeffreys AJ, MacLeod A, Tamaki K, Neil DL, Monckton DG. Minisatellite repeat coding as a digital approach to DNA typing. Nature. 1991;354:204–209. doi: 10.1038/354204a0. [DOI] [PubMed] [Google Scholar]
- Jeffreys AJ, Tamaki K, MacLeod A, Monckton DG, Neil DL, Armour JAL. Complex gene conversion events in germline mutation at human minisatellites. Nat Genet. 1994;6:136–145. doi: 10.1038/ng0294-136. [DOI] [PubMed] [Google Scholar]
- Jin L, Chakraborty R. Population structure, stepwise mutations, heterozygote deficiency and their implications in DNA forensics. Heredity. 1995;74:274–285. doi: 10.1038/hdy.1995.41. [DOI] [PubMed] [Google Scholar]
- Krings M, Stone A, Schmitz RW, Krainitzki H, Stoneking M, Pääbo S. Neanderthal DNA sequences and the origins of modern humans. Cell. 1997;90:19–30. doi: 10.1016/s0092-8674(00)80310-4. [DOI] [PubMed] [Google Scholar]
- Lucotte G, Hazout S. Y chromosome DNA haplotypes in Basques. J Mol Evol. 1996;42:472–475. doi: 10.1007/BF02498642. [DOI] [PubMed] [Google Scholar]
- Manzano C, Aguirre AI, Iriondo M, Martin M, Osaba L, de la Rua C. Genetic polymorphisms of the Basques from Gipuzkoa: Genetic heterogeneity of the Basque population. Ann Hum Biol. 1996a;23:285–296. doi: 10.1080/03014469600004522. [DOI] [PubMed] [Google Scholar]
- Manzano C, Orue JM, de la Rua C. The ′basqueness′ of Alava: A reappraisal from a multidisciplinary perspective. Am J Phys Anthropol. 1996b;99:249–258. doi: 10.1002/(SICI)1096-8644(199602)99:2<249::AID-AJPA3>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- May C, Jeffreys AJ, Armour JAL. Mutation rate heterogeneity and the generation of allele diversity at the human minisatellite MS205 (D16S309) Hum Mol Genet. 1996;5:1823–1833. doi: 10.1093/hmg/5.11.1823. [DOI] [PubMed] [Google Scholar]
- Mellars PA. Archaeology and the population-dispersal hypothesis of modern human origins in Europe. Phil Trans R Soc Lond B. 1992;337:225–234. doi: 10.1098/rstb.1992.0100. [DOI] [PubMed] [Google Scholar]
- ————— . The Upper Paleolithic revolution. In: Cunliffe B, editor. Prehistoric Europe. An illustrated history. Oxford, UK: Oxford University Press; 1998. pp. 42–78. [Google Scholar]
- Mourant AE. The blood groups of the Basques. Nature. 1947;160:505–506. doi: 10.1038/160505b0. [DOI] [PubMed] [Google Scholar]
- Nauta MJ, Weissing FJ. Constraints on allele size at microsatellite loci: Implications for genetic differentiation. Genetics. 1996;143:1021–1032. doi: 10.1093/genetics/143.2.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M. Molecular evolutionary genetics. New York, NY: Columbia University Press; 1987. [Google Scholar]
- Page RDM. TreeView: An application to display phylogenetic trees on personal computers. Comput Appl Biosci. 1996;12:357–358. doi: 10.1093/bioinformatics/12.4.357. [DOI] [PubMed] [Google Scholar]
- Perez-Lezaun A, Calafell F, Seielstad M, Mateu E, Comas D, Bosch E, Bertranpetit J. Population genetics of Y chromosome short tandem repeats in humans. J Mol Evol. 1997;45:265–270. doi: 10.1007/pl00006229. [DOI] [PubMed] [Google Scholar]
- Relethford JF. Mutation rate and excess African heterozygosity. Hum Biol. 1997;60:785–792. [PubMed] [Google Scholar]
- Richards M, Côrte-Real H, Forster P, Macaulay V, Wilkinson-Herbort H, Demaine D, Papiha S, Hedges R, Bandelt HJ, Sykes B. Paleolithic and Neolithic lineages in the European mitochondrial gene pool. Am J Hum Genet. 1996;59:185–203. [PMC free article] [PubMed] [Google Scholar]
- Richards M, Macaulay V, Sykes B, Pettitt P, Hedges R, Forster P, Bandelt HJ. Reply to Cavalli-Sforza and Minch. Am J Hum Genet. 1997;61:251–254. [Google Scholar]
- Rogers AR, Jorde LB. Ascertainment bias in estimates of average heterozygosity. Am J Hum Genet. 1996;58:1033–1041. [PMC free article] [PubMed] [Google Scholar]
- Royle NJ, Armour JAL, Webb M, Thomas A, Jeffreys AJ. A hypervariable locus D16S309 located at the distal end of 16p. Nucleic Acids Res. 1992;20:1162. doi: 10.1093/nar/20.5.1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajantila A, Pääbo S. Language replacement in Scandinavia. Nat Genet. 1995;11:359–360. doi: 10.1038/ng1295-359. [DOI] [PubMed] [Google Scholar]
- Sajantila A, Lahermo P, Anttinen T, Lukka M, Sistonen P, Savontaus M, Aula P, Beckman L, Tranebjaerg L, Gedde-Dahl T, et al. Genes and languages in Europe: An analysis of mitochondrial lineages. Genome Res. 1995;5:42–52. doi: 10.1101/gr.5.1.42. [DOI] [PubMed] [Google Scholar]
- Scozzari R, Cruciani F, Malaspina P, Santolamaza P, Ciminelli BM, Torroni A, Modiano D, et al. Differential structuring of human populations for homologous X and Y microsatellite loci. Am J Hum Genet. 1997;61:719–733. doi: 10.1086/515500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semino O, Passarino G, Brega A, Fellous M, Santachiara-Benerecetti AA. A view of the Neolithic demic diffusion in Europe through two Y chromosome-specific markers. Am J Hum Genet. 1996;59:964–968. [PMC free article] [PubMed] [Google Scholar]
- Shriver MD, Jin L, Boerwinkle E, Deka R, Ferrell RE, Chakraborty R. A novel measure of genetic distance for highly polymorphic tandem repeat loci. Mol Biol Evol. 1995;12:914–920. doi: 10.1093/oxfordjournals.molbev.a040268. [DOI] [PubMed] [Google Scholar]
- Slatkin M, Ranala B. Estimating the age of alleles by use of intraallelic variability. Am J Hum Genet. 1997;60:447–458. [PMC free article] [PubMed] [Google Scholar]
- Thioulouse J, Chessel D, Doldec S, Olivier JM. ADE-4: A multivariate analysis and graphical display software. Stat Comput. 1997;7:75–83. [Google Scholar]