

Abstract
Targeted α-particle therapy offers the potential for more specific tumor cell killing with less damage to surrounding normal tissue than β-emitters because of the combination of short path length (50–80 μm) with the high linear energy transfer (100 KeV μm−1) of this emission. These physical properties offer the real possibility of targeted (pre-targeted) α-therapy suitable for the elimination of minimal residual or micrometastatic disease. Targeted and pre-targeted radioimmunotherapy (RIT) using α–emitters such as 212Bi (T1/2 = 1.01 h) and 212Pb (T1/2 =10.6 h) has demonstrated significant utility in both in vitro and in vivo model systems. 212Pb, a promising α-particle emitting source, is the longer-lived parent nuclide of 212Bi, and serves as an in vivo generator of 212Bi. The radionuclide has been successfully used in RIT and pre-targeted RIT and demonstrated the enhanced therapeutic efficacy in combination with chemotherapeutics, such as gemcitabine and paclitaxel. The following perspective addresses the modes of radionuclide production, radiolabeling and chelation chemistry, as well as the application of 212Pb to targeted and pre-targeted radiation therapy.
Introduction
The better understanding of the molecular differences between cancer and normal cells has led to the development of therapies that directly target cancer cells, including the use of monoclonal antibodies (mAbs) directed at tumor-associated antigen. Cancer therapy represents one major area in which mAbs have been successful. Although such antibody therapies have shown significant successes in cancer treatment, strategies to increase their efficacy are urgently needed. One such strategy is to link antibodies against tumor-associated antigens to highly toxic radionuclides, which brings to bear the killing power of these radionuclides directly onto tumor cells. Specially, targeted, cytocidal radionuclides (β- and α-particle emitters) can be localized in malignant tissue for therapeutic applications via the use of appropriate targeting vectors. These vectors include tumor antigen binding mAbs and their variants, or cell surface receptor binding peptides. Towards this end, FDA approval for two radiolabeled anti-CD20 mAbs, 90Y-labeled ibritumomab tiuxetan (Zevalin) in 2002, and 131I-labeled Bexxar in 2003 were landmark events in the developmental history of therapeutic radiolabeled mAbs.1 The radiolabeled antibody is also recognized for its potential efficacy as both a monotherapy and for its enhanced efficacy when used in combination therapy.
New radioimmunotherapy (RIT) approaches incorporating α-particle emitters have been considered and have led to the development of both chelating agents and execution of pre-clinical studies. The α-particle has a very short path length (<100 μm), but a very high linear energy transfer (LET),2 with typical energy deposition of ~100 keV/μm compared to 0.2 keV/μm typically for a β-particle. The relative biological effectiveness of high LET radiation exhibits no dose rate dependence and is effective even under hypoxic conditions.3 The α-particle, a He nucleus, is quite relatively large compared to a β-particle, and the emission is associated with discrete high energies and a dense ionization track that is also associated with a high probability of inflicting irreparable and cytocidal DNA double strand breaks.4–8 An individual cancer cell can in theory be killed by interaction with only a single α-particle traversing the nucleus of a cell.9–11 The fundamental physics and radiobiology of a β-particle emitter provides a poor tumor to normal tissue dose ratio for treatment of single cell disease. On the other hand, delivery of an α-emitting radio nuclide to the cell membrane is sufficient to kill malignant cells, requiring only a few α-particle decays at the cell membrane due to 3 dimensional emission geometry considerations, to effect a 99.99% level of cell kill with correspondingly low normal tissue toxicity.12 Consequently, α-emitters are well suited for hematologic disease, micrometastatic disease, and tumor cells near the surface of cavities. High homogeneity of antigenic expression is required for the complete destruction of micrometastases with high LET. Conversely, with radiations of low LET with a longer path length, the cross-fire of the β-emitters may make up for the non-homogeneity of antigen expression. A number of pre-clinical studies have concluded that α-emitters may be more effective than β-emitters administered at comparable doses in RIT.13,14 However, high cost and/or limited or unresolved availability are major obstacles that have limited the clinical evaluation of mAbs radiolabeled with α-emitters. With the elimination of many obstacles and a better understanding of inherent imitations of mAbs, the active targeting and delivery vector of the radiation, many radiolabeled mAbs have been, or currently are being evaluated.
Although there are more than 100 α-particle emitting radionuclides, the majority of these radionuclides have half-lives that are either too short or too long for any meaningful or realistic therapeutic use, their production is not economically viable, or no viable chemistry for their use presently exists. The most widely studied α-particle candidates for therapy (Table 1) are 211At (T1/2 = 7.2 h), 212Bi (T1/2 = 61 min), 212Pb (T1/2 = 10.2 h), 213Bi (T1/2 = 46 min), and 225Ac (T1/2 = 10 days). Clearly, opportunities continue to exist in select areas of both coordination chemistry and conjugation chemistry. 212Pb and 212Bi are both promising α-particle emitting sources that have well-described radiochemistry for antibody linkage and are readily obtained from a 224Ra generator.15 212Pb is actually a β-emitter and is the immediate parental radionuclide of 212Bi. Therefore, one strategy that has been devised has been to label a mAb with 212Pb to serve then as an in vivo generator for the production of 212Bi, thereby effectively extending the short half-life of 212Bi. The major advantage of targeting 212Pb to the tumor instead of 212Bi is that 212Pb delivers greater than 10 times the dose per unit of administered activity compared to 212Bi alone or the α-emitter 213Bi.16 Therefore, the 10.6 h half-life of 212Pb also makes dose preparation and administration easier, and permits all operations to be executed more efficiently than with the short half-life 212Bi. This review describes the uses and strategies for 212Pb as a potential radiotherapeutic, the use of 212Pb radiolabeled mAbs directed approaches, the requisite and current chemistry, and discusses pre-clinical trials, with an emphasis on the development of 212Pb towards clinical translation.
Table 1.
Selected Therapeutic Radionuclides
| Radionuclide | Half-life (h) | Imaging decay | RIT decay | Emax (MeV) | Production |
|---|---|---|---|---|---|
| 212Pb | 10.64 | γ | β− EC |
0.57 | 224Ra/212Pb generator |
| 212Bi | 1.01 | γ | α β− EC |
6.09 6.05 |
228Pb/212Pb generator |
| 213Bi | 0.76 | γ | α β− EC |
5.87 5.55 |
228Th/212Pb generator |
| 225Ac | 240.00 | γ | α EC |
5.83 5.79 |
n-capture of 232Th→225Ac or 226Ra(p,2n)225Ac |
| 211At | 7.21 | γ | α EC |
5.87 | 209Bi (α, 2n)211At |
Production of 212Pb -- Generators
Radionuclide generators are systems wherein a longer-lived parent radionuclide is used to continuously generate, by radioactive decay, a shorter-lived daughter radionuclide of interest and whereby that desired radionuclide can be selectively separated and obtained by chemical means. 212Pb\212Bi and 213Bi are members of decay chains of the long-lived parents 232Th and 233U, respectively, and can therefore be produced by generators. 212Pb is produced from the decay chain of 228Th and can be available from a 224Ra generator that facilitates the on-site production of 212Bi or 212Pb, which may be selectively eluted by controlling the acid strength of HCl or HI eluant from that same ion-exchange based generator system and then used for radiolabeling mAbs, peptides, or other vectors conjugated with suitable bifunctional chelating agents.17
The original generator used 228Th, which has a 1.9 y half-life, deposited on Na2TiO3 on which the 228Th and its immediate daughter radionuclide 224Ra are absorbed.18 The 212Pb and the other decay daughters (Figure 1) were separated by maintaining a flow of water over the parent to wash away the 220Rn daughter, which has a half-life of 55s. The generator was operated by eluting 220Rn with water into a reservoir, waiting a few minutes for the radon gas to decay to 212Pb, followed by passing that solution through an organic cation-exchanger to absorb the 212Pb. A theoretical maximum yield of 212Pb (ca. 80 %) with a breakthrough level of ~2 × 10−4 Bq of 228Th or 224Ra per Bq of 212Pb was reported. At radioactivity levels greater than 37 MBq (1 mCi), radiolytic breakdown of the ion-exchanger support caused increasing back pressure and decreasing yields.15 All decays of the 212Pb and 212Bi lead to α-emissions, either directly, or through their daughter, 212Po (T½ = 0.3 μs). This generator based on 228Th experienced problems with radiolytic damage in the resin with consequent diminished yield and was also a serious radiation safety problem.
Figure 1.
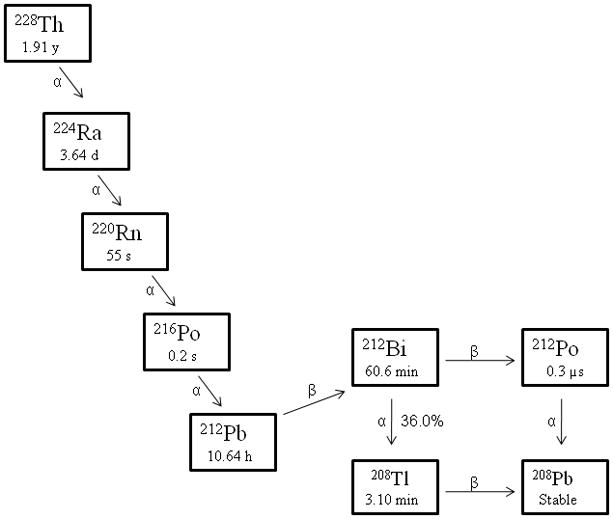
Decay Schemes for the Production of 212Pb
Evaporation (emanation) based generator systems were also developed to overcome radiolytic effect limitations. This type of generator also based on the principle of collecting 220Rn, however as a gas emanating from [229Th] barium stearate, and accommodates a low radioactivity to mass ratio to avoid the destructive effects of radiolysis.19 A 50 MBq (1.4 mCi) generator was evaluated where such a source could be moved into, or out from a collection chamber. When the source was inside the chamber, the decay product of 220Rn, 212Pb, was then deposited on the walls of a polyethylene bottle. The 212Pb could be washed off the plastic surface with aqueous solutions without detection of any 228Th (<10−9 Bq of 228Th per Bq of 212Pb) breakthrough or other long-lived parent nuclides. The emanation yield was only 50 % initially and decreased gradually due to radiolytic damage of the barium stearate support.
An improved generator construct based on the same principle, but with a different method for collecting the 220Rn and decay products has also been suggested.20 In this construct, the amount of 228Th doped barium stearate has been increased to lessen the destructive effects of radiolytic damage to the emanation ability. The collected yields of 212Pb concomitantly increased to approximately 70 %. So far, characterization of the properties of emanation generators has only been possible with tracer levels of radioactivity due to limited availability of 228Th.
To avoid problems originating from 228Th-based generators, another generator based on 224Ra (T½ = 3.7 d) was designed.15,21 224Ra is separated from 228Th by absorbing 228Th as the nitrate complex onto an anion-exchanger, while 224Ra elutes through the column. The 224Ra is then absorbed on to the macroporous organic cation ion-exchange resin (AG-MP-50) which then serves as the source for either a 212Bi or 212Pb. 212Bi can selectively be eluted from the generator with low acid concentrations of HI (0.05–0.2 M). At higher acid concentrations of either HCl or HI (1–6 M), a mixture of both 212Pb and 212Bi can be eluted. In our preparation, 212Pb was first eluted from the 224Ra/212Pb with 2 M HCl. The 212Pb eluate was then diluted to 0.1 M HCl and loaded onto a small AG-50 × 4 resin and the 212Bi eluted from the resin with 0.2 M HI. These generators have been available at source strength of ~0.7 GBq (0.02 Ci). Breakthrough of 224Ra and 228Th from these generators has been found to be 4 × 10−4 Bq of 224Ra per Bq of 212Pb and 10−6 Bq of 228Th per Bq of 212Pb. This generator has given good yields of 212Bi and its parent nuclide 212Pb, but it must be regenerated after 1–2 weeks because of the short half-life of 224Ra.
Chemistry of radiolabeling
The stable sequestration of the radionuclide in vivo is considered as one of the major aspects of targeted radiation therapy. It allows the delivery of radiation to tumor to be maximized while minimizing toxicity.22 Continued interest exists in the area of synthesis of bifunctional chelating agents, their conjugation to mAbs and peptides, and their subsequent use for sequestering radioactive metal ions for use in RIT and radioimmunoimaging (RII) applications. A variety of technologies are used to conjugate radioisotopes to antibodies, dependent on the chemical nature of the radionuclide. One of the fundamental requirements is that the conjugation of a radionuclide to a mAb or the conditions imposed by the radiolabeling protocols must maintain the affinity/avidity of the mAb for its target antigen.
Choosing an appropriate bifunctional chelating agent that forms an adequately stable complex of the metallic radionuclide of choice and within the context of the application is a critical factor. Acylic diethylenetriaminepentaacetic acid (DTPA) and macrocyclic 1,4,7,10-tetraazacyclododecane tetraacetic acid (DOTA)-based chelators represent the most commonly utilized classes of agents used in RIT. Generally, DTPA and other acyclic chelates exhibit fast complex association rates, whereas DOTA derivatives and other macrocyclic chelates have slower complex dissociation rates. A sampling of bifunctional chelating agents derived from DTPA include the cyclic dianhydride derivative,23 1B4M-DTPA24 and a family of trans-cyclohexyl derivatives that include the specific stereoisomer, CHX-A″DTPA. CHX-A″ DTPA is an effective chelator for 111In, 90Y, and 177Lu.25–27
Numerous bifunctional analogs of the macrocycle DOTA (Figure 2) have been used effectively for labeling antibodies with 111In, 90Y, 177Lu, 212Pb, and 212Bi.22,28–32 DOTA complexes for lanthanides and other metal ions tend to yield eight coordinate square anti-prisms that exists in an equilibrium between isomeric arrangements for carboxylate arms and ring twists forms that saturate the coordination spheres about Bi(III) and Pb(II). A number of lead and bismuth complexes has been prepared and evaluated for unique in situ generator system.33,34 Their favorable nuclear properties and availability make them well suited for in vivo assessment of new antitumor immunotherapeutic techniques. A problem with the clinical use of 212Bi or 212Pb radioimmunoconjugates (RICs) is the potential for radiotoxicity as a consequence of either premature release of the metal by the chelating agent or metabolic catabolism of the RIT releasing the radiometal.
Figure 2.
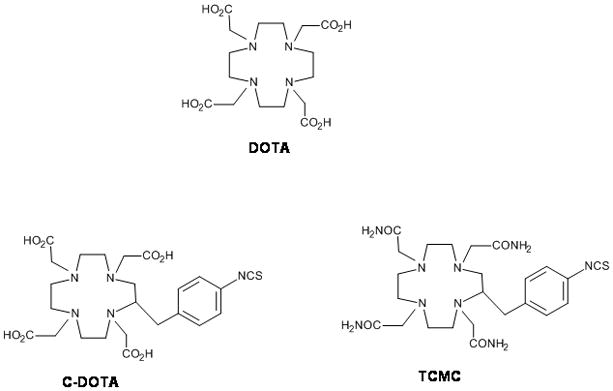
Selected Structure of DOTA and its analogues in RIT
Studies to evaluate the potential usefulness of a C-functionalized DOTA have led to conflicting results. Fundamental studies of the stability of the complex indicated that both the DOTA[Bi(III)] and DOTA[Pb(II)] complex were exceedingly stable and that a suitably stable complexes for in vivo applications formed.34 Indeed, the complex thus formed with a C-functionalized DOTA was confirmed as stable in vivo; however, significant slow complex formation rates are a hindrance to the use of DOTA.35 Functionalized DOTA ligands have also been reported to be highly sensitive to the presence of M(II) ion contamination of the radionuclide.36 Thus, despite forming a kinetically inert complex with Bi(III), DOTA was not found suitable due to slow complex formation rates versus the constraints of the half-lives of 212Bi, and, even more so, for the shorter half-life 213Bi.
However, DOTA was also shown to be an adequately stable in vivo chelator for Pb(II),34 which allows for conjugating and delivery of 212Pb, the precursor of 212Bi in targeted therapy. The ability to exploit the longer lived 212Pb effectively allows time for tumor targeting of the α-emitter, 212Bi. Key to its use, however, is the ability to form a stable chelate of 212Pb that controls the in vivo localization through the decay event formation of the 212Bi. In theory, the decay of 212Pb should not present a problem in retention of the 212Bi. Conservation principles dictate that the recoil energy of the Bi nucleus is only about 0.5 eV. This is not sufficient to break a chemical bond, which requires ~10 eV. However, the γ-ray emitted when 212Pb decays is internally converted over 30 % of the time.37 The resulting cascade of conversion electrons makes that percentage of the metal–chelate complex unstable. Because the free highly energetic radio-bismuth that is ejected during this process poorly re-associates with DOTA, toxicity occurs as 212Bi accumulates in or is formed within organ binding sites.
One of the challenges to the use of the Pb(II) radionuclides is the afore noted issue of acid catalyzed dissociation of the radiometal from DOTA, posing an additional obstacle to maintaining the 212Pb-DOTA complex after 212Pb-mAb internalization. Loss of 212Pb post-internalization of mAb delivery to cells has been reported as a source of marrow toxicity when using a DOTA conjugate wherein the dissociated 212Pb was free to be transported to the bone and subsequently then decay to 212Bi.35 Chappell et al. demonstrated that the Pb(II)[4-NCS-Bz-TCMC] complex was more stable compared to Pb(II)[-DOTA] complex, with increased resistance to complex dissociation under lower pH conditions.38 The TCMC ligand (Figure 2) also showed many other advantages over the DOTA ligand including more efficient conjugation reaction to mAb CC49 resulting in a conjugation product with higher numbers of chelate conjugated while retaining antigen binding and immunoreactivity as well as greater radiolabeling yields. Thus, these combined advantages promote use of TCMC over use of DOTA mAb immunoconjugates for immunotherapeutic and imaging studies when using Pb(II) radionuclides.
Maumela et al. reported the remarkable stability of the Pb(II) complex formed with the N,N,N,N-tetraamide analog of DOTA, as this complex retained its integrity even at significantly high acidic conditions, e.g., 0.5 M HCl.39 Additionally, the binding affinity of this ligand for Pb(II) strongly differs from that observed for the 1,4,8,11-tetraazacyclotetradecane tetraacetic acid (TETA) analog wherein that Pb(II) complex is less stable by > 10 log units.40 Hancock et al. reported a crystal structure of the Pb(II) complex of the N,N,N,N-tetraamide analog of DOTA that showed a water molecule bound to the metal center in such a way that a weak interaction between one water hydrogen atom and the Pb(II) was possible.41
The impact of choosing the correct macrocyclic platform for assembling a bifunctional chelating agents is well demonstrated by the differences in the succeptibility to acidic conditions demonstrated by these macrocyclic complexes. The macrocyclic bifunctional ligand, TCMC, which has been very efficiently radiolabeled with Pb(II) radionuclides has been found to be less labile at pH 3.5 than the corresponding DOTA complex, conferring enhanced resistance to acid-catalyzed dissociation within the cell.38 Hence, TCMC continues to be used for sequestration of 212Pb as opposed to DOTA.
More recently, Cuenot et al. have reported their studies of the Pb(II) complex of 1,4,7,10-tetrakis(carbamoylmethyl)-1,4,7,10-tetraaza-cyclododecane (TCMC), also known as DOTAM.40 They related that the reaction between Pb(II) and TCMC produced a mononuclear complex, even under mild acidic conditions, which corroborates the radiolabeling experience of this ligand with 212Pb as being quite rapid and efficient under similar conditions. Their crystal structure of the Pb(II)-TCMC complex revealed that the Pb(II) was fully encapsulated inside the TCMC with the eight-coordinate sphere saturated by the four ring nitrogens and the four amide oxygen atoms. Lastly, and most importantly, helical geometry of the four amide arms leaves no gap in the coordination sphere of the metal. This arrangement is indicative of a stereochemically inactive lone pair for Pb(II). This result is also highly supportive of this complex being extremely stable for in vivo sequestration of Pb(II) and supports the suitability of TCMC for the potential clinical applications being reviewed here.
Dosimetry
The simple definition of absorbed dose is the energy absorbed in a particular volume divided by the mass of the volume; tumor response and normal tissue damage are a function of dose.42 Generally, short half-lives, short range, high LET, and the complicated decay pathways of α-particle emitters differentiates their dosimetry from that of β-emitters. In targeted radionuclide therapy, dosimetry is complicated by several factors: heterogeneous radionuclide distribution, short-range particulate radiation, and few actual radioactive incidents per cell. A number of biological and chemical variables,43,44 e.g., heterogeneous radionuclide conjugation, heterogeneous target expression, antibody avidity, tumor vascularity, and interstitial pressure in the tumor have to be accounted for through model systems to estimate in vivo dose. Physical characteristics and the amount of the radionuclide are also factors.
The Medical standard approach to dosimetry calculations has been described by the Medical Internal Radionuclide Dose (MIRD)45 and have been applied for in vivo experiments for dose determination after α-particle radiation therapy. Given the high energy of α-particles delivered over their short range, conventional MIRD calculations and models may not always yield biologically meaningful information. To estimate the absorbed dose coming from α-particles decaying through multiple unstable daughters, with their own intrinsic biodistribution, prediction of absorbed dose and potential toxicity, Hamacher and Sgouros reported on three models to estimate normal organ absorbed doses for the following parent radionuclides: 225Ac, 212Pb, 211At, 223Ra, and 213Bi. Comparing doses in the case of a 0.1 g rapidly accessible tumor to those of a 10 g solid tumor, parent radionuclides with a short half-life yielded a higher dose burden to normal organs than longer lived radionuclides. At 20 Gy, the corresponding absorbed dose to a rapidly accessible 0.1 g tumor would be 1.7 and 0.9 Gy in liver and kidney from 225Ac, 0.4 and 0.3 Gy to bone and small intestines from 223Ra, and 2.2 and 3.0 Gy in small intestines from 212Pb and 213Bi, respectively.46
Thus, microdosimetry is typically required for α-emitter dosimetry for the analysis of cell culture experiments involving low concentrations of α-emitting nuclides. Such computations may be difficult to perform due to unknown microdistribution of the radionuclide.47–49 Average cell survival probabilities derived from macroscopic dosimetry experiments also may not reflect the true cell survival probabilities. Evidence of bystander effects and delayed cell death has demonstrated that cells not hit or traversed by α-particles may express or communicate a decreased survival.50–52 The impact of bystander effects related to the dose of 212Pb has yet to be reported, however, papers relating bystander effects originating from 211At studies are available and one would predict that similar effects would be generated with 212Pb.53,54 Likewise, RBE values reported from in vitro and in vivo experiments should be interpreted with caution. More accurate cell survival probabilities might be better obtained through in vitro experiments with absolute determination of the number of α-particle traversals of subcellular compartments.9
Surprisingly as yet, there are no dosimetry data results associated with actual 212Pb radiolabeled mAbs thus far in the literature. This deficiency provides an area of opportunity for study that will have to be addressed as this radionuclide moves closer to translation to clinical trial evaluation.
Pre-clinical studies of Non-Targeted 212Pb
The use of 212Pb without a targeting vehicle as a therapeutic agent has been reported. In the study, 212Pb in the form of a sulfur colloid was used to treat ovarian carcinoma after i.p. injection. A dose dependent survival was demonstrated. At doses of 70 μCi, death occurred as a result of gastrointestinal injury. Histologically, 212Pb in the sulfur colloid caused extensive tumor necrosis compared to colloid alone. However, the use of the sulfur colloid may be limited as a carrier for 212Pb because the colloid resulted in uneven peritoneal distribution of radionuclide to the bowel surface resulting in gastrointestinal toxicity at the higher doses.55
Rotmensch et al. used ferrous hydroxide as a colloidal carrier for 212Pb because of higher retention time in the peritoneal cavity than ferric sulfide or hydroxide.56 In this application, 212Pb prolonged the mean survival time in a dose dependent manner after i.p. injection in the treatment of an ascites producing tumor. The radiosensitivity and chromosomal aberrations of cells increased with 212Pb. However, no major side effects or toxicity was found with administration of up to 2.6 mCi of ferrous hydroxide-212Pb.
Pre-clinical studies of Targeted 212Pb
As noted previously, the limitations of the short half-life α-particle emitter, 212Bi, may be effectively extended by using its in vivo generator parental radionuclide, 212Pb, sequestered in a chelating agent conjugated to the carrier molecule. This increases uptake ratios between tumor and normal organs. However, as also noted, 36% of the 212Bi formed could be lost from the DOTA complex on the α-decay of 212Pb due to the electronic effects of the one of the decay pathways (Figure 1), potentially causing toxicity. A study from Ruble et al. demonstrated the efficacy of 212Pb-labeled mAb 103A in treating the Rauscher leukemia virus (RVB3) resulting in histological cure in all animals. The animals showed no evidence of splenic tumor foci with a dose of just 0.74 MBq (20 μCi), but all of the animals died of bone marrow toxicity.57 No other organ showed any sign of radiotoxicity. The radioactivity in the bone (3.2 % ID/g) was twice the level of that in the tissue of control animals treated with 212Pb-B3 (1.6 % ID/g). The high bone marrow toxicity observed in this study sharply contrasted with the lack of marrow toxicity in the animals treated with 212Bi-103A-mAb. These results show that 212Bi lost from 212Pb-mAb increased marrow toxicity as compared to 212Bi-103A-mAb. Additionally, the study utilized bifunctional DOTA, whose complex begins to exhibit some measurable lability at the pH that would be encountered in the lyzosomes post-internalization of the radioimmunoconjugate. As a result of that acidic environment, 212Pb itself could also have dissociated from the complex and then exited the tumor cells. Pb(II) at these very low concentrations is known to traffic in the blood and become transported to bone where it can bind to the bone, decay to 212Bi, and be a source of toxicity. Attempts at limiting hematological and marrow toxicities by administration of heavy metal chelators such as 2,3-dimercapto-1-propanesulfonic acid (DMPS) and meso-2,3-dimercaptosuccinic acid (DMSA) have been explored albeit not in any great depth for 212Pb.58 While both were found to be effective in improving whole body clearance of both 212Bi and 212Pb as well as bone deposition levels, only DMPS was noted to have any impact on reducing renal accumulation.
HER2, expressed in a variety of epithelial cancers, is proving to be an ideal target for radioimmunotherapy.59,60 Horak et al. evaluated the efficacy of 212Pb-AE1-mAb for targeting HER2 on ovarian tumors in nude mice.61 Transient bone marrow toxicity and lengthy renal toxicity were observed after i.v. injection of 0.93 MBq (25 μCi); doses of 1.48 MBq (40 μCi) resulted in a cellular bone marrow toxicity and subsequent death of all animals. However, treatment of three days post-inoculation s.c. tumors with 0.37–0.74 MBq (10–20 μCi) of 212Pb-AE1 resulted in 100 % tumor free survival for 180 days, with all control animals developing tumors by day 20. In additional studies, the growth of small, more established tumors (15 mm3) was modestly inhibited, while the growth of larger tumors (146 mm3) was unaffected following administration of 212Pb-DOTA-AE1. The poor therapeutic efficacy of the larger tumors could be explained by long blood residence time, slow tumor targeting, and perhaps poor tumor penetration, resulting in low tumor/blood ratios for the 212Pb-AE1-mAb.
Targeted α-radiation therapy with mAb, which binds to tumor-associated antigen, may be efficacious and more appropriately employed in a coordinated strategy for the treatment and management of disseminated peritoneal disease. There are, however, inherent limitations associated with their use for targeting and treatment of solid tumors.62 212Pb-labeled trastuzumab for the treatment disseminated peritoneal disease has been suggested.63 A pilot radioimmunotherapy experiment tested mice bearing LS-174T intraperitoneal (i.p.) xenografts as a model. The study established a maximum tolerated dose (MTD) of 0.74 – 1.48 MBq (20–40 μCi) for the mice. The median survival of animals receiving 0.37 MBq (10 μCi) increased from 19 to 56 days (p = 0.008). A multi-dosing regimen of 212Pb-TCMC-trastuzumab administered at monthly intervals (up to 3 monthly doses of 212Pb-TCMC-trastuzumab) increased median survival of mice bearing 3 d LS-174T i.p. xenografts to 110 days.
Approaches to increase the therapeutic efficacy of targeted radiation therapy have been explored. These include harnessing the potential of synergistic cytotoxicity of targeted α-particle radiation therapy with chemotherapeutics and radiosensitizers. The combination of gemcitabine (GEM) with RIT using β-emitters has been extensively studied.64,65 For targeted α-particle therapy using 212Pb-TCMC-trastuzumab has been evaluated in combination with gemcitabine (GEM) for treating disseminated peritoneal disease.66 Treatment using mice bearing i.p. LS-174T xenografts with gemcitabine (GEM) followed 24–30 hr later by either 0.19 or 0.37 MBq (5 or 10 μCi) of 212Pb-TCMC-trastuzumab resulted in improvement of median survival; from 31 to 51 days in the absence or presence of GEM with 0.19 MBq (5 μCi) of 212Pb-TCMC-trastuzumab, respectively, and from 45 up to 70 days at the 0.37 MBq (10 μCi) dose in the absence or presence of GEM, respectively, compared to 16 days for untreated animals. Three weekly doses of gemcitabine in conjunction with 212Pb resulted in a median survival of 90 days vs 21 days for the untreated group of animals. Treatment with two cycles of 10 μCi 212Pb-TCMC-trastuzumab with two doses of GEM resulted in the greatest therapeutic efficacy with a median survival of 196.5 days. This therapeutic regimen combining chemotherapeutics and high LET radioimmunotherapy may have tremendous potential in cancer treatment, particularly in the context of microscopic and residual disease post-surgical resection.
Radioimmunotherapy using β-emitters combined with paclitaxel has been well studied for treatment of lymphoma in pre-clinical studies and found to be synergistic. The regimen is also sensitive to administration order and timing.67,68 Milenic et al. have recently evaluated the ability of paclitaxel to potentiate the therapeutic efficacy of HER-2 targeting α-emitting high LET 213Bi-CHX-A″-trastuzumab and 212Pb-TCMC-trastuzumab in a multimodality regimen for the management of disseminated i.p. disease.69 Combination treatment of paclitaxel administered 24 hr before, concurrently with, or 24 hr after 212Pb-TCMC-trastuzumab (10 μCi) was studied to elucidate the optimal administration order of the two therapeutics. An enhanced therapeutic efficacy of 212Pb-TCMC-trastuzumab was observed in the group that received 600 μg of paclitaxel 24 hr before the RIT. Nearly a 4-fold increase in the median survival, from 44 to 171 days, in this group was observed as compared with the group that received 212Pb-TCMC-trastuzumab alone. The response appears to be quite specific to the 212Pb-TCMC-trastuzumab compared to 212Pb-TCMC-HuIgG, a labeled non-specific human immunoglobulin.
Other Applications of Targeted 212Pb
Liposomes carrying isotopes might also act as mediators of in vivo targeted radiotherapy at low total doses to the organism. Rosenow et al. described the properties of liposomes containing 212Pb by encapsulation.70 Liposomes incorporating 212Pb remained at least partially intact in vivo. The potential of this tool for in vivo radioimmunotherapy lies in the possibility of maintaining cytotoxic activity in the circulation and in various organs for perfusion therapy of neoplasms or immune suppression.
A study with 212Pb/212Bi-ethylenediamine tetra-methylenephosphonic acid (EDTMP) demonstrated the intriguing possibility for therapy of osteosarcoma and bone metastases. EDTMP is a chelating agent with a high affinity for bone and has also been used to direct other radionuclides to bone for palliation therapy of bone lesions. However, EDTMP was found to be an unsatisfactory chelator for 212Bi and 212Pb due to the instability, resulting in very high kidney uptake values and lower bone uptake values compared to 212Pb/212Bi-DOTAMP.71,72
Hassfjell et al. proposed the use of 212Pb and 212Bi chelated to the bone-seeking ligand, 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraphosphonic acid (DOTMP) for therapy of osteosarcomas, or bone metastases from breast, prostate, and lung cancers.72 Both radiometal tetraphosphonate complexes localized rapidly in the bone matrix of mice, especially in regions with high bone turnover, a condition frequently observed in osteosarcomas and bone metastases. After 30 minutes, femur uptake of 16 % ID/g of 212Bi-DOTMP and 10 % ID/g 212Pb-DOTMP were reached. At this time point, the radioactivity in the blood was 0.6 % ID/g for both agents, and decreased to 0.06 % ID/g for 212Bi and 0.02 % ID/g for 212Pb, respectively, at later time points. Approximately one-third of the in vivo generated 212Bi was lost from 212Pb-DOTMP, similar to that reported for the 212Pb-DOTA complex.
Diener et al. proposed a unique potential role for 212Pb in radionuclide therapy by more stably encapsulating radionuclides inside of fullerenes, especially where conventional chelation chemistry is inadequate due to physical and/or chemical properties of radionuclide.73 212Pb@C60 and its malonic ester derivatives allowed the 212Pb detivative to be generated in situ from the decay of the parental 224Ra. The 212Pb appeared to recoil into C60 following α-decay from its parent. A preliminary biodistribution study in mice demonstrated that 212Pb did not accumulate in bone after being administrated as an endohedral fullerene, in contrast to results with polyhydroxylated radiofullerenes and conventional polyaminocarboxylate chelators for 212Pb, but showed rather slow clearance. Only ~2 % ID/g accumulated in the bone using the 212Pb@C60 malonate. Studies to actively target this intriguing construct have surprisingly not appeared as yet.
Peptides, as opposed to mAb targeted α-therapy, have also been recently investigated to take advantage of both rapid targeting with cellular internalization combined with rapid clearance pharmacokinetics. Miao et al evaluated the therapeutic efficacy in the B16/F1 mouse melanoma animal model of a unique melanoma-targeting peptide radiolabeled with 212Pb.74 Treatment of melanoma-bearing mice with 50, 100, and 200 μCi of 212Pb-DOTA-Re(Arg11)CCMSH extended their mean survival to 22, 28, and 49.8 days, respectively, when compared with 14.6-day mean survival of the untreated control group. Forty-five percent of the animals receiving 200 μCi doses of a 212Pb-DOTA-Re(Arg11)CCMSH survived the study disease-free. The advantage of administering 212Pb-DOTA-Re(Arg11)CCMSH is that the radiolabeled peptide will circulate, target melanoma tumor cells and be cleared from the body as the 212Pb-labeled peptide within the time frame of the radionuclide half-life. Only minimal amounts of the α-emitting 212Bi compound will exist thereby minimizing normal tissue exposures from any “free” 212Bi. Peptide-targeted 212Pb, rapidly internalized and then retained by tumor cells decays to the α-particle emitting 212Bi, localizing the highly toxic short-ranged α-radiation within the tumor cells. Once internalized and retained, the close proximity to the nucleus would facilitate a greater opportunity of traversing the nucleus and concomitantly increase the odds of cell death. Finally, the 10.6 h half-life of 212Pb makes dose preparation and administration easier and more convenient than the short half-life (T1/2 = 60.6 min) 212Bi. No difference was detected in the biodistribution of 212Pb and 212Bi during the 48 hr study period, demonstrating that no significant amounts of 212Bi were escaping the 212Pb-DOTA-Re(Arg11)CCMSH molecule and redistributing in vivo. The rapid clearance combined with the rapid targeting and internalization of 212Pb-DOTA-Re(Arg11)CCMSH likely prevented measurable amounts of 212Bi from being released.
Pre-clinical studies of Pre-Targeted 212Pb
Antibody pre-targeting is a process that addresses optimal delivery of radiation to tumors.75 Su et al. evaluated an antibody pre-targeting system with mAb-steptavidin, clearing agent and DOTA-biotin for solid tumor radiotherapy using the in vivo 212Pb/212Bi generator.76 Compared to its γ-emitting analogues, 212Pb-DOTA-biotin was not stable and as with previous studies more than 30 % of the 212Bi formed was released from 212Pb-DOTA. However, the pre-targeting of 212Pb/212Bi provided good tumor uptake, tumor-to-blood ratios and normal non-target tissue/blood ratios with the exception of kidney, the primary biological deposition site for Bi(III). In addition, the dosimetry calculation of 212Pb in the mouse xenograft model showed that the system provided a tumor dose of 93 rad/μCi and that the ratio of tumor to marrow and tumor to kidney was 386:1 and 12:1, respectively.
Applications of 203Pb
One challenge associated with performing pre-clinical experiments with 212Pb is the execution of accurate biodistribution and targeting assays of a 212Pb-radiolabeled mAb. One viable option is to employ 203Pb as a surrogate nuclide. 203Pb has a favorable half-life (T1/2 = 52 h) and decays with 80.1 % emission of γ-rays at 279 keV that is compatible with single photon emission computerized tomography (SPECT). This makes the radionuclide ideally suited as a matched radionuclide tracer 212Pb targeted radionuclide therapy. The nuclide is potentially useful for imaging, tissue distribution studies, dosimetry data acquisition, as well as chemical exchange studies. 203Pb can be easily produced via the 203Tl(d, 2n)203Pb reaction by irradiating natural Tl2O3 or an enriched Tl2O3 (203Tl) target with 13.7 MeV deuterons from a cyclotron. Purified 203Pb has been used to label trastuzumab, shown to be immunoreactive and demonstrated favorable biodistribution properties in vivo, indicating the suitability and feasibility of 203Pb-labeled biomolecules to target cellular antigens.77 Imaging and biodistribution studies performed with 203Pb-DOTA-B72.3 in nude mouse bearing LS-174T tumors, showed no major accumulation of lead in the bone and other organs. Clear and distinct γ-camera images of LS-174T tumors were obtained by injection of 203Pb-DOTA-B72.3 (Figure 3).78 Miao et al. also evaluated DOTA-Re(Arg11)CCMSH radiolabeled with 203Pb as a matched pair imaging agent for 212Pb- DOTA-Re(Arg11)CCMSH.79 203Pb- DOTA-Re(Arg11)CCMSH exhibited high melanoma uptake and a biodistribution pattern similar to that of 212Pb-DOTA-Re(Arg11)CCMSH, highlighting its potential as a matched pair imaging probe for 212Pb-DOTA-Re(Arg11)CCMSH (Figure 4).
Figure 3.

Image with 203Pb-B72.3-DOTA in athymic mice bearing LS-174T tumors.
Figure 4.
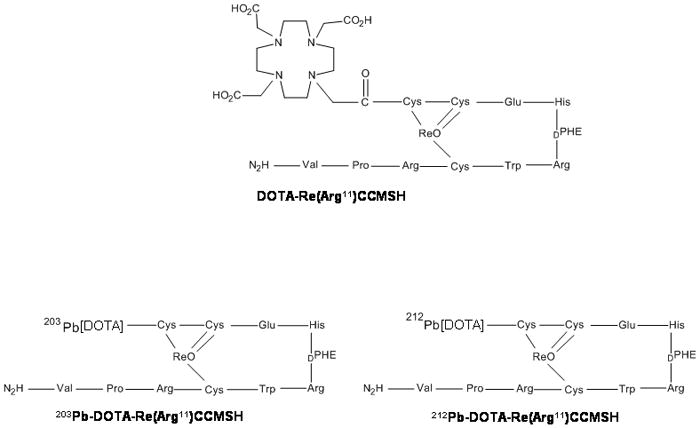
Structure of 212Pb-DOTA-Re(Arg11)CCMSH and 203Pb-DOTA-Re(Arg11)CCMSH
Potential Prospects and Conclusion
The high LET of α-particle radiation and short path length, although not ideal for large burden disease, has been proposed as ideal for the treatment of smaller tumor burdens, micrometastatic disease, and disseminated disease. Furthermore, far greater cancer killing probabilities are often achievable with α-particle radiation than with alternative strategies. Despite the favorable properties of α-particle radiation, the development of α-particle RIT has been limited by the poor availability, as a result of limited amounts or economic limitations, or by the actual physical characteristics of α-emitting radionuclides.
Early RIT studies using α-emitting radioisotopes were performed with 212Bi, in large part because of the availability of 224Ra. The short half-life of 212Bi creates the same limitations associated with even the shorter half-life of 213Bi. 212Pb, which decays to 212Bi, offers a means to utilize the α–particles from 212Bi decay for targeted therapy. Towards this end, a mAb radiolabeled with 212Pb serves as an in vivo generator of 212Bi thereby extending the delivery time for the 212Bi daughter for actually arrive and impact target tumor tissues. The process also has the effect of reducing the dose that is needed to effect therapy. As noted above, a dose of 10 μCi of 212Pb was equi-effective as a 500 μCi injected dose of 213Bi in the identical model system.69 Conservation principles dictate that the recoil energy of the Bi nucleus is only about 0.5 eV, which is not adequate by itself to break a chemical bond; however, the internal conversion process of one of the decay pathways of 212Pb does provide a mechanism for loss of the 212Bi daughter. Appropriate therapy strategies for 212Pb exist that deal with this loss of 212Bi within the context of the environment and disease presentation, e.g., the targeting of intracavitary metastatic or micrometastatic malignancies are considered reasonable.
While both pre-clinical and early clinical studies appear promising, several obstacles obstruct the pathway to widespread acceptance and use of targeted α-therapy. Enhanced therapeutic efficacy can be attained through selective dose delivery to radiosensitive areas of tumors. To improve current dosimetry models, more accurate determination of the radionuclide microdistribution should be provided. Along with improvement of dosimetry for the clinical situation, cell survival probabilities after given numbers of α-particle traversals require a more accurate determination.
Chelation and linking chemistry remains a challenge for the multiple decay pathway radionuclides. Earlier studies have noted the inadequacy of DOTA in maintaining a stable complex during decay from 212Pb to 212Bi. Additionally, the DOTA-Pb(II) complex is modestly acid labile, which may be a source of toxicity when internalized and metabolized as a radioimmunoconjugate. Use of the TCMC-Pb(II) complex obviates the pH lability associated with DOTA, but the loss from the decay process remains unsolved and limits the choices of appropriate therapeutic applications. Recently, a possible application for α-particle RIT using coordination with Bi(III) and mAbs Pb(II) has been suggested.80 A single-strapped analogue to porphyrin 5 coordinated with both Bi(III) and Pb(II) is stable leading to a possible application of a 212Pb/212Bi in vivo generator for medical applications. Despite having traversed many chelation challenges, more efficient conjugation and radiolabeling protocols remain to be developed to produce more consistent products with higher specific activities to optimize therapeutic potentials.
The clinical advantages and increases in efficacy obtained using combination therapies are becoming more evident. Chemotherapy in conjunction with α-particle RIT using the appropriate targeting vehicle would lead to efficient therapy following procedures such as cytoreductive surgery or peritoneal external beam radiation therapy. 212Pb is a promising α-particle emitting source, providing alternative options in the treatment and management of cancer. The recent advances from our laboratory and others demonstrate the tremendous potential of combination therapy studies using high linear energy transfer RIT with 212Pb and chemotherapeutics such as gemcitabine and paclitaxel to treat disseminated peritoneal disease as well as other appropriately scaled disease.
Furthermore, the utilization of the matched pair approach using 203Pb, which shows favorable pharmacokinetic and imaging properties, highlight new potential in therapy and imaging with mAbs, or peptides radiolabeled with 212Pb. However, mAb based molecular imaging and RIT have yet to reach their full potentials in both the pre-clinical and clinical domains. Continued efforts to refine and optimize all of the components to improve efficacy and minimize toxicity along with carefully planned pre-clinical investigation and improved targeting strategies will facilitate translation into clinical evaluation to move the field forward.
Acknowledgments
This research was supported [in part] by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Biographies
Kwon Joong Yong received his B.A. in 1992 and M.S. in 1994 from the Kangwon National University, Republic of Korea. Under the supervision of Professor Okot-Kotber, he obtained his Ph.D. in 2004 from Kansas State University. He worked to develop methods for the purification and enhanced production of phytase from plant source. He was a Post-doctoral fellow at Georgetown University Medical Center in 2004–2009. He studied characterization of HDAC isoforms in breast cancer and developed novel HDAC inhibitors. He is presently in the research group of Dr. M. W. Brechbiel at the National Cancer Institute.
Martin W. Brechbiel received a B.A. in 1979 from Gettysburg College and a M.S. in 1982 from the U. of Delaware under the guidance of Professor Harold Kwart. After working for FMC Corp, he joined the National Cancer Institute in 1983. Thereafter, he worked to develop novel bifunctional chelating agents for sequestering radionuclides and their conjugation to immunoproteins under the direction of Dr. Otto A. Gansow while simultaneously obtaining a Ph.D from American U. in 1988 with Professor Thomas Cantrell. He remained with the NCI and in 2001 was appointed as the Section Chief of the Radioimmune & Inorganic Chemistry Section. His research group’s activities span the range of continuing development of novel chelating agents for radionuclides, the development of contrast media for MRI, EPR, and CT imaging, SPECT and PET imaging agents, and the development of rationally designed targeted α-therapy regimens.
References
- 1.Sharkey RM, Burton J, Goldenberg DM. Expert Rev Clin Immunol. 2005;1:47–62. doi: 10.1586/1744666X.1.1.47. [DOI] [PubMed] [Google Scholar]
- 2.Behr TM, Behe M, Stabin MG, Wehrmann E, Apostolidis C, Molinet R, Strutz F, Fayyazi A, Wieland E, Gratz S, Koch L, Goldenberg DM, Becker W. Cancer Res. 1999;59:2635–2643. [PubMed] [Google Scholar]
- 3.Hall EJ. Radiology for the Radiologist. 4. J. B. Lippincott Company; Philadelphia: 1994. [Google Scholar]
- 4.Allen BJ, Raja C, Rizvi S, Li Y, Tsuji W, Zhang D, Song EY, Qu CF, Kearsley J, Graham P, Thompson J. Phys Med Biol. 2004;49:3703–3712. doi: 10.1088/0031-9155/49/16/016. [DOI] [PubMed] [Google Scholar]
- 5.Blakely EA, Kronenberg A. Radiat Res. 1998;150:126–145. [PubMed] [Google Scholar]
- 6.Howell RW, Goddu SM, Narra VR, Fisher DR, Schenter RE, Rao DV. Radiat Res. 1997;147:342–348. [PMC free article] [PubMed] [Google Scholar]
- 7.Azure MT, Archer RD, Sastry KSR, Rao DV, Howell RW. Radiat Res. 1994;140:276–283. [PMC free article] [PubMed] [Google Scholar]
- 8.Goodhead DT. Int J Radiat Biol. 1994;65(1):7–17. doi: 10.1080/09553009414550021. [DOI] [PubMed] [Google Scholar]
- 9.Soyland C, Hassfjell SP. Int J Radiat Biol. 2000;76:1315–1322. doi: 10.1080/09553000050151583. [DOI] [PubMed] [Google Scholar]
- 10.Pugliese M, Durante M, Grossi GF, Montforti F, Orlando D, Ottolenghi A, Scampoli P, Gialanella G. Int J Radiat Biol. 1997;72:397–407. doi: 10.1080/095530097143176. [DOI] [PubMed] [Google Scholar]
- 11.Hei TK, Wu L, Liu S, Vannais D, Waldren CA, Randers-Pehrson G. Proc Natl Acad Sci USA. 1997;94:3765–3770. doi: 10.1073/pnas.94.8.3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Humm JL, Cobb LM. J Nucl Med. 1990;31:75–83. [PubMed] [Google Scholar]
- 13.Aurlien E, Kvinnsland Y, Larsen RH, Bruland OS. Int J Radiat Biol. 2002;78(2):1333–1342. doi: 10.1080/09553000110094788. [DOI] [PubMed] [Google Scholar]
- 14.Behr TM, Sgouros G, Stabin MG, Behe M, Angerstein C, Blumenthal RD, Apostolidis C, Molinet R, Sharkey RM, Koch L, Goldenberg DM, Becker W. Clin Cancer Res. 1999;5:3031–3043. [PubMed] [Google Scholar]
- 15.Atcher RW, Friedman AM, Hines JJ. Appl Radiat Isot. 1988;39:283–286. doi: 10.1016/0883-2889(88)90016-0. [DOI] [PubMed] [Google Scholar]
- 16.Leichner PK. Nucl Med. Mosby; St. Louis: 1996. pp. 558–562. [Google Scholar]
- 17.Mirzadeh S. Appl Radiat Isot. 1998;4:345–349. doi: 10.1016/s0969-8043(97)00287-x. [DOI] [PubMed] [Google Scholar]
- 18.Zucchini GL, Friedman AM. Int J Nucl Med Biol. 1982;9:83–84. doi: 10.1016/0047-0740(82)90082-1. [DOI] [PubMed] [Google Scholar]
- 19.Hassfjell SP, Hoff P. Apply Radiat Isot. 1994;45:1021–1025. [Google Scholar]
- 20.Hassfjell SP. Apply Radiat Isot. 2001;55:433–439. doi: 10.1016/s0969-8043(00)00372-9. [DOI] [PubMed] [Google Scholar]
- 21.Atcher RW, Hines JJ, Friedman AM. J Radioanal Nucl Chem Letter. 1987;117:155–162. [Google Scholar]
- 22.Milenic DE, Brady ED, Brechbiel MW. Nat Rev Drug Discovery. 2004;3:488–498. doi: 10.1038/nrd1413. [DOI] [PubMed] [Google Scholar]
- 23.Macklis RM, Kinsey BM, Kassis AI, Ferrara JL, Atcher RW, Hines JJ, Coleman CN, Adelstein SJ, Burakoff SJ. Science. 1988;240:1024–1026. doi: 10.1126/science.2897133. [DOI] [PubMed] [Google Scholar]
- 24.Brechbiel MW, Gansow OA. Bioconjugate Chem. 1991;2:187–194. doi: 10.1021/bc00009a008. [DOI] [PubMed] [Google Scholar]
- 25.Brechbiel MW, Gansow OA. J Chem, Soc, Perkin Trans. 1992;1:1173–1178. [Google Scholar]
- 26.Wu C, Kobayashi H, Sun B, Yoo TM, Paik CH, Gansow OA, Carrasquillo JA, Pastan I, Brechbiel MW. Bioorg Med Chem. 1997;5:1925–1934. doi: 10.1016/s0968-0896(97)00130-2. [DOI] [PubMed] [Google Scholar]
- 27.Milenic DE, Garmestani K, Chappell LL, Dadachova E, Yordanov A, Ma D, Schlom J, Brechbiel MW. Nucl Med Biol. 2002;29:431–442. doi: 10.1016/s0969-8051(02)00294-9. [DOI] [PubMed] [Google Scholar]
- 28.Parker D. Chem Soc Rev. 1990;19:271–291. [Google Scholar]
- 29.Chappell LL, Rogers BE, Khazaeli MB, Mayo MS, Buchsbaum DJ, Brechbiel MW. Bioorg Med Chem. 1999;7:2313–2320. doi: 10.1016/s0968-0896(99)00171-6. [DOI] [PubMed] [Google Scholar]
- 30.Chappell LL, Ma D, Milenic DE, Garmestani K, Beitzel MP, Brechbiel MW. Nucl Med Biol. 2003;30:581–595. doi: 10.1016/s0969-8051(03)00033-7. [DOI] [PubMed] [Google Scholar]
- 31.McMurry TJ, Brechbiel MW, Kumar K, Gansow OA. Bioconjgate Chem. 1992;3:108–117. doi: 10.1021/bc00014a004. [DOI] [PubMed] [Google Scholar]
- 32.Hassfjell S, Brechbiel MW. Chem Rev. 2001;101:2019–2036. doi: 10.1021/cr000118y. [DOI] [PubMed] [Google Scholar]
- 33.Gansow OA, Brechbiel MW, Pippin CR, McMurry TJ, Lambrecht R, Colcher D, Schlom J, Roselli M, Strand M, Huneke RB, Ruegg CL. Antibody Immunoconj Radiopharm. 1991;4:413–425. [Google Scholar]
- 34.Pippin CG, McMurry TJ, Brechbiel MW, McDonald M, Lambrecht R, Milenic D, Roselli M, Colcher D, Gansow OA. Inorg Chim Acta. 1995;239:43–51. [Google Scholar]
- 35.Ruegg CL, Anderson-Berg WT, Brechbiel MW, Mizadeh S, Gansow OA, Strand M. Cancer Res. 1990;50:4221–4226. [PubMed] [Google Scholar]
- 36.Delgago R, Fausto da Silva JJR. Talanta. 1982;29:815–822. doi: 10.1016/0039-9140(82)80251-8. [DOI] [PubMed] [Google Scholar]
- 37.Mirzadeh S, Kumar K, Gansow OA. Radiochim Acta. 1993;60:1–9. [Google Scholar]
- 38.Chappell LL, Dadachova E, Mienic DE, Garmestani K, Wu C, Brechibiel MW. Nucl Med Biol. 2000;27:93–100. doi: 10.1016/s0969-8051(99)00086-4. [DOI] [PubMed] [Google Scholar]
- 39.Maumela H, Hancock RD, Carlton L, Reibenspies JH, Wainwright KP. J Am Chem Soc. 1995;117:6698–6707. [Google Scholar]
- 40.Cuenot F, Meyer M, Espinosa E, Guilard R. Inorg Chem. 2005;44:7895–7910. doi: 10.1021/ic0508019. [DOI] [PubMed] [Google Scholar]
- 41.Hancock RD, Reibenspies JH, Maumela H. Inorg Chem. 2004;43:2981–2987. doi: 10.1021/ic030277a. [DOI] [PubMed] [Google Scholar]
- 42.Zanzonico PB. J Nucl Med. 2000;41:297–308. [PubMed] [Google Scholar]
- 43.Buchsbaum DJ, Langmuir VK, Wessels BW. Med Phys. 1993;20:551–567. doi: 10.1118/1.597142. [DOI] [PubMed] [Google Scholar]
- 44.Adams GP, Schier RJ. Immunol Methods. 1999;231:249–260. doi: 10.1016/s0022-1759(99)00161-1. [DOI] [PubMed] [Google Scholar]
- 45.Howell RW, Wessels BW, Loevinger RJ. J Nucl Med. 1999;40:3–10. [Google Scholar]
- 46.Hamacher KA, Sgouros G. Med Phys. 2001;28:1857–1874. doi: 10.1118/1.1395026. [DOI] [PubMed] [Google Scholar]
- 47.Roeske JC, Stinchcomb TG. J Nucl Med. 1997;38:195–201. [PubMed] [Google Scholar]
- 48.Stinchcomb TG, Roeske JC. Med Phys. 1999;26:1960–1971. doi: 10.1118/1.598701. [DOI] [PubMed] [Google Scholar]
- 49.Roeske JC, Stinchcomb TG. Radiat Res. 2000;153:16–22. doi: 10.1667/0033-7587(2000)153[0016:tcpmfa]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 50.Belyakov OV, Prise KM, Trott KR, Michael BD. Int J Radiat Biol. 1999;75:985–993. doi: 10.1080/095530099139746. [DOI] [PubMed] [Google Scholar]
- 51.Prise KM, Belyakov OV, Folkard M, Michael BD. Int J Radiat Biol. 1998;74:793–798. doi: 10.1080/095530098141087. [DOI] [PubMed] [Google Scholar]
- 52.Mothersill C, Seymour C. Int J Radiat Biol. 1997;71:421–427. doi: 10.1080/095530097144030. [DOI] [PubMed] [Google Scholar]
- 53.Zalutsky MR, Reardon DA, Pozzi OR, Vaidyanathan G, Bigner DD. Nucl Med Biol. 2000;34(7):779–785. doi: 10.1016/j.nucmedbio.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mairs RJ, Fullerton NE, Zalutsky MR, Boyd M. Int J Dose Response. 2007;5(3):204–213. doi: 10.2203/dose-response.07-002.Mairs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rotmensch J, Atcher RW, Schlenker R, Hines J, Grdina D, Block BS, Press MF, Herbst AL, Weichselbaum R. Gynecol Oncol. 1987;32:236–239. doi: 10.1016/s0090-8258(89)80040-x. [DOI] [PubMed] [Google Scholar]
- 56.Rotmensch J, Atcher RW, Hines J, Grdina D, Schwartz JS, Toohil M, Herbst AL. Am J Obstet Gynecol. 1989;160:789–797. doi: 10.1016/0002-9378(89)90293-7. [DOI] [PubMed] [Google Scholar]
- 57.Ruble G, Wu C, Squire RA, Gansow OA, Strand M. Int J Radiat Oncol Biol Phys. 1996;34:609–616. doi: 10.1016/0360-3016(95)02119-1. [DOI] [PubMed] [Google Scholar]
- 58.Jones SB, Tiffany LJ, Garmestani K, Gansow OA, Kozak RW. Nucl Med Biol. 1996;23:105–113. doi: 10.1016/0969-8051(95)02006-3. [DOI] [PubMed] [Google Scholar]
- 59.Agus DB, Bunn PA, Franklin W, Garcia M, Ozols RF. Semin Oncol. 2000;27:53–63. [PubMed] [Google Scholar]
- 60.Ross JS, Mckenna BJ. Cancer Invest. 2001;19:554–568. doi: 10.1081/cnv-100103852. [DOI] [PubMed] [Google Scholar]
- 61.Horak E, Hartmann F, Garmestani K, Wu C, Brechbiel MW, Gansow OA, Hartmann NF, Waldmann TA. J Nucl Med. 1997;38:1944–1950. [PubMed] [Google Scholar]
- 62.Nahum A. Phys Med Biol. 1996;41:1957–1972. doi: 10.1088/0031-9155/41/10/008. [DOI] [PubMed] [Google Scholar]
- 63.Milenic DE, Garmestani K, Brady ED, Albert PS, Ma D, Abdulla A, Brechbiel MW. Cancer Biother Radiopharm. 2005;20:557–568. doi: 10.1089/cbr.2005.20.557. [DOI] [PubMed] [Google Scholar]
- 64.Gold DV, Schutsky K, Modrak D, Cardillo TM. Clin Cancer Res. 2003;9:3929–3937. [PubMed] [Google Scholar]
- 65.Gold DV, Modrak D, Schutsky K, Cardillo TM. Int J Cancer. 2004;109:618–626. doi: 10.1002/ijc.20004. [DOI] [PubMed] [Google Scholar]
- 66.Milenic DE, Garmestani K, Brady ED, Abdulla A, Flynn J, Brechbiel MW. Clin Cancer Res. 2007;13:1926–1935. doi: 10.1158/1078-0432.CCR-06-2300. [DOI] [PubMed] [Google Scholar]
- 67.Burke PA, Denardo SJ, Miers LA, Kukis DL, Denardo GL. Cancer. 2002;94:1320–1331. doi: 10.1002/cncr.10303. [DOI] [PubMed] [Google Scholar]
- 68.Denardo SJ, Kukis DL, Kroger LA, O’Donnell RT, Lamborn KR. Proc Natl Acad Sci USA. 1997;94:4000–4004. doi: 10.1073/pnas.94.8.4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Milenic DE, Garmestani K, Brady ED, Baidoo KE, Albert PS, Wong KJ, Flynn J, Brechbiel MW. Clin Cancer Res. 2008;14:5108–5115. doi: 10.1158/1078-0432.CCR-08-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rosenow MK, Xucchini GL, Bridwell PM, Stuart FP, Fridman AM. Int J Nucl Med Biol. 1983;10:189–197. doi: 10.1016/0047-0740(83)90078-5. [DOI] [PubMed] [Google Scholar]
- 71.Hassfjell SP, Hoff P, Bruland OS, Alstad J. J Label Compd Radiopharm. 1994;34(8):717–734. [Google Scholar]
- 72.Hassfjell SP, Bruland OS, Hoff P. Nucl Med Biol. 1997;24:231–237. doi: 10.1016/s0969-8051(97)00059-0. [DOI] [PubMed] [Google Scholar]
- 73.Diener MD, Alford JM, Kennel SJ, Mirzadeh S. J Am Chem, Soc. 2007;129:5131–5138. doi: 10.1021/ja068639b. [DOI] [PubMed] [Google Scholar]
- 74.Miao Y, Hylarides M, Fisher DR, Shelton T, Moore H, Wester DW, Fritzberg AR, Winkelmann CT, Hoffman T, Quinn TP. Clin Cancer Res. 2005;11:5616–5621. doi: 10.1158/1078-0432.CCR-05-0619. [DOI] [PubMed] [Google Scholar]
- 75.Theodore LJ, Fritzberg AR, Schultz JE, Axworthy DB. Radioimmunotherapy of cancer. NeoRx Corporation; Seattle, Washington: 2000. pp. 195–221. [Google Scholar]
- 76.Su FM, Beaumier P, Axworthy D, Atcher R, Fritzberg A. Nucl Med Biol. 2005;32:741–747. doi: 10.1016/j.nucmedbio.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 77.Garmestani K, Milenic DE, Brady ED, Plascjak PS, Brechbiel MW. Nucl Med Biol. 2005;32:301–305. doi: 10.1016/j.nucmedbio.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 78.Milenic DE, Roselli M, Brechbiel MW, Pippin CG, McMurray TJ, Carrasquillo JA, Colcher D, Lambrecht R, Gansow OA, Schlom J. Eur J Nucl Med. 1998;25:471–480. doi: 10.1007/s002590050246. [DOI] [PubMed] [Google Scholar]
- 79.Miao Y, Figueroa SD, Fisher DR, Shelton T, Moore H, Testa RF, Hoffman TJ, Quinn TP. Melanona Imag. 2008;49:823–829. doi: 10.2967/jnumed.107.048553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Halime Z, Lachkar M, Boitrel B. Biochimie. 2009;91:1318–1320. doi: 10.1016/j.biochi.2009.03.001. [DOI] [PubMed] [Google Scholar]