

Abstract
Deletion of phenylalanine residue 508 (ΔF508) in the cystic fibrosis (CF) transmembrane conductance regulator protein (CFTR) is a major cause of CF. Small molecule ‘correctors’ of defective ΔF508-CFTR cellular processing hold promise for CF therapy. We previously identified and characterized bithiazole CF corrector 1 and s-cis-locked bithiazole 2. Herein, we report the regiodivergent synthesis of Nγ and Nβ isomers of thiazole-tethered pyrazoles with improved hydrophilicity compared to bithiazoles. We synthesized a focused library of fifty-four pyrazolylthiazoles 3, which included examples of both regioisomers 4 and 5. The thiazole-tethered pyrazoles allowed incorporation of property-modulating functionality on the pyrazole ring – ester, acid, and amide – while retaining ΔF508-CFTR corrector activity (EC50) of under 1 μM. The most active pyrazolylthiazole (14h) has an experimentally determined logP of 4.1, which is 1.2 log units lower than bithiazole CF corrector 1.
Introduction
Cystic fibrosis (CFa), a serious disease afflicting ~1 in 2,500 in the Caucasian population,1 is caused by inherited mutations in the CF transmembrane conductance regulator (CFTR) gene.2 The most common disease-causing mutation of CFTR is ΔF508, a deletion of phenylalanine at position 508, which accounts for ~70% of all CF alleles such that ~90% of CF patients carry at least one copy of ΔF508-CFTR allele.3 The ΔF508 mutation alters CFTR biosynthesis resulting in a misfolded, rapidly degraded protein that is poorly trafficked out of the endoplasmic reticulum to the cell surface.4 Without physiological CFTR chloride channel functioning at epithelia cell membranes in lung, pancreas and intestine, water and salt secretion are compromised, causing chronic respiratory infections, airway obstruction by viscous mucous, pancreatic exocrine insufficiency, etc.5
Small-molecule therapy for ΔF508-caused CF is thought to have considerable promise.6 In our previous work, bithiazole lead compound 1 (Figure 1) was identified as a ‘corrector’ capable of partially restoring the targeting of ΔF508-CFTR to the cell membrane in transfected cells and primary cultures of bronchial epithelial cells from ΔF508 patients.7 In follow-up work, nanomolar corrector potency of the bithiazole class was obtained by locking the thiazole core of 1 into an s-cis conformation (see 2 in Figure 1).8 Although s-cis-locked bithiazole corrector 2 affords improved potency compared to the original corrector 1, its high hydrophobicity (clogP 6.65) is a concern for further development. Therefore, our focus has been to identify a new chemotype, based on the original bithiazole scaffold, that would allow for incorporation of property-modulating functionality while retaining corrector activity. Because of the lack of useful crystal structure data for ΔF508-CFTR, we embarked on a ligand-based compound design strategy wherein many of the core structural features of our previously identified bithiazole CF correctors were retained while simultaneously incorporating property-modulating modifications with the aim of improving hydrophilicity relative to 1. Target compounds were synthesized and screened for corrector activity. This effort led us to a pyrazolylthiazole core (3) and the production of a focused library of pyrazolylthiazoles, including regioisomers 4 and 5. As Figure 1 illustrates, pyrazole 3 retains many aspects of the core framework of 1 but replaces the right-hand thiazole with a pyrazole ring. Compared to the thiazole of 1, the pyrazole ring accommodates the attachment of extra functionality, such as a C5 (see R3 in 4) or C3 (see R3 in 5) amide, to modulate properties such as logP.
Figure 1.

SAR around early bithiazole lead 1 led to s-cis-locked bithiazole 2, but these compounds have poor hydrophilicity. The structural features of these bithiazoles inspired the design of pyrazolylthiazole libraries based on differentially Nβ- and Nγ-substituted analogs 4 and 5, respectively.
One vexing drawback with the bithiazole core of 1 is that it essentially precludes modification. For example, consider the right-hand thiazole ring of 1 (red substructure): positions S1, C2, N3, and C4 are fixed and addition of substituents at C5 forces the two thiazole rings to adopt an orthogonal posture that is quite different from the conformation of s-cis-locked 2 – our best bithiazole corrector. In contrast, pyrazoles, which are considered a privileged heterocyclic structure with many reports of biological activity,9 allow for modification at C5 (see 3), Nβ (see 4), and Nγ (see 5). As illustrated in Figure 1, we reasoned that, as a starting point, retaining the N-(4-methylthiazol-2-yl)pivalamide on the left-hand thiazole ring and the 5-chloro-2-methoxyaryl on the right-hand thiazole ring (see bithiazole 1) might impart corrector activity to pyrazolylthiazole 3 while allowing for the introduction of property-modulating functionality “X” at C5 (see 3) of the pyrazole ring. However, there are two important differences caused by the thiazole → pyrazole modification. First, the aniline moiety of 1 is replaced with a benzylic moiet in 3; this might cause the loss of a possible hydrogen bonding interaction with the target protein. Second, the thiazole “S” is replaced with a pyrazole “C–X” (X = carboxylic acid derivative in this work); this might cause non-productive interaction with the target protein. On the other hand, the X-moiety could be exploited to improved drug properties, such as logP in this work, and might also lead to alternative productive interactions with the target protein. These two thiazole → pyrazole implications are highlighted by the ellipses in 1 versus 3 as well as the red substructures in 4 and 5 (Figure 1). To investigate the consequences of these structural changes vis-à-vis the discovery of a new CF corrector chemotype, we designed a focused library based on the pyrazolylthiazole scaffold wherein the substituents adorning the pyrazole substructure are varied.
Results and Discussion
Chemistry
Pyrazoles can be synthesized through a Knorr cyclocondensation between a 1,3-diketone and hydrazine or a substituted hydrazine.10 This general method often suffers from lack of regioselectivity in the production of substituted pyrazoles.11 There are only sporadic reports addressing the synthesis of pyrazolylthiazoles by this condensation reaction.12 However, none of these methods involve diversification of this heteroatom-rich bisazole or delineate routes to acquire both N-regioisomers of the pyrazole from a common intermediate. Our discovery efforts led us to explore general and integrated synthetic methods to access a series of functionally and regioisomerically diversified pyrazolylthiazoles; the results are reported herein.
This synthetic effort began with conversion of the amino group of 6 into amide 7 (Scheme 1; two variants – pivaloyl and benzoyl – were prepared based on our bithiazole work) through a CDI-mediated coupling reaction, which occurred in greater than 79% yield. Both of these amides demonstrated corrector activity in prior bithiazole studies.7 Amide 7 was then subjected to Claisen reaction with diethyl oxalate in the presence of 2.1 equivalents of LHMDS, giving 1,3-diketone 8 in excellent yield (>90%). With the 2,4-dioxo-4-(thiazol-5-yl)butanoate 8 in hand, we turned to pyrazole formation and expected both pyrazole regioisomers, 9 and 10, in the reaction with mono-substituted hydrazines. In fact, the condensation reaction between 8 and substituted hydrazines proceeds with excellent selectivity to deliver the Nγ regioisomers 9 in good yield (>87%; 9:10>10:1). A minor by-product was found to be 7, indicating that the 2,4-dioxobutanoate moiety is susceptible to a reverse Claisen reaction in the presence of hydrazine nucleophiles. The excellent regioselectivity of this condensation reaction reflects the fact that the two carbonyl groups in 8 have very different electrophilicity. From pyrazolylthiazole 9, the final diversification in this series was achieved by converting the ester moiety into the corresponding amide (→11) via aminolysis in good yield (>73%) or acid (→ 12) by saponification. Thirty-nine Nγ-substituted pyrazolylthiazoles were prepared (see Table 1) by varying all three diversity points (two R1 inputs, four R2 inputs, and four NR3R3′ inputs). The X-ray crystal structure determined for one Nγ-substituted analog (9b) is shown in Figure 2.
Scheme 1.

Synthesis of Nγ-substituted pyrazolylthiazoles.
Reagents and conditions: (a) R1CO2H, CDI, DMF, 85 °C (b) Diethyl oxalate, LHMDS, THF, −78 °C (c) R2NHNH2, EtOH (d) R3R3′NH, AlMe3, DCM, 0 °C (e) NaOH, THF/H2O (f) Ethanol amine, MW, 160 °C, 30 Min
Table 1.
Nγ-Substituted pyrazolylthiazole analogs.
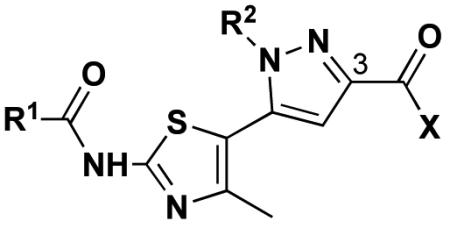
R1 = C(CH3)3, C6H5
R2 = CH2CH=CH2, C6H4(4-Br), CH2C6H5, (CH2)2OH
X = OEt, NHC6H4(4-OMe), NHCH2C6H5, NH(CH2)2OH, N(CH2CH2)2O, OH
| Compd | R1 | R2 | X |
|---|---|---|---|
| 9a | C(CH3)3 | CH2CH=CH2 | OEt |
| 9b | C(CH3)3 | C6H4(4-Br) | OEt |
| 9c | C(CH3)3 | CH2C6H5 | OEt |
| 9d | C(CH3)3 | CH2CH2OH | OEt |
| 9e | C6H5 | CH2CH=CH2 | OEt |
| 9f | C6H5 | C6H4(4-Br) | OEt |
| 9g | C6H5 | CH2C6H5 | OEt |
| 9h | C6H5 | CH2CH2OH | OEt |
| 11a | C(CH3)3 | CH2CH=CH2 | NHC6H4(4-OMe) |
| 11b | C(CH3)3 | CH2CH=CH2 | NHCH2C6H5 |
| 11c | C(CH3)3 | CH2CH=CH2 | N(CH2CH2)2O |
| 11d | C(CH3)3 | C6H4(4-Br) | NHC6H4(4-OMe) |
| 11e | C(CH3)3 | C6H4(4-Br) | NHCH2C6H5 |
| 11f | C(CH3)3 | C6H4(4-Br) | N(CH2CH2)2O |
| 11g | C(CH3)3 | CH2C6H5 | NHC6H4(4-OMe) |
| 11h | C(CH3)3 | CH2C6H5 | NHCH2C6H5 |
| 11i | C(CH3)3 | CH2C6H5 | N(CH2CH2)2O |
| 11j | C(CH3)3 | CH2CH2OH | NHC6H4(4-OMe) |
| 11k | C(CH3)3 | CH2CH2OH | NHCH2C6H5 |
| 11l | C(CH3)3 | CH2CH2OH | N(CH2CH2)2O |
| 11m | C6H5 | CH2CH=CH2 | NH(CH2)OH |
| 11n | C6H5 | C6H4(4-Br) | NH(CH2)OH |
| 11o | C6H5 | CH2C6H5 | NH(CH2)OH |
| 11p | C6H5 | CH2CH2OH | NH(CH2)OH |
| 11q | C6H5 | CH2CH=CH2 | NHC6H4(4-OMe) |
| 11r | C6H5 | C6H4(4-Br) | NHC6H4(4-OMe) |
| 11s | C6H5 | CH2C6H5 | NHC6H4(4-OMe) |
| 11t | C6H5 | CH2CH2OH | NHC6H4(4-OMe) |
| 11u | C6H5 | CH2CH=CH2 | N(CH2CH2)2O |
| 11v | C6H5 | C6H4(4-Br) | N(CH2CH2)2O |
| 11w | C6H5 | CH2C6H5 | N(CH2CH2)2O |
| 11x | C6H5 | CH2CH2OH | N(CH2CH2)2O |
| 12a | C(CH3)3 | CH2CH=CH2 | OH |
| 12b | C(CH3)3 | C6H4(4-Br) | OH |
| 12c | C(CH3)3 | CH2C6H5 | OH |
| 12d | C6H5 | CH2CH=CH2 | OH |
| 12e | C6H5 | C6H4(4-Br) | OH |
| 12f | C6H5 | CH2C6H5 | OH |
| 12g | C6H5 | CH2CH2OH | OH |
Figure 2.

X-ray crystallographic structure of Nγ-substituted pyrazolylthiazole 9b (note: the S1′-C5′-C5-N1 dihedral angle is 62.4°).
Since the Nβ-substituted pyrazole regioisomer could not be accessed in practical yield through the direct condensation of 8 with mono-substituted hydrazines, an alternate route was explored to synthesize these compounds (Scheme 2). First, 8 was reacted with hydrazine monohydrate to deliver pyrazole 13 in 63% yield. The Nβ position of 13 was then selectively alkylated, giving mainly 10 (R = H) in 59-66% yield when R1 = pivaloyl (10:9 = 8:1). The major by-product of this reaction is N-alkylation of the amide moiety leading to formation of bis-alkylated product (10: R = R2). This complication was so severe with the benzamide analog of 13 (R1 = C6H5) that the major product was, in fact, the bisalkylation product (10: R = R2); the desired mono-alkylated product (10: R = H) was obtained in only 20% yield. Therefore, final diversification on the Nβ-substituted isomers was mainly focused on pivaloyl amide analogs of 10 and consisted of hydrolysis or aminolysis to deliver 14 and 15, respectively. Unlike the relatively easy aminolysis of 9 (Scheme 1) that proceeds at 0 °C, initial attempts at the aminolysis of 10 (0 °C in DCM; Scheme 2) failed to deliver 14. X-ray crystallographic analysis of 10a (R1 = tBu/R2 = allyl; Figure 3) reveals that the R2 substituent imparts significant steric hindrance at the carboethoxy and retards amidyl formation at the pyrazole C5 position. This challenge was overcome by employing microwave irradiation in the AlMe3-mediated aminolysis, delivering various amides and completing pyrazole diversification. In total, fifteen Nβ-substituted pyrazolylthiazoles were prepared for this focused library (see Table 2).
Scheme 2.

Synthesis of Nβ-substituted pyrazolylthiazole.
Reagents: (a) NH2NH2·H2O, EtOH (b) R2Br, K2CO3, Acetone, 60 °C (c) R3R3′NH, AlMe3, DCM, MW, 100 °C (d) NH2(CH2)2OH, EtOH, MW 180 °C (e) NaOH, THF/H2O.
Figure 3.

X-ray crystallographic structure of Nβ-substituted pyrazolylthiazole 10a (note: the S1′-C5′-C3-N2 dihedral angle is 2.6°).
Table 2.
Nβ-Substituted pyrazolylthiazole analogs.
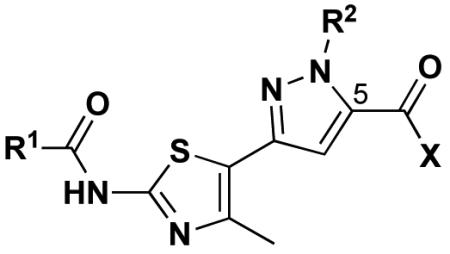
R1 = C(CH3)3
R2 = CH2CH=CH2, C6H4(4-Br), CH2C6H3(2-OMe/4-Cl)
X = OEt, NHC6H4(4-OMe), NHCH2C6H5, N(CH2CH2)2O, NH(CH2)2OH, OH
| Compd | R1 | R2 | X |
|---|---|---|---|
| 10a | C(CH3)3 | CH2CH=CH2 | OEt |
| 10b | C(CH3)3 | CH2C6H5 | OEt |
| 10c | C(CH3)3 | CH2C6H3(2-OMe/4-Cl) | OEt |
| 14a | C(CH3)3 | CH2CH=CH2 | NHC6H4(4-OMe) |
| 14b | C(CH3)3 | CH2CH=CH2 | NHCH2C6H5 |
| 14c | C(CH3)3 | CH2CH=CH2 | N(CH2CH2)2O |
| 14d | C(CH3)3 | CH2C6H5 | NHC6H4(4-OMe) |
| 14e | C(CH3)3 | CH2C6H5 | NHCH2C6H5 |
| 14f | C(CH3)3 | CH2C6H5 | N(CH2CH2)2O |
| 14g | C(CH3)3 | CH2C6H3(2-OMe/4-Cl) | NHC6H4(4-OMe) |
| 14h | C(CH3)3 | CH2C6H3(2-OMe/4-Cl) | N(CH2CH2)2O |
| 14i | C(CH3)3 | CH2C6H3(2-OMe/4-Cl) | NHCH2C6H5 |
| 14j | C(CH3)3 | CH2C6H3(2-OMe/4-Cl) | NH(CH2)2OH |
| 15a | C(CH3)3 | CH2CH=CH2 | OH |
| 15b | C(CH3)3 | CH2C6H5 | OH |
Structure-Activity Relationships
The Nγ- and Nβ-regioisomeric pyrazolylthiazoles were assayed for ΔF508-CFTR corrector activity. Our established cell-based corrector assay was used in which I− influx was measured in FRT cells coexpressing human ΔF508-CFTR and the I−-sensitive fluorescent sensor YFP-H148Q/I152L.13 Following 24 h incubation with test compounds, I− influx was determined from the kinetics of YFP-H148Q/I152L quenching in response to I− addition in cells treated with a cAMP agonist and the potentiator genistein. Out of the fifty-four compounds tested, eight had significant corrector activity as judged by concentration-dependent increases in I− influx as exemplified in Figure 4 for 14g and 14h.
Figure 4.

Dose-response relation for increased I− influx in ΔF508-CFTR cells treated with pyrazolylthiazoles: 14g (in black) and 14h (in red).
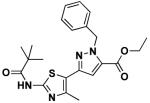
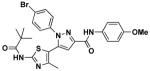
Structures for these compounds as well as their EC50 and Vmax values from ion influx data are summarized in Table 3. Their EC50 values range from 0.93 to 8.5 μM while increasing I− influx up to 7.3 μM/s (note: increased I− influx is a quantitative measure of effective ΔF508-CFTR corrector activity). As illustrated in Table 3, pyrazolylthiazole 14h is the best corrector among the eight hits and of comparable potency to our previously reported bithiazole 1. Of the eight active compounds, 10b, the only ester, and 11d, the only Nγ-substituted pyrazole, are the least effective pyrazolylthiazole correctors. Considering that ester-containing pyrazolylthiazoles 9 and 10 are all inactive except for 10b, the carboxamide group at C5 of the pyrazole ring seems to be an important determinant of activity. Another observation from Table 3 is that the majority of active pyrazolylthiazoles are Nβ-substituted pyrazole isomers; there is only one active Nγ-substituted corrector (11d) and it has relatively low activity.
Table 3.
Pyrazolylthiazole DF508-CFTR corrector activity.
| corrector structure |
corrector number |
EC50 (μM)a | Vmax (μM/s)b |
|---|---|---|---|
|
10b | 8.5 | 2.6 |
|
11d | 3.4 | 2.5 |
|
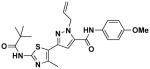
14a | 0.93 | 0.5 |
|
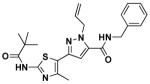
14b | 3.0 | 1.4 |
|
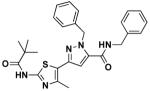
14e | 0.75 | 1.0 |
|
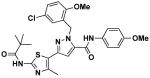
14g | 1.0 | 6.1 |
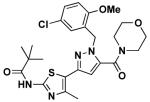
|
14h | 1.0 | 7.26 |
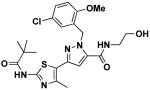
|
14j | 3.0 | 2.4 |
Concentration where the increased I− influx is 50% of Vmax.
Vmax is maximum increase in I− influx due to compound effect.
An understanding of the active geometries of our compounds is necessary to develop insights regarding the ΔF508-CFTR binding site. Molecular models were constructed based on the X-ray crystal structures of bisazoles 9b (Figure 2) and 10a (Figure 3) to examine these various geometries). Image A in Figure 5 depicts initial lead compound 1 in its effective s-cis conformation.8 In this conformation, the bithiazole rings are nearly coplanar, which we believe positions the aniline moiety to extend into an important binding pocket.8 Our least active compound, 11d (image B), is forced to adopt a non-planar geometry about the pyrazolylthiazole core with a tethering S-C-C-N dihedral geometry of 62.4°. This conformational change (planar to nearly orthogonal) relative to 1 is the consequence of steric interactions between the Nγ 4-bromophenyl substituent on the pyrazole ring and the neighboring thiazole ring; the X-ray crystal structure of 9b (the ester precursor to amide 11d; Figure 2) reflects this same conformational predisposition. As discussed in other published work,8 we speculate that a planar arrangement of the bis-heterocycle core is vital for corrector activity and the orthogonal posture of Nγ-substituted analogs may explain why most of these pyrazolylthiazoles are inactive. These structural insights are further supported by conformational analysis of 14g – one of our most potent pyrazolylthiazole (image C). Nβ substitution on the pyrazole ring of 14g allows the two heterocycles to adopt a nearly coplanar conformation without confronting steric congestion. Indeed, the bisazole dihedral angle in crystalline Nβ-substituted pyrazole analog 10a is 2.6° (e.g., nearly planar; see also the X-ray crystal structure of 10a in Figure 3). Image C shows the s-cis conformation of bisazole 14g in which the Nβ benzyl moiety can extend into the important binding pocket discussed above (in the context of 11d).8 In contrast, the s-trans conformation of 14g (image D), obtained by an ~180° rotation around the tethering thiazole-pyrazole bond, allows the C5 carboxamide moiety to occupy this binding pocket. These possibly “dually active conformations” of 14g may account for some of the corrector activity of bisazole 14g compared to, for example, bisazole 14j.
Figure 5.

Bisazole molecular models. A: 1 – s-cis bithiazole conformation (active conformation; see s-cis-locked bithiazole 2 in Figure 1). B: 11d – Nγ substituted pyrazole ring nearly orthogonal to the thiazole ring. C: 14g – Nβ substituted s-cis pyrazolylthiazole → places Nβ benzyl moiety in the same region of space as the aniline moiety in 1. D: 14g – Nβ substituted s-trans pyrazolylthiazole → places the C5 amide moiety in the same region of space as the aniline moiety in 1.
As various carboxamide-substituted Nβ-substituted pyrzoles are active (see 14a/b/e/g/h/j) while the majority of the corresponding esters are not, the carboxamide group at C5 of the pyrazole seem to play a significant role in effecting corrector activity. Noting that 14h, our most active pyrazolylthiazole, has an amide derived from a secondary amine while all other active pyrazolylthiazoles incorporate amides derived from primary amines, suggests that the carboxamide might function as hydrogen bond acceptor (as opposed to functioning as an H-bond donor). As discussed above and illustrated in Figure 6a, we also note that free rotation about the thiazole—pyrazole C,C-tether of Nβ-substituted pyrazolylthiazoles places the Nβ benzyl moiety “B” of this heterocycle, in its s-cis configuration, in nearly the same relative position as the aniline moiety “A1” of the active8 s-cis conformer bithiazole (see left and center structures in Figure 6a, respectively). This conformational homology is not possible in Nγ-substituted pyrazolylthiazoles.
Figure 6.

a) Pyrazolylthiazole (s-cis/s-trans) vs. s-cis-locked bithiazole conformational interplay where the Nβ benzyl (B) moiety can overlay the C2 thiazole aniline (A1) moiety or the C5 pyrazole amide (A2) moiety can overlay the C2 thiazole aniline (A1) moiety. b) Superimposed wireframe models of bithiazole 1 and thiazolylpyrazole 14a.
A superimposition of s-cis bithiazole 1 and s-trans conformer pyrazolylthiazole 14a (see Figure 6b) illustrates another interesting point. Specifically, the s-trans conformer pyrazolylthiazole can place its C5 amide moiety (A2 on the pyrazole) in a similar position as the aniline (e.g., A1) moiety of the s-cis bithiazole conformer. Consequently, Nβ substituted pyrazoles can also, apparently, be active by placing the C5 amide moiety (A2) in the binding site addressed by the aniline moiety (A1) of bithiazoles 1 or 2 (see right and center structures in Figure 6a, respectively).
It is most interesting to note that Nβ-substituted pyrazolylthiazole 14j, which presents a 5-chloro-2-methoxybenzyl substituent at Nβ and a C5 amide derived from 2-aminoethanol, retains corrector activity. As illustrated in Figure 6a, this intriguing result suggests that perhaps the active presentation of 14j is its s-cis conformation (compare the s-cis pyrazolylthiazole and s-cis-locked bithiazole conformations). In contrast, pyrazolylthiazoles 14a and 14b may be active through their s-trans conformation (compare the s-trans pyrazolylthiazole and the s-cis-locked bithiazole conformations). The implication here is that pyrazolylthiazoles 14e, 14g, and 14h – with aryl-containing benzyl and amide moieties – may be active through either (or both) their s-cis or their s-trans conformations. Studies employing additional pyrazolylthiazole analogs are underway to probe/validate these initial insights.
LogP measurements
LogP represents a compound’s partition coefficient log value determined from octanol versus water, where a smaller logP correlates with better water solubility. LogP is a well-established parameter for the ADME profiling as it has implications in solubility, absorption, distribution, metabolism, and excretion14 – which are important for orally administered drugs15 – and, according Lipinski’s rule of 5,14 should generally be <5 for good bioavailability.
LogP can be related to the experimentally determined capacity factor k by measuring the retention time of the compound using reverse-phase HPLC.16, 17 (see Supporting Information for ClogP values). A standard calibration curve is constructed using compounds with known logP values and experimentally determined logk values (see diamond data points in Figure 7); this standard calibration curve is then used to correlate logk (determined from measured retention time; see Supporting Information) with logP for each pyrazolylthiazole corrector. These data for 11b and 14a/b/e/g/h/j are depicted in Figure 7. Most of the active pyrazolylthiazole correctors (squares in Figure 7) have logP values of less than 5, while bithiazole 1 has a logP greater than 5. The most active pyrazolylthiazole, 14h, has a logP value of 4.1, which is ~1.2 logP units better than bithiazole 1, and 14j has a logP of 3.5.
Figure 7.

Standard calibration curve correlating experimentally determined capacity factor k with logP. Data are shown for reference compounds (◆: A = 4-chlorophenol, B = 2,4-dichlorophenol, C = 3,4,5-trichlorophenol, D = pentachlorophenol, E = p,p’-DDT), bithiazole 1, and pyrazolylthiazole 11d/14a/14b/14e/14g/14h/14j.
Conclusions
Synthetic access to both Nγ- and Nβ-substituted regioisomeric pyrazolylthiazoles was achieved by regiodivergent synthesis from the common intermediate 8. Of the fifty-four pyrazolylthiazoles prepared, eight demonstrated significant ΔF508-CFTR corrector activity. This new class of bisazole correctors was designed by replacing one thiazole ring of our previously reported bithiazole correctors with a pyrazole ring. The resulting pyrazolylthiazole correctors diversify the bisazole corrector family, function as a new chemotype with corrector activity, and allow for incorporation of additional functionality to modulate drug properties such as logP. The work reported here led to the discovery of new correctors with improved hydrophilicity and with a structural core that accommodates diversification. Ongoing studies will capitalize on the structural, conformational, and logP insights reported here.
Experimental Section
All purchased starting materials and regents were used without further purification. Product purification was performed either on an automated flash chromatography system (Combiflash by Teledyne: 35 min of elution with linear gradient from 100% hexane to 100% EtOAc solvent) with silica gel columns or on an HPLC system [Waters:15 mL/min flow rate, linear gradient elution with 0.1% TFA-containing H2O/MeCN from 5-95% MeCN in 20 min. Xterra Prep MS C18 OBD column (19 mm × 100 mm) and dual wavelength absorbance detector]. NMR spectra (1H at 600 MHz; 13C at 151 MHz) were recorded in CDCl3 solvent on a Varian 600. Chemical shifts are expressed in parts per million relative to internal TMS or solvent. Coupling constants are expressed in units of hertz (Hz). Splitting patterns are designated as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), and bs (broad singlet). LC/MS (Waters Micromass ZQ) specifications are as follows: electrospray (+) ionization, mass ranging from 100 to 900 Da, 20-V cone voltage. LC: Xterra MS C18 column (2.1 mm × 50mm × 3.5 μm), 0.2 mL/min water/acetonitrile (containing 0.1% TFA), 30 min linear gradient 0-100% acetonitrile. The LC/MS UV detector is a diode array with 200-400nm wave length range. Purity is based on the peak area percentage of the UV diode array signals. Compound purities by RP-HPLC were ≥95%.
Δ508-CFTR corrector activity assay
FRT epithelial cells stably coexpressing human ΔF508-CFTR and the high-sensitivity halide-sensing fluorescent protein YFP-H148Q/I152L18 were used as described previously.6 Cells were grown at 37° C (95% air / 5% CO2) for 24 h and then incubated for 16 - 20 h with 50 μL of medium containing the test compound. At the time of the assay, cells were washed with PBS and then incubated with PBS containing forskolin (20 μM) and genistein (50 μM) for 20 min. Measurements were carried out using aFLUOstar fluorescence plate reader (Optima; BMG LABTECH Gmbh) equipped with 500 ± 10 nm excitation and 535 ± 15 nm emission filters (Chroma Technology Corp.). Each well was assayed individually for I− influx by recording fluorescence continuously (200 ms per point) for 2 s (baseline) and then for 12 s after rapid (<1 second) addition of 165 μL PBS in which 137 mM Cl− was replaced by I−. Initial I− influx rate was computed exponential regression. All experiments contained negative control (DMSO vehicle) and positive control [N-(2-(5-chloro-2-methoxyphenylamino)-4′-methyl-4,5′-bithiazol-2′-yl)benzamide].
1-(2-Amino-4-methylthiazol-5-yl)ethanone HCl (6)
Thiazole 6 was prepared as described in the literature.19
General procedure A: Preparation of 8 via CDI-mediated Amide Formation
Carboxylic acid (1.75 equiv) was dissolved in DMF (3.3 mL/mmol of carboxylic acid) and carbonyldiimidazole (CDI; 1.75 equiv) was added slowly to manage CO2 evolution. After all the CDI had been added, the solution was stirred for an additional 15 min at which point thiazole HCl salt 6 (1 equiv). The reaction mixture was warmed to 85 °C and stirred for 20 h. After the reaction was complete, the reaction mixture was cooled to room temperature and poured into water (17 mL/mmol of carboxylic acid) to precipitate the product, which was then collected by filtration, washed with water (3×200ml), and dried under vacuum at 100°C for 18 h to deliver 7.
N-(5-acetyl-4-methylthiazol-2-yl)pivalamide (7a)
Pivalic acid (5.0 g, 49 mmol) was reacted with the thiazole·HCl 6 (5.4 g, 28 mmol) by general procedure A and 7a was obtained as an off-white solid (5.3 g, 79%). 1H NMR (600 MHz, CDCl3) δ 9.06 (s, 1H), 2.64 (s, 3H), 2.51 (s, 3H), 1.34 (s, 9H); 13C NMR (151 MHz, CDCl3) δ 190.66, 176.46, 158.72, 155.21, 125.30, 39.29, 30.44, 27.16 18.08; LC/MS: cal. [M+H+]= 241.10, found 241.12.
(Z)-Ethyl 2-hydroxy-4-(4-methyl-2-pivalamidothiazol-5-yl)-4-oxobut-2-enoate (8a)
To a solution of LHMDS (1M; 50.3 mL, 50.3 mmol) in THF (50 mL) cooled to −78°C was slowly added 7a (5.5 g, 22.9 mmol) in dry THF (100 mL) via a syringe and the mixture was stirred for 30 min. Diethyl oxalate (3.7 mL, 27.4 mmol) was added quickly in one portion and the resulting mixture was stirred at −78°C for 2 h before being being allowed to warm to room temperature for another 2 h. When the reaction was completed, water (50 mL) and 1N aq. HCl (50 mL) were sequentially added to the reaction mixture and the product was extracted with EtOAc (3 × 100 mL). The combined organic extract was washed with brine twice and dried over MgSO4. Filtration and solvent removal under vacuum delivered the crude product which was purified on a silica gel column with automated flash chromatography (solvent system: gradient hexane/ethyl acetate) to give 8 as a yellow solid (7.22 g, 92%). 1H NMR (600 MHz, CDCl3) δ 9.08 (s, 1H), 6.70 (s, 1H), 4.39 (q, J = 6, 2H), 2.71 (s, 3H), 1.40 (t, J = 6, 3H), 1.35 (s, 9H); 13C NMR (151 MHz, CDCl33) δ 186.24, 176.56, 165.29, 162.01, 160.00, 157.46, 123.06, 101.63, 62.57, 39.33, 27.08, 18.60, 14.12; LC/MS: ESI-MS, cal. [M+H+]= 341.12, found 341.04.
General Procedure B: Preparation of Nγ-Substituted Pryazoles, 9 via Cyclocondensation with 8
A mixture of substituted hydrazine (or hydrazine hydrochloride; 1.05 equivalent) and 8 in absolute ethanol (3.3 mL/mmol of 8) was stirred at room temperature for 18 h. After the starting material was consumed as indicated by TLC, the solvent was removed by rotoevaporation and the concentrate was extracted with ethyl acetate (3.3 mL/mmol of 8) and washed with water. The ethyl acetate layer was dried over Na2SO4, filtered, and concentrated under reduced pressure to deliver the crude Nγ-substituted pyrazole product, which was then purified by silica gel column chromatography (Combiflash).
Ethyl 1-(4-bromophenyl)-5-(4-methyl-2-pivalamidothiazol-5-yl)-1H-pyrazole-3-carboxylate (9b)
Para-bromophenylhydrazine (0.14 g, 0.64 mmol) was reacted with 8a (0.217 g, 0.63 mmol) by general procedure B. After purification, an off-white product was obtained (0.27 g, 87%). 1H NMR (600 MHz, CDCl3) δ 8.80 (s, 1H), 7.50 (d, J=6, 2H), 7.27 (d, J= 6, 2H), 7.02 (s, 1H), 4.46 (q, J = 6, 2H), 2.01 (s, 3H), 1.42 (t, J = 6, 3H), 1.33 (s, 9H); 13C NMR (151 MHz, CDCl3) δ 176.30, 162.19, 158.25, 147.48, 144.95, 138.29, 135.34, 132.52, 126.59, 122.64, 113.14, 112.69, 61.58, 39.34, 27.37, 15.90, 14.61; LC/MS: cal. [M+H+] and [M+2+H+] =491.08 and 493.08, found 490.97 and 492.86.
General Procedure C: Preparation of 11a-I via Aminolysis of Nγ-substituted Pyrazole Ester 9
Pyrazolylthiazole 9 (1 equiv) was dissolved in dry DCM (15 mL/mmol of 9) and cooled to 0 °C for 10 min. AlMe3 (2.0 equiv) in hexane (1.0 M) was added and the resulting mixture was stirred at room temperature for 12 h. When the reaction was complete, water was added followed by 0.1N aq. HCl to neutralized the mixture which was then extracted with EtOAc. The EtOAc layer was dried over Na2SO4, filtered, and concentrated under reduced pressure to give the crude amide 10, which was then purified by silica gel column chromatography with Combiflash.
1-(4-Bromophenyl)-N-(4-methoxyphenyl)-5-(4-methyl-2-pivalamidothiazol-5-yl)-1H-pyrazole-3-carboxamide (11d)
Pyrazolylthiazole 9b (100 mg, 0.20 mmol) was reacted with anisidine (28 mg, 0.22 mmol) by general procedure C and an off-white solid product was obtained (85 mg, 73% yield). 1H NMR (600 MHz, CDCl3) δ 8.84 (s, 1H), 8.64 (s, 1H), 7.61 (d, J = 9.0, 2H), 7.54 (d, J = 8.8, 2H), 7.28 (d, J = 8.8, 2H), 7.11 (s, 1H), 6.91 (d, J = 9.0, 2H), 3.82 (s, 3H), 2.05 (d, J = 5.6, 3H), 1.32 (s, 9H); 13C NMR (151 MHz, CDCl3) δ 176.29, 159.15, 158.35, 156.63, 147.90, 147.53, 138.28, 136.09, 132.67, 131.03, 126.38, 122.64, 121.74, 114.46, 112.81, 111.50, 77.43, 77.22, 77.01, 55.71, 39.34, 27.36, 15.93; LC/MS: cal. [M+H+] = 568.10, found 568.10.
Synthesis of ethyl 3-(4-methyl-2-pivalamidothiazol-5-yl)-1H-pyrazole-5-carboxylate (13)
2,4-Dioxo-4-(thiazol-5-yl)butanoate 8 (2.0 g, 5.9 mmol) and hydrazine hydrate (0.40 mL, 6.4 mmol) were dissolved in absolute ethanol (20 mL) and the mixture was stirred at room temperature for 20 h. When the reaction was over, the reaction mixture was concentrated to half volume by rotoevaporation and the resulting solid was collected by filtration. This solid residue was washed with cold ethanol (x3), dried under vacuum, and used in the next step without further purification. A small portion of product remained in the ethanol filtrate, which was concentrated under reduced pressure and the resulting solid collected. The two portions of product were combined and weighed (1.66 g, 83%).1H NMR (600 MHz, DMSO+CDCl3) δ 13.79 and 11.99 (s and s, 1H), 11.77 (s, 1H), 6.97 and 6.85 (s and s, 1H), 4.34 and 4.29 (q and q, J = 7.1, 2H), 2.46 and 2.37 (s and s, 3H), 1.31 (triplet overlapping, J = 7.1, 3H), 1.23 (s, 9H); 13C NMR (151 MHz, DMSO+CDCl3) δ 176.83(minor) and 176.53, 161.81(minor) and 158.79, 156.90(minor) and 156.00, 144.96 (minor) and 144.87, 143.11(minor) and 143.07, 135.30(minor) and 134.57, 116.84, 106.91and 106.22(minor), 60.93, and 60.15(minor), 40.05(minor) and 38.74, 26.59, 16.38 and 15.92(minor), 14.24 (minor) and 14.15; LC/MS: cal. [M+H+] = 337.13, found 337.05.
General procedure D: Preparation of 10 via Alkylation of the Pyrazolylthiazole 13
Pyrazolylthiazole 13 (1 equiv), K2CO3 (0.75~0.85 equiv) and an alkylating agent were dissolved in acetone (3.3 mL/mmol of 13). The mixture was then refluxed (60 °C) for 24 h. When the reaction was complete, the reaction mixture was cooled to room temperature and concentrated under reduced pressure. Water was added to the resulting mixture, which was then extracted with EtOAc (3x). The organic extracts were combined, dried over MgSO4, filtered, and concentrated by rotoevaporation. The resulting crude product was then purified by silica gel chromatography (Combiflash; Hexane/EtOAc gradient elution).
Ethyl 1-allyl-3-(4-methyl-2-pivalamidothiazol-5-yl)-1H-pyrazole-5-carboxylate (10a, R=H, R2=Allyl)
Allyl bromide (0.172 mL, 1.98 mmol) was reacted with pyrazolylthiazole 13 (0.5 g, 1.49 mmol) by general procedure D. Product 10a was obtained after purification (0.33 g, 59% yield). 1H NMR (600 MHz, CDCl3) δ 8.71 (s, 1H), 6.95 (s, 1H), 6.04 (m, 1H), 5.19 (m, 3H), 5.14 (dd, J = 1.1, 17.0, 1H), 4.37 (q, J = 7.1 2H), 2.52 (s, 3H), 1.39 (t, J = 7.1, 3H), 1.33 (s, 9H); 13C NMR (151 MHz, CDCl3) δ 175.75, 159.36, 155.65, 143.29, 143.19,133.10, 133.09, 118.20, 117.74, 109.61, 61.21, 54.15, 39.11, 27.21, 16.35, 14.23; LC/MS: cal. [M+H+]=377.17, found 377.13.
Ethyl 1-benzyl-3-(4-methyl-2-pivalamidothiazol-5-yl)-1H-pyrazole-5-carboxylate (10a, R=H, R2=Bn)
Benzyl chloride (0.12 mL, 0.96 mmol) was reacted with pyrazolylthiazole 13 (0.27 g, 0.80 mmol) by general procedure D. Product 10b was obtained after purification (0.23 g, 66%). 1H NMR (600 MHz, CDCl3) δ 8.74 (s, 1H), 7.31 (m, 5H), 6.96 (s, 1H), 5.76 (s, 2H), 4.33 (q, J = 7.1, 2H), 2.51 (t, J = 7.1 3H), 1.33 (s, 9H); 13C NMR (151 MHz, CDCl3) δ 175.98, 159.62, 155.77, 143.82, 143.61, 137.08, 133.27, 128.89, 128.70, 128.46, 128.06, 128.05, 127.96, 118.51, 109.99, 61.40, 60.59, 55.22, 39.31, 27.43, 21.26, 16.73, 14.41, 0.21; LC/MS: cal. [M+H+] = 427.18, found 427.08.
Ethyl 1-(5-chloro-2-methoxybenzyl)-3-(4-methyl-2-pivalamidothiazol-5-yl)-1H-pyrazole-5-carboxylate (10a, R=H, R2=CH2(2-MeO-5-Clphenyl))
2-(Bromomethyl)-4-chloro-1-methoxybenzene (0.37 g, 1.57 mmol) was reacted with 13 (0.5 g, 1.49 mmol) by general procedure D. Product 10c was obtained after purification (0.37 g, 51%). 1H NMR (600 MHz, CDCl3) δ 8.70 (s, 1H), 7.17 (dd, J = 2.6, 8.7, 1H), 7.02 (s, 1H), 6.79 (d, J = 8.7, 1H), 6.59 (d, J = 2.6, 1H), 5.76 (s, 2H), 4.32 (q, J = 7.1, 2H), 3.86 (s, 3H), 2.53 (s, 3H), 1.34 (t, J = 7.1, 3H), 1.32 (s, 9H); 13C NMR (151 MHz, CDCl3) δ 175.90, 159.47, 155.74, 155.25, 144.02, 143.96, 134.06, 128.38, 128.00, 127.27, 125.79, 118.39, 111.59, 110.01, 61.48, 56.01, 50.28, 39.31, 27.45, 16.75, 14.36; LC/MS: purity and Calculated [M+H+] and [M+2+H+] =491.15 and 493.14, found 491.10 and 493.08.
General Procedure E: Preparation of 14a-i via Aminolysis of 10 with non-alcoholic amines
To a solution of ethyl ester 10 (1 equiv) in dry DCM (10 mL/mmol of 10), which was chilled at 0 °C in a sealed microwave reaction vessel, was added 2.0 M trimethyl aluminum (1.2 equiv) in hexanes. The resulting mixture was stirred at 0 °C for 10 min and then the amine reactant (1.2 equiv) in dry DCM (1 mL/mmol of 10) was injected into the mixture. The reaction tube was then mounted to the microwave reactor and irradiated with microwave at 100 °C for 40 min. After colling, water and 0.1N aq. HCl were added to the reaction mixture sequentially to neutralize the solution. DCM extraction (3x), drying over MgSO4, filtration, and rotoevaporation gave a residue which purified by silica gel chromatography (CombiFlash).
1-Allyl-N-(4-methoxyphenyl)-3-(4-methyl-2-pivalamidothiazol-5-yl)-1H-pyrazole-5-carboxamide (14a)
Ethyl ester 10a (100 mg, 0.27 mmol) was reacted with anisidine (40 mg, 33 mmol) by general procedure E and gave 14a (45 mg, 67%). 1H NMR (600 MHz, CDCl3) δ 8.80 (s, 1H), 7.83 (s, 1H), 7.50 (d, J = 8.7, 2H), 6.91 (d, J = 9.0, 2H), 6.73 (s, 1H), 6.08 (ddd, J = 5.8, 10.9, 16.1, 1H), 5.23-5.10 (m, 4H), 3.81 (s, 3H), 2.51 (s, 3H), 1.32 (s, 9H); 13C NMR (151 MHz, CDCl3) δ 176.02, 157.72, 157.19, 155.66, 143.92, 143.35, 136.48, 133.56, 130.21, 124.50, 122.59, 118.31, 118.09, 114.53, 105.41, 77.46, 77.25, 77.04, 55.74, 54.19, 39.33, 27.44, 16.71; LC/MS: cal. [M+H+] = 454.19, found 454.16.
1-allyl-N-benzyl-3-(4-methyl-2-pivalamidothiazol-5-yl)-1H-pyrazole-5-carboxamide (14b)
Ethyl ester 10a (100 mg, 0.27 mmol) was reacted with benzylamine (36 uL, 33 mmol) by general procedure E and gave 14b (85 mg, 91% yield). 1H NMR (600 MHz, CDCl3) δ 8.93 (s, 1H), 7.30 (m, 4H), 6.87 (t, J = 5.6, 1H), 6.64 (s, 1H), 6.02 (ddt, J = 5.7, 11.3, 17.0, 1H), 5.12 (m, 4H), 4.59 (d, J = 5.8, 2H), 2.46 (s, 3H), 1.29 (t, J = 2.9, 9H); 13C NMR (151 MHz, CDCl3) δ 176.09, 159.69, 155.66, 143.74, 143.20, 137.89, 136.22, 133.66, 128.98, 127.98, 127.89, 118.34, 117.82, 105.52, 54.04, 43.75, 39.28, 27.38, 16.63; LC/MS: cal. [M+H+]=438.20, found 438.13.
Synthesis of N,1-dibenzyl-3-(4-methyl-2-pivalamidothiazol-5-yl)-1H-pyrazole-5-carboxamide (14e)
Ethyl ester 10b (100 mg, 0.23 mmol), benzylamine (0.51 mL, 4.7 mmol), and sodium cyanide (5.3 mg, 0.1 mmol) were mixed in MeOH (8 mL) and refluxed for 18 h. Upon cooling, the methanol was removed by rotoevaporation and the residue was taken up in EtOAc (50 mL), wahed with water (50 mL; 3x) and 0.1N aq. HCl (50 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The concentrate was purified by silica gel chromatography (Combiflash), giving pure product of 14e (31.9 mg, 34.41%). 1H NMR (600 MHz, CDCl3) δ 8.71 (s, 1H), 7.37-7.23 (m, 10H), 6.58 (s, 1H), 6.24 (t, J = 5.6, 1H), 5.79 (s, 2H), 4.58 (d, J = 5.8, 2H), 2.50 (s, 3H), 1.32 (s, 9H); 13C NMR (151 MHz, CDCl3) δ 175.94, 159.67, 155.63, 143.79, 143.36, 137.70, 137.31, 136.12, 129.07, 128.71, 128.33, 128.00, 127.99, 127.94, 118.46, 105.32, 54.98, 43.80, 39.31, 27.44, 16.74; LC/MS: cal. [M+H+]= 488.21, found 488.20.
1-(5-chloro-2-methoxybenzyl)-N-(4-methoxyphenyl)-3-(4-methyl-2-pivalamidothiazol-5-yl)-1H-pyrazole-5-carboxamide (14g)
Ethyl ester 10c(30 mg, 0.061 mmol) was reacted with anisidine (12 mg, 0.098 mmol) by gerenal procedure E to give 14g (26 mg, 75%). 1H NMR (600 MHz, CDCl3) δ 8.83 (s, 1H), 7.76 (s, 1H), 7.48 (d, J = 8.5, 2H), 7.16 (dd, J = 2.6, 8.7, 1H), 6.89 (d, J = 9.0, 2H), 6.79 (d, J = 2.5, 1H), 6.75 (d, J = 8.7, 2H), 5.78 (s, 2H), 3.80 (s, 3H), 3.78 (s, 3H), 2.51 (s, 3H), 1.32 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 176.00, 157.68, 157.18, 155.73, 155.47, 144.06, 143.84, 130.26, 128.52, 128.15, 127.91, 125.76, 122.42, 118.27, 114.53, 111.67, 105.38, 55.97, 55.71, 49.90, 39.32, 27.43, 16.71; LC/MS: cal. [M+H+]=568.18, found 568.17.
N-(5-(1-(5-chloro-2-methoxybenzyl)-5-(morpholine-4-carbonyl)-1H-pyrazol-3-yl)-4-methylthiazol-2-yl)pivalamide (14h)
Ethyl ester 10c (30 mg, 0.061 mmol) was reacted with morpholine (7 uL, 0.091 mmol) by gerenal procedure E to give 14h (28 mg, 87%). 1H NMR (600 MHz, CDCl3) δ 8.74 (s, 1H), 7.19 (dd, J = 2.6, 8.7, 1H), 6.86 (d, J = 2.6, 1H), 6.77 (d, J = 8.7, 1H), 6.38 (d, J = 9.6, 1H), 5.50 (s, 2H), 3.79 (d, J = 12.0, 3H), 3.66 (d, J = 24.8, 4H), 3.45 (s, 4H), 2.50 (s, 3H), 1.31 (s, 9H); 13C NMR (151 MHz, CDCl3) δ 175.95, 160.92, 155.76, 155.55, 143.95, 143.31, 136.09, 128.96, 128.83, 127.45, 125.67, 118.40, 111.95, 105.67, 66.82, 56.16, 49.29, 39.31, 27.43, 16.72; LC/MS: cal. [M+H+] = 532.18, found 532.14.
General procedure F for Preparation of amide 11m-p and 14j: Aminolysis of 9 or 10 with alcoholic amine
Pyrazolylthiazole 9 or 13 was dissolved in dry ethanol (4 mL) in a 10 mL microwave reaction tube. Ethanolamine (20 equivalent) was added to the solution, the tube was sealed, placed in a microwave reactor, and the reaction mixture was heated at 180 °C for 30 minutes. After the tube had cooled, ethanol was removed under reduced pressure, aq. NH4Cl was added, and the mixture was extracted with chloroform (x3). The chloroform extracts were combined and the solvent removed under reduced pressure to give a crude product, which was purified with HPLC.
1-(5-chloro-2-methoxybenzyl)-N-(2-hydroxyethyl)-3-(4-methyl-2-pivalamidothiazol-5-yl)-1H-pyrazole-5-carboxamide (14j)
Following general procedure F, 14j was obtained in 33% yield. 1H NMR (600 MHz, CDCl3) δ 7.19 (dd, J = 2.6, 8.7, 1H), 6.80 (d, J = 8.8, 1H), 6.73 (d, J = 2.6, 1H), 6.72 (s, 1H), 6.52 (s, 1H), 5.76 (s, 2H), 3.85 (s, 3H), 3.82 (t, J = 5.4, 2H), 3.59 (dd, J = 5.5, 10.3, 2H), 2.60 (s, 3H), 1.37 (s, 9H); 13C NMR (151 MHz, CDCl3) δ 178.26, 160.36, 159.63, 155.31, 140.97, 137.49, 134.35, 128.57, 127.81, 127.17, 125.52, 117.96, 111.65, 104.79, 77.35, 77.22, 77.01, 76.80, 61.75, 55.85, 49.93, 42.12, 39.94, 26.56, 13.43; LC/MS: cal. [M+H+] = 506.16, found 506.15.
Supplementary Material
Acknowledgements
The authors thank the Tara K. Telford Fund for Cystic Fibrosis Research at UC Davis, the National Institutes of Health (DK072517 and GM076151), and the National Science Foundation [CHE-0614756, CHE-0443516, CHE-0449845, CHE-9808183 (NMR spectrometers)] for their generous support.
Footnotes
Abbreviations: CFTR, cystic fibrosis transmemberane conductance regulator; SAR, structure-activity relationship; CF, cystic fibrosis; ER endoplamic reticulum
Supporting Information Available: CCDC information for 9b and 10a X-ray crystallographic structures, calculated and extrapolated logP values of active pyrazolylthiazoles 11d/14a/14b/14e/14g/14h/14j and the bithiazole 1, logk and logP values of reference compounds (used to establish the logP vs. logk trendline) and pyrazolylthiazole correctors, experimentals for the preparation of pyrazolylthiazole carboxylic acids of both regioisomers of N-substituted pyrazoles, and HPLC/mass spectral data for active pyrazolylthiazoles 11d/14a/14b/14e/14g/14h/14j. This material is available free of charge via the Internet at http://pubs.acs.org.
References & Notes
- 1.Bobadilla JL, Macek M, Fine JP, Farrell PM. Cystic fibrosis:a worldwide analysis of CFTR mutations. Correlation with incidence data and application to screening. Hum. Mutat. 2002;19:575–606. doi: 10.1002/humu.10041. [DOI] [PubMed] [Google Scholar]
- 2.Snouwaert JN, Brigman KK, Latour AM, Malouf NN, Boucher RC, Smithies O, Koller BH. An animal model for cystic fibrosis made by gene targeting. Science. 1992;257:1083–1088. doi: 10.1126/science.257.5073.1083. [DOI] [PubMed] [Google Scholar]
- 3.Sharma M, Benharouga M, Hu W, Lukacs GL. Conformational and temperature-sensitive stability defects of the delta F508 cystic fibrosis transmembrane conductance regulator in postendoplasmic reticulum compartments. J. Biol. Chem. 2001;276:8942–50. doi: 10.1074/jbc.M009172200. [DOI] [PubMed] [Google Scholar]
- 4.Skach WR. Defects in processing and trafficking of the cystic fibrosis transmembrane conductance regulator. Kidney Int. 2000;57:825–831. doi: 10.1046/j.1523-1755.2000.00921.x. [DOI] [PubMed] [Google Scholar]
- 5.Zeiher BG, Eichwald E, Zabner J, Smith JJ, Puga AP, McCray PB, Jr., Capecchi MR, Welsh MJ, Thomas KR. A mouse model for the delta F508 allele of cystic fibrosis. J. Clinic. Invest. 1995;96:2051–2064. doi: 10.1172/JCI118253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pedemonte N, Lukacs GL, Du K, Caci E, Zegarra-Moran Ol., Galietta Luis J. V., Verkman AS. Small-molecule correctors of defective ΔF508-CFTR cellular processing identified by high-throughput screening. J. Clinic. Invest. 2005;115:2564–2571. doi: 10.1172/JCI24898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoo CL, Yu GJ, Yang B, Robins LI, Verkman AS, Kurth Mark J. 4′-Methyl-4,5′-bithiazole-based correctors of defective ΔF508-CFTR cellular processing. Bioorg. Med. Chem. Lett. 2008;18:2610–2614. doi: 10.1016/j.bmcl.2008.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu GJ, Yoo CL, Yang B, Lodewyk MW, Meng L, El-Idreesy TT, Fettinger JC, Tantillo DJ, Verkman AS, Kurth MJ. Potent s-cis-Locked Bithiazole Correctors of ΔF508 Cystic Fibrosis Transmembrane Conductance Regulator Cellular Processing for Cystic Fibrosis Therapy. J. Med. Chem. 2008;51:6044–6054. doi: 10.1021/jm800533c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Tanitame A, Oyamada Y, Ofuji K, Terauchi H, Kawasaki M, Wachi M, Yamagishi J. Synthesis and antibacterial activity of a novel series of DNA gyrase inhibitors: 5-[(E)-2-arylvinyl]pyrazoles. Bio. Med.Chem. Lett. 2005;15:4299–4303. doi: 10.1016/j.bmcl.2005.06.103. [DOI] [PubMed] [Google Scholar]; (b) Li Q, Zu Y, Shi R, Yao L, Fu Y, Yang Z, Li L. Synthesis and antitumor activity of novel 10-substituted camptothecin analogues. Bioorg. Med. Chem. 2006;14:7175–7182. doi: 10.1016/j.bmc.2006.06.061. [DOI] [PubMed] [Google Scholar]; (c) Keter FK, Nell Margo J., Guzei Ilia A., Omondi Bernard, Darkwa James. Anticancer activities of bis(pyrazol-1-ylthiocarbonyl)disulfides against HeLa cells. J. Chem. Res. 2009:322–325. [Google Scholar]; (d) Dai H, Li Y, Du D, Qin X, Zhang X, Yu H, Fang J. Synthesis and Biological Activities of Novel Pyrazole Oxime Derivatives Containing a 2-Chloro-5-thiazolyl Moiety. J. Agri. Fd. Chem. 2008;56:10805–10810. doi: 10.1021/jf802429x. [DOI] [PubMed] [Google Scholar]; (e) De Morin FF, Chen JJ, Doherty EM, Hitchcock S, Huang Q, Kim JL, Liu G, Nixey T, Paras NA, Petkus J, Retz DM, Smith AL, Tasker A, Zhu J. Preparation of nitrogen-containing bicyclic heteroaryl compounds as antitumor agents. PCT Int. Appl. 2007, WO 2007076092 A2 20070705 [Google Scholar]
- 10.Knorr L. Action of ethyl acetoacetate on phenylhydrazine. I. Ber. 1883;16:2597–2599. [Google Scholar]
- 11.(a) Elguro J. Pyrazoles and Their Benzo Derivatives. Compr. Hetero Chem. 1984;5:167–303. [Google Scholar]; (b) Makino K, Kim HS, Kurasawa Y. Synthesis of pyrazoles. J. Heterocycl. Chem. 1998;35:489–497. [Google Scholar]; (c) Silva VLM, Silva AMS, Pinto DCGA, Cavaleiro JAS, Elguero J. 3(5)-(2-Hydroxyphenyl)-5(3)-styrylpyrazoles: Synthesis and Diels-Alder alder transformations. Eur. J. Org. Chem. 2004:4348–4356. [Google Scholar]; (d) Heller ST, Natarajan SR. 1,3-Diketones from Acid Chlorides and Ketones: A Rapid and General One-Pot Synthesis of Pyrazoles. Org. Lett. 2006;8:2675–2678. doi: 10.1021/ol060570p. [DOI] [PubMed] [Google Scholar]
- 12.(a) Yanborisov TN, Zhikina IA, Andreichikov, Yu S, Milyutin AV, Plaksina AN. Synthesis and pharmacological activity of heteroylpyruvic acids and their derivatives. Khimiko-Farmatsevticheskii Zhurnal. 1998;32:26–28. [Google Scholar]; (b) Dinges J, Akritopoulou-Zanze I, Arnold LD, Barlozzari T, Bousquet PF, Cunha GA, Ericsson AM, Iwasaki N, Michaelides MR, Ogawa N, Phelan KM, Rafferty P, Sowin TJ, Stewart KD, Tokuyama R, Xia Z, Zhang HQ. Hit-to-lead optimization of 1,4-dihydroindeno[1,2-c]pyrazoles as a novel class of KDR kinase inhibitors. Bioorg. Med. Chem. Lett. 2006;16:4371–4375. doi: 10.1016/j.bmcl.2006.05.052. [DOI] [PubMed] [Google Scholar]
- 13.Yang H, Shelat AA, Guy RK, Gopinath VS, Ma T, Du K, Lukacs GL, Taddei A, Folli C, Pedemonte N, Galietta LJV, Verkman AS. Nanomolar Affinity Small Molecule Correctors of Defective ΔF508-CFTR Chloride Channel Gating. J. Bio. Chem. 2003;278:35079–35085. doi: 10.1074/jbc.M303098200. [DOI] [PubMed] [Google Scholar]
- 14.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development. Adv. Drug. Del. Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 15.(a) Kerns EH, Di L. Physicochemical profiling: overview of the screens. Drug Discovery Today. 2004;1:343–348. doi: 10.1016/j.ddtec.2004.08.011. [DOI] [PubMed] [Google Scholar]; (b) Wang J, Krudy G, Hou T, Zhang W, Holland G, Xu X. Development of Reliable Aqueous Solubility Models and Their Application in Druglike Analysis. J. Chem. Info. Model. 2007;47:1395–1404. doi: 10.1021/ci700096r. [DOI] [PubMed] [Google Scholar]; (c) Dressman JB, Thelen K, Jantratid E. Towards quantitative prediction of oral drug absorption. Clinic. Pharmacokin. 2008;47:655–667. doi: 10.2165/00003088-200847100-00003. [DOI] [PubMed] [Google Scholar]
- 16.Braumann T. Determination of hydrophobic parameters by reversed-phase liquid chromatography: theory, experimental techniques, and application in studies on quantitative structure-activity relationships. J. Chromatogr. 1986;373:191–225. doi: 10.1016/s0021-9673(00)80213-7. [DOI] [PubMed] [Google Scholar]
- 17.Vrakas D, Tsantili-Kakoulidou A, Hadjipavlou-Litina D. Exploring the consistency of logP estimation for substituted coumarins. QSAR & Comb. Sci. 2003;22:622–629. [Google Scholar]
- 18.Galietta LJ, Haggie PM, Verkman AS. Green fluorescent protein-based halide indicators with improved chloride and iodide affinities. FEBS Lett. 2001;499:220–224. doi: 10.1016/s0014-5793(01)02561-3. [DOI] [PubMed] [Google Scholar]
- 19.Hantzsch A. Thiazoles from thiamides. Justus Liebigs Ann. Chem. 1889;250:257–73. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.