

Abstract
The rexinoid bexarotene represses cyclin D1 by causing its proteasomal degradation. The epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) erlotinib represses cyclin D1 via different mechanisms. We conducted a preclinical study and two clinical/translational trials (a window-of-opportunity and phase II) of bexarotene plus erlotinib. The combination repressed growth and cyclin D1 expression in cyclin-E– and KRAS/p53–driven transgenic lung cancer cells. The window-of-opportunity trial in early-stage non-small-cell lung cancer (NSCLC) patients (10 evaluable) repressed cyclin D1 (in tumor biopsies and buccal swabs) and induced necrosis and inflammatory responses including in cases with KRAS mutations. The phase II trial in heavily pre-treated, advanced NSCLC patients (40 evaluable; a median of two prior relapses per patient [range, 0–5]; 21% with prior EGFR-inhibitor therapy) produced three major clinical responses in patients with prolonged progression-free survival (583, 665, and 1460-plus days). Median overall survival was 22 weeks. Hypertriglyceridemia was associated with an increased median overall survival (P = 0.001). Early PET response did not reliably predict clinical response. The combination was generally well tolerated, with toxicities similar to those of the single agents. In conclusion, bexarotene plus erlotinib was active in KRAS-driven lung cancer cells, was biologically active in early-stage mutant-KRAS NSCLC, and was clinically active in advanced, chemotherapy-refractory mutant-KRAS tumors in this study and previous trials. Additional lung cancer therapy or prevention trials with this oral regimen are warranted.
Keywords: rexinoid, epidermal growth factor receptor-tyrosine kinase inhibitor, cyclin D1, lung cancer
Introduction
As the leading cause of cancer-related mortality in the United States (1), lung cancer needs improved treatment and effective prevention (2). Understanding the biology of lung cancer should help in developing better clinical strategies to control the disease (2,3). Uncontrolled cancer cell proliferation is associated with cell-cycle alterations (4). Cyclin D1 and cyclin E regulate the G1/S transition; prior work revealed that these cyclins are often aberrantly expressed in lung carcinogenesis (5). G1 cyclin expression is also a negative prognostic factor in lung cancer (6–8). Given these data, the cell cycle is a promising lung cancer target (9).
The epidermal growth factor receptor (EGFR) and its ligands regulate normal and neoplastic cell growth (10). Increased EGFR expression frequently occurs in lung carcinogenesis (11). The EGFR also can regulate the cell cycle since EGF induces cyclin D1 expression through distinct mechanisms (12–18). Treatment with the EGFR tyrosine kinase inhibitor (TKI) erlotinib causes G1 arrest and inhibits induction of cyclin D1 in immortalized and NNK-transformed human bronchial epithelial (HBE) and non-small cell lung cancer (NSCLC) cell lines (9,19). Erlotinib prolongs survival in some previously treated NSCLC patients (20). Lung cancers harbor activating EGFR mutations are especially clinically responsive to EGFR TKIs, but EGFR wild-type tumors, especially those with KRAS mutations, are resistant (21–24). There is a need for improved treatments of NSCLC with or without KRAS or EGFR mutations. We previously reported results from a mechanistic window-of-opportunity trial, where we obtained pre- and post-erlotinib treatment biopsies from aerodigestive tract cancers (19). Cyclin D1 expression was a biomarker of response when therapeutic intratumoral drug levels were achieved and even in tumors with wild-type EGFR (19).
Retinoids (natural and synthetic derivatives of vitamin A) exert anti-proliferative, differentiation-inducing, and pro-apoptotic effects (reviewed in ref. (25). Retinoid X receptor (RXR) agonists (rexinoids) and classical retinoic acid receptor (RAR) agonists activate distinct nuclear receptors, but can engage similar pathways (9,25). Rexinoids are able to bypass RARβ repression, a frequent NSCLC alteration, which contributes to resistance to classical retinoids that activate RARs (2,25). In premalignant HBE cells and in some lung cancer cell lines, the rexinoids can inhibit growth and reduce EGFR, phospho-EGFR, and cyclin D1 expression (25–30). Bexarotene causes cyclin D1 repression via proteasomal degradation of cyclin D1 that confers G1 arrest (26,27). A bexarotene window-of-opportunity trial in resected NSCLC cases (30) established a non-linear correlation between plasma and tumor concentrations of bexarotene (30). Repression of cyclin D1, EGFR, phospho-EGFR, and proliferation was associated with high intratumoral levels of bexarotene (30). These and other findings indicate that cyclin D1 is both a biomarker and therapeutic target (9,25). These data also suggest that a combination regimen that confers cyclin D1 repression would have activity against lung cancer.
Furthermore, cyclin D1 levels were much higher in NSCLC cell lines harboring EGFR mutations than in NSCLC cell lines expressing wild-type EGFR and mutant KRAS (31). This finding implied a link between high cyclin D1 levels and reliance on the EGFR pathway. Other pathways could cause high cyclin D1 expression, suggesting the promise of combining EGFR inhibition with a rexinoid that targets cyclin D1 for repression via a separate pathway. Cooperation between retinoid and EGFR pathways in vitro was reported (28,32). Based on this cooperation, we conducted a phase I clinical trial combining erlotinib with bexarotene for advanced aerodigestive tract tumors (33). The drugs were safely combined clinically at full single-agent doses. Cyclin D1 was repressed in post-treatment versus pre-treatment buccal swabs of most patients on this trial, confirming the expected biomarker response in surrogate tissues (33). Encouraging clinical results included activity in patients unlikely to benefit from erlotinib alone because their lung cancers lacked activating EGFR mutations (33). Trial patients who had major objective responses included non-Asian men and smokers (both of whom typically do not respond well to EGFR TKIs (33).
The results we report here come from new studies of bexarotene plus erlotinib in transgenic lung cancer cell lines driven by some of the same changes that are linked to clinical lung cancer formation (e.g., KRAS mutations) and from two clinical/translational trials of bexarotene plus erlotinib, one a window-of-opportunity trial in patients with early-stage NSCLC scheduled for resection, the other a phase II trial in patients with advanced-stage, refractory NSCLC. Clinical/translational endpoints included changes in cyclin D1 expression and other biomarkers (total EGFR, phospho-EGFR, and Ki-67 expression) in pre-treatment versus post-treatment biopsies; correlations of these changes with tumor EGFR or KRAS mutations, necrosis or inflammation in post-treatment versus pre-treatment biopsies; cyclin D1 expression in post-treatment versus pre-treatment buccal swabs (window trial); and clinical tumor response rate (including data from early positron emission tomography [PET]), overall and progression-free survival, and toxicity (including hypertriglyceridemia and rash; phase II trial).
Materials and Methods
Cell culture, proliferation assays, and transgenic models
The ED1 (cyclin Ehigh) cell line was derived from a transgenic mouse that developed lung cancer after expressing wild-type cyclin E under control of the human surfactant C promoter (34,35). The 393P lung cancer cell line was derived from KrasLA1/+; p53R172HΔG/+ transgenic mice (36). ED1 and 393P cells were each cultured in RPMI 1640 media supplemented with 10% fetal bovine serum (FBS) and 1% antibiotic and antimycotic solution at 37°C in 5% CO2 and in a humidified incubator. For proliferation assays, ED1 (4.5 × 103) and 393P (5 × 103) cells were independently seeded per well of six-well tissue culture plates in triplicates and treated 24 hours later with the indicated agents. Triplicate replicate experiments were performed. Logarithmically growing cells were assayed 72 hours post-treatment using the CellTiter-Glo assay (Promega) and established methods (19). Erlotinib (Genentech Inc, South San Francisco, CA; OSI Pharmaceuticals, Melville, NY) and bexarotene (Ligand, Inc., San Diego, CA; Eisai Pharmaceuticals, New York, NY) were each dissolved in the vehicle, dimethyl sulfoxide (DMSO). Transgenic mice were sacrificed following an Institutional Animal Care and Use Committee (IACUC)-approved protocol. Harvested lung tissues were formalin-fixed, paraffin-embedded, and sectioned for hematoxylin and eosin (H & E) and immunohistochemical staining, as before (34).
Immunoblot and immunohistochemical assays
ED1 and 393P cells were independently treated with erlotinibalone, bexarotene alone, combinations of these agents, orvehicle alone. In brief, cells were plated at the same respective densities as used in the proliferation assays. Cells were harvested 72 hours post-treatments and subjected to immunoblotanalyses, as before (19). Antibodies werepurchased that recognized cyclin D1 (M-20; Santa Cruz Biotechnology, Santa Cruz, CA) or actin (C-11; Santa Cruz Biotechnology). Immunoblot assays assessed cyclin D1 and actin expression (33,34). H & E staining was performed as in prior work (30,34). Immunohistochemical assays for cyclin D1, ki-67, total EGFR and phosho-EGFR were each used (19,30,34) and scored (as was the histopathology) by a pathologist (V.M.), who was unaware of treatment assignments. The percent change in expression of each biomarker was scored. The percentages of tumor volumes with necrosis in post-treatment versus pre-treatment biopsies were provided. The scoring system used for assessments of acute (neutrophilic infiltrate) or chronic (lymphoplasmacytic infiltrate) inflammatory responses were: (0) no inflammation; (1+) 1–10% of tumor volume; (2+) 11–20% of tumor volume; and (3+) >20% of tumor volume.
Patients
Eligible patients had a histologic or cytologic diagnosis of NSCLC. For the advanced stage phase II trial, eligibility included NSCLC stage IV or stage IIIB with malignant effusion, without curative treatment options, Karnofsky performance status of 60 or greater, and age older than 18 years. Prior chemotherapy or radiotherapy was allowed. Fasting triglycerides had to be below the upper limit of normal (ULN). For the window of opportunity trial, eligible patients had a pathologic diagnosis of NSCLC (with at least 5 unstained slides required from the pre-treatment biopsy), clinical stages I, II or IIIA, were older than 18 years and were candidates for resection. Prior chemotherapy or radiotherapy was not allowed. Effective contraception or sexual abstinence was required for female patients of childbearing potential or male patients with female partners of childbearing potential.
Exclusion criteria were hepatic dysfunction with either bilirubin greater than the ULN, or transaminase (AST or ALT) greater than 2.5 times ULN or more than 5 times ULN if there were known liver metastases; renal dysfunction (creatinine clearance < 30 mL/min); and a serious uncontrolled medical disorder, or active infection, which would impair ability to receive study treatment. Patients with dementia or altered mental status prohibiting understanding or rendering of informed consent and compliance with the protocol were excluded. Prior therapeutic use of bexarotene or concurrent use of other anti-cancer approved or investigational agents were each not allowed. Previous treatment with other EGFR inhibitors was permitted.
Inclusion and exclusion criteria were assessed within 14 days before initiation of therapy with the exception of radiographic studies, which were performed within 28 days of screening. These clinical trials were conducted after approval by the Committee for the Protection of Human Subjects at Dartmouth College and the Institutional Review Board. Informed consent was obtained from each patient before enrollment onto these studies.
Study drugs
An IND application for the combination of erlotinib and bexarotene was submitted to the US Food and Drug Administration (FDA) and was granted to the Principal Investigator (K.H.D.). Bexarotene capsules 400 mg/m2/day orally and erlotinib 150 mg orally were taken at the same time daily. These doses were established from a phase I study (33). Treatment continued until progression of disease, unacceptable adverse effects, or withdrawal of informed consent. Atorvastatin was started for abnormal elevated fasting triglycerides or cholesterol levels. Dose modifications were allowed for toxicities from each agent. For triglyceride levels 400 – 800 mg/dL, the statin dose was increased up to the maximum as per the package insert of the agent, followed by the addition of fenofibrate. Bexarotene dose was reduced for a fasting serum triglyceride level > 800 mg/dL. Bexarotene dose suspensions were made for fasting serum triglyceride levels >1200 mg/dL. Once triglyceride levels fell below 800 mg/dL, bexarotene was restarted, provided the interruption was < 21 days.
Clinical evaluations
Patient evaluations for toxicities included physical examination and laboratory studies (CBC, comprehensive metabolic profile, and lipid profile) performed bi-weekly during the first month on therapy, then monthly while on treatment, along with monthly assessment of thyroid function (TSH, T3Q, and T4). Additional tests were as clinically indicated. Radiographic evaluations with chest computed tomography (CT) were performed every 8 weeks; responses were assessed using the RECIST criteria. For a subset of patients, a PET-CT scan was performed within 4 weeks before treatment initiation, at 10 (+/− 2) days and at 8 weeks of study therapy. PET response was based on the European Organization for Research and Treatment of Cancer criteria for PET metabolic response. An assessment was made between early response by PET and radiographic response at 2 months.
In the window-of-opportunity trial, patients received bexarotene 400 mg/m2/day orally and erlotinib 150 mg as single daily oral doses for 7 to 9 days before surgical resection and on the day of surgery. No dose modifications were permitted. Hypolipidemic therapy was not administered.
Buccal mucosal specimens
In the window of opportunity trial, buccal specimens were harvested on day 1 (before treatment) and on the day of surgery by swabbing the buccal mucosa, as previously described (33) except for lysis performed with the Laemmli sample buffer (BioRad, Hercules, CA). Immunoblot analyses were performed with densitometry to determine changes in cyclin D1 expression relative to actin expression as a loading control and to confirm integrity of the protein.
EGFR and KRAS mutational analyses
Genomic DNA was isolated from paraffin-embedded tissue sections of NSCLCs using the Gentra PureGene Blood Kit Plus (Qiagen, Valencia, CA) and the manufacturer’s recommended procedures. Samples were screened for mutations in EGFR exons 19 and 21 using polymerase chain reaction (PCR) and restriction digestion followed by capillary electrophoresis. The seven most common KRAS mutations were detected using allelic discrimination probes with real time PCR. Sanger sequencing confirmed the results, as in prior work (19,22,23,33,37,38).
Statistical analyses
In vitro changes in growth and in cyclin D1 expression were assessed using the two-sample t-test with significance defined as a two-sided P < 0.05 using Microsoft Excel software. Time to progression and overall survival were determined using the Kaplan and Meier method. The prespecified endpoints of response to treatment in the window of opportunity trial were defined as a 50% decrease in cyclin D1, EGFR, phospho-EGFR, or Ki-67 immunohistochemical expression (post-treatment versus pre-treatment specimens). The prespecified number of NSCLC cases for the window of opportunity trial was 12. No observed responses observed in 12 patients would have excluded the possibility of a response rate higher than 20% with a P value < 0.05 (39). The prespecified primary endpoint of the advanced stage phase II trial was radiographic response rates (including PET responses) of the combination using RECIST criteria. The prespecified secondary endpoints were median survival, time to progression, toxicities, and EGFR mutational analysis of the responding cases. The planned sample size for the advanced stage NSCLC trial was 40. No observed responses in 40 patients would have excluded the probability of a response rate of 7% or higher with a P value < 0.05 (39). Associations between survival and triglyceride levels and rash were independently determined using the log-rank test performed with STATA statistical suite version 10.0 (Stata Corp, College Station, TX). Significance was defined as a P < 0.05.
Results
Preclinical results
KRAS mutations at codons 12, 13, or 61 were not detected in DNA sequence analysis of genomic DNA isolated from ED1 cells (data not shown), confirming that ED1 cells lacked KRAS activation. Drug dosages were chosen for each cell line to search for cooperative effects when bexarotene was combined with erlotinib. Significant growth repression occurred in both ED1 and 393P lung cancer cell lines after combining bexarotene with erlotinib (Fig. 1A). This repression was associated with cooperative repression of cyclin D1 protein expression in both ED1 and 393P cells (Fig. 1B; representative immunoblots). Figure 1C displays the intensities of the signals shown in Figure 1B. Results from three independent replicate experiments were pooled. These pooled experiments confirmed the representative results shown in Figure 1B and established a statistically significant repression of cyclin D1 (data not shown).
Figure 1.

Growth response and induced changes in cyclin D1 immunoblot expression following individual or combined bexarotene and erlotinib treatments of genetically-defined murine lung cancer cells. (A) Individual and combined growth response of murine lung cancer cell lines driven by cyclin E (ED1 cells, cyclin Ehigh) or by KRAS/p53 (393P cells, K-rasLA1/+/p53R172HΔG/+) when independently treated with the indicated dosages of bexarotene, erlotinib, or the combination. (B) Immunoblot changes in expression of cyclin D1 (relative to actin as a loading control) of a representative experiment following treatment with bexarotene, erlotinib, or the combination. (C) The intensities of the signals in panel B (relative to actin) were determined and displayed in this panel showing cooperative inhibitory effects on cyclin D1 expression. The symbols * and ** refer respectively to statistically significant changes, P < 0.05 and P < 0.01.
The ED1 cell line was derived from transgenic mice expressing in the lung wild-type cyclin E; these mice develop pre-malignant and malignant lung lesions that recapitulate many changes found in human lung carcinogenesis (34, 35). Whether cyclin D1 was aberrantly expressed in this murine lung carcinogenesis model was examined because cyclin D1 expression is deregulated in human lung carcinogenesis (5). Cyclin D1 immunohistochemical expression increased during the progression of pre-malignant lung lesions to invasive or metastatic lung lesions in this murine transgenic model expressing wild-type cyclin E in the lung (Supplemental Fig. 1). This finding is consistent with a role for cyclin D1 as a biomarker and/or pharmacologic target for lung carcinogenesis. Clinical trials were conducted to explore these possibilities.
Window-of-opportunity clinical trial
Patient characteristics
We enrolled 14 early-stage lung cancer patients into this trial; 12 received study treatment, 11 underwent lung cancer resections between December 23, 2005, and June 16, 2009, and 10 were evaluable. Important features of these 10 patients included the following characteristics (Table 1).
Table 1.
Patient histopathology and biomarker characteristics in the window-of-opportunity clinical trial
| Patients | Age | Gender | Smoking Status | Histology | EGFR | KRAS | % Reduction* |
Necrosis % | Chronic Inflammation† | Acute Inflammation† | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cyclin D1 | EGFR | pEGFR | ||||||||||
| 1 | 63 | Female | Current | Squamous cell carcinoma | wt | wt | 65 | 30 | 90 | 5‡ | 1+ | 1+ |
| 2 | 52 | Male | Never | Adenocarcinoma | exon 21 | wt | 40 | 30 | 0 | 30 | 2+ | 2+ |
| 3 | 68 | Female | Former | Adenosquamous carcinoma | wt | mut | 0 | 0 | 0 | 0 | 0 | 0 |
| 4 | 71 | Female | Former | Adenocarcinoma | wt | wt | 0 | 0 | 100 | 10 | 3+§ | 3+ |
| 5 | 61 | Female | Former | Adenocarcinoma | wt | mut | 80 | 0 | 0 | 40 | 1+ | 3+ |
| 6 | 59 | Female | Current | Bronchioloalveolar carcinoma | wt | mut | 50 | 0 | 95 | 0 | 0 | 0 |
| 7 | 52 | Female | Current | Adenocarcinoma | wt | mut | 0 | 0 | 0 | 40 | 3+ | 3+ |
| 8 | 63 | Male | Former | Basaloid carcinoma | wt | wt | 0 | 0 | 100 | 30 | 1+ | 3+ |
| 9 | 61 | Female | Former | Adenocarcinoma | wt | mut | 40 | 65 | 80 | 20 | 3+ | 1+ |
| 10 | 63 | Male | Former | Adenocarcinoma | NA | NA | 80 | 0 | 75 | 5 | 2+ | 0 |
Abbreviations: EGFR, epidermal growth factor receptor; pEGFR, phosphorylated EGFR, wt, wild-type, mut, mutant and NA, not assessed.
The indicated percentages reflect the proportion of biopsies with the indicated changes in the post-treatment versus pre-treatment biopsies.
The scoring system used to assess chronic and acute inflammatory changes in biopsies is described in Materials and Methods.
Minimal necrosis (5%) without change in the post-treatment versus pre-treatment biopsy.
Chronic inflammation (3+) without change in the post-treatment versus pre-treatment biopsy.
Histopathological, biomarker, and molecular genetic results in tumors
Ten patients had adequate pre-treatment and post-treatment tissues harvested for analyses of histopathological and biomarker responses and molecular genetic alterations (Table 1). Of these 10 patients, 1 had an activating EGFR mutation at exon 21, and 5 had KRAS codon 12 mutations in their lung cancers; 8 had decreased cyclin D1, total EGFR, or phospho-EGFR immunohistochemical expression in post-treatment versus pre-treatment biopsies; and 6 had reduced cyclin D1 expression, 6 had a decline in phospho-EGFR expression, and 3 had reduced EGFR expression in immunohistochemical assays Basal Ki-67 immunohistochemical expression was reduced in these cases making comparisons of changes in pre-treatment versus post-treatment biopsies inconclusive (data not shown). Of note, 8 of 10 post-treatment biopsies exhibited necrosis and evidence of chronic or acute inflammatory responses (Table 1). A single biopsy pair (patient 1) showed minimal necrosis (5%) without change in the post-treatment versus pre-treatment biopsy. No pre-treatment biopsies displayed acute inflammatory responses (data not shown). A single case (patient 4) had evidence of chronic inflammation in the pre-treatment biopsy, but no change in this degree of inflammation was found in the post-treatment biopsy.
Biomarker responses were analyzed for associations with EGFR and KRAS mutations (Table 1). A decline of cyclin D1 and EGFR immunohistochemical expression was detected in the NSCLC case with an activating EGFR mutation. Of the 5 cases with KRAS mutations, 3 had a decrease in cyclin D1, 1 had reduced EGFR, and 2 had repressed phospho-EGFR immunohistochemical expression in post-treatment versus pre-treatment biopsies. This high proportion of cases with biomarker and histopathological changes (necrosis, chronic or acute inflammation) after combined bexarotene and erlotinib treatments exceeded the proportion in our prior window-of-opportunity trials of each agent alone (19, 30). Of note, these responses occurred whether or not EGFR or KRAS mutations were detected in the NSCLCs (Table 1). Repression of cyclin D1, total EGFR, and phospho-EGFR expression is shown in representative post-treatment versus pre-treatment tumor biopsies (Fig. 2A). As shown in Fig. 2B, no histopathological evidence of necrosis (left panel) or evidence of necrosis (arrow, middle panel) or inflammatory responses (right panel, arrows) is apparent in representative photomicrographs of post-treatment lung cancer biopsies with these features.
Figure 2.
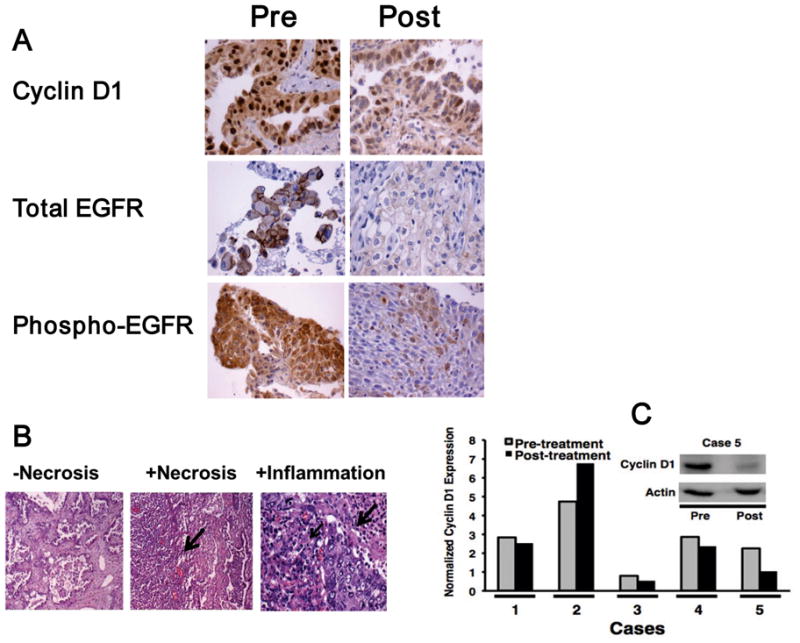
Biomarker, histopathological and buccal swab treatment effects in the window of opportunity trial combining bexarotene with erlotinib. (A) Reduced immunohistochemical expression of cyclin D1 (patient 5), total EGFR (patient 9), and phospho-EGFR (patient 1) in post-treatment “Post” versus pre-treatment “Pre” lung cancer biopsies (200×). (B) No evidence of necrosis (−Necrosis, left panel), presence of necrosis (+Necrosis, arrow, middle panel) and inflammatory response (+inflammation, small arrow shows chronic inflammatory infiltrate and large arrow shows acute inflammatory infiltrate surrounding a lung cancer, right panel) in representative post-treatment lung cancer biopsies (200×). (C) Cyclin D1 immunoblot expression in pre-treatment versus post-treatment buccal mucosal swabs. Normalized cyclin D1 protein signals (relative to actin expression) declined in the majority of the displayed post-treatment versus pre-treatment buccal mucosal swabs. The cyclin D1 immunoblot is shown for a representative case (case 5) relative to actin expression as a loading and protein integrity control.
Cyclin D1 expression in buccal swabs
Cyclin D1 also was assessed by immunoblot analyses of pre-treatment (day 1) versus paired post-treatment (day of surgery) buccal swabs. Specimens were obtained from 6 patients, and 5 swabs were evaluable based on adequate actin expression. Normalized cyclin D1 protein expression was repressed in 4 post-treatment versus pre-treatment swabs (Fig. 2C). The window of opportunity trial was well tolerated and without clinical toxicity.
Phase II clinical trial
Forty-two patients were enrolled into the advanced-stage phase II trial, of which 40 received study treatment (between October 13, 2005, and February 14, 2008). Clinical characteristics of these patients (Table 2) include the following findings: 52% women; 67% with adenocarcinoma histopathology; median age of 67 years (range, 46–77); 14% current smokers and 17% never smokers; a median of 2 prior chemotherapy regimens per patient (range, 0–5); and 21% had prior EGFR inhibitor therapy.
Table 2.
Clinical characteristics of patients in the phase II clinical trial (N = 42, N = 40 treated)
| Median age | 67 years (range, 46–77) |
| Gender | 22 Females (52%), 20 males (48%) |
| Race | 42 White non-Hispanic (100%) |
| Disease stage | 42 Stage IV (100%) |
| Tumor histology | 28 Adenocarcinomas (67%) 5 Bronchioloalvealor type (12%) 2 Squamous cell carcinomas (5%) 10 Non-small cell carcinomas not otherwise specified (24%) 1 Mixed histology (2%) 1 Non-small cell carcinoma with sarcomatoid features (2%) |
| Prior anti-EGFR therapy | 9 (21%) |
| Prior chemotherapies | 2 (median; range 0–5) |
| Smoking status | 7 Never (17%), 6 current (14%), 29 former (69%) |
Radiographic and metabolic responses
We evaluated radiographic responses in 19 patients, who remained on study treatment for two or more months. There were 2 objective responses (1 complete and 1 partial) at two months and a third objective response (partial) after two months (15.8% response rate; 95% confidence interval, 3.4%–40%). Six patients had stable disease, including 1 patient with prior gefitinib therapy (35 weeks on study). Fluorodeoxyglucose (FDG) PET-CT scans were performed at baseline, at 8–12 days, and at 2 months of therapy in 14 patients with clinical characteristics similar to those of the entire trial cohort (data not shown). Radiographic responses at 2 months in these patients were as follows: One complete response (early PET [at 8–12 days] showing a metabolic response), 1 partial response (early PET showing metabolic progression), 2 patients with stable disease (early PET showing stable disease in 1 and progression in 1), and 10 patients with disease progressions (early PET showing stable disease in 5 and progression in 5).
Time to progression and overall survival
Patients received a median of 43 days of therapy (range, 5–1460-plus days) and were assessed for survival in an intent-to-treat analysis. Median time to progression was 7 weeks. Median overall survival (Fig. 3A) for all analyzed patients was 22 weeks (range, 1–274-plus weeks); 22 weeks (range, 2–274-plus weeks) for the 36 patients who received more than 2 weeks of therapy; and 23 weeks (range, 4–274-plus weeks) for the 26 patients who received study treatment for at least one month.
Figure 3.

Kaplan-Meier survival estimates and effects of hypertriglyceridemia or rash on survival in the phase II bexarotene and erlotinib lung cancer trial. (A) Overall survival. (B) Improved survival after appearance of hypertriglyceridemia (P = 0.001) or (C) rash (P = 0.03).
Association of survival with triglyceridemia
Patients who developed hypertriglyceridemia in the first 4 weeks of erlotinib plus bexarotene treatment (without anti-lipid treatment) had an increased median overall survival (24 weeks) versus patients who did not (16 weeks). There was a statistically significant association between triglyceride levels and increased overall survival (P = 0.001; Fig. 3B).
Association between survival, skin rash, and clinical characteristics
Patients who developed a skin rash during therapy with erlotinib plus bexarotene had an increased median overall survival (25 weeks) versus patients who did not (14 weeks; P = 0.03; Fig. 3C). Combined effects of hypertriglyceridemia and rash on survival could not be assessed because of the small number of cases with normal triglycerides that did not develop rash (3 patients) versus patients with hypertriglyceridemia who developed rash (18 patients). There was no statistically significant association between triglyceride levels and the presence or absence of rash. Patients with adenocarcinoma had a longer median overall survival (22 weeks) versus patients with other histopathologic diagnoses (7 weeks; P = 0.02). There was no association between survival and smoking status, number of prior chemotherapies, gender, prior anti-EGFR therapy, cholesterol level, presence of diarrhea, or LDH level. Patients with anemia or low albumin had decreased survival.
EGFR and KRAS mutations
We analyzed tumor samples from the three cases (two females, one male) having the longest progression-free survival (PFS). The longest PFS was 1460-plus days, which occurred for a female patient who was a never smoker, had adenocarcinoma treated with second-line therapy, had no prior EGFR inhibitor treatment, had a partial response, and is still receiving bexarotene plus erlotinib treatment for NSCLC with wild-type EGFR and wild-type KRAS. The other female patient of this subgroup had a complete response and PFS of 583 days; she was a never smoker, had first-line therapy for bronchioloalveolar carcinoma, and had an activating EGFR mutation at exon 21 without a KRAS mutation in her NSCLC. She recurred after 583 days, was biopsied at this point, and did not have the resistance mutation EGFR T790M. The male patient had a partial response and PFS of 665 days; he was a former smoker, had no prior EGFR inhibitor treatment, and had two prior therapies for adenocarcinoma with wild-type EGFR and mutant KRAS. This patient never progressed and died of a cause unrelated to lung cancer.
Toxicity
Clinical toxicities and laboratory abnormalities are summarized in Table 3. Most patients had hypertriglyceridemia, as expected after bexarotene treatment. There were no pancreatitis cases. Skin toxicity, manifested as dry skin, non-specific rash or acneiform rash, was common, generally mild, and did not require treatment interruption. There were no cases of severe diarrhea or interstitial lung disease. No cumulative toxicities were noted. Four patients died on study: none with evidence of lung cancer progression, two of cardiac causes, and two of sepsis/multiorgan failure. Treatment was discontinued for grade 3 pulmonary hemorrhage (1 patient), mouth sores (1 patient), cough (1 patient), and abdominal pain (1 patient). One patient, who had one prior chemotherapy regimen, developed eosinophilia and fatigue at 2 weeks of treatment on bexarotene plus erlotinib. These symptoms did not improve when treatment was interrupted; the patient discontinued study participation and was diagnosed with a hypereosinophilic syndrome within a month of stopping study treatment. This syndrome did not respond to therapy, including imatinib. This patient had received erlotinib plus bexarotene six months previously in the window-of-opportunity trial without adverse events and with evidence of a biomarker response (decreased cyclin D1 protein) in the resected lung cancer, which had mutant KRAS. Six patients had bexarotene dose reductions (4 for hypertriglyceridemia and 2 for headache). Two patients had erlotinib dose reductions for rash.
Table 3.
Adverse events
| Adverse Event | Grade 1/2 (%) | Grade 3/4 (%) |
|---|---|---|
| Rash | 55 | 6 |
| Triglycerides | 74 | 13 |
| Diarrhea | 31 | 0 |
| Cholesterol | 32 | 0 |
| Thyroxine (T4) | 38 | 0 |
| T3 elevated | 60 | 0 |
| ALT | 27 | 3 |
| AST | 30 | 3 |
| Alkaline phosphatase | 24 | 0 |
| Total bilirubin | 8 | 3 |
| Leukopenia | 14 | 3 |
| Hypokalemia | 5 | 0 |
| Hyponatremia | 11 | 0 |
| Eosinophilia | 0 | 3 |
| Hemoptysis | NA | 10 |
| Dyspnea | NA | 10 |
| Pneumonia (post-obstructive) | NA | 10 (8) |
| Pleural effusion | NA | 8 |
| Confusion | NA | 10 |
| Abdominal pain | NA | 3 |
| Acute renal failure | NA | 3 |
Abbreviations: ALT, aspartate aminotransferase; AST, alanine aminotransferase; NA, not assessed.
Discussion
The three closely inter-related studies we report here--a preclinical study comprising several analyses in transgenic models and two clinical/translational trials--enhanced our understanding of using bexarotene plus erlotinib for treating and, potentially, preventing lung cancer. The combination repressed growth and cyclin D1 expression in cyclin-E– and KRAS-mutation–driven transgenic non-small-cell lung cancer (NSCLC) cells. The window-of-opportunity trial in early-stage NSCLC patients repressed cyclin D1 and induced necrosis and inflammatory responses including in patients with KRAS mutations. The phase II trial in heavily pre-treated, advanced NSCLC patients produced major clinical responses (including of tumors with KRAS or EGFR mutations) with prolonged survival in three patients. Hypertriglyceridemia or rash significantly increased median overall survival. In summary, bexarotene plus erlotinib was active in KRAS-driven NSCLC cells, was biologically active in early-stage mutant-KRAS NSCLC, and was clinically active in advanced, chemotherapy-refractory mutant-KRAS tumors in this study and previous trials including the recently reported BATTLE trial (40), which was reported after submission of this study.
Derived from transgenic mice with lung cancers driven by high levels of cyclin E or by mutant KRAS/p53, the genetically defined murine lung cancer cell lines studied recapitulate common changes found in human lung cancers. Transgenic cyclin E expression also conferred aberrant expression of cyclin D1 in the lung carcinogenesis lesions that arose in these mice. This observation mimics aberrant cyclin D1 expression found in human lung carcinogenesis (5–9). We examined individual and combined treatment effects of bexarotene plus erlotinib on cyclin D1 expression and growth in these lines. Intriguingly, erlotinib plus bexarotene was significantly more active than erlotinib (or bexarotene) alone in lung cancer (19,30,33). When coupled with the novel findings reported here in the KRAS-driven transgenic lung cancer model, this is a clinically-relevant result. Responsive lung cancers to this regimen included those with KRAS mutations. Thus, this regimen appeared to broaden the clinical activity of this EGFR-TKI.
Whether the clinical activity of bexarotene plus erlotinib is associated with a decline in cyclin D1 expression (or with histopathological changes) in NSCLC with or without EGFR or KRAS mutations has not been addressed previously. The EGFR TKI erlotinib is active against advanced-stage NSCLC and prolongs survival in patients, who relapse after chemotherapy, especially when rash occurs (20,21). Bexarotene treatment also can produce clinical responses in advanced-stage lung cancer (30). An early trial of bexarotene plus chemotherapy was encouraging (41). Subsequent work did not show an overall survival benefit with this regimen (42), but did show a survival advantage for patients who developed hypertriglyceridemia (42).
The studies reported here now address the link between cyclin D1 expression and effects of bexarotene plus erlotinib. The combination repressed cyclin D1 expression and induced necrotic and inflammatory changes in tumors in the window-of-opportunity trial. Cyclin D1 repression occurred in tumors with or without EGFR or KRAS mutations (Table 1). The biologic activity of the combination in this trial was better than that of a previous trial of bexarotene alone in similar patients (30), where, in addition, necrosis induction did not occur. Patients with advanced stage NSCLC had objective responses in the phase II trial. Our phase II study did not include cyclin D1 analyses, but the recently reported BATTLE trial including an arm of bexarotene plus erlotinib in similar patients did (40). The BATTLE trial found that the combination was active in mutant-KRAS tumors and that cyclin D1 overexpression was predictive of its clinical activity (40). The clinical activity of the same regimen that was reported here was significantly associated with hypertriglyceridemia or skin rash (Fig. 3). Toxicities in the phase II trial were similar to those previously reported for single-agent bexarotene or erlotinib (19,30). The objective responses to bexarotene plus erlotinib in heavily pre-treated NSCLC patients are notable because of the rarity of clinical responses in patients with multiple prior relapses after chemotherapy (43, 44).
Prior studies showed that early FDG-PET can monitor tyrosine kinase inhibitor responses (45,46). In our phase II trial, an early decline in FDG-PET activity predicted radiographic response in some cases, but we did not find an association between unchanged or increased FDG-PET activity and radiographic response at two months. The early increase in metabolic activity detected in a partial responder was likely a flare that was indicative of this clinical response. A larger cohort of responding cases is needed to define precisely the relationship between changes in early FDG-PET activity and response of NSCLC to bexarotene plus erlotinib.
Treatment was third line or higher for many of the NSCLC patients entered onto the phase II trial. Based on historical controls, the expected overall survival in similar patients treated with chemotherapy is four or fewer months (43, 44). With a median overall survival of 5.5 months for all treated patients and of 6 or more months for patients who developed hypertriglyceridemia or rash (Fig. 3), our present phase II trial provides evidence for the clinical activity of bexarotene plus erlotinib. The combined results from this and our prior phase I trial of the same regimen (33) show that patients with elevated triglycerides had a median survival of 24 weeks (range, 1–274-plus) versus 21 weeks (range, 4–72) for patients with normal triglycerides (P = 0.008). Future work should explore the mechanistic basis for this beneficial association.
The lung cancer patients in the phase II trial with the longest PFS and overall survival had features that differed intriguingly from patients who responded in prior NSCLC EGFR-TKI trials, where EGFR-TKI treatment increased survival of patients with activating EGFR mutations in their NSCLC (22,23,47). Of the three longest survivors in our present trial, only one had an activating EGFR mutation in the tumor. Another of the longest survivors had NSCLC with a KRAS mutation, which is linked to a poor response to EGFR inhibition in lung and many other cancers (24,47,48). The third longest-term survivor had neither an EGFR nor a KRAS mutation in the lung tumor. With these collective findings of activity in NSCLC with or without EGFR or KRAS mutations, bexarotene plus erlotinib potentially can address the medical need of NSCLC patients with KRAS mutations and wild-type EGFR, whose need is not met by EGFR inhibitors or other chemotherapies.
Our recent work revealed that cyclin-dependent kinase-2 (CDK-2) inhibition is active against NSCLC cells even with RAS mutations (49). Perhaps incorporating a CDK-2 inhibitor into a cyclin D1–targeting regimen would enhance clinical activity against lung cancer including those with KRAS mutations. Future clinical work should definitively examine whether combining bexarotene with erlotinib has activity against KRAS-mutant lung cancer.
Often deregulated in lung carcinogenesis, cyclin D1 and EGFR are targets for lung cancer therapy and chemoprevention (9,25). Erlotinib and bexarotene are taken orally, tolerable in combination, and thus attractive candidates for clinical lung cancer chemoprevention within optimized populations. Before launching large, costly clinical cancer chemoprevention trials of the combination, however, investigators need to confirm that pathways engaged in vitro are also activated in relevant in vivo models. Relevant pathway activation has been seen in mechanistic studies of the effects of EGFR-TKI or rexinoid treatments in preventing carcinogen-induced lung cancer in mice (50). These findings should be extended to use of this combination regimen in clinically predictive mouse lung cancers models. Activity in these models would provide a further rationale to use the same regimen for clinical lung cancer chemoprevention in the optimal patient population.
The encouraging activity of this regimen includes a) suppression of cyclin D1 and cell growth in KRAS-driven lung cancer cells, b) suppressed cyclin D1 expression and induced necrosis in short-term therapy of early-stage NSCLC (including patients with KRAS mutations), and c) activity in advanced chemotherapy-refractory NSCLC in our prior phase I trial, current phase 2 trial, and the recently reported BATTLE trial. These translational research results warrant further study of bexarotene plus erlotinib for lung cancer therapy or chemoprevention.
Supplementary Material
Acknowledgments
Grant Support
This work was supported by Community Clinical Oncology Program (CCOP) grant CA37447-21 (K.H.D.), by the National Institutes of Health and the National Cancer Institutes grants RO1-CA087546 (E.D.) and R01-CA111422 (E.D.), a Samuel Waxman Cancer Research Foundation award (E.D.), an American Lung Association grant (X.L.), and a Prouty Grant from the Norris Cotton Cancer Center (E.D.). A.M. Busch was supported by a National Research Service Award from the National Institutes of Health (T32-CA009658). Additional funding was provided by Ligand, Eisai (Woodcliff Lake, NJ) and Genentech (K.H.D.). E. Dmitrovsky is an American Cancer Society Clinical Research Professor supported by a generous grant from the F.M. Kirby Foundation.
Footnotes
Note: Presented in part at the 102nd Annual Meeting of AACR, Orlando, April 2–6, 2011.
Disclosure of Potential Conflicts of Interest
Funding support for these clinical trials was provided by Ligand, Eisai and Genentech to K.H.D.
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2020;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Dragnev KH, Stover D, Dmitrovsky E. Lung cancer prevention: the guidelines. Chest. 2003;123:60S–71S. doi: 10.1378/chest.123.1_suppl.60s. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 4.Sherr CJ. Cell cycle control and cancer. Harvey Lect. 2000;96:73–92. [PubMed] [Google Scholar]
- 5.Lonardo F, Rusch V, Langenfeld J, Dmitrovsky E, Klimstra DS. Overexpression of cyclins D1 and E is frequent in bronchial preneoplasia and precedes squamous cell carcinoma development. Cancer Res. 1999;59:2470–2476. [PubMed] [Google Scholar]
- 6.Betticher DC, Heighway J, Hasleton PS, et al. Prognostic significance of CCND1 (cyclin D1) overexpression in primary resected non-small-cell lung cancer. Br J Cancer. 1996;73:294–300. doi: 10.1038/bjc.1996.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Betticher DC, White GR, Vonlanthen S, et al. G1 control gene status is frequently altered in resectable non-small cell lung cancer. Int J Cancer. 1997;74:556–562. doi: 10.1002/(sici)1097-0215(19971021)74:5<556::aid-ijc14>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 8.Ratschiller D, Heighway J, Gugger M, et al. Cyclin D1 overexpression in bronchial epithelia of patients with lung cancer is associated with smoking and predicts survival. J Clin Oncol. 2003;21:2085–2093. doi: 10.1200/JCO.2003.03.103. [DOI] [PubMed] [Google Scholar]
- 9.Freemantle SJ, Liu X, Feng Q, et al. Cyclin degradation for cancer therapy and chemoprevention. J Cell Biochem. 2007;102:869–877. doi: 10.1002/jcb.21519. [DOI] [PubMed] [Google Scholar]
- 10.Rusch V, Mendelsohn J, Dmitrovsky E. The epidermal growth factor receptor and its ligands as therapeutic targets in human tumors. Cytokine Growth Factor Rev. 1996;7:133–141. doi: 10.1016/1359-6101(96)00016-0. [DOI] [PubMed] [Google Scholar]
- 11.Rusch V, Klimstra D, Linkov I, Dmitrovsky E. Aberrant expression of p53 or the epidermal growth factor receptor is frequent in early bronchial neoplasia and coexpression precedes squamous cell carcinoma development. Cancer Res. 1995;55:1365–1372. [PubMed] [Google Scholar]
- 12.Sah JF, Eckert RL, Chandraratna RAS, Rorke EA. Retinoids suppress epidermal growth factor-associated cell proliferation by inhibiting epidermal growth factor receptor-dependent ERK1/2 activation. J Biol Chem. 2002;277:9728–9735. doi: 10.1074/jbc.M110897200. [DOI] [PubMed] [Google Scholar]
- 13.Masuda M, Suzui M, Yasumatu R, et al. Constitutive activation of signal transducers and activators of transcription 3 correlates with cyclin D1 overexpression and may provide a novel prognostic marker in head and neck squamous cell carcinoma. Cancer Res. 2002;62:3351–3355. [PubMed] [Google Scholar]
- 14.Lin SY, Makino K, Xia W, et al. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol. 2001;3:802–808. doi: 10.1038/ncb0901-802. [DOI] [PubMed] [Google Scholar]
- 15.Kijima T, Niwa H, Steinman RA, et al. STAT3 activation abrogates growth factor dependence and contributes to head and neck squamous cell carcinoma tumor growth in vivo. Cell Growth Differ. 2002;13:355–362. [PubMed] [Google Scholar]
- 16.Buerger C, Nagel-Wolfrum K, Kunz C, et al. Sequence-specific peptide aptamers, interacting with the intracellular domain of the epidermal growth factor receptor, interfere with Stat3 activation and inhibit the growth of tumor cells. J Biol Chem. 2003;278:37610–37621. doi: 10.1074/jbc.M301629200. [DOI] [PubMed] [Google Scholar]
- 17.Song L, Turkson J, Karras JG, Jove R, Haura EB. Activation of Stat3 by receptor tyrosine kinases and cytokines regulates survival in human non-small cell carcinoma cells. Oncogene. 2003;22:4150–4165. doi: 10.1038/sj.onc.1206479. [DOI] [PubMed] [Google Scholar]
- 18.Park OK, Schaefer TS, Nathans D. In vitro activation of Stat3 by epidermal growth factor receptor kinase. Proc Natl Acad Sci USA. 1996;93:13704–13708. doi: 10.1073/pnas.93.24.13704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petty WJ, Dragnev KH, Memoli VA, et al. Epidermal growth factor receptor tyrosine kinase inhibition represses cyclin D1 in aerodigestive tract cancers. Clin Cancer Res. 2004;10:7547–7554. doi: 10.1158/1078-0432.CCR-04-1169. [DOI] [PubMed] [Google Scholar]
- 20.Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353:123–132. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 21.Linardou H, Dahabreh IJ, Kanaloupiti D, et al. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol. 2008;9:962–972. doi: 10.1016/S1470-2045(08)70206-7. [DOI] [PubMed] [Google Scholar]
- 22.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 23.Paez JG, Jänne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 24.Pao W, Wang TY, Riely GJ, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2:e17. doi: 10.1371/journal.pmed.0020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Freemantle SJ, Spinella MJ, Dmitrovsky E. Retinoids in cancer therapy and chemoprevention: promise meets resistance. Oncogene. 2003;22:7305–7315. doi: 10.1038/sj.onc.1206936. [DOI] [PubMed] [Google Scholar]
- 26.Dragnev KH, Pitha-Rowe I, Ma Y, et al. Specific chemopreventive agents trigger proteasomal degradation of G1 cyclins: implications for combination therapy. Clin Cancer Res. 2004;10:2570–2577. doi: 10.1158/1078-0432.ccr-03-0271. [DOI] [PubMed] [Google Scholar]
- 27.Langenfeld J, Kiyokawa H, Sekula D, Boyle J, Dmitrovsky E. Posttranslational regulation of cyclin D1 by retinoic acid: a chemoprevention mechanism. Proc Natl Acad Sci USA. 1997;94:12070–12074. doi: 10.1073/pnas.94.22.12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lonardo F, Dragnev KH, Freemantle SJ, et al. Evidence for the epidermal growth factor receptor as a target for lung cancer prevention. Clin Cancer Res. 2002;8:54–60. [PubMed] [Google Scholar]
- 29.Boyle JO, Langenfeld J, Lonardo F, et al. Cyclin D1 proteolysis: a retinoid chemoprevention signal in normal, immortalized, and transformed human bronchial epithelial cells. J Natl Cancer Inst. 1999;91:373–379. doi: 10.1093/jnci/91.4.373. [DOI] [PubMed] [Google Scholar]
- 30.Dragnev KH, Petty WJ, Shah SJ, et al. A proof-of-principle clinical trial of bexarotene in patients with non-small cell lung cancer. Clin Cancer Res. 2007;13:1794–1800. doi: 10.1158/1078-0432.CCR-06-1836. [DOI] [PubMed] [Google Scholar]
- 31.Kobayashi S, Shimamura T, Monti S, et al. Transcriptional profiling identifies cyclin D1 as a critical downstream effector of mutant epidermal growth factor receptor signaling. Cancer Res. 2006;66:11389–11398. doi: 10.1158/0008-5472.CAN-06-2318. [DOI] [PubMed] [Google Scholar]
- 32.Rubin Grandis J, Zeng Q, Tweardy DJ. Retinoic acid normalizes the increased gene transcription rate of TGF-alpha and EGFR in head and neck cancer cell lines. Nat Med. 1996;2:237–240. doi: 10.1038/nm0296-237. [DOI] [PubMed] [Google Scholar]
- 33.Dragnev KH, Petty WJ, Shah S, et al. Bexarotene and erlotinib for aerodigestive tract cancer. J Clin Oncol. 2005;23:8757–8764. doi: 10.1200/JCO.2005.01.9521. [DOI] [PubMed] [Google Scholar]
- 34.Ma Y, Fiering S, Black C, et al. Transgenic cyclin E triggers dysplasia and multiple pulmonary adenocarcinomas. Proc Natl Acad Sci USA. 2007;104:4089–4094. doi: 10.1073/pnas.0606537104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Freemantle SJ, Dmitrovsky E. Cyclin E transgenic mice: discovery tools for lung cancer biology, therapy and prevention. Cancer Prev Res. 2010;3:1513–1518. doi: 10.1158/1940-6207.CAPR-10-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gibbons DL, Lin W, Creighton CJ, et al. Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev. 2009;23:2140–2151. doi: 10.1101/gad.1820209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lièvre A, Bachet JB, Boige V, et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol. 2008;26:374–379. doi: 10.1200/JCO.2007.12.5906. [DOI] [PubMed] [Google Scholar]
- 38.Pan Q, Pao W, Ladanyi M. Rapid polymerase chain reaction-based detection of epidermal growth factor receptor gene mutations in lung adenocarcinomas. J Mol Diagn. 2005;7:396–403. doi: 10.1016/S1525-1578(10)60569-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clopper C, Pearson ES. The use of confidence or fiducial limits illustrated in the case of the binomial. Biometrika. 1934;26:404–413. [Google Scholar]
- 40.Kim ES, Herbst RS, Wistuba, et al. The Battle trial: personalizing therapy for lung cancer. Cancer Discovery. doi: 10.1158/2159-8274.CD-10-0010. published ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khuri FR, Rigas JR, Figlin RA, et al. Multi-institutional phase I/II trial of oral bexarotene in combination with cisplatin and vinorelbine in previously untreated patients with advanced non-small-cell lung cancer. J Clin Oncol. 2001;19:2626–2637. doi: 10.1200/JCO.2001.19.10.2626. [DOI] [PubMed] [Google Scholar]
- 42.Blumenschein GR, Khuri FR, von Pawel J, et al. Phase III trial comparing carboplatin, paclitaxel, and bexarotene with carboplatin and paclitaxel in chemotherapy-naive patients with advanced or metastatic non-small-cell lung cancer: SPIRIT II. J Clin Oncol. 2008;26:1879–1885. doi: 10.1200/JCO.2007.12.2689. [DOI] [PubMed] [Google Scholar]
- 43.Edelman MJ. Second-line chemotherapy and beyond for non-small-cell lung cancer. Clin Adv Hematol Oncol. 2004;2:373–378. [PubMed] [Google Scholar]
- 44.Massarelli E, Andre F, Liu DD, et al. A retrospective analysis of the outcome of patients who have received two prior chemotherapy regimens including platinum and docetaxel for recurrent non-small-cell lung cancer. Lung Cancer. 2003;39:55–61. doi: 10.1016/s0169-5002(02)00308-2. [DOI] [PubMed] [Google Scholar]
- 45.Su H, Bodenstein C, Dumont RA, et al. Monitoring tumor glucose utilization by positron emission tomography for the prediction of treatment response to epidermal growth factor receptor kinase inhibitors. Clin Cancer Res. 2006;12:5659–5667. doi: 10.1158/1078-0432.CCR-06-0368. [DOI] [PubMed] [Google Scholar]
- 46.Sunaga N, Oriuchi N, Kaira K, et al. Usefulness of FDG-PET for early prediction of the response to gefitinib in non-small cell lung cancer. Lung Cancer. 2008;59:203–210. doi: 10.1016/j.lungcan.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 47.Mok TS, Wu Y, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 48.Massarelli E, Varella-Garcia M, Tang X, et al. KRAS mutation is an important predictor of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. Clin Cancer Res. 2007;13:2890–2896. doi: 10.1158/1078-0432.CCR-06-3043. [DOI] [PubMed] [Google Scholar]
- 49.Galimberti F, Thompson SL, Liu X, et al. Targeting the cyclin E-Cdk-2 complex represses lung cancer growth by triggering anaphase catastrophe. Clin Cancer Res. 2010;16:109–120. doi: 10.1158/1078-0432.CCR-09-2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liby K, Black CC, Royce DB, et al. The rexinoid LG100268 and the synthetic triterpenoid CDDO-methyl amide are more potent than erlotinib for prevention of mouse lung carcinogenesis. Mol Cancer Ther. 2008;7:1251–1257. doi: 10.1158/1535-7163.MCT-08-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.