

Abstract
Blunt chest trauma resulting in pulmonary contusion is a common but poorly understood injury. We previously demonstrated that lung contusion activates localized and systemic innate immune mechanisms and recruits neutrophils to the injured lung. We hypothesized that the innate immune and inflammatory activation of neutrophils may figure prominently in the response to lung injury. To investigate this, we used a model of pulmonary contusion in the mouse that is similar to that observed clinically in humans and evaluated postinjury lung function and pulmonary neutrophil recruitment. Comparisons were made between injured mice with and without neutrophil depletion. We further examined the role of chemokines and adhesion receptors in neutrophil recruitment to the injured lung. We found that lung injury and resultant physiological dysfunction after contusion was dependent upon the presence of neutrophils in the alveolar space. We show that CXCL1, CXCL2/3, and CXCR2 are involved in neutrophil recruitment to the lung after injury, and that ICAM-1 is locally expressed and actively participates in this process. Injured gp91phox deficient mice showed improved lung function, indicating that oxidant production by neutrophil NADPH oxidase mediates lung dysfunction after contusion. These data suggest that both neutrophil presence and function are required for lung injury after lung contusion.
Keywords: Neutrophil, pulmonary contusion, inflammation, chemokine, cytokine, mouse model
INTRODUCTION
Pulmonary contusion is a relatively common injury seen after blunt trauma that is associated with significant morbidity, organ dysfunction (ALI/ARDS), and is potentially lethal1. Increasing evidence suggests that the maladies seen after lung contusion are not simply a mechanical phenomenon but are inflammatory in nature2, 3. Following lung contusion, in addition to structural damage to the lung, an inflammatory cell infiltrate composed primarily of neutrophils (PMNs) ensues. Various inflammatory mediators are produced, leading to the breakdown of pulmonary capillary basement membranes, hypoxia, increased pulmonary capillary resistance, myocardial dysfunction, production of toxic oxygen metabolites, and alterations in inflammatory cell function.
The pathogenesis of acute lung injury is an incompletely understood process. In a number of animal models of lung injury and in human studies, neutrophil accumulation in the lung is a key event in the early development of acute lung injury and ARDS4, 5. Activation, localization and extravasation of neutrophils from the circulation to site of injury is a complex process that is thought to be dependent upon early response cytokine expression (IL-1β, TNF-α); the production of chemotactic molecules, such as chemokines, complement 5a (C5a), and leukotriene B4 (LTB4); and the upregulation of cell surface-adhesion molecules such as ICAM-16. The accumulation of activated neutrophils in the interstitium and alveolar space, results in the production of reactive oxygen species and the release of proteolytic enzymes leading to acute inflammation. Although required for clearance of invading pathogens, activated neutrophils may independently contribute to pulmonary damage and dysfunction. However, recent evidence also suggests that apoptosis of type 2 epithelial cells may also contribute to acute lung injury7, 8. Inflammatory processes at the airway/epithelial cell interface can cause impaired gas exchange, decreased lung function, and release of proapoptotic mediators into the alveolar space. Thus mechanisms in addition to neutrophil accumulation in the lung may also participate in acute injury.
To further understand the role of neutrophils in acute lung injury, we previously showed that pulmonary contusion activates local innate immune mechanisms in a TLR2 and TLR4 dependent manner resulting in the recruitment of neutrophils to the lung. Furthermore, neutrophil recruitment after lung contusion is associated significant impairment in lung function after injury. Consistent with others’ studies, our data indicated that lung contusion initiates an inflammatory response culminating with neutrophil recruitment to the lung and resulting in an acute lung injury. We further demonstrated localized expression of early response cytokines, elevated systemic levels of CXC chemokines, upregulated pulmonary ICAM-1 and neutrophil CD11b expression, and a robust pulmonary neutrophilia associated with a clinically significant reduction in the PaO2/FiO2 (P/F) ratio following lung contusion9-11.
Based upon these findings, we hypothesize that neutrophil recruitment to the lung is in part responsible for the pulmonary dysfunction seen after lung contusion and is dependent upon CXC chemokine and ICAM-1 expression. As an extension of our previous work, in these studies, we sought to determine: 1) Whether neutrophil recruitment to the lung is an essential component of the lung injury seen after contusion; 2) The mechanisms responsible for neutrophil recruitment after lung injury; and 3) The manner in which neutrophils might injure the lung. This results from this study show that neutrophils are primarily responsible for pulmonary dysfunction after lung contusion. We also demonstrate that neutrophil recruitment to the injured lung is dependent on expression of CXC chemokines, the CXCR2 receptor, and ICAM-1. Finally, we show that neutrophil NADPH oxidase mediates lung injury after contusion.
METHODS
Animals
Age-matched wild-type (WT) C57/BL6 mice and gp91phox- mice were utilized in this study. WT mice were maintained were bred and maintained at the animal facility at Wake Forest University Health Sciences. The gp91phox- mice were obtained from Jackson Laboratories (B6.129S6-Cybbtm1Din/J, Bar Harbor, ME). Animals were bred and maintained under specific pathogen-free conditions at the animal facility at Wake Forest University Health Sciences. The protocol used in this study was approved by the Animal Care and Use Committee (#A07-246). Blunt Chest Injury Model: Injury was induced using the Cortical Contusion Impactor (CCI) as described previously11. Briefly, mice are anesthetized with 2% isoflurane at a flow rate of 1L/min. The mouse is positioned left lateral decubitus and during inspiration, the right chest is struck with the CCI along the posterior axillary line, 1cm above the costal margin. Control animals receive anesthetic alone. Mice were followed for various times after injury as specified in the Figure Legends. Serum and tissue samples were collected at the time of death by isoflurane overdose and cervical dislocation.
Bronchoalveolar lavage (BAL)
At 24H after injury, BAL was performed by cannulation of the trachea and lavage was performed using 4ml of phosphate buffered saline (PBS, Sigma Biochemical, St. Louis, MO) at 4°C. BAL was centrifuged at 300 × g, 4°C for 10 minutes and supernatant collected and stored at −70°C until use. The cell pellet was counted and differentiated as previously described11.
Arterial Blood Gas
Arterial blood gas (ABG) samples at 24H were obtained from the dorsal tail artery. Animals were induced with isoflurane anesthesia (3%) and then maintained on an admixture of 1.5% isoflurane with 100% oxygen at a flow rate of 1L/min. After 5 minutes, the dorsal tail artery was identified, severed, and blood was collected and used to determine the partial pressure of arterial oxygen (PaO2). PaO2 was measured in each sample using a Stat Profile pHOx Blood gas Analyzer (Nova Biomedical, Waltham, MA) according to the manufacturer’s instructions.
Neutralization/blocking studies
Neutrophils were depleted_using the functional grade RB6-8C5 monoclonal antibody (eBioscience, San Diego, CA) that reacts with mouse Ly6G. Twenty-four hours prior to lung injury mice were given 100ug in PBS of α-Ly6G or an IgG isotype control by intraperitoneal injection. Neutrophil depletion was confirmed by flow cytometry and evaluation of peripheral blood smear with differential staining. Blocking of the CXCR2 receptor was performed using the hexapeptide antileukinate, Ac-RRWWCR-NH2 (AnaSpec, Inc., Fremont, CA), as previously described4, 12. Mice were given a 52mg/kg dose of antileukinate or PBS subcutaneously 30 minutes prior to injury. Neutralization of CXCL1 (KC) and CXCL2/3 (MIP-2) chemokine activity was performed using anti-KC and anti-MIP-2 monoclonal antibodies (R&D Systems, Minneapolis, MN). Mice were given 25ug of α-KC and/or 25ug of α-MIP-2 in a single dose intravenously 30 minutes prior to injury. Isotype controls were administered in a similar fashion. Neutralization of ICAM-1 was performed using α-ICAM-1 monoclonal antibody (R&D Systems, Minneapolis, MN). Mice were given 50mcg of α-ICAM-1 30 minutes prior to injury. Isotype controls were administered in a similar fashion.
ICAM-1 Immunohistochemistry
Paraffin embedded sections were deparaffanized in three changes of xylenes for 5 minutes each and hydrated through three changes for 3 minutes each of 100% EtOH, 95% EtOH, and 80% EtOH. Endogenous peroxidase was quenched with 3% H2O2 in PBS for 10min. An antigen retrieval step was performed by heating sections at 95°C in 10mM citrate buffer at pH6.0 for 25min. Sections were blocked for non-specific binding with 10% FBS in PBS for 60 minutes. A three-step staining procedure was performed using a 1:100 dilution of hamster α-mouse ICAM-1 antibody (BD Biosciences, San Jose, CA), a 1:100 dilution of a biotinylated mouse α-hamster secondary antibody (BD Biosciences, San Jose, CA) and Streptavidin HRP (BD Biosciences, San Jose, CA). The ICAM-1 staining was visualized with DAB substrate for peroxidase (Vector Laboratories, Burlingame CA). Sections were counterstained with Mayer’s Hematoxylin Solution (Sigma-Aldrich, St. Louis, MO) and coverslips were mounted using VectaMount Mounting Medium (Vector Laboratories, Burlingame, CA).
IL-6 and CXCL1 assay
Serum levels of IL-6 and CXCL1 at 3H after injury were measured using commercially available ELISA kits (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions. Samples were assayed in duplicate.
Statistical analysis
Data are reported using GraphPad Prism (v 4.03, San Diego, CA) and expressed as the mean ± SE of independent observations as indicated in the Figure legends. One-way analysis of variance (1way ANOVA) with multiple comparison post test (Bonferroni) was used to compare the means between injury groups. A p-value <0.05 was considered to be significant.
RESULTS
Neutrophil depletion prior to lung contusion improves post-injury pulmonary function
We have previously reported an association between reduced pulmonary neutrophilia and improved pulmonary function in injured TLR 2 and 4 deficient animals10, 11. To further understand the role of neutrophils in pulmonary dysfunction after contusion, we used antibodies to inhibit neutrophil recruitment to the lung after injury. Mice were treated with α-Ly6G, a monoclonal antibody with specificity for neutrophils, 24hrs prior to lung injury. As confirmed by peripheral blood smear and flow cytometry, α-Ly6G treatment resulted in depletion of greater than 90% of circulating neutrophils with no effect on the circulating monocyte population(data not shown). Despite no significant difference in CXCL1 serum levels induced by injury in neutrophil depleted mice (Figure 1A), but consistent with neutrophil depletion, BAL PMN counts were significantly diminished at 24hrs after lung injury when treated with α-Ly6G (Figure 1B). In neutropenic animals, pulmonary function improved significantly as evidenced by a marked increase in P/F ratios (Figure 1C) that correlated with a significant decrease in serum IL-6 (Figure 1D). Injured mice treated with IgG isotype control antibody showed PMN infiltrate in the BAL and P/F ratios that were not significantly different from injured mice without pretreatment. These data correlate PMN in the BAL with lung dysfunction and is similar to what can be observed clinically in patients with pulmonary contusion.
Fig. 1. Neutrophil depletion improves post-injury pulmonary function.
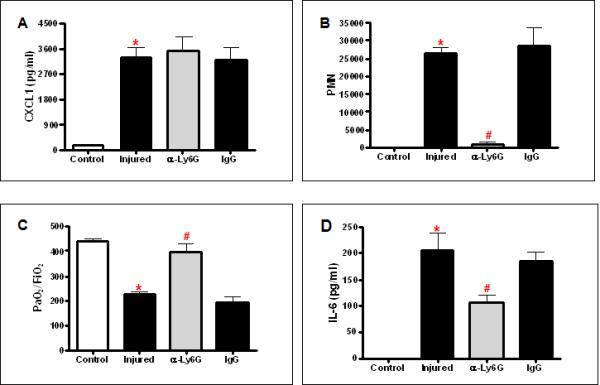
Mice were pre-treated with anti-Ly6G (α-Ly6G) or IgG at 24H prior to injury. Samples from uninjured (Control, open bars) and injured animals (filled bars) were isolated after injury. (A) Serum was assayed by ELISA for CXCL1 as described in methods. There is a significant increase in CXCL1 in injured mice (*p<0.001). CXCL1 levels in α-Ly6G pretreated, neutropenic injured mice were not significantly different from untreated injured mice. N=7,7,7,5 for experimental groups, respectively, as shown. (B) The BAL cell pellet was counted and differentiated as described in methods. There is a significant increase in PMN in the BAL in injured mice (*p<0.001). α-Ly6G pretreated, neutropenic injured mice showed a significant decrease in PMN (#p<0.001). There is no significant difference in PMN between injured and IgG treated mice. n=6,6,6,3 for experimental groups, respectively, as shown. (C) Blood gas analysis showed significantly decreased PaO2:FiO2 in injured animals (*p≤0.001). α-Ly6G pretreated, neutropenic injured mice showed significantly better PaO2:FiO2 (#p<0.001). There is no significant difference in PaO2:FiO2 between injured and IgG treated mice. n=5 for all experimental groups. (D) Serum was assayed by ELISA for IL-6 as described in methods. There is a significant increase in IL-6 in injured mice (*p<0.001). IL-6 levels in α-Ly6G pretreated, neutropenic injured mice were significantly decreased compared with untreated injured mice (#p<0.001). IgG pretreatment did not show a significant difference in IL-6 levels compared with untreated injured mice. N=7,7,7,5 for experimental groups, respectively, as shown.
CXC chemokines and the CXC chemokine receptor 2 (CXCR2) receptor mediate neutrophil recruitment to the injured lung after lung contusion
Having demonstrated a causal relationship between of pulmonary neutrophilia and postinjury pulmonary dysfunction, we next sought to determine the mechanisms that mediate neutrophil recruitment to the injured lung. The CXC chemokines, CXCL1 and CXCL2/3, have been shown to be involved in PMN recruitment in models of acute lung injury13. We have previously reported increased systemic levels of CXCL1 at 3hrs after lung contusion. Based upon these findings, we hypothesized that CXCL1 and possibly CXCL2/3 were involved with neutrophil recruitment to the lung after lung contusion10, 11. As both CXC chemokines act through a common receptor, CXCR2, we used antileukinate, a hexapeptide inhibitor of CXCR2, to inhibit CXCR2 signaling in our model of lung contusion4. Treatment with antileukinate resulted in a significant reduction in BAL neutrophils at 24hrs after lung contusion (Figure 2A). The decrease in PMN in the BAL correlated with an improvement in pulmonary function after injury (Figure 2B).
Fig. 2. CXCR-2 dependent neutrophil recruitment to the injured lung after lung contusion.
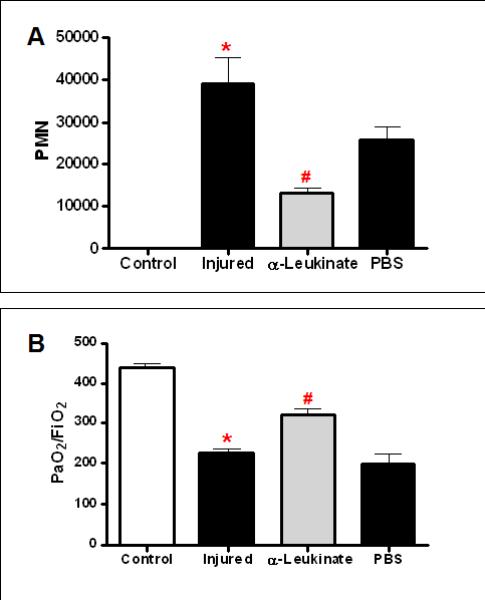
Mice were pre-treated with anti-leukinate (α-leukinate) or vehicle (PBS) at 30min. prior to injury. BAL and arterial blood from uninjured (Control, open bars) and injured animals (filled bars) were isolated at 24H after injury. (A) The cell pellet was counted and differentiated as described in methods. There is a significant increase in PMN in the BAL in injured mice (*p<0.001). α-Leukinate treated injured mice showed a significant decrease in PMN (#p<0.001). There is no significant difference in PMN between injured and PBS treated mice. n=6,6,6,3 for experimental groups, respectively, as shown. (B) Blood gas analysis showed decreased PaO2:FiO2 in injured animals (*p≤0.001). α-Leukinate treated injured mice showed significantly better PaO2:FiO2 (#p<0.05). There is no significant difference in PaO2:FiO2 between injured and IgG treated mice. n=5,5,5,3 for all experimental groups, respectively, as shown.
We next sought to determine the roles of CXCL1 and CXCL2/3 after lung contusion using blocking antibodies. Inhibition of either CXCL1 or CXCL2/3 alone had no significant effect on PMN counts in the BAL. However, when both CXCL1 and CXCL2/3 were inhibited, there was a significant reduction in BAL PMN numbers (Figure 3). Collectively, these data suggest that CXC chemokines mediate neutrophil trafficking to the lung after lung contusion, but other complimentary mechanisms exist outside the CXC family of chemokines.
Fig. 3. CXC chemokine dependent neutrophil recruitment to the injured lung after lung contusion.
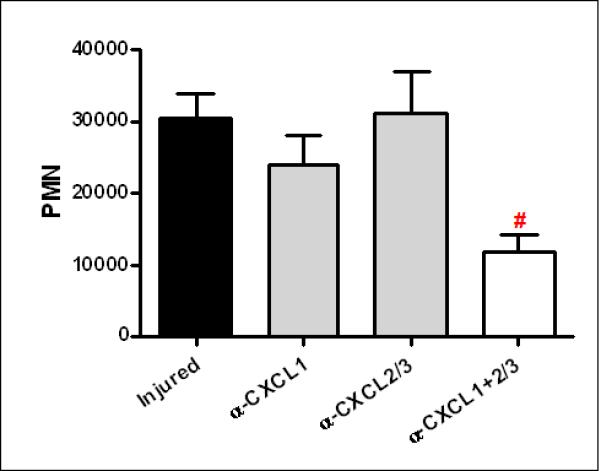
Mice were pre-treated with α-CXCL1, α-CXCL2/3 or both (α-CXCL1±2/3) at 30 min. prior to injury. BAL from injured animals was isolated at 24H after injury. The cell pellet was counted and differentiated as described in methods. There was no significant difference in PMN the BAL in injured mice or injured mice treated with either antibody alone. In contrast, injured mice treated with both antibodies showed a significant decrease in PMN (#p<0.05) when compared with untreated injured mice. n=6 for all experimental groups.
ICAM-1 expression is induced by lung injury and mediates neutrophil accumulation
ICAM-1 interacts with the β2 integrin, CD11b/18, on the surface of the neutrophil and mediates firm adhesion, an essential step for neutrophil diapedesis into areas of inflammation14. We have previously demonstrated increased CD11b expression on the surface of circulating neutrophils shortly after lung contusion, leading us to hypothesize that ICAM-1 is involved in this inflammatory process9, 10. We initially sought to determine whether lung contusion induces expression of ICAM-1 in the injured lung. As shown in Figure 4A, there is a marked increase in pulmonary ICAM-1 expression at 3hrs after lung injury.
Fig. 4. ICAM-1 dependent neutrophil recruitment to the injured lung after lung contusion.
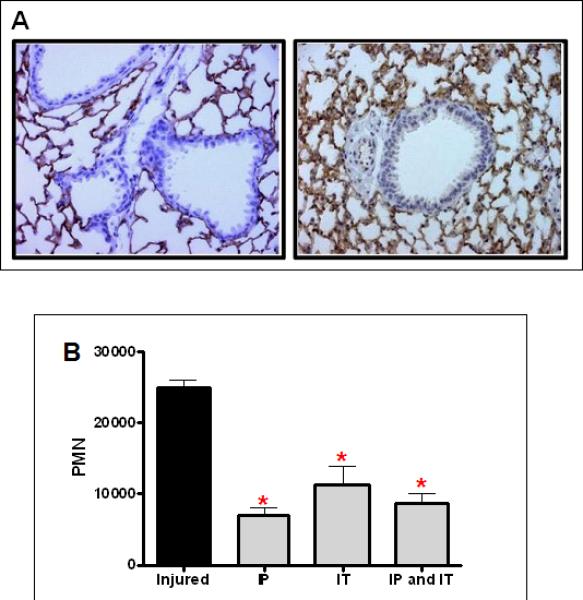
(A) Immunostaining for ICAM-1 in lungs from uninjured (left panel), and injured (right panel) animals at 3H after injury (40X magnification). Specimens are representative of immunohistochemistry for least 3 animals. (B) Mice were treated with α-ICAM-1 antibody either intraperitoneally (IP), intratracheally (IT), or both routes (IP and IT) at 30min. prior to injury. BAL was counted and differentiated as described in methods. There is a significant decrease in PMN in the BAL between injured mice with and without treatment (*p<0.001). There is no significant difference in decreased PMN level with different routes of administration. n=6 for experimental groups.
Studies administering blocking antibodies were then performed to determine whether ICAM-1 participated in neutrophil accumulation in the lung. In order to reach the alveolar space, neutrophils must leave the vascular space and traverse the pulmonary interstitial space. These steps require interaction with endothelial and epithelial cells and ICAM-1 has been shown to be involved with both of these processes15. Therefore, we administered blocking antibody intraperitoneally and intratracheally to assess the endothelial and epithelial contributions to this process, respectively (Figure 4B). Our results indicate that ICAM-1 is an active participant in neutrophil recruitment to the lung after contusion. Interestingly, the route of administration made little difference as no additive or synergistic response was seen.
PMN gp91phox expression mediates lung dysfunction after contusion
The NADPH oxidase plays an essential role in host defense and innate response to infection through the production of superoxide anion16. We tested whether NADPH oxidase also participates in the response to non infectious injury. As shown in Figure 5A, mice deficient in gp91phox showed an enhanced PMN recruitment to the injured lung. There is a marked increase in serum CXCL1 levels at 3hrs after lung injury (Figure 5B), consistent with the increased neutrophils in the BAL of gp91phox deficient mice. Despite the observed increase in PMN, lung function, as measured by PaO2/FiO2, was not significantly different from control, uninjured animals. These results suggest that oxidant injury mediated by NADPH oxidase, contributes to lung dysfunction after pulmonary contusion. Taken together, these results support the hypothesis that the mechanisms resulting in acute lung injury after lung contusion are similar to the innate immune response to infection.
Fig. 5. gp91phox dependent lung dysfunction after injury.
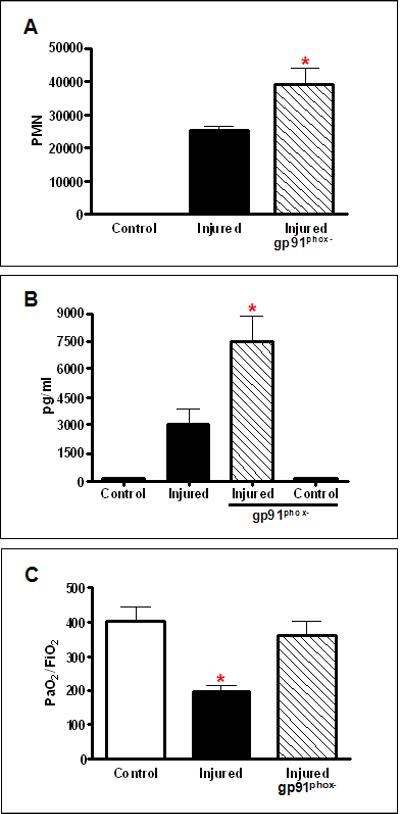
(A) BALs from injured WT (closed) and Injured gp91phox deficient (gp91phox-, crosshatched) mice were analyzed for neutrophil infiltration (PMN) at 24H. Injured gp91phox- mice show a significant increase (*p≤.01) in PMN compared with injured WT mice. (B) CXCL1 was measured in the serum from Control and Injured mice at 3H after injury. There is a significant increase (*p<0.001) in CXCL1 in the serum of Injured gp91phox deficient (gp91phox-, crosshatched) compared with Injured WT mice. Control uninjured serum CXCL1 levels are shown for comparison. (C) Blood gas analysis showed decreased PaO2:FiO2 in Injured WT mice (*p≤0.01). Injured gp91phox deficient (gp91phox-, crosshatched) showed no significant difference in PaO2:FiO2 from uninjured Control mice. n=5-6 mice for all experiments.
DISCUSSION
We found that lung injury and resultant dysfunction after contusion was dependent upon the presence of neutrophils in the alveolar space. We also demonstrate that acute lung injury and lung dysfunction is caused, at least in part, by oxidant activity associated with the presence of neutrophils in the microvasculature and alveolar space of the lung. Upon activation and arrival to the lung, neutrophils release cytotoxic substances including reactive oxygen species, eicosanoids, cationic proteins, and proteolytic enzymes6. During infection, these substances play an important role in host defense, but these same mediators potentially can damage pulmonary parenchyma giving rise to the theory that neutrophils are central to the pathogenesis of lung injury. This has been demonstrated in human and animals studies6, 15, 17. In patients with ARDS, the degree of pulmonary neutrophilia is directly proportional to mortality17. In animal models of ischemia/reperfusion, immune complex alveolitis, and endotoxin mediated lung injury, the inhibition of neutrophils and their function improves outcome, lung function, and prevents lung injury (reviewed18).
However, the remains some controversy surrounding the theory that neutrophils mediate acute lung injury. For example, acute lung injury has been demonstrated to occur after bleomycin and hyperoxic induced lung injuries in neutropenic animals and in the absence of pulmonary neutrophilia7. Enhanced apoptosis of type 2 alveolar epithelial cells was postulated to be central to the pathogenesis of lung injury. Others have shown that inhibition of Fas/FasL signaling using of siRNA, Fas deficient animals, and Fas neutralizing antibody results in reduced pulmonary inflammation in models of direct and indirect lung injury8, 19, 20. At present, we have not evaluated alveolar epithelial apoptosis in our model of lung injury. Although a direct relationship between lung dysfunction and pulmonary neutrophilia is demonstrated, it is possible that neutrophils may influence epithelial cells through cell to cell interactions or through the release of inflammatory mediators that activate intrinsic or extrinsic apoptotic pathways that ultimately result in lung injury. In the present study we show that neutrophil mediated oxidant injury is responsible, at least in part, for posttraumatic lung injury. It appears that neutrophils play a salient role as an initiator of lung physiological dysfunction.
There are four major families of chemokines, CXC, CC, C, and CXC3, that behave as potent chemotactic factors for leukocytes21. CXC chemokines are primarily chemotactic for neutrophils and have been demonstrated to be involved in neutrophil recruitment to the lung in several models of lung injury22. CXCL1 and CXCL2/3 are the principle CXC chemokines in mice and are homologues of IL-8 in man. IL-8 is perhaps the most extensively studied chemokine in man. It has been demonstrated to be elevated in the BAL in patients with ARDS, and has been independently associated with organ dysfunction, organ failure, and death after injury23. Although CXCL1 and CXCL2/3 act through a shared receptor, CXCR2, they have different receptor affinities, levels of expression and differentially effect neutrophil migration, apoptosis, respiratory burst, and phagocytosis. For example, only CXCL1 is selectively transported to the blood whereas CXCL2/3 is retained in the pulmonary compartment24. CXCL1 systemically primes circulating neutrophils to migrate to the lung in response to MIP-2. Endothelial cells, alveolar macrophages and alveolar epithelial cells produce CXC chemokines and express CXCR2 on their surface25.
In our model of lung injury, we have previously reported elevated levels of CXCL1, and suggested that CXCL1, CXCL2/3, and CXCR2 were involved in neutrophil recruitment to the lung after pulmonary contusion10, 11. The studies herein confirm and extend this observation. We also show that inhibition of CXCR2 did not completely abrogate neutrophil accumulation in the lung and that inhibition of CXCL1 or CXCL2/3 alone did not significantly affect pulmonary neutrophilia. These data support that other signaling pathways and mechanisms are involved in neutrophil recruitment to the injured lung. Candidate chemotactic mediators for neutrophils in this model include LTB4, C5a, C3a, CC chemokines (MIP-1a), and other members of the CXC chemokine family (CXCL5 and CXCL15) (reviewed26). Alveolar macrophages and type 2 alveolar epithelial cells express these mediators and may participate in the injury response to pulmonary contusion.
PMN recruitment from the blood into the lung occurs at the postcapillary venules27. The initial steps of this cascade include E-, P-, and L-selectin mediated rolling of neutrophils along the surface of the endothelial cell. Subsequent to this, firm adhesion occurs as a result of interaction between β2 integrins on the surface of the neutrophil and members of the immunoglobulin superfamily of intracellular adhesion receptors on the endothelial cell. The CD11b/18 integrin complex on neutrophils interacts primarily with ICAM-1 on the surface of the endothelial cell. The importance of this interaction in neutrophil recruitment to areas of inflammation in the lung has been demonstrated in other models of lung injury where deficiency or inhibition of either CD11b/18 or ICAM-1 reduced neutrophil migration to the lung14. ICAM-1 is involved in neutrophil movement across the endothelial cell into the interstitium and the epithelial cell into the alveolar space. Crossing the epithelial barrier has been shown to be pivotal for inducing lung injury28.
We previously demonstrated increased CD11b expression in circulating neutrophils after lung contusion and suggested that ICAM-1 was likely involved in neutrophil migration to the lung9 (and our unpublished observations). Here we show that ICAM-1 is locally expressed and actively participates in neutrophil recruitment to the lung after contusion. As either route of administration (IP or IT) was effective at reducing neutrophil infiltration, is appears that ICAM-1 expression on endothelial and epithelial cells equally participate in this process. Furthermore, similar to CXCL1 and CXCL2/3, it appears that other mediators in addition to ICAM-1 are involved in neutrophil recruitment. Neutrophil migration to the lung has been described to be mediated by CD11b/18 dependent and independent pathways depending on the stimulus. Pulmonary neutrophilia due to Gram negative bacteria or IL-1 is CD11b/18 dependent while that due to gram positive infection and C5a are CD11b/18 independent18, 29. Additionally, other members of the immunoglobulin superfamily of receptors aside from ICAM-1 such as VCAM-1 and PECAM-1 may be involved in this process30.
NADPH oxidase is indispensable to host defense response as it is the principle pathway neutrophils utilize for killing invading pathogens. As oxidant production in models of infectious and noninfectious lung injury has been shown to figure prominently in the inflammatory response, we used mice lacking functional NADPH oxidase complex to determine that oxidant production was an essential contributor to lung injury after contusion. Gp91phox- mice had improved lung function after contusion; however, oxidant production via alternative pathways such as nitric oxide synthase, molybdenum hydroxylases (xanthine oxidase), myeloperoxidase, and those generated through mitochondrial respiration cannot be entirely excluded based upon our findings. Despite improved lung function, gp91phox- mice had increased systemic levels of CXCL1 and PMN migrating into lung. Similar findings have been reported by others who have indicated that impaired oxidant production in gp91phox- mice can promote increased leukocyte infiltration to sites of inflammation with minimal affect on microvascular injury31, 32. NADPH oxidase-derived superoxide may have a regulatory role in the activation of NF-κB and various NF-κB dependent genes such as CXCL1 and CXCL2/332. In our model, a negative feedback loop might exist whereby superoxide anion normally has an inhibitory effect on CXCL1 transcription, and in its absence, CXCL1 expression is increased resulting in enhanced PMN migration to the lung. Nevertheless, our data demonstrate that neutrophils play a salient role as an initiator of lung physiological dysfunction.
In summary, we sought to determine whether: 1) pulmonary neutrophilia was responsible for reduced pulmonary function; 2) to determine the mechanisms responsible for neutrophil recruitment to the lung and 3) to determine if neutrophil oxidant activity mediates lung dysfunction after contusion. We used blocking techniques to test the effect of various mediators on neutrophil recruitment. Our results showed that neutrophil recruitment to the injured lung is dependent upon CXC chemokines, the CXCR2 receptor, and localized expression of ICAM-1. We found that pulmonary neutrophilia and oxidant production via NADPH oxidase is correlated with lung dysfunction after lung contusion.
Acknowledgments
This work was supported, in part, by the WFUSM Venture Fund, the American College of Surgeons C. James Carrico Faculty Research Fellowship, ALA RG-52711-N, GM083154 and Clowes ACS/AAST/NIGMS Mentored Clinical Scientist Award (JJH), AI057770 (EMH), RR023570 (CEM) and AI065791 (CEM, BKY).
Footnotes
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Ciesla DJ, Moore EE, Johnson JL, Burch JM, Cothren CC, Sauaia A. A 12-year prospective study of postinjury multiple organ failure: has anything changed? Arch Surg. 2005;140:432–438. doi: 10.1001/archsurg.140.5.432. [DOI] [PubMed] [Google Scholar]
- 2.Gebhard F, Kelbel MW, Strecker W, Kinzl L, Bruckner UB. Chest trauma and its impact on the release of vasoactive mediators. Shock. 1997;7:313–317. doi: 10.1097/00024382-199705000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Keel M, Ecknauer E, Stocker R, et al. Different pattern of local and systemic release of proinflammatory and anti-inflammatory mediators in severely injured patients with chest trauma. J Trauma. 1996;40:907–912. doi: 10.1097/00005373-199606000-00008. [DOI] [PubMed] [Google Scholar]
- 4.Lomas-Neira JL, Chung CS, Grutkoski PS, Miller EJ, Ayala A. CXCR2 inhibition suppresses hemorrhage-induced priming for acute lung injury in mice. J Leukoc Biol. 2004;76:58–64. doi: 10.1189/jlb.1103541. [DOI] [PubMed] [Google Scholar]
- 5.Raghavendran K, Davidson BA, Woytash JA, et al. The evolution of isolated bilateral lung contusion from blunt chest trauma in rats: cellular and cytokine responses. Shock. 2005;24:132–138. doi: 10.1097/01.shk.0000169725.80068.4a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abraham E, Carmody A, Shenkar R, Arcaroli J. Neutrophils as early immunologic effectors in hemorrhage- or endotoxemia-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1137–L1145. doi: 10.1152/ajplung.2000.279.6.L1137. [DOI] [PubMed] [Google Scholar]
- 7.Jiang D, Liang J, Fan J, et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11:1173–1179. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 8.Neff TA, Guo RF, Neff SB, et al. Relationship of acute lung inflammatory injury to Fas/FasL system. Am J Pathol. 2005;166:685–694. doi: 10.1016/S0002-9440(10)62290-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoth JJ, Stitzel JD, Gayzik FS, et al. The pathogenesis of pulmonary contusion: an open chest model in the rat. J Trauma. 2006;61:32–44. doi: 10.1097/01.ta.0000224141.69216.aa. [DOI] [PubMed] [Google Scholar]
- 10.Hoth JJ, Wells JD, Brownlee NA, et al. Toll-like receptor 4-dependent responses to lung injury in a murine model of pulmonary contusion. Shock. 2009;31:376–381. doi: 10.1097/SHK.0b013e3181862279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoth JJ, Hudson WP, Brownlee NA, et al. Toll-like receptor 2 participates in the response to lung injury in a murine model of pulmonary contusion. Shock. 2007;28:447–452. doi: 10.1097/shk.0b013e318048801a. [DOI] [PubMed] [Google Scholar]
- 12.Hayashi S, Yatsunami J, Fukuno Y, Kawashima M, Miller EJ. Antileukinate, a hexapeptide inhibitor of CXC-chemokine receptor, suppresses bleomycin-induced acute lung injury in mice. Lung. 2002;180:339–348. doi: 10.1007/s00408-002-0106-7. [DOI] [PubMed] [Google Scholar]
- 13.Guo RF, Ward PA. Mediators and regulation of neutrophil accumulation in inflammatory responses in lung: insights from the IgG immune complex model. Free Radic Biol Med. 2002;33:303–310. doi: 10.1016/s0891-5849(02)00823-7. [DOI] [PubMed] [Google Scholar]
- 14.Moreland JG, Fuhrman RM, Pruessner JA, Schwartz DA. CD11b and intercellular adhesion molecule-1 are involved in pulmonary neutrophil recruitment in lipopolysaccharide-induced airway disease. Am J Respir Cell Mol Biol. 2002;27:474–480. doi: 10.1165/rcmb.4694. [DOI] [PubMed] [Google Scholar]
- 15.Ward PA. Acute lung injury: how the lung inflammatory response works. European Respiratory Journal. 2003;22:22S–23S. doi: 10.1183/09031936.03.00000703a. [DOI] [PubMed] [Google Scholar]
- 16.Nauseef WM. Biological roles for the NOX family NADPH oxidases. J Biol Chem. 2008;283:16961–16965. doi: 10.1074/jbc.R700045200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weiland JE, Davis WB, Holter JF, Mohammed JR, Dorinsky PM, Gadek JE. Lung neutrophils in the adult respiratory distress syndrome. Clinical and pathophysiologic significance. Am Rev Respir Dis. 1986;133:218–225. doi: 10.1164/arrd.1986.133.2.218. [DOI] [PubMed] [Google Scholar]
- 18.Reutershan J, Ley K. Bench-to-bedside review: acute respiratory distress syndrome - how neutrophils migrate into the lung. Crit Care. 2004;8:453–461. doi: 10.1186/cc2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matute-Bello G, Liles WC, Frevert CW, et al. Blockade of the Fas/FasL system improves pneumococcal clearance from the lungs without preventing dissemination of bacteria to the spleen. J Infect Dis. 2005;191:596–606. doi: 10.1086/427261. [DOI] [PubMed] [Google Scholar]
- 20.Perl M, Chung CS, Lomas-Neira J, et al. Silencing of Fas, but not caspase-8, in lung epithelial cells ameliorates pulmonary apoptosis, inflammation, and neutrophil influx after hemorrhagic shock and sepsis. Am J Pathol. 2005;167:1545–1559. doi: 10.1016/S0002-9440(10)61240-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luster AD. Chemokines--chemotactic cytokines that mediate inflammation. N Engl J Med. 1998;338:436–445. doi: 10.1056/NEJM199802123380706. [DOI] [PubMed] [Google Scholar]
- 22.Puneet P, Moochhala S, Bhatia M. Chemokines in acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol. 2005;288:L3–15. doi: 10.1152/ajplung.00405.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adams JM, Hauser CJ, Livingston DH, Lavery RF, Fekete Z, Deitch EA. Early trauma polymorphonuclear neutrophil responses to chemokines are associated with development of sepsis, pneumonia, and organ failure. J Trauma. 2001;51:452–456. doi: 10.1097/00005373-200109000-00005. [DOI] [PubMed] [Google Scholar]
- 24.Quinton LJ, Nelson S, Zhang P, et al. Selective transport of cytokine-induced neutrophil chemoattractant from the lung to the blood facilitates pulmonary neutrophil recruitment. Am J Physiol Lung Cell Mol Physiol. 2004;286:L465–L472. doi: 10.1152/ajplung.00153.2003. [DOI] [PubMed] [Google Scholar]
- 25.Vanderbilt JN, Mager EM, Allen L, et al. CXC chemokines and their receptors are expressed in type II cells and upregulated following lung injury. Am J Respir Cell Mol Biol. 2003;29:661–668. doi: 10.1165/rcmb.2002-0227OC. [DOI] [PubMed] [Google Scholar]
- 26.Olson TS, Ley K. Chemokines and chemokine receptors in leukocyte trafficking. Am J Physiol Regul Integr Comp Physiol. 2002;283:R7–28. doi: 10.1152/ajpregu.00738.2001. [DOI] [PubMed] [Google Scholar]
- 27.Reutershan J, Morris MA, Burcin TL, et al. Critical role of endothelial CXCR2 in LPS-induced neutrophil migration into the lung. J Clin Invest. 2006;116:695–702. doi: 10.1172/JCI27009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Q, Park PW, Wilson CL, Parks WC. Matrilysin shedding of syndecan-1 regulates chemokine mobilization and transepithelial efflux of neutrophils in acute lung injury. Cell. 2002;111:635–646. doi: 10.1016/s0092-8674(02)01079-6. [DOI] [PubMed] [Google Scholar]
- 29.Burns AR, Smith CW, Walker DC. Unique structural features that influence neutrophil emigration into the lung. Physiol Rev. 2003;83:309–336. doi: 10.1152/physrev.00023.2002. [DOI] [PubMed] [Google Scholar]
- 30.Su WH, Chen HI, Jen CJ. Differential movements of VE-cadherin and PECAM-1 during transmigration of polymorphonuclear leukocytes through human umbilical vein endothelium. Blood. 2002;100:3597–3603. doi: 10.1182/blood-2002-01-0303. [DOI] [PubMed] [Google Scholar]
- 31.Gao XP, Standiford TJ, Rahman A, et al. Role of NADPH oxidase in the mechanism of lung neutrophil sequestration and microvessel injury induced by Gram-negative sepsis: studies in p47phox-/- and gp91phox-/- mice. J Immunol. 2002;168:3974–3982. doi: 10.4049/jimmunol.168.8.3974. [DOI] [PubMed] [Google Scholar]
- 32.Zhang WJ, Wei H, Frei B. Genetic deficiency of NADPH oxidase does not diminish, but rather enhances, LPS-induced acute inflammatory responses in vivo. Free Radic Biol Med. 2009;46:791–798. doi: 10.1016/j.freeradbiomed.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]