

Abstract
Redox homeostasis governs a number of critical cellular processes. In turn, imbalances in pathways that control oxidative and reductive conditions have been linked to a number of human disease pathologies, particularly those associated with aging. Reduced glutathione is the most prevalent biological thiol and plays a crucial role in maintaining a reduced intracellular environment. Exposure to reactive oxygen or nitrogen species is causatively linked to the disease pathologies associated with redox imbalance. In particular, reactive oxygen species can differentially oxidize certain cysteine residues in target proteins and the reversible process of S-glutathionylation may mitigate or mediate the damage. This post-translational modification adds a tripeptide and a net negative charge that can lead to distinct structural and functional changes in the target protein. Because it is reversible, S-glutathionylation has the potential to act as a biological switch and to be integral in a number of critical oxidative signaling events. The present review provides a comprehensive account of how the S-glutathionylation cycle influences protein structure/function and cellular regulatory events, and how these may impact on human diseases. By understanding the components of this cycle, there should be opportunities to intervene in stress- and aging-related pathologies, perhaps through prevention and diagnostic and therapeutic platforms. Antioxid. Redox Signal. 15, 233–270.
I. Introduction
The production of reactive oxygen (ROS) and nitrogen (RNS) species occurs under normal physiological conditions. Cells have evolved elaborate mechanisms to maintain redox homeostasis. There exists a significant and growing body of literature that links human disease pathologies with altered redox metabolism and homeostasis in cells and organisms. In particular, cancer and neurological disorders are characterized by alterations in a variety of pathways involving reduced glutathione (GSH). Redox-mediate dysregulation of signal transduction pathways leads to uncontrolled cell growth (cancer) and cell death (neurodegenerative disorders). As a tripeptide of glutamic acid, cysteine, and glycine, GSH represents one of the most prevalent and important thiol buffers in the cell. The ratio of GSH (reduced) and its disulfide, GSSG (oxidized), contributes to the redox potential of the cell and thereby contributes to redox homeostasis (Fig. 1). Oxidative or nitrosative stress induced by physiological or pathological conditions leads to a decreased ratio of GSH/GSSG. In addition to the level of GSH and GSSG in cellular compartments, redox homeostasis is defined by the GSH content that is utilized by proteins themselves. The disulfide proteome has been described as having two subproteomes, a structural group and a redox-sensitive group (300). The redox-sensitive proteome can be post-translationally modified through disulfide linkages between GSH and redox-sensitive cysteine residues within proteins, for example, S-glutathionylated (P-SSG). Regulation through S-glutathionylation has been ascribed to a large number of proteins that fall into the following clusters: cytoskeletal, glycolysis/energy metabolism, kinase and signaling pathways, calcium homeostasis, antioxidant enzymes, and protein folding (271). This review focuses on the molecular mechanisms that govern the redox-sensitive disulfide proteome and highlights proteins that are dysregulated in diseases of aging, specifically cancer and neurodegenerative disorders.
FIG. 1.

Glutathione as a biological redox buffer. The ratio of GSH/GSSG reflects the redox capacity of the cell. The ratio is kept in balance through oxidation/reduction reactions involving GSH peroxidase and GSH reductase. Reactive oxygen species-/reactive nitrogen species-induced changes that decrease GSH lead to cell death via apoptosis or necrosis. GP, GSH peroxidase; GR, GSH reductase; GSH, reduced glutathione; GSSG, oxidized glutathione.
A. Glutathione homeostasis
Intracellular GSH concentrations affect various components of the S-glutathionylation cycle. Therefore, the enzymes that regulate intracellular GSH can contribute, though indirectly to redox signaling events. Glutathione, initially described as “philothion” or love of sulfur was crystallized, determined to contain sulfur and renamed glutathione (125). It is found in most prokaryotes and all eukaryotes and in humans that the usual range of plasma GSH is 10–30 μM and it has a flow rate to the kidney of 1 l/min, whereby ∼60 μmol GSH is delivered to the lumen of the proximal tubule where concentrations can approach 3 mM with a turnover half-life of 20 min. Turnover of renal GSH suggests that a further 2.4 μmol/h is delivered to the tubule lumen. Since the concentration in urine is 1–3 μM, this is an excretion rate of <1 nmol/h, implying that 99% of the GSH is reabsorbed.
Intracellular GSH levels are governed by the rate of de novo GSH synthesis and export. GSH cannot freely cross the cell membrane and as such there are GSH transporters in some tissues and enzymes that facilitate catalysis and uptake. In general, GSH must be catabolized via a salvage pathway and the constitutive amino acids brought into the cell for subsequent de novo synthesis. To date, only one enzyme is known to facilitate this process. Gamma-glutamyltransferase (GGT) is a cell surface heterodimeric glycoprotein expressed at high levels in kidney tubules, biliary epithelium, and brain capillaries (112). GGT catalyzes the degradation of extracellular GSH, hydrolyzing the γ-glutamyl bond between glutamate and cysteine and releasing the product cysteinyl-glycine (CG) and glutamate. CG is cleaved by the membrane-bound dipeptidase to cysteine and glycine (Fig. 2). The resulting cysteine can be transported into the cell. This salvage supplements the constitutive amino acids required for de novo synthesis, achieved by sequential catalytic steps involving γ-glutamylcysteine synthetase (γ-GCS) and glutathione synthetase. The γ-GCS reaction represents the committed step in the biosynthesis and is subject to feedback inhibition by GSH, but cysteine is rate limiting. Reactions of GSH can be subdivided into those involving the γ-glutamyl and those of the sulfhydryl of cysteine. The amino terminal peptide bond is formed through the γ-carboxyl of the glutamate residue. This bond is resistant to degradation by serum proteolytic enzymes and this allows interorgan transport of GSH.
FIG. 2.

Regulation of GSH metabolism by GGT. GGT hydrolyzes extracellular GSH and releases glutamic acid and cysteinyl-glycine ○1. Cysteinyl-glycine is cleaved by the membrane-bound dipeptidase to cysteine and glycine, and the products are transported into the cells ○2. The γ-glutamyl moiety is transferred to the acceptors and transported into the cells ○3. The metabolic components participate in the de novo GSH synthesis catalyzed by GCS and GCL ○4. Intracellular GSH is exported out of cells through MRP ○5. The dashed lines indicate the upregulation of GCS, GCL, GGT, and MRP by oxidative stress. GCL, glutamate cysteine ligase; GCS, γ-glutamylcysteine synthetase; GGT, gamma-glutamyltransferase; MRP, multidrug resistant protein.
B. Proximal donors for S-glutathionylation reactions
Although both ROS and RNS can lead to S-glutathionylated proteins, there is no precise understanding of which chemical moieties might act as proximal donors for the post-translational modification. Chemically, nucleophilicity is provided by the thiol group of cysteine, and with a pKa of 9.65 (free) and 8.5 (GSH), it is basic enough to imbue important chemical properties. The pKa of cysteine within a protein is determined by the neighboring protein environment but can have an aliphatic thiol that provides proteins with nucleophilic sites for a number of diverse post-translational modifications. Under oxidative conditions the thiol can be oxidized to a thiyl radical (RS.) that has strong reactivity with oxygen, leading to higher levels of cysteine oxidation (Fig. 3). Moreover, disulfide bonds between vicinal thiols can have critical implications for three-dimensional protein structures. Dependent upon the steric properties and surrounding environment of the cysteine, lipidation can occur through S-isoprenylation, S-farnesylation, S-geranylgeranylation, or S-palmitoylation. These additions provide significant bulk with resultant impact upon protein structure, function, and subcellular compartmentalization. For example, modifications of the cysteine residues in the ras oncogene can influence cellular transformation pathways. The direct addition of GSH to low pK cysteines creates an S-glutathionylated residue (Fig. 3), with an increase in MW of 305 and a net increase in negative charge (from the glycine residue of GSH). Nitrosylation of cysteines can also occur upon exposure to nitrosative stress. Of relevance, conversion of these residues to S-glutathionylated cysteines can occur, indicating that products of nitric oxide have significant capacity to cause S-glutathionylation. Identification of those active metabolite(s) that might act as proximal donors for this modification remains indeterminate. There is debate in the literature as to which GSH species could facilitate the reaction. Low levels of oxidative stress induced by H2O2 or diamide do not generally produce high levels of S-glutathionylation. Nitrosylated cysteine residues can transduce to S-glutathionylated, potentially through the following scheme, implicating S-nitrosoglutathione (GSNO) as an intermediate.
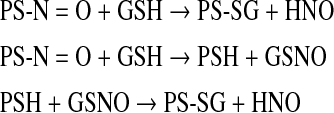 |
FIG. 3.

S-glutathionylation cycle. Cysteine residues on proteins that have a low pKa are targets for redox modulation under conditions of oxidative or nitrosative stress. The cysteine residue within proteins can be oxidized to form sulfenic, sulfinic, and sulfonic acids. Both sulfenic and sulfinic acids of proteins can be reduced or conjugated to GSH to form S-glutathionylated proteins via glutathione S-transferases, Grx, or nonenzymatically. The post-translational modification can be reversed by Grx and/or sulfiredoxin. Grx, glutaredoxin.
In this example, GSNO can react with cysteine residues through trans-nitrosylation followed by thiol disulfide exchange and thus can act as a plausible donor in the S-glutathionylation reaction. Other possible species from the interaction of nitric oxide and GSH may lead to S-glutathionylation. Nitroxyl (HNO) may have biological importance and pharmacological properties. For example, the degree of electrophilic selectivity of HNO for thiols is higher than for other nucleophiles (thiol>amine>oxygen nucleophiles) and it can react with GSH to give N-hydroxysulfenamide (GS–NH–OH), which can rearrange to generate a sulfinamide (GS(O)NH2). Reaction of GS(O)NH2 with GSH can produce glutathione disulfide-S-oxide (GS(O)SG), a product that also has the potential to act as the proximal donor in S-glutathionylation reactions. A sulfinic acid (GS(O)OH) and NH3 are possible hydration products from sulfinamide. Key intermediates leading to the synthesis of GS(O)SG are the sulfinamides (GS(O)NH2 and GS(O)–NH–SG). The reaction of GS(O)SG or GS. with a cysteine residue (R-SH) can lead to the formation of the S-glutathionylated mixed disulfide. Thus, while GSH itself may not be an ideal proximal donor in S-glutathionylation, candidates that may be involved include GSSG, GS–O–SG, and GS–O–O–SG (126, 266). NO can also produce oxidized intermediates that lead to the highly nitrosative species such as N2O3, which reacts with GSH to produce GSNO. The latter can then react with cysteine through trans-nitrosylation followed by thiol-disulfide exchange resulting in S-glutathionylation. Nitrosative species such as nitrogen dioxide can also interact directly with GSH to lead to S-glutathionylated cysteine residues. Some, or all, of these reactions may produce products that lead to the post-translational modification.
Oxidation and reduction reactions play an essential role in numerous cell-signaling cascades, including those associated with proliferation, inflammatory responses, apoptosis, and senescence. Oxidative stress can be defined as the result of the imbalance between the production of oxidants and their removal by antioxidants and can play an important role in (i) the origination, progression, and malignancy of a number of cancers and (ii) a number of diseases of progressive dementia. For example, high levels of ROS and/or RNS can lead to cell senescence and/or death. Recent studies have identified certain types of ROS as second messengers in signaling pathways, thus implicating them in the regulation of cell phenotype by acting as effector molecules. Understanding the origin of various ROS/RNS, and their roles in disease initiation and progression, as well as in the cell signaling pathways involved in these could facilitate prognosis, diagnosis, and the development of therapies that might act to prevent and/or counteract such human pathologies. As a component of this, a more detailed understanding of those features of S-glutathionylation that regulate cellular events may help to enhance our understanding of the interface of the pathways involved.
II. Detection of S-Glutathionylation
Although the importance of cysteine post-translational modifications has been appreciated for many years, progress in the field has been limited by the restricted sensitivity of many of the methods used to measure, what can be, very low levels of products. More advanced proteomic approaches, together with optimization of other detection methods, have allowed investigators greater flexibility in experimental design and interpretation. Contingent upon endpoint, each of the following approaches has been utilized effectively to yield publishable results.
A. Antibody detection of S-glutathionylation
Antibodies have been developed to detect protein S-glutathionylation. Antibody detection is a blunt instrument that provides information on global S-glutathionylation status, unless preceded by immunoprecipitation of a specific protein of interest. Table 1 summarizes the commercially available antibodies along with the specific antigen used to generate the reagent. S-glutathionylation of proteins is predominantly detected using antibodies generated against protein-glutathione adducts by employing methods such as western blotting, ELISA or immunohistochemistry/immunocytochemistry. In addition, S-glutathionylation mouse monoclonal antibodies with a fluorescent DyLight 488 or DyLight 549 label are available for fluorescent applications (Arbor Assays, MI). GSH can be conjugated to bovine serum albumin (BSA), keyhole limpet hemocyanin (KLH), and human lysozyme (LZM) to generate an immunogen for antibody generation in rabbits, rats, and mice or using phage display libraries (123). However, only highly abundant S-glutathionylated proteins are detected and it is not possible to determine the identity of the modified proteins. Recent approaches have shown that it is possible to generate peptide-based antibodies against the GS-binding domain of proteins such as mitochondrial complex I and II and to determine domain specific functionality (139). Specifically, two peptides of the S-glutathionylated domains of, respectively, the 51 kDa, 200GAGAYIC(GSH)GEETALIESIEGK219, and 75 kDa, 361VDSDTLC(GSH)TEEVFPTAGAGTDLR382, subunit of complex I were used to generate distinct polyclonal antibodies in rabbits. In addition, two non-S-glutathionylated peptides were used as chimeric isotopes for control antibodies. Upon binding, each antibody generated from the S-glutathionylated peptides decreased superoxide production by 37% and 57%, respectively, in isolated bovine mitochondrial Complex I (36, 139). Further, these antibodies have been used successfully in ELISA, Western blotting, and immunoprecipitation applications (36). Such reports have demonstrated that antibodies can be used not only in immunodetecting methods but also as tools to study biological samples.
Table 1.
Commercially Available Antibodies Directed Toward S-Glutathionylated Moieties
| Company | Origin | Clone | Immunogen | Applications |
|---|---|---|---|---|
| Virogen | Mouse | D8 | Unknown | WB |
| Abcam | Mouse | D8 | Glutathione conjugated to Keyhole Limpet Hemocyanin (KLH) | ELISA, ICC/IF, IHC-P, IP, WB |
| AbD Serotec | Rabbit | Glutathione-glutaraldehyde-BSA | ELISA | |
| Abnova | Mouse | D8 | Glutathione-protein complexes | WB |
| Rat | Glutathione conjugated to BSA | ELISA, IHC-P | ||
| Thermo | Mouse | D8 | Glutathione | ELISA, WB, IP |
| Advanced targeting systems | Rabbit | Glutathione-glutaraldehyde-BSA | ELISA, IHC-P | |
| Rat | Glutathione-glutaraldehyde-BSA | ELISA, IHC-P | ||
| Arbor assays | Mouse | L4H | Glutathione conjugated to KLH | ELISA, WB, IP |
| Mouse | L4H | Glutathione conjugated to KLH, DyLight 488 label | Fluorescence | |
| Mouse | L4H | Glutathione conjugated to KLH, DyLight 549 label | Fluorescence | |
| Enzo | Mouse | D8 | Unknown | ELISA, WB, IP |
| Cellsciences | Rabbit | Glutathione conjugated to BSA | ELISA, WB, IHC | |
| Rat | Glutathione-glutaraldehyde-protein | ELISA | ||
| Fritzgerald | Mouse | D8 | Unknown | Unknown |
| Genway | Rat | Glutathione-glutaraldehyde-BSA | ELISA | |
| Kamiya | Mouse | D8 | Glutathione conjugated to KLH | ELISA, WB, IP, IHC |
| Lifespan | Mouse | Lv022 | Glutathione | ELISA, WB, IP |
| Mouse | 2q68 | Glutathione | WB, IHC | |
| Mouse | D8 | Unknown | WB | |
| Mouse | GF5 | Unknown | WB | |
| Rabbit | Glutathione-glutaraldehyde-BSA | ELISA, IHC | ||
| Rat | Glutathione-glutaraldehyde-BSA | ELISA | ||
| Chemicon | Mouse | D8 | Glutathione | WB, IHC |
| Rabbit | Glutathione-glutaraldehyde | IHC | ||
| Rat | Glutathione-glutaraldehyde-BSA | IHC | ||
| Santa Cruz | Mouse | 1v022 | Glutathione | WB, IP |
| Mouse | D8 | Glutathione | WB, IP | |
| Mouse | 2Q68 | Glutathione | WB, IC | |
| US Biological | Rabbit | Glutathione-glutaraldehyde-BSA | ELISA | |
| Rabbit | Glutathione-glutaraldehyde | ELISA, IHC |
B. Analytical detection and quantification of P-SSG
Analytical methodologies provide the platform for quantitative analysis of specific S-glutathionylation, as well as information on which cysteine residues are targets for modification. Traditionally, derivatization (157, 228) and metabolic labeling (30, 96) together with, or without, affinity chromatography (37, 192) have been used to study S-glutathionylation, but advances in proteomic technologies and mass spectrometry have made the detection, quantification, and high-throughput analysis more tractable (109, 227, 273). Proteomic approaches coupled with analysis of protein structure and function (273, 274) were utilized to combine identification of specific cysteine residues in vitro with structural and functional characteristics of the target proteins. Specifically, glutathione S-transferase pi (GSTP) and GSTP-SSG were analyzed by circular dichroism and intrinsic fluorescence analysis to show that the alpha-helical content is decreased in GSTP-SSG and this translated into a decrease in enzyme activity and loss of protein:protein interactions. Similar technology was utilized to identify and characterize redox modification of endoplasmic reticulum (ER) chaperones (274).
Incorporation of radioactively labeled 35S-cysteine in combination with the protein synthesis inhibitor, cycloheximide, can provide estimates of S-glutathionylation after in vitro exposure to oxidative stress (231). However, the technique requires that further S-glutathionylation during the solubilization or extraction procedure is blocked with N-ethylmaleimide. In addition to radioactive labeling, biotinylated cell-permeable GSH ethyl esters have been used (263). As an example, subjecting HeLa cells, preloaded with biotinylated GSH ethyl ester to oxidative stress through tumor necrosis factor α (TNFα) stimulation, led to the incorporation of the marker and subsequent binding of S-glutathionylated proteins to streptavidin-agarose beads (263). In-gel digestion and Edman sequencing confirmed that two of the S-glutationylated proteins were thioredoxin peroxidase II and annexin II. Combining mass spectrometry with affinity purification methods has proved beneficial in identification and quantification protocols. A combination of two-dimensional sodium dodecyl sulfate–polyacrylamide gel electrophoresis and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry was applied to show that GSTP is S-glutathionylated on Cys47 and Cys101 after oxidative and nitrosative stress (273). Other studies using sensitive electrospray ionization-LTQ-Orbitrap mass spectrometry determined that hemoglobin could be S-glutathionylated through a cysteine sulfenic intermediate on the Cys β93 residue (227). Lind et al. (157) developed an S-glutathionylation-specific labeling protocol through modification of the biotin switch method (133) by substituting ascorbate with glutaredoxin 3 (Grx3) (C14S,C65Y mutant) as the reducing agent. This labeling procedure together with biotin-streptavidin enrichment and mass spectrometry should be highly adaptable to the accurate determination of S-glutathionylated proteins under basal and experimental conditions (Fig. 4). Conjugation of GSH and GSSG with fluorescein allows labeling of S-glutathionylated proteins (149) under situations where optical fluorescence microscopy and fluorescent immunoblotting application are utilized.
FIG. 4.

Biotin labeling of sulfhydryls to detect P-SSG. Labeling of S-glutathionylated proteins as described by Lind et al. (157). Free sulfhydryls are alkylated with N-ethylmaleimide followed by reduction of S-glutathionylated proteins with Grx3. Reduced SH groups are labeled with biotin-maleimide and purified with streptavidin agarose. Purified proteins are separated on a two-dimensional sodium dodecyl sulfate–polyacrylamide gel electrophoresis gel, and spots are excised and digested with trypsin. The resultant tryptic peptides are spotted on a MALDI plate and subjected to MALDI-TOF mass spectrometry followed by database searching using the MASCOT algorithm. 2D SDS-PAGE, two-dimensional sodium dodecyl sulfate–polyacrylamide gel electrophoresis. MALDI-TOF, matrix-assisted laser desorption/ionization time-of-flight. (To see this illustration in color the reader is referred to the web version of this article at www.liebertonline.com/ars).
It has been suggested that protein thiols may be a more accurate indicator of total cellular redox status, since they represent a potentially larger redox active pool than free glutathione (113). Using the fluorescent thiol-specific probe ThioGlo-1tm, the glutathione disulfide mimetic, NOV-002, was shown to modifiy free thiols in a variety of tissues and to cause S-glutathionylation of serum and liver proteins (Fig. 5) (273). In keeping with the total cellular environment, an approach has recently been developed for derivatization and detection of sulfenic acids after thiol oxidation with H2O2 (245). Modification of sulfenic acid by dimedone yields an antibody-specific hapten epitope that allows the determination of cellular thiol oxidation status. This method is specific for sulfenic acids and allows discrimination over sulfinic and sulfonic acid modifications and has been successfully applied in several biological systems, including cancer cell lines (245).
FIG. 5.

Fluorescent detection of modified sulfhydryls. The thiol reactive compound ThioGlo-1 binds to free sulfhydryls on cysteine residues and can be used to semiquantitatively determine modified cysteines by fluorescence.
Advances in proteomics combined with advances in mass spectrometry instrumentation will undoubtedly enhance capabilities in detection and perhaps more importantly quantification of S-glutathionylated proteins. Currently, there exists a bottleneck because of the large size of the data files generated; however, the rapid development of higher computing power and better database search algorithms will certainly lessen such limitations. This will also reduce the number of false-positives, but additional conformational studies using alternative methodologies will still need to be developed.
III. Enzymes That Catalyze the S-Glutathionylation Cycle
Like phosphorylation, S-glutathionylation is a regulated post-translational modification. Numerous in vitro studies with ROS/RNS and GSH at high levels show that redox-sensitive proteins can be S-glutathionylated nonenzymatically. Within cells, several proteins are now recognized to promote and remove S-glutathionylation participating in the S-glutathionylation cycle.
A. Proteins with S-glutathionylase activity
It is generally accepted in the signal transduction field that cyclical phosphorylation is governed by kinases and phosphatases, either of which can have generalist or specialist functions. Our understanding of the S-glutathionylation cycle is presently limited. However, a handful of proteins are players and as such it is possible to construct a scheme for the forward and reverse steps of the S-glutathionylation cycle. S-glutathionylation can serve to regulate directly the structure/function of a quite diverse range of proteins and also serves to prevent the sequential oxidation of thiol groups to sulfenic, sulfinic, and sulfonic acids; the latter is generally irreparably damaged and leads to the proteosomal degradation of the protein. Reversibility of S-glutathionylation provides a switch to control response to changes in redox conditions and cells have evolved a degree of functional redundancy in regulation of this reversible reaction.
1. Glutathione S-transferases
Historically, glutathione S-transferases (GSTs) have been described as a family of ubiquitously expressed Phase II detoxification enzymes that catalyze GSH conjugation to electrophilic compounds through thioether linkages. The family is composed of members that can be present in the cytosolic, mitochondrial, and membrane-bound microsomal compartments. The cytosolic GSTs are divided into seven classes: Alpha, Mu, Omega, Pi, Sigma, Theta, and Zeta. There is >60% sequence identity between family members and functionally they are similar and have overlapping substrate specificities (167, 173).
In the 1990s the role of GSTs in redox-mediated kinase signaling emerged. It was discovered that the pi class of GST plays a regulatory role in cellular stress response, apoptosis, and proliferation through direct protein:protein interactions with c-Jun N-terminal kinase (JNK) (3, 94, 292). The list of involvement with kinase interactions has expanded and to-date includes JNK, apoptosis signal-regulating kinase 1 (ASK1), TNF receptor-associated factor 2 (TRAF2), and GSK as well as other GST family members [for review see ref. (271)]. It has been shown that GSTP can potentiate the forward reaction of the S-glutathionylation cycle (165, 273). The kinetics and magnitude of protein S-glutathionylation is significantly greater in GSTP1P2 wild-type mice than those from knockout animals (273). Distinct from the protein:protein interactions described above, the catalytic activity is required for protein S-glutathionylation. In response to both ROS and RNS, cells expressing mutants of GSTP that lack the catalyitically active tyrosine residue have diminished capacity to S-glutathionylate proteins (273). There appears to be promiscuity with respect to substrate proteins. In cell models of nitrosative stress resistance a decrease in S-glutathionylated proteins was concurrent with reduced GSTP levels (130). In terms of specificity, GSTP does play a necessary role in the S-glutathionylation of 1-cys peroxiredoxin (1-cysPrx) (165, 194, 220). Oxidation of the catalytic cysteine of 1-cysPrx to a stable cys-sulfenic acid has been associated with loss of peroxidase activity. However, heterodimerization of 1-cysPrx with GSTP mediates the S-glutathionylation of the previously oxidized cysteine, followed by subsequent spontaneous reduction of the mixed disulfide, thus restoring its peroxidase activity (165).
It is well established that kinases are auto-regulated (i.e., phosphorylated) as a means of feedback regulation. Interestingly, GSTP is itself also subject to S-glutathionylation on Cys47 and Cys101, thereby suppressing its catalytic properties and even its protein interactions with JNK, instead promoting GSTP multimerization (Fig. 6). It is not known whether S-glutathionylation of GSTP is a trigger for dissociation from the kinase complex. However, Cys47 and Cys101 reside in two distinct effector domains that are required for direct interaction with JNK, whereas Cys47 is critical to ligand binding with Prx1. It is worth noting that those proteins with which GSTP has been previously shown to have protein:protein interactions are also targets for S-glutathionylation (53, 165).
FIG. 6.

The interplay of protein phosphorylation and protein S-glutationylation pathways in cell signaling. Under basal conditions, GSTP forms heterodimers with JNK and TRAF2, resulting in kinase inactivation. Oxidative or nitrosative stress induces complex dissociation and results in S-glutathionylation and/or phosphorylation and activation of c-jun, JNK, and TRAF2. S-glutathionylation of GSTP leads to oligomerization and enzyme inactivation. These processes are reversed when stress is removed. JNK, c-Jun N-terminal kinase, GSTP, glutathione S-transferase pi. (To see this illustration in color the reader is referred to the web version of this article at www.liebertonline.com/ars).
The impact of GST polymorphisms on disease susceptibility, particularly when associated with ROS/RNS, has been reviewed elsewhere (173). Four active, functionally different polymorphisms of GSTP (GSTP1*A–D) have been identified (173) (Table 2). To date, any evaluations of these differences have not considered the context of their impact on S-glutathionylation or variation in an individual's ability to respond to oxidative or nitrosative stress. Future and ongoing studies in this arena could provide new insights into pharmacogentic response to drugs as well as the disease susceptibilities.
Table 2.
Polymorphisms of Glutathione S-transferase Pi
| Allele | Nucleotide | Variability | Activity |
|---|---|---|---|
| GSTP1*A | Ile105 | Ala114 | Reference |
| GSTP1*B | Val105 | Ala114 | Decrease |
| GSTP1*C | Val105 | Val114 | Decrease |
| GSTP1*D | Ile105 | Val114 | No change |
GSTP1*B and *C have decreased catalytic activity (173).
2. Gamma-glutamyl transpeptidase
GGT catalyses the breakdown of GSH into glutamate and CG. Both GSH and CG have been shown to form mixed disulfides with cysteine residues in redox-sensitive proteins. In a previous study it was reported that S-glutathionylation is responsible for up to 85% of cases of S-thiolation (244). It was reported that activation of GGT resulted in a marked increase of CG bound to cellular and extracellular proteins and the effect was largely prevented by acivicin, a GGT inhibitor (52). Protein-bound CG levels were much lower than those of S-glutathionylation, yet increased levels of protein-bound CG led to a decrease in the levels of protein S-glutathionylation, and the reverse was also true (52). Active GGT on the cell surface can act as a GSH sink, removing it from the local environment and affecting the intracellular GSH/GSSG redox balance.
Due to the low pKa of the cystein in CG, it can easily interact with metal cations, promoting a redox-cycling process that results in the production of ROS and stimulation of oxidative reactions (Fig. 2). Stark et al. first suggested that CG could cause the reduction of ferric Fe(III) to ferrous Fe(II) ion (262). In agreement with this, Paolicchi et al. demonstrated that in systems including ADP-Fe (III) complex, GSH can reduce some iron but the reaction rate increases significantly when GGT or CG is included (202). Moreover, in histiocytic lymphoma U937 cells overexpressing GGT or addition of CG directly stimulates extracellular production of H2O2, which can be suppressed by noncell permeant iron chelators, emphasizing the importance of extracellular iron in the reaction (70). The GSH/GGT-mediated prooxidants significantly affect the signaling cascades through the reversible S-glutathionylation of the critical cysteine residues of the target molecules, resulting in dysregulation of the proliferative/apoptotic balance in cells. GGT as a cell surface protein may also play a significant role in the S-glutathionylation of cell surface or extracellular proteins.
3. Grx1 and Grx2
Grx is a thiol disulfide oxidoreductase and member of the thioredoxin superfamily. A primary function of Grx is to facilitate the removal of GSH from S-glutathionylated proteins, S-deglutathionylation [for review see refs. (178, 250)], but somewhat unexpectedly Grx also has some activity in the forward reaction. Specifically, Grx1 and Grx2 were shown to promote S-glutathionylation of GAPDH, actin, and PTP1B using GS· as the proximal donor (90). The role of Grx as an S-glutathionylase appears limited. It is interesting that GAPDH was identified as the target for Grx-mediated S-glutathionylation since it is among a subset of proteins that is S-glutathionylated despite the fact that the target cysteine is not in a basic environment and thus lacks the characteristic low pKa (210). To date, other known substrates for Grx-mediated S-glutathionylation have not been identified. With GSTP (and likely other GSTs) redox regulation of molecular targets may be determined by the context of the proteins, rendering some degree of specificity.
B. Proteins with deglutathionylase activity
The protein S-glutathionylation cycle is initiated under conditions of oxidative or nitrosative stress and is reversed when a reducing environment is restored. As such, the GSH moiety may be removed (deglutathionylated) by Grx and/or sulfiredoxin (Srx). The role of Grx in deglutathionylation has been extensively studied and is proposed to occur through direct thiol/disulfide exchange reactions in three steps: (i) Grx reacts with P-SSG to release P-SH and form the covalent intermediate, Grx-SSG (178); (ii) Grx-SSG is reduced by GSH, yielding reduced Grx, and GSSG (90, 177); (iii) GSSG is reduced by glutathione reductase to restore GSH levels. The role of Grx in this cycle and its impact on various clinical implications has been reviewed (177). However, a role for Srx in the S-gluthionylation cycle is emerging. Findlay et al. reported that Srx can act as a general deglutathionylase for a number of proteins, including PTP1B and actin (79, 80). S-glutathionylation of PTP1B inhibits phosphatase activity. However, Srx restores the activity of PTP1B via deglutathionylation. Park et al. demonstrated that deglutathionylation of 2-Cys peroxiredoxin (Prx1) is specifically catalyzed by sulfiredoxin (203). In these studies, the affinity of Srx and Grx1 toward Prx1 was evaluated and it was determined that Srx has a greater binding affinity for Prx1. The biological relevance was supported in cultured tumor cells after exposure to oxidative stress.
The tissue distribution, subcellular localization, and substrate specificity of Grx and Srx differ, suggesting distinctive roles in redox regulation. Grx1 and Grx2 are localized in the cytosol and mitochondria/nucleus, respectively (128). Grx1 has also been identified in the inner membrane space of the mitochondria and has been suggested to translocate to the nucleous; however, definite evidence for nuclear localization of either Grx1 or Grx2 is limited [see review (178)]. Srx contains a single cysteine residue, whereas Grx contains two within a conserved CXXC motif. Consequently, the molecular mechanism of deglutathionylase activity will undoubtedly differ during disulfide exchange reactions. More importantly, any substrate specificity for these enzymes for target proteins has yet to be defined. There exist a growing number of disease states (described in subsequent sections) that are associated with S-glutathionylated proteins. A better understanding of deglutathionylation pathways in the context of both Grx and Srx could potentially provide druggable targets that may be a platform for disease treatments.
IV. Redox Regulation of Kinase Signaling Pathways
A. S-glutathionylation and modulation of mitogenic signaling
S-glutathionylation of several kinases, phosphatases and transcription factors can impact signal transduction pathways, particularly in cancer cells. For example, Figure 7 depicts the Ras-MEK-extracellular signal-regulated kinase (ERK) pathway and its key regulators, many of which contribute to the regulation of cell growth.
FIG. 7.

S-glutathionylation as a regulator for Ras-mitogen-activated protein kinase pathways in cancer. This figure depicts several sites in the Ras-mitogen-activated protein kinase pathway in cancer cells where reversible glutathionylation may serve as a potential regulatory mechanism. Ras, PKA, PKC, SHP-1, SHP-2, PTP1B, SERCA, RyR, and S100 proteins are highlighted, indicating that they are the targets of this modification.
1. Ras-MEK-ERK pathway
Ras is an essential component of the signaling pathway that underlies growth factor-induced cell proliferation, differentiation, or survival. Ras is a member of the small GTPase superfamily, functioning as regulated GDP/GTP switches between inactive GDP-bound and active GTP-bound states. The three human ras alleles encode H-Ras, N-Ras, and K-Ras proteins. The Ras GDP/GTP cycle is controlled by two classes of regulatory proteins: guanidine nucleotide exchange factors that stimulate the formation of Ras-GTP, and GTPase-activating proteins (GAPs) that stimulate hydrolysis of the bound GTP, thereby returning Ras to the GDP-bound form. Mutated ras alleles are found in 30% of human cancers, occurring at particularly high frequencies in colon and pancreatic carcinomas. For example, K-Ras mutations can be detected in 30%–40% of all patients with colorectal cancer (182). The mutations render Ras insensitive to GAPs and persistently activated in the absence of external stimuli. Such gain-of-function mutations of Ras are thought to be involved in the process of cell transformation. S-glutathionylation of H-Ras on Cys118, a reactive thiol that has been identified in the GTP-binding region of H-Ras, increases its activity and leads to p38 and Akt phosphorylation, which in turn contributes to the induction of protein synthesis (1). As part of this pathway, it has been speculated that modification of the critical thiol in the Ras GTP-binding domain may create a conformational change that favors GTP hydrolysis.
2. Protein tyrosine phosphatases
Protein tyrosine phosphatases (PTPs) are generally considered to be negative regulators of tyrosine phosphorylation-involved signaling pathways and are implicated in cancer development through a somewhat obscure process of fine-tuning the intensity and/or duration of signaling through phosphorylation. Several groups have shown that either RNS or epidermal growth factor (EGF) can reversibly inactivate PTP1B by causing its S-glutathionylation (272). In these situations, the active site Cys215 is the residue subject to oxidation to sulfenic acid, reacting further with GSH to form the mixed disulfide (15).
In the protein tyrosine phosphatase family, a subgroup of cytoplasmic PTPs containing two SH2 N-terminal domains and a C-terminal PTP domain and are referred to as Src-homology protein tyrosine phosphatases (SHPs). SHP-1 and SHP-2 are intimately involved in regulation of cell growth, regulating mitogen-activated protein kinase (MAPK), and chemotactic response pathways (44). SHP-1 and SHP-2 each promote the activation of the Ras-MAPK signaling pathway. SHP-1 activity is Ras-dependent (146), whereas SHP-2 dephosphorylates phosphotyrosine sites on growth factor receptors that recruit p120 Ras-GAP, thus preventing the inhibition of Ras activation by p120 Ras-GAP (4). Activating mutations of PTPN11 (human SHP-2 gene) resulting in a loss of auto-inhibition of PTP activity have been identified in individuals with Noonan syndrome, a human developmental disorder sometimes associated with juvenile myelomonocytic leukemia. Further, somatic mutations of PTPN11 are associated with pediatric leukemias (170). SHP-1 and SHP-2 were susceptible to S-glutathionylation by GSSG in vitro, but these observations have not been replicated in vivo (230). Nevertheless, these phosphatases are functionally quite important and as a consequence their regulation through thiol modifications could prove to be physiologically significant and pertinent to disease pathologies. As a consequence, further studies are merited.
3. Protein kinase A
Protein kinase A (PKA) is the main intracellular receptor of the second messenger cyclic adenosine monophosphate and upon activation phosphorylates a number of substrates that regulate such cellular functions as glycogen metabolism, cell proliferation, and differentiation. Possible cross-talk between cyclic adenosine monophosphate/PKA and MAPK pathways has recently been reviewed (95). The catalytic subunit of PKA contains two cysteines at positions 199 and 343. Humphries et al. reported that both cysteines are susceptible to S-glutathionylation, but it was the modification of Cys199 that led to kinase inactivation (129). In light of the pleiotropic impact of PKA on a number of critical signaling pathways (some of which have disease relevance), the functional importance of this modification may have quite broad significance.
B. Phosphatidylinositol 3-kinase-Akt-p53 pathway
Akt/protein kinase B is a Ser/Thr kinase with functional significance in cell survival pathways. Akt is recruited to the membrane by phosphatidylinositol 3-kinase (PI3K) through generation of phosphoinositol triphosphate, where it is activated by phosphorylation at Thr308 and Ser473 by kinases, including PDK (phosphoinositol-dependent kinase) and protein kinase C (PKC). The function of PI3K is opposed directly by the tumor suppressor phosphatase and tensin homolog deleted from chromosome 10 (PTEN). PTEN is an important target for redox regulation. It was reported that S-nitrosothiols oxidatively modified PTEN, leading to reversible inhibition of its phosphatase activity and that the oxidized species was a mixed disulfide (311). In macrophages, ATP-induced ROS inactivated PTEN by S-glutathionylation, leading to PI3K/Akt pathway activation and a subsequent equilibrium shifted toward cell survival (54). The dephosphorylation of Akt is mediated by the Ser/Thr protein phosphatase 2A (PP2A). It was demonstrated that H2O2 inhibited PP2A activity in human colon carcinoma Caco-2 cells, and GSSG was able to inhibit the activity of isolated PP2A, indicating that PP2A is a target for regulation by S-glutathionylation (222). In general, the phosphatase family members with cysteine residues that are critical to their catalytic activity are subject to regulation by S-glutathionylation.
The p53 tumor suppressor is a potent transcription factor that, in response to a variety of cellular stresses, including DNA damage, hypoxia, chemotherapeutic drugs, oxidative stress, and many aberrant growth signals, controls the expression of a wide array of genes involved in cell cycle control, DNA repair, differentiation, and apoptosis (286). Approximately 30%–50% of human cancers contain mutations in p53. Inactivation of the p53 gene or disruption of p53-regulated pathways eliminates p53 function in many cancers. Murine double minute 2 (Mdm2) is an E3 ubiquitin ligase that plays a key role in maintaining p53 at critical physiological levels by targeting it for proteasome-mediated degradation in normal, unstressed cells. Expression of the Mdm2 gene is p53-dependent and thus p53 and Mdm2 cooperate in a negative feedback loop. Akt phosphorylates Mdm2, enhances nuclear localization of Mdm2, and increases ubiquitination and degradation of p53 (176). p53 binds to its consensus DNA sequence as a homo-tetramer. In human malignant glioblastoma and colon carcinoma cells, p53 was S-glutathionylated and mass spectral analysis identified the modification sites as Cys124, Cys141 and Cys182 in the proximal DNA-binding region (283). As a consequence of S-glutathionylation, p53 lost the ability to recognize its consensus DNA sequence. S-glutathionylation at these sites also interfered with p53 oligomerization, another important step in p53-targeted gene transactivation (283). Precisely how redox control of the multiple p53 mediated pathways relates to cancer is not clear. Nevertheless, the very fact that p53 can be influenced by S-glutathionylation implies an important cross-talk between these pathways.
C. I kappa B kinase-nuclear factor kappa B pathway
Nuclear factor kappa B (NFκB) is an inducible transcription factor that plays a role in the expression of over 100 genes involved in immunity, inflammation, proliferation, and in defense against apoptosis (140). It can tip the balance between apoptosis and proliferation and malignant growth of tumor cells. NFκB belongs to the Rel family of proteins that includes five members: RelA (p65), RelB, c-Rel, NFκB1 (p50/p105), and NFκB2 (p52/p100). The inactive form of NFκB is localized in the cytosol as p65/p50 (most abundant) or p50/c-Rel heterodimers through interaction with IκB repressor proteins. Upon stimulation, IκB is phosphorylated by I kappa B kinase (IKK) and degraded via the ubiquitin-proteasome pathway, allowing NFκB to translocate into the nucleus, bind to DNA and activate transcription. The NFκB survival pathway is regulated by S-glutathionylation at multiple steps. Therapeutic strategies that selectively enhance S-glutathionylation of those components could be effective therapeutic approach in oncology and drug discovery efforts with interference of this modification as an endpoint could be a viable approach.
IKK, when phosphorylated by active Akt, phosphorylates IκB and relieves the inhibition of NFκB. It was reported that in alveolar epithelial cells, IKKβ activity was suppressed by H2O2-induced S-glutathionylation of Cys179, a process that occurred through a sulfenic intermediate precursor (229). S-glutathionylation of IKKβ hence dampened TNFα-induced NFκB activation. NFκB itself is also a direct target for S-glutathionylation. One group reported that p50-NFκB was S-glutathionylated at Cys62, a residue located in the DNA-binding domain and caused a reversible inhibition of its DNA-binding ability (212). Moreover, these studies showed that the formation of a protein sulfenate at the same residue contributed much less to the inhibition of p50-NFκB DNA-binding. The implication from this observation is quite important since it implies that it is not just any modification at Cys62, but a mixed disulfide that changes protein conformation and interrupts the p50-NFκB-DNA interaction. Under normal conditions, increased ROS prevents NFκB activation by cytokines, whereas hypoxia has been shown to promote both degradation of IκBα and p65-NFκB nuclear translocation and binding to DNA and transactivation in mouse and human cells (218). The cysteine precursor N-acetyl-L-cysteine (NAC) enhanced hypoxic apoptosis in cells. This occurred at the level of abrogating hypoxia-induced p65-NFκB binding to DNA and subsequent NFκB-dependent expression of survival genes (218). S-glutathionylation of p65 was diminished under hypoxic conditions in cell in which Grx was knocked down (217). It was proposed that Grx may serve as a catalyst for S-glutathionylation of p65 under the GSH-thiyl-radical generating conditions of hypoxia and NAC treatment. Under normoxic conditions, NAC is proposed to restore GSH levels and redox homeostasis, which would disfavor S-glutathionylation reactions. As such, delivery of NAC to the hypoxic core of tumors could provide an effective means of cell killing.
D. JNK-c-Jun pathway
c-Jun N-terminal kinase (JNK) is a stress-activated protein kinase implicated in pro-apoptotic signaling in cancer and contributes to the mediation of the cytotoxicity of a variety of chemotherapeutic agents. Activator protein 1 is a homo- or heterodimeric transcription factor consisting of Jun (c-Jun, JunB, and Jun D) and Fos (c-Fos, FosB, and Fra-1), which regulates a large number of genes involved in cancer cell signaling. Both subunits interact with each other via their basic leucine-zipper domain. JNK phosphorylates c-Jun and activates c-Jun transcriptional activity. A decrease in the cellular GSH/GSSG ratio provided an alteration in the redox potential that led to the oxidation of c-Jun thiols by mechanisms that include both S-glutathionylation and intermolecular disulfide bridge formation (144). Specifically, S-glutathionylation of c-Jun occurs at Cys269, but not at the disulfide bridge between c-Jun subunits, sterically blocking DNA binding (144).
MAPK/ERK kinase kinase 1 (MEKK1) is the first characterized JNK kinase that activates JNK through phosphorylation of mitogen-activated protein kinase kinase 4. Activation of MEKK1 is known to transmit a cell survival signal. Cross and Templeton reported that either N-ethylmaleimide or menadione directly inhibited MEKK1 activity in human prostate cancer cells and that the inhibition involved S-glutathionylation at a single unique cysteine residue, Cys1238, in the ATP-binding domain of MEKK1 (53). They also confirmed that modification by S-nitrosylation or oxidation of cysteine to sulfenic acid was not an effective way of achieving the inhibition. ASK1 is also able to phosphorylate mitogen-activated protein kinase kinase 4 on the same sites and to activate JNK. In contrast to MEKK1, activation of ASK1 mediates TNFα-induced apoptosis. ASK1 was activated under oxidative stress (53), and its kinase activity was regulated by thioredoxin (Trx), a redox regulatory protein known to protect cells from TNFα-induced cytotoxicity (171). Trx bound to the N-terminus of ASK1 and inhibited its activity, whereas oxidation of Trx under oxidative conditions disrupted this interaction and subsequently activated ASK1 (238). S-glutathionylation of Trx1 has been demonstrated in model cell systems from plants through humans (108). In each case, the modification does not occur at cysteines spanning the redox regulatory domain, but at residue Cys73, located in the flexible α3–β4 loop of the protein. S-glutathionylation of Trx1 leads to a reduction in the enzymatic activity, but whether the mixed disulfide alters its affinity for other potential interacting proteins or even its subcellular localization remains to be shown.
The intracellular regulatory control of JNK by GSTP was the first example of direct binding of a small redox protein with a kinase (3). Subsequently, it seems that there are other examples where GSTs play a regulatory role in cellular signaling through interaction with key kinases involved in cellular responses to stress, apoptosis, and proliferation. In unstressed cells, c-Jun, JNK, and GSTP form a protein complex that serves to sequester JNK in an inactive state. However, under oxidative or nitrosative stress, the complex is disrupted and all three components are subject to S-glutathionylation. When the GSTP-JNK complex is disrupted the released JNK is able to activate c-Jun and results in subsequent activation of downstream effectors. Under these conditions, the liberated GSTP forms oligomers (271). GSTP is S-glutathionylated at Cys47 and Cys101, both identified as critical residues for the interaction with JNK (273). GSTP has also been implicated in control of TNFα-induced apoptosis at levels upstream of JNK. GSTP can form a heterodimer with the TRAF2 and inhibit TRAF2-mediated JNK activation (301). Moreover, through this process, GSTP can also interfere with the interactions between TRAF2 and ASK1, thereby inhibiting the auto-phosphorylation of ASK1 (301).
GSTP upregulation has been observed in many tumors compared with the surrounding normal tissues and in various cancer cell lines resistant to anticancer agents (268), but precisely why such high GSTP levels are maintained in tumor cells has never really been clear. There are now many examples where GSTP expression levels have been experimentally manipulated in order to try to understand the importance of this GST to cells. For example, forced expression of GSTP in NIH3T3 fibroblasts increased the activation of p38 and ERK MAPK pathways (310). However, GSTP-deficient (GSTP1P2−/−) mice present a phenotype that is partially characterized by higher levels of circulating blood cells (all three lineages) than wild-type animals (235). Pharmacological manipulation of the GSTP inhibitor TLK199 also increased the number of peripheral white blood cells in wild-type mice but not in GSTP1P2−/− animals. Thus, both the genetics and pharmacology support the principle that GSTP has a role in regulation of myeloproliferation (235). Subsequently, it was reported that the enhanced myeloproliferation in GSTP1P2−/− mice was associated with increased JNK activation in bone marrow cells (94). The discrimination between the survival and apoptotic functions of JNK seems to correlate with the level and duration of the enzyme activation. A strong and sustained activation is associated with apoptosis, whereas a weaker and transient phosphorylation is correlated with proliferation. This is presumably tissue specific, and increased JNK activation observed in GSTP1P2−/− bone marrow cells could well be a contributory factor in the observed increased levels of proliferation of these cells.
The peroxiredoxin (Prx) family of redox regulatory proteins also has an impact on JNK activity and also directly associates with GSTP to prevent oxidative damage to membranes (165). Under oxidative stress conditions, Prx can be S-glutathionylated on its active site cysteine (194). The S-glutathionylation mediated by GSTP through the formation of the heterodimer between Prx and GSTP is an intermediate step in the regeneration of Prx catalytic activity (165). Overexpression of Prx, primarily through interaction with GSTP-JNK complex, prevents JNK release form the complex and causes resistance to radiation and suppression of JNK activation and apoptosis in lung cancer cells (143). Historically, GSTs were originally identified as ligandins or binding proteins, and more recent studies suggest that such proteins that are known to bind to GSTP are also targets for S-glutathionylation. One possibility is that the protein:protein proximity interactions might facilitate this post-translational modification. Because the role of GSTP in catalyzing S-glutathionylation reactions is recently reported, the relative balance of GSTP function between ligand binding (i.e., protein:protein interactions), protein S-glutathionylation, and catalytic detoxification is yet to be characterized. It does seem that GSTP is established as a versatile and multifunctional protein, the existence of which may be contingent upon the long-term selective pressures of convergent evolution.
V. S-Glutathionylation and Modulation of Survival/Apoptosis
Under normal physiological conditions, the number of cells within an organ remains constant with a balanced equilibrium of cell death and mitosis. However, ROS/RNS-induced changes in redox potential can produce disease pathologies and can lead cells to apoptosis or necrosis and disrupt this equilibrium. In general, the extent of cell death is proportional to severity of the disease. Low levels of ROS/RNS can lead to activation of JNK/NFκB pathways and induce apoptotic death pathways. In contrast, high levels of oxidants promote necrotic cell death through inhibition of caspase activity that is required to modulate apoptosis. Apoptosis or programmed cell death is a ubiquitous homeostatic process and its deregulation is widely regarded as either a cause or consequence of distinct pathologies including cancer and autoimmune and neurodegenerative diseases. A number of signaling pathways leading to the progression of apoptosis have been extensively documented. However, recent reports serve to highlight how changes in the redox environment might influence the efficient activation of the cell death machinery (85). In particular, GSH depletion and S-glutathionylation can be specific regulators of apoptosis triggered by a wide variety of stimuli, including activation of death receptors, stress, environmental agents, and cytotoxic drugs (85). It is possible to reformulate these emerging paradigms into our current understanding of known cell death mechanisms. Figure 8 depicts some key signal transduction pathways critical to survival/apoptosis in which regulation by reversible S-glutathionylation of signaling intermediates has been implicated.
FIG. 8.

Apoptosis/survival signaling pathways that are regulated by reversible S-glutathionylation. This figure demonstrates key signal transduction pathways in which regulation by reversible S-glutathionylation of signaling intermediates has been implicated. Kinases (PKC, IKK, JNK, and MEKK1), phosphatases (phosphatase and tensin homolog deleted from chromosome 10, and protein phosphatase 2A), transcription factors (p53, nuclear factor kappa B, and c-Jun), redox proteins (GSTP, Trx, and Prx), and death molecules (Fas and caspase 3) are highlighted, indicating that they are the targets of this modification. PP2A, protein phosphatase 2A; PTEN, phosphatase and tensin homolog deleted from chromosome 10.
A. S-glutathionylation of death receptors
Apoptotic cell death results from transduction of extracellular death signals coupled with developmentally controlled activation of endogenous execution programs. Induction of apoptosis through external signals may be triggered by activation of death receptors, such as Fas by Fas ligand (FasL), DR4, DR5 by TNF-related apoptosis-inducing ligand (TRAIL), and TNFR1 by TNFα. Activation of Fas, DR4, and DR5 leads to the formation of the death-inducing signaling complex through the recruitment of the Fas-associated death domain (FADD) and caspase-8 and -10. Initiator caspase-8 is activated and further amplifies the apoptotic cascade by activation of executioner caspases-3, -6, and -7. In contrast, TNFα-induced signaling results in the recruitment of TNFR-associated death domain protein (TRADD) and TRAF2. This complex mediates pro-apoptotic signaling cascades, including the activation of ASK1 and JNK, leading to the transcriptional/post-transcriptional regulation of apoptotic genes. TNF is a cytokine that leads to apoptosis when TNF binds to TNF-receptor. TNFα-induced apoptosis can correlate with increased protein S-glutathionylation, a process that can be inhibited by overexpression of B-cell lymphoma 2 (Bcl-2) (263).
Another transmembrane protein of the TNF family is Fas, a death receptor expressed on the surface of a variety of normal and malignant lymphoid cells as well as nonlymphoid tumors and tumor cell lines. In particular in the immune system, its primary function is to trigger apoptosis when Fas ligand binds. Recently, S-glutathionylation of Fas receptor at Cys294 has been linked to Fas ligand-induced apoptosis. Evidence suggests that addition of the mixed disulfide is a critical regulatory event that promotes Fas aggregation and enhances binding of Fas ligand to Fas, thereby amplifying the apoptotic signaling cascades (10).
Because tumor cells have an altered redox status they are generally more resistant to oxidative stress. Increased levels of ROS are found in Ras-transformed NIH3T3 fibroblasts (132). Clement and Stamenkovic also demonstrated that increasing the intracellular superoxide anion (O2−) concentration in human melanoma cells abrogated Fas-mediated apoptosis (50). Conversely, Fas-resistant bladder tumor and osteosarcoma cells were rendered sensitive to Fas signal by decreasing their intracellular superoxide levels (50). Thus, it was proposed that increased intracellular superoxide levels are responsible for the resistance of tumor cells to Fas-induced apoptosis. The underlying mechanism could be due to oxidative inactivation of the caspases.
B. S-glutathionylation of caspases
The caspases belong to a family of at least 12 cysteine proteases, which play an integral part in the execution phases of apoptosis. Caspases are present in an inactive form in the cytoplasm. Pro-caspases become activated during apoptosis by proteolytic processing at specific sites, followed by assembly of the active form. The active enzyme can cleave a number of defined substrates and lead to the eventual dismantling of the cell. Hence, regulation of caspase activity is important in cancer signaling.
Since their activities rely on the catalytic cysteines, the caspases clearly represent potential targets for regulation by S-glutathionylation. Pan et al. found that caspase-3 was S-glutathionylated in human umbilical vein endothelial cells (HUVECs) under basal conditions and became deglutathionylated by Grx upon the TNFα-induced apoptosis (201). Small interference RNA knockdown of Grx significantly inhibited TNFα-induced endothelial cell death due to the attenuated caspase-3 cleavage concomitant with increased caspase-3 S-glutathionylation. Cysteine-to-serine mutations (C163S, C184S, and C220S) of caspase-3 that were predicted to prevent S-glutathionylation showed increased cleavage compared with wild-type caspase-3. This inverse correlation between caspase-3 S-glutathionylation and cleavage was further confirmed by the observation that in vitro S-glutathionylation of caspase-3 inhibited its cleavage with recombinant caspase-8. Apart from the inhibition of caspase-3 cleavage, Huang et al. reported that after activation with actinomycin D when caspase proteins were incubated with GSSG, the activity of caspase 3 was inhibited in a dose- and time-dependent manner and that the process was reversed by thiol-specific reducing reagents. When biotin-labeled GSSG was incubated with recombinant caspase-3, the biotin label was found associated with both p12 and p17 subunits of active caspase-3. Matrix-assisted laser desorption ionization mass spectrometric analysis of GSSG-treated recombinant caspase-3 identified the specific S-glutathionylation sites as Cys135 of the p17 protein (equivalent to Cys163 of caspase-3) and Cys45 of the p12 protein (equivalent to Cys220 of caspase-3) (127). This provides a novel mechanism-based, regulatory role for S-glutathionylation for the possible control of caspase activity in apoptosis. Of note, in the early stages of apoptosis high levels of ROS actually block apoptosis by downregulating caspase activity (110), a fact that could account for the resistance of tumor cells to ordinarily lethal drug challenges.
VI. Redox Regulation of Calcium-Dependent Proteins
Calcium (Ca) is an alkali earth metal essential to a number of cellular processes. Intracellular calcium ions (Ca2+) are stored in the ER, mitochondria, and some proteins and mobilization and/or release from these stores is a trigger for many signaling events that mediate a multitude of functions, including cell contraction and movement, the control of ion pumps, and critical signaling pathways. Calcium efflux can be induced by both ROS and RNS; in fact, many redox switches are calcium dependent. In addition, modification of Cys residues on calcium transporters can alter calcium homeostasis (see below).
A. Protein kinase C
PKC is a family of serine/threonine kinases that play a pivotal role in signal transduction. The activity of cPKC (PKCα, PKCβ1, and PKCβ2) is calcium dependent and, through phosphorylation of downstream targets, modulates transcription, proliferation, and cognition. When activated, PKCs translocate to the plasma membrane and have long-term activation. The PKC family is involved in tumor promotion and progression with roles in regulating mitogenesis, cell adhesion, apoptosis, angiogenesis, invasion, and metastasis (33, 174, 241). Elevated PKC levels are associated with progressive malignancy and drug resistance of breast, lung, and colon carcinomas. Human PKC isozymes contain 16–28 Cys residues including one or two Cys-rich zinc finger regions in the regulatory domain and 5–8 Cys in the catalytic domain (104). The thiol-specific oxidant diamide can induce S-glutathionylation of seven PKC isozymes (α, β1, β2, γ, δ, ɛ, and ζ). Diamide-induced S-glutathionylation of PKC inactivated the kinase activity of those isozymes, whereas PKCδ was unique in its resistance to inactivation (295) (48). Within the range of diamide concentrations that resulted in the inactivation of other isozymes, PKCδ activity was potentiated. Further evidence indicated that S-glutathionylation of the Cys residue in the catalytic domain (e.g., Cys499 of PKCα) inactivates PKCs and modification of Cys in the regulatory domain stimulates PKCδ activity by provoking Zn2+ release from the regulatory domain (47). Much work remains to establish the physiological importance of these modifications and whether there are any connections with disease pathologies. As discussed above, thiol-mediated regulation of phosphatases now seems to be partnered with a similar control of kinase activities. Such congruence serves as an interesting example of the confluence of phosphorus and sulfur-based biochemistry. Understanding the evolution and consequences of these cross-regulatory events will prove to be quite educational.
B. Sarco/ER calcium ATPase
Cellular calcium levels are regulated by transport systems in the ER, which, to a large extent, include the sarcoplasmic endoplasmic reticulum calcium ATPase (SERCA) and the ryanodine receptor (RyR). SERCA is the ER calcium uptake system that transports cytosolic Ca2+ into the sarcoplasmic reticulum, thereby quenching cytoplasmic Ca2+-regulated signals. As a consequence of nitrosative stress, Ca2+ uptake by SERCA2 is activated once it is S-glutathionylated at Cys674 (2). In direct contrast to the function of SERCA, RyR is an ER calcium release channel, releasing Ca2+ from the ER into the cytosol and producing a Ca2+ signal amplification. NAPDH oxidase activity stimulates S-glutathionylation of RyR1 and RyR2, which would contribute to faster calcium release from isolated triads (120). Multiple cysteine residues in RyR1 are substrates for S-glutathionylation, nitrosylation, or disulfide oxidation. However, S-glutathionylation of Cys3635, the only cysteine so far characterized as involved in RyR-regulated calcium release, does not result in the oxidative enhancement of channel activity (12). Since RyR has 100 cysteine residues, the oxidative targets that are functionally relevant to the redox-sensing properties of the channel need further identification.
C. Nitric oxide synthase
Cellular levels of NO are controlled by several isoforms of nitric oxide synthase (NOS): neuronal (nNOS, NOS1), inducible (iNOS, NOS2), and endothelial (eNOS, NOS3). While each isoform is a product of a distinct gene, both nNOS and eNOS are constitutively expressed and primarily found in neurons and endothelial cells, respectively. NO generation by these enzymes is controlled by the elevation of intracellular Ca2+ and the consequent activation of calmodulin (CaM). iNOS is not constitutively expressed and is not calcium dependent. The active form of eNOS is a homodimer with zinc ions tetrahedrally coordinated to two pairs of symmetrical cysteines. These cysteines are in a basic environment and, as a consequence, have a low pK and may be subject to S-glutathionylation. It has been shown that S-nitrosylation of some of these cysteines results in dissociation of homodimers into inactive monomers (224). eNOS can also be palmitoylated and consequently attached to the inner part of the plasma membrane. Its activation results in an NO burst close to the plasma membrane where NADPH oxidase and a chloride ion channel-3 (CIC-3) are also located. Ca2+ fluxes can activate NADPH oxidase and superoxide-radical generated on the exterior of the cell from either oxidative or nitrosative stress can influx through CIC-3 channels. When spatially close, the eNOS and CIC-3 channels may generate ONOO−, which together with excess GSH can induce eNOS S-glutathionylation. Thus, the effect of NO on Ca2+/NO homeostasis can start as an extracellular NO-mediated surface protein-thiol modification.
A glutathione metabolite of the preclinical drug O2-{2,4-dinitro-5-[4-(N-methylamino)benzoyloxy]phenyl}1-(N,N-dimethylamino)diazen-1-ium-1,2-diolate (PABA/NO) inhibits SERCA, perhaps, at the same site as thapsigargin initiating intracellular Ca2+ increase, activating CaM and consequently eNOS with the resultant NO burst. Our published report has shown that PABA/NO causes intracellular NO levels to rise above a certain threshold through eNOS activation with a subsequent link between S-nitrosylation and S-glutathionylation (166). Indeed, there is evidence to suggest that two distinct pools of S-nitrosylated proteins exist, one that is GSH stable and another that is GSH labile and subject to rapid conversion to a S-glutathionylated product. Two possible mechanisms of NO-mediated protein S-glutathionylation can be envisioned: through a GSNO (activated thiol) formation and its consequent reaction with protein-thiol (179) or through an intermediate protein-thiol nitrosylation (activated protein-thiol: analog of sulfenic acid) and its consequent reaction with GSH. The exact mechanism of eNOS modification is unknown but in vivo experiments have shown that eNOS activation in aortas and iNOS transgenetic expression in mouse heart both result in NO-induced protein S-glutathionylation. This dynamic modification may serve to physiologically downregulate eNOS by NO under normal conditions. Conversely, eNOS deglutathionylation can result in eNOS upregulation, maintaining physiological NO levels. Under normal physiological conditions the NO increase might be controlled by S-nitrosylation/glutathionylation of eNOS as an immediate response or by similar modification/activation of SERCA in steady-state regulation.
VII. S-Glutathionylation and Ubiquitin-Proteasome Pathway
A critical strategy to control the magnitude and duration of signal amplification in cancer is through ubiquitin/proteasome-dependent downregulation. Protein targeted for degradation is tagged with a highly conserved 8.5 kDa protein, ubiquitin. The attachment of ubiquitin to substrate proteins involves three separate enzymatic reactions (Fig. 9). First, ubiquitin is activated by ubiquitin-activating enzyme (E1) through a thioester bond between the conserved thiol group of E1 and the C-terminal glycine of ubiquitin, in an ATP-dependent manner. Second, the activated ubiquitin is conjugated to a cysteine residue in an ubiquitin-conjugating enzyme (E2) via a thioester bond. Lastly, the ubiquitin is transferred from the E2 enzyme to a lysine residue in the protein by ubiquitin ligase (E3). Proteins bearing polyubiquitin chains are targeted to the proteasome for proteolytic cleavage. It was revealed by mass spectrometry that the E2 protein was a target for S-glutathionylation under oxidative stress (86). The cysteines in the active sites of E1 and E2 enzymes actively participate in the ubiquitination process; thus, they need to be maintained in a reduced state. The activities of E1 and E2 enzymes in retinal pigment epithelial cells were inhibited by a decrease in the GSH/GSSG ratio, which was consistent with the suppressed proteolytic cleavage (197). In PC12 cells, inhibition of GSH synthesis results in decreased ubiquitin conjugation to the E1 enzyme (134). E3 ubiquitin ligase also relies on the active cysteines to catalyze the conjugation of ubiquitin to the target proteins. However, there is no evidence that E3 enzymatic activity is subject to redox regulation. Besides the E1 and E2 enzymes, the proteasome is also a target for S-glutathionylation. Demasi et al. reported that the 20S proteasome is subject to S-glutathionylation in intact cells and that S-glutathionylation differentially regulates proteasome proteolytic activity (65).
FIG. 9.

S-glutathionylation in the ubiquitin-proteosome pathway. This diagram outlines the sequential steps involved in ubiquitination/proteasome-mediated protein degradation. S-glutathionylation steps for E1, E2, and proteasome (-SSG) have been indicated. This redox-regulated post-translational modification leading to inhibition of protein degradation could differentially influence cancer development and cancer prevention, depending on the function of the substrate protein in specific signaling pathways.
S-glutathionylation of the components involved in the ubiquitin pathway shuts down the proteasomal degradation process. On the one hand, this could be cancer promoting if the target protein mediates a proliferative (e.g., MAPK pathway) or pro-apoptotic signal (e.g., JNK pathway). On the other hand, if protein degradation is required as a survival signal (e.g., activation of NFκB pathway) this could favor cancer prevention.
VIII. S-Glutathionylation and Unfolded Protein Response
A. Signaling pathways in the unfolded protein response
Secretory and trans-membrane proteins are processed in the ER. Such processing includes a series of post-translational modifications, notably glycosylation and disulfide bond formation. In contrast to the reducing conditions of the cytosol (where the GSH:GSSG ratio is ∼100:1), protein disulfide bond formation depends on the highly oxidizing conditions within the ER compartment (GSH:GSSG ∼3:1). This unique environment also provides a platform to sense oxidative and nitrosative stress. Stress upon the ER results in the accumulation of misfolded proteins, leading to cellular deployment of the unfolded protein response (UPR) (Fig. 10) (271). The UPR imparts three primary functions: (i) initially restore normal function of the cell by halting protein translation, (ii) activate the signaling pathways that lead to increased production of molecular chaperones involved in protein folding, and (iii) trigger the degradation of terminally misfolded proteins (271). If these objectives are not achieved within a certain time frame, or the disruption is prolonged, the UPR will initiate apoptosis. In mammalian cells, it is well established that ER stress and the UPR are components of hypoxic stress response in tumors (77).
FIG. 10.

The UPR and pro-apoptotic pathways. This figure depicts the UPR signaling cascades and UPR-related pro-apoptotic pathways. During homeostasis, three endoplasmic reticulum membrane signaling molecules, pancreatic ER kinase, IRE1, and ATF6, are negatively regulated through associations with BiP. Oxidative (reactive oxygen species) and nitrosative (reactive nitrogen species) stress leads to S-glutathionylation of PDI that blunts isomerase activity. As a consequence, protein folding is dysregulated and leads to the accumulation of unfolded proteins. BiP triggers the UPR by disassociating from the membrane signaling molecules, thereby promoting transcriptional and translational regulation of gene expression and signals pro-apoptotic pathways. ATF6, activating transcription factor 6; ER, endoplasmic reticulum; PERK, pancreatic ER kinase; UPR, unfolded protein response.
The ER contains several key chaperone proteins that catalytically mediate protein folding and prevent aggregation of proteins as they undergo maturation. Multiple canonical pathways ensure the quality control in protein folding within the ER (Fig. 9) [reviewed in ref. (271)]. Specifically, the ER membrane harbors three signal-transducing proteins that modulate the UPR: (i) pancreatic ER kinase, (ii) activating transcription factor 6, and (iii) inositol-requiring enzyme 1. Regulation of these three proteins is contingent upon interactions with binding immunoglobulin protein (BiP) also known as 78 kDa glucose-regulated protein (GRP78). The accumulation of misfolded proteins results in the dissociation of BiP and elicits the UPR. Translational attenuation occurs during hypoxia through the activation of pancreatic ER kinase and phosphorylation of eukaryotic initiation factor 2α (25). Different mechanisms contribute to ER-induced apoptosis triggered by ROS and RNS. The first involves dissociation of the inhibitor GSTP from TRAF2-ASK1-JNK complex and activation of JNK. Multiple proteins within this cascade are targets of S-glutathionylation and have been discussed in prior sections. A second pathway involves transcriptional activation of the gene that encodes the C/EBP homologous protein (CHOP). Overexpression of CHOP has been reported to activate inositol-requiring enzyme 1 through pro-apoptotic Bcl-2 member BAX and BAK (118). CHOP can be phosphorylated by the p38 MAP kinase, a process that leads to cell cycle arrest (294). The third ER-induced apoptotic pathway involves caspase activation, including caspase 3, and this has also been discussed in previous sections.
B. Protein disulfide isomerase
The most abundant chaperone in the lumen of the ER is protein disulfide isomerase (PDI). PDI is the first and most well-characterized member of the PDI subfamily that belongs to the thioredoxin superfamily with two catalytically inactive and two active thioredoxin domains (115). PDI is organized into five domains (a, b, b′, a′, and c) and the C-terminal KDEL sequence retains it to the ER. The crystal structure of yeast PDI suggests that four thioredoxin domains (a, b, b′, and a′) form a twisted U shape and/or an alternative boat conformation with the catalytic domains facing each other and an internal hydrophobic surface that interacts with misfolded proteins (270). Similar to GSTP, PDI has both enzymatic and protein binding functions. As a PDI, it contains two active sites in the a and a′ thioredoxin domains, each having two conserved cysteine residues that cycle between oxidized (disulfide) and reduced (dithiol) states to facilitate the folding and correct disulfide bond formation of its protein substrates (115). As a chaperone, PDI functions as a subunit of prolyl-4-hydroxylase and as a microsomal triglyceride transfer protein (115).
PDI is regulated by the ER oxidase 1 (Ero1), which restores reduced PDI to an oxidized state through disulfide exchange with Ero1. Ero1 activity is attenuated under oxidizing conditions in the ER through modulation of noncatalytic cysteine residues, which differ in yeast and human (246). Post-translational modifications that alter PDI function have recently been described. Nitrosylation of catalytic cysteine residues in the a and a′ domains of PDI occurs in the brains of patients with sporadic Parkinson's and Alzheimer's disease (AD) (280), both of which involve ER stress and activation of the UPR. Nitrosylated PDI fails to function properly and thereby leads to the accumulation of misfolded proteins. In addition, our group reported that PDI undergoes S-glutathionylation upon treatment with the anticancer agent PABA/NO (274) and that S-glutathionylation occurred at two of the four active-site cysteine residues (Cys53 or Cys56, or Cys397 or Cys400), resulting in enzyme inactivation and activation of the UPR and cancer cell death. Collectively, these studies indicate that redox regulation of PDI is an upstream signaling event in the UPR (Fig. 10), deregulation of which can lead to tissue injury, and may even have relevance for cancer therapeutics (271, 274).
PDI occurs abundantly on the surface of cancer cells (252). A study with human breast ductal carcinoma tissue and histologically normal tissue concluded that a subset of ∼30 proteins, including PDI, were characteristic of epithelial neoplasia (20). However, a role for PDI in the cancer phenotype is not well characterized and any kind of clinical correlation requires further work. In addition, proteomic analyses in a wide range of cancer cell lines have revealed alterations in the expression pattern of other PDI family members that correlate with UPR and various pathologies and they are listed in Table 3.
Table 3.
Mammalian Protein Disulfide Isomerase Family Members Implicated in Unfolded Protein Response and Pathologies
| Protein | Cellular localization | ↑ In unfolded protein response | Implication in pathologies |
|---|---|---|---|
| PDI | ER, surface, nucleus, cytosol, secreted | Yes | See text (251, 270, 273, 279) |
| ERp5 | ER, surface | Yes | See text (251) |
| PDIp | ER, cytosol | unknown | Upregulation in Parkinson's disease |
| ERp29 | ER, surface | Yes | Unknown |
| ERdj5 | ER | Yes | Unknown |
| HAG-2 | ER, surface, cytosol | Unknown | Hormone responsive breast tumor growth (20, 251) |
| HAG-3 | |||
The involvement of other protein disulfide isomerase family members in the unfolded protein response, cancer, and neurodegenerative disorders has not been reported to date, so they are not listed in this table.
ER, endoplasmic reticulum.
IX. Redox Regulation of Cell Migration and Mobilization
Cell migration is a central process in the development and maintenance of multicellular organisms. Orchestrated movement of cells in particular directions to destined locations are required for proper tissue formation during embryonic development, wound healing, and immune responses. Errors within this process frequently result in serious consequences, such as mental disorders, vascular disease, tumor formation, and metastasis. Knowledge of the mechanisms regulating cell migration may lead to the development of novel therapeutic strategies for controlling, for example, invasion of tumor cells. Here we focus on S-glutathionylation of cytoskeletal, cytosolic, and surface proteins all of which to some extent play roles in cell migration, bone marrow mobilization, and tumor metastasis.
A. S-glutathionylation of cytoskeletal proteins
The most prevalent S-glutathionylated cytoskeletal component is actin, an abundant protein involved in maintaining the infrastructure of the cytoplasmic matrix. S-glutathionylation of actin alters the ratio of soluble:polymerized protein, and consequently results in changes in the cellular architecture and membrane ruffling with concomitant changes in intracellular trafficking of many types of molecules. Wang et al. reported that S-glutathionylation at Cys374 of actin was prevalent in unstimulated cells; when cells were stimulated with growth factors (EGF, FGF), Grx-mediated deglutathionylation of actin led to actin filament polymerization and cytoskeletal rearrangement (289, 291). The S-glutathionylation of actin also plays a key role in cell spreading and in disassembly of actinomyosin complex during cell adhesion (78). Regarding protein–protein interactions, the modified actin, relative to the unmodified protein, manifests a weaker affinity for tropomyosin. Dalle-Donne (57, 58, 101, 233) and colleagues have carried out a series of studies to identify those residues that are critical in the S-glutathionylation of actin and the corresponding alterations in structure/function relationships. They showed that S-glutathionylation increased exposure of hydrophobic regions of the protein surface, increased ATP exchange, and decreased susceptibility to proteolysis. Structural changes of one of the loop domains were judged to be influence general actin polymerization kinetics. Table 4 summarizes the identified cytoskeletal proteins that are targets of S-glutathionylation in vitro or in cell culture. Some of their functions are subject to regulation by this modification.
Table 4.
Cytoskeletal Proteins Susceptible to S-Glutathionylation
Accumulating evidence has revealed the correlation between S-glutathionylation of cytoskeletal proteins and neurodegenerative diseases that will be discussed in subsequent sections. For example, S-glutathionylation of actin was increased in fibroblasts of patients with Friedreich's ataxia (FRDA) (204). The aberrant polymerization of S-glutathionylated Tau into Alzheimer-associated filament was also reviewed (68). Taken together, documentation of the redox regulation of cytoskeletal proteins may help in understanding the pathogenesis of such diseases and provide possible therapeutic directions.
B. Redox regulation of bone marrow mobilization
Serpins are a broadly distributed family of protease inhibitors that cause a conformational change to inhibit target enzymes. Humans have 36 serpin-like genes, out of which 27 encode inhibitory serpins (150). For example, SerpinA1 (antitrypsin) inhibits neutrophil elastase, and SerpinA3 (antichymotrypsin) inhibits cathepsin G. Accumulating evidence has suggested that SerpinA1 and SerpinA3 play an unexpected role in regulating the bone marrow hematopoietic microenvironment as well as influencing the migratory behavior of hematopoietic precursors. Hematopoietic stem and progenitor cells normally reside in the bone marrow but can be mobilized into the peripheral blood after treatment with cytokine or chemotherapy (153). Winkler et al. reported that SerpinA1 and SerpinA3 were downregulated in bone marrow during cytokine-induced hematopoietic progenitor mobilization (299). It was further demonstrated that radiation-induced SerpinA1 upregulation in bone marrow was responsible for radiation-induced inhibition of hematopoietic stem and progenitor cell mobilization (281). Thus, inhibition of SerpinA1 and SerpinA3 expression or activity in bone marrow might provide means to improve the outcome of clinical stem cell transplantation. However, mutations in SerpinA1 result in insoluble aggregates accumulated in the ER and lead to cirrhosis; the deficiency of SerpinA1 or SerpinA3 results in emphysema (160). The noninhibitory serpins function as hormone transporters, molecular chaperones, or tumor suppressors (150). For instance, SerpinB5 (Maspin) has been linked to inhibition of mammary carcinoma cell metastasis (316).
The inactivation of serpins involves spontaneous conformational transition to latency (150), and mutations that result in polymerization, one molecule docking into another to form an inactive long-chain serpin polymer (73, 160). It was previously demonstrated that a marked decrease in elastase-inhibitory properties of SerpinA1 occurred after modification of the protein with a number of thiol-specific reagents, including GSSG, and that this decrease in activity was correlated with changes in spectral properties suggestive of a protein conformational change (279). Removal of the thiol modifications with DTT restored the inhibitory activity, strongly suggesting that the altered properties of the derivatives were due to thiol modification alone (279). There is a single cysteine residue in SerpinA1 that has been localized at position 232. SerpinA1 and SerpinA3 were shown to be S-glutathionylated in plasma from mice treated with a redox modulating agent that mitigates myeloproliferation (273). The sites of modification and impact on functionality have yet to be determined.
X. Cancer and Redox Homeostasis
A. Energy metabolism
Under normal physiological conditions, cells maintain a redox buffer with GSH/GSSG at a ratio of 100:1 to minimize the oxidative action of ROS/RNS. Perturbation of this redox state will lead to oxidative stress that is a significant contributing factor for carcinogenesis (99). Cancer cells differ from normal cells as they exhibit resistance to oxidative stress, increased metabolic activity, mitochondrial dysfunction coupled with uncontrolled cell growth and abnormal balance of cell survival versus apoptosis. Upregulation of GGT as described in the previous section is a major contributing factor to the altered redox state of cancer cells. Here we describe some of the clusters of S-glutathionylated proteins in the context of cancer biology.
Malignant, rapidly growing tumor cells typically have glycolytic rates that are significantly higher than those of their normal tissues of origin (Warburg effect), which results in decreased mitochondrial ATP formation (177). Some tumor cells can survive without functional mitochondria. The Warburg effect suggests that mitochondria dysfunction may be an adaptive response in cancer cells that diminishes apoptosis and enables cell survival under hypoxic conditions. This hypothesis is also interpreted as the consequence of poor blood supply to tumors causing the hypoxia environment that makes glycolysis the viable metabolic pathway for energy production. It was demonstrated that the embryonic M2 splice isoform of pyruvate kinase is exclusively present in cancer cells and is essential for the shift in cellular metabolism to aerobic glycolysis that promotes tumorigenesis (46). Interference with energy metabolism by abnormal ROS/RNS levels is probably a critical event in carcinogenesis.
A recent series of observations has implicated S-glutathionylation of the glyoxalase system as critical in influencing intermediary metabolism. Glyoxalase 1 and 2 constitute a detoxification system that protects against two oxo-aldehydes and in particular, methylglyoxal. Methylglyoxal is a product of catalytic degradation of both triose phosphates and amino acid metabolism. Detoxification of these molecules proceeds through a hemithioacetal intermediate (GSH conjugation to the methylglyoxal) to D-lactate and free GSH. As a consequence of enterobacterial metabolism, high levels of methylglyoxal are particularly prevalent in the gastrointestinal tract. Although the primary sequence of glyoxalase 1 has been appreciated for some time (221), there has been considerably less information about whether the protein is subject to post-translational modifications or how enzymatic activity may be modulated. Reversible inactivation of glyoxalase 1 has been documented after nitrosylation of cysteine 139 of the enzyme (63). This result predicts that this and/or other cysteine residues may have importance in achieving post-transcriptional regulation of enzymatic activity. A recent study used mass spectrometry to identify four post-translational products (21). These included removal of N-terminal methionine, N-terminal acetylation, a vicinal disulfide bridge at Cys19/20, and S-glutathionylation at Cys139 (21).
S-glutathionylation of Glyoxalase1 effectively inhibited enzyme activity at physiologically relevant concentration of GSH (21). This study implies that direct regulation of the enzyme can occur in response to changes in cellular redox state. Since deregulation of the glyoxalase system has been implicated in a number of human pathologies (particularly diabetes and cancer), regulatory control by S-glutathionylation may be a critical component worthy of further studies. In addition, a number of key enzymes that actively participate in energy metabolism have been identified by mass spectrometry as targets for ROS/RNS-induced S-glutathionylation and their enzymatic activities are reversibly inhibited by S-glutathionylation; for review see refs. (177, 271). Figure 11 summarizes these enzymes and the critical steps involved in energy metabolism (i.e., glycolysis, citric acid cycle, and electron transport chain) are indicated. It does not seem unreasonable to suggest that S-glutathionylation of some of the enzymes of intermediary metabolism may be functionally associated with uncontrolled growth, the Warburg effect, and/or dysregulation of proliferation/apoptotic pathways in tumor cells.
FIG. 11.

Enzymes involved in energy metabolism that are regulated by S-glutathionylation. This figure summarizes the key participants in energy metabolism, of which regulation by reversible S-glutathionylation (-SSG) has been implicated. The steps of glycolysis, citric acid cycle, and electron transport chain are indicated.
B. S-glutathionylation and tumor metastasis
Cancer is more than just a mass of transformed cells, and 90% of deaths from solid tumors can be attributed to metastasis. Several discrete steps are discernible in the biological cascade of tumor cell metastasis: loss of cellular adhesion, increased invasiveness and entry into the blood stream (intravasation), survival in the circulation, exit into new tissues (extravasation), and eventual colonization of a distant site. Despite extensive studies, how metastasis occurs is still poorly understood.
Several metastasis suppressor genes have been confirmed, among which, the nucleoside diphosphate kinase (Nm23) was first identified and characterized by its reduced RNA levels in tumor cells exhibiting high metastatic potential (232). Overexpression of cellular Nm23 correlated with decreased metastatic potential of breast, melanoma, colon, and oral squamous cell carcinoma. Mutations of Nm23 observed in some cancers, including colorectal carcinoma, were associated with increased tumor aggressiveness. In response to H2O2 treatment in vitro and in vivo, Nm23-H1 was oxidized to various states that included S-glutathionylation, intra- and interdisulfide bonds, and sulfonic acid. Either oxidation or S-glutathionylation of the Cys109 of Nm23-H1 inhibited its enzymatic activity and accordingly its metastatic suppressor activity (151). This report points out that altered redox regulation in cancer cells may favor tumor metastasis by inhibiting metastasis suppressors.
Proteins facilitating metastasis could be therapeutic targets for redox modulation. Specifically, ERp5, another PDI family member, is a plausible candidate. ERp5 is expressed on the cell surface (Table 3) and is upregulated in invasive clinical breast cancer specimens, and its overexpression enhanced breast cancer cell metastasis through activation of the EGF receptor/PI3K and downstream signaling molecules, including Akt and RhoA (107). ERp5 overexpression is not limited to breast cancer. ERp5 was also upregulated during full-blown multiple myeloma and promoted efficient MHC class I chain-related protein A shedding, an event associated with immune suppression and progression of multiple myeloma (136). Importantly, the enzyme activity of ERp5 was required for MHC class I chain-related protein A shedding. S-glutathionylation of PDI shunts isomerase activity. The active site of ERp5 contains the WCGHC motif (like PDI) that contains the target cysteine within a basic region of the protein for S-glutathionylation. As such, this may prove to be a possible therapeutic targeting opportunity in cancer metastasis and immune response.
C. S100 proteins in cancer and leukocyte migration
The S100 proteins constitute a multigene calcium-binding family comprising 20 known human members. They are commonly upregulated in tumors and this is often associated with tumor progression. The expression pattern of different members of the family in various solid tumors has been extensively reviewed and how detection may be useful for diagnosis, monitoring and as possible therapeutic targets has been discussed (239). Cysteine residues have been suggested to play a pivotal role in the regulation of biological activity of the S100 proteins. Among the 20 recognized S100 sequences, only three do not contain cysteine residues. However, redox regulation of the S100 proteins is not extensively understood. As such, this section focuses mainly on the family members that are targets of S-glutathionylation and oxidation, namely, S100B, S100A8, and S100A9.
1. S100B
Serum levels of S100B increase in a stage-dependent manner in patients with melanoma and are associated with the presence of metastases, allowing S100B to be used as a diagnostic marker for the staging of malignant melanoma in a clinical setting. S100B contains a conserved cysteine, Cys84, in the C-terminus. GSNO and GSSG induce nitrosylation and S-glutathionylation at Cys84 of purified S100B and result in a global alteration of protein structure (315). The activity of the S100 proteins strongly depends on their metal ion binding. The accessibility of Cys84 was shown to increase upon calcium binding in human S100B (258), and S-glutathionylation of Cys84 by GSSG was observed only for the calcium-bound S100B. Since these modifications so far have only been shown in vitro, the physiological relevance of S100B nitrosylation and S-glutathionylation need to be explored further.
2. S100A8 and S100A9
S100A8 and S100A9 are upregulated in many cancers and are also implicated in the metastatic process. Vascular endothelial growth factor A, transforming growth factor β, and TNFα secreted by primary tumors induce expression of S100A8 and S100A9 in myeloid and endothelial cells within the lung before tumor metastasis (219). S100A8 and S100A9 are thus considered as chemoattractants to facilitate the homing of tumor cells to premetastatic sites within the lung parenchyma. S100A8 and S100A9 also increase the motility of circulating cancer cells by p38 MAPK-mediated activation of tumor cell pseudopodia (122). In addition, high or elevated levels of expression in activated leukocytes and their extracellular roles in leukocyte migration have linked them with various acute/chronic inflammatory disorders. Therefore, S100A8 and S100A9 could be viable therapeutic targets in the goal to prevent tumor cell metastases and/or inflammatory diseases.
S100A8 and S100A9 are readily oxidized by ROS/RNS that leads to structural changes that alter their functions. S100A8 and S100A9 can be nitrosylated by GSNO in vitro; S100A8 is more susceptible than S100A9 and nitrosylated S100A8 suppresses mast cell-mediated inflammation by reducing leukocyte adhesion and extravasation in the rat mesenteric microcirculation (156). While GSSG and GSNO can lead to S-glutathionylation of S100A8 and S100A9 in vitro, only the S-glutathionylated S100A9 was detected in cytosol of activated neutrophils (156). S-glutathionylation of S100A9 reduces its capacity to form heterocomplexes with S100A8 and abrogates S100A9-mediated neutrophil adhesion to endothelial cells. Combined with the functional changes reported for nitrosylated S100A8, particular oxidative modifications of these proteins may regulate the magnitude of neutrophil migration and limit tissue damage in acute inflammation.
XI. Redox Dysregulation in Pathophysiology
ROS/RNS-induced redox alterations can lead to cell death through dysregulation of mitogenic and apoptotic pathways previously described. Cell death is a crucial event that leads to the clinical pathology of many diseases that will be highlighted in this section.
A. Liver injury
Hepatotoxicity may result from exposure to any one of a number of chemical or biological stresses. Drugs, alcohol, and viruses are foremost in this regard and one common theme to the inflicted damage is the intermediate involvement of ROS and RNS. As a consequence of its function as a detoxification organ, the liver has high cellular levels of GSH and commensurately high expression patterns of Phase I and II detoxification enzymes, particularly GSTs. Although species- and sex-dependent expression patterns exist, GST alpha and mu isozyme activities are the most prevalent.
Death of hepatocytes is crucial in causing the clinical manifestations of hepatotoxicity. Changes in the redox balance after exposure to agents that deplete GSH induce apoptosis and necrosis in hepatocytes. At least one cause:effect relationship implicates redox dysregulation of the many proteins that modulate cell survival and death, including, NF-κB, JNK, PKC, and cysteine-dependent caspases. As discussed in other sections of this review, many of these proteins are subject to regulation or inactivation by nitrosylation or S-glutathionylation. High concentrations of damaging agents can lead to relatively large shifts in redox homeostasis resulting in necrosis of primary hepatocyte cultures. On the other hand, more modest redox alterations do not seem to promote apoptosis in hepatocytes. Any external insult that depletes GSH will alter overall intracellular redox homeostasis and regulate the response of liver cells to TNFα or Fas-induced apoptosis. Thus, redox alterations in hepatocytes may be important in regulating the cell death pathways that mediate a broad range of different liver pathologies, independent of the causative agent. As a general cytokine, TNF can regulate inflammation in the liver, but can also promote liver injury during the inflammatory process. Liver damage caused by exogenous agents (e.g., carbon tetrachloride and chronic alcohol) is markedly decreased in mice deficient in the expression of the TNF receptor. In this regard, sustained elevated levels of TNF may be responsible for promoting apoptosis and necrosis in hepatocytes and this may contribute directly to liver pathologies such as liver ischemia, alcoholic liver disease, and liver toxicity caused by agents like carbon tetrachloride. There are some indications that the precise role of TNF in controlling the response of liver to oxidative/nitrosative damage may be subject to a threshold effect. For example, Han et al. (111) suggest that moderate changes in cellular redox potential caused by sublethal levels of agents that cause depletion of cytoplasmic GSH levels can sensitize cells to TNFα-induced apoptosis through interference with NF-kB signaling. Larger redox changes producing substantially decreased GSH levels cause necrosis in hepatocytes, regardless of the presence of TNF. They imply from such observations that a certain critical redox threshold exists and, once passed, results in irreversibly high levels of cell necrosis, but below such a threshold, the redox changes may actually sensitize cells to TNF-induced apoptosis.
Acetaminophen overdose is lethal in humans and mortality has been linked with severe GSH depletion in hepatocytes. The condition can be reversed if GSH is replenished through timely treatments with NAC. This serves to emphasize how critical GSH is to liver cell survival. However, it is possible that drug-induced alterations, in addition to GSH depletion, may be important in the pathology and/or prognosis of this condition. For example, it has been suggested that GSH depletion may result in the activation of redox-regulated proteins such as JNK and that this has a direct role in mediating hepatocyte death caused by acetaminophen (111). As discussed earlier, kinases such as JNK and ASK1 are quite susceptible to inhibition by GSTP or Prx. S-glutathionylation of JNK will cause the breakdown of the protein:protein interactions necessary to keep the GSTP:JNK complex together. This will activate JNK that in hepatocytes could push the cells toward apoptotic pathways. Moreover, most protein tyrosine phosphatases, including JNK phosphatase, have low pK cysteines subject to regulation through S-glutathionylation, inhibiting these enzymes and enabling the persistence of activated kinases. The involvement of these pathways (activated by the S-glutathionylation) in the steps leading to liver damage is intriguing and merits further study.
B. Diabetes mellitus
Approximately 90% of diabetic patients are of the type 2 diabetes mellitus phenotype characterized by insulin resistance at target tissues and impaired insulin secretion. Type 1 diabetes mellitus results from an inflammatory autoimmune response that can lead to destruction of β-cells of Langerhans and characteristic insulin insufficiency. One complication associated with diabetes is increased in oxidative stress (28) and it has been shown that patients with type 1 or type 2 diabetes mellitus have depleted levels of GSH in erythrocytes (61, 265). In addition, hyperglycemia can lead to the generation of free radicals and increased lipid peroxidation, which together with a decrease in antioxidants such as vitamin E and glutathione peroxidase (GPX), further impacts cellular defenses against oxidative stress induced damage (297). Given the decrease in plasma and erythrocyte GPX (297), a natural assumption may be made that increasing these enzymes might be a plausible treatment strategy for diabetes. However, GPX1-overexpressing mice develop insulin resistance compared with wild-type animals (172). In addition, they have a 30%–70% reduction in insulin-induced phosphorylation of the liver insulin receptor and are fatter and heavier than wild type (172). At least one interpretation of these data is that antioxidant levels may play a role in the balance of the insulin resistance effects of high ROS (243) and that ROS may be involved in the regulation of insulin sensitivity (159). While ROS contribute to the disease phenotype, mitochondrial dysfunction has been established as an etiologic factor in diabetic pathophysiology. During hyperglycemic episodes, excessive glucose oxidation can overload the mitochondrial electron transport chain and increase superoxide production, generated by mitochondrial complexes I and III (28, 307). Superoxide in turn is converted to H2O2 by manganese superoxide dismutase (MnSOD). H2O2 is scavenged by GPX by catalyzing GSSG formation through interaction with GSH. GSSG can be reduced to GSH by GSH reductase at the expense of NADPH and completes the redox cycle (Fig. 1). It is clear from this that GSH is involved in mitochondrial dysfunction and may thus influence the diabetic condition. The lower erythrocyte GSH levels in diabetic patients (49, 61, 184, 265) are accompanied by increases in GSSG with lower γ-GCS and glutathione reductase activities, but no change in glutathione synthetase (184). Taken together, the increase in GSSG and decrease in glutathione reductase activity could produce a more oxidized cellular environment, whereas the lower GCS activity could impair GSH synthesis. In general, erythrocyte proteins would be exposed to an oxidizing environment with less redox buffering capacity, with a consequent potential for protein degradation. It remains unclear if the redox state of erythrocytes merely reflects oxidative protection capacity or might result in additional impairment of insulin regulation. NAC treatment did not increase GSH concentration in adolescents with type 1 diabetes (60), suggesting that more aggressive treatments are needed.
The shift toward a more oxidizing cellular environment could create conditions conducive to higher levels of S-glutathionylated proteins. In fact, patients with type 2 diabetes with microangiopathy have increased S-glutathionylated hemoglobin and lower GSH levels compared to controls (240), implying altered redox signaling as a characteristic of the disease. Although other studies have also identified increased S-glutathionylated hemoglobin in type 2 diabetic patients (186, 193), there is at least one example where no differences were found (124). Incubation of rat retinal Muller cells with diabetic concentrations of glucose does lead to an increase in Grx1, translocation of NF-κB to the nucleus, and induction of intercellular adhesion molecule-1 (ICAM-1) (249). In addition, Grx was also increased in retinal homogenates of diabetic rats (249). In follow-up studies the same authors found that inhibition of IκB kinase prevented the increase in ICAM-1 induced by glucose or Grx1 overexpression (248). Interestingly, under nonstressed conditions, IκB kinase is S-glutathionylated on Cys-179, suggesting that deglutathionylation by Grx1 may have some role in diabetes (248). GSTA4 catalyzes thioether bond formation of α- and β-unsaturated aldehydes with GSH for detoxification (247). GSTA4 is downregulated in adipose tissue of obese insulin resistant mice and in subcutaneous adipose tissue of obese insulin-resistant patients (56). Such data support the contention that S-glutathionylation is evident in diabetes and redox regulation could impact diabetic pathophysiology (Table 5).
Table 5.
S-Glutathionylated Proteins Observed in Diabetes Mellitus Models
C. Cardiovascular disease
Cardiovascular disease (CVD) is the leading cause of death in the United States, accounting for 34.3% of all deaths. Given that diabetes mellitus is a cardiovascular risk factor for CVD (196, 255), it seems reasonable to assume that oxidative stress is a major contributor to CVD. In fact, there is documented evidence that ROS, thioredoxin, Grx, and glutathione each have contributory roles in the pathophysiology of CVD (5, 102, 200). ROS-activating enzymes are found in vascular endothelial cells and ROS production causes oxidation of LDL, which in turn causes expression of vascular adhesion molecule-1 (VCAM-1) and monocyte chemotactic protein-1 (MCP-1), both redox-sensitive proteins (91). Treatment with niacin (nicotinic acid), which reduces myocardial infarction, reverses ROS-induced increases in these redox-sensitive proteins and also increases GSH and NADPH (91). Increases in GPX can help overcome deleterious effects of excess levels of ROS. Transgenic mice overexpressing GPX show lower levels of thiobarbituric acid-reactive substances in the left ventricular area after myocardial infarction compared to wild type (253). In addition, they show an increase in survival rate during the infarction period and lower left ventricular end-diastolic pressure (253), suggesting that redox regulation through GPX is involved in myocardial infarction. While excessive ROS levels may eventually induce apoptosis, Grx has an antiapoptotic effect in cells treated with ROS. For instance, it has been shown that ROS induces activation of Akt through PI-3 kinase, and its activation is inhibited by PI-3 kinase inhibitors (211). The Cys297 and Cys311 residues of Akt can be S-glutathionylated during oxidative stress and reduced by Grx1. The reduction would inhibit Akt association with PP2A and subsequent dephosphorylation of Akt leads to continuous Akt activation (185). Further evidence for the role of ROS and Grx1 in CVD comes from ischemic preconditioning studies where Grx1 expression is increased after adaptation to ischemia (164). In addition, transgenic mice overexpressing Grx1 showed improved ventricular recovery and smaller infarct size after ischemia, whereas Grx1 knockout mice had decreased functional recovery and larger infarct size (164). This was accompanied by decreased ROS production in hearts overexpressing Grx1 and increased ROS production in the knockout animals (164). A critical role player in vascular smooth muscle cell hypertrophy, angiotensin II, increases production of ROS (1). Induction of hypertrophy by angiotensin II leads to the S-glutathionylation of Ras on Cys118 and activation of p38 and Akt (1); however, it is inhibited by Grx1 overexpression. The same authors determined that SERCA activity is increased by S-glutathionylation and prevented by mutation of Cys674 (2). This is interesting since SERCA is responsible for the mobilization of Ca2+ and regulates cardiac muscle relaxation. Recently, treating endothelial cells with cinnamaldehyde induced p65 S-glutathionylation of NF-κB and inhibited TNFα-induced p65 nuclear translocation (155). In addition, p65 S-glutathionylation is prevented by treatment with the GSH synthesis inhibitor, buthionine sulfoximine (BSO) (155). Given the importance of oxidative stress and ROS in CVD, S-glutathionylation seems to have roles with differential outcomes in CVD protection, whereas Grx1 has mainly beneficial effects. It has been recently shown that diminished Grx1 levels in elderly rats contributed to increased apoptotic susceptibility of cardiomyocytes via regulation of NFκB function (89).
GSTP has been found in mouse (305), rat (190), and human (276) cardiac tissue. Biotransformation of the nitrovasodilator, glycerin trinitrate is accomplished by rat (190) or human (276) GSTM isoforms, and in the former, this isozyme is downregulated in the left ventricle and aortic vessel of the spontaneously hypertensive rat (313), GSTP, still has an important role in CVD. In fact, GSTP1P2−/− mice show an increase in endothelial dysfunction after exposure to tobacco smoke (51) and acrolein (risk factors for CVD) compared to wild types. The acrolein-induced endothelial dysfunction is prevented by pretreatment with NAC, demonstrating the involvement of redox regulation in GSTP protection of CVD.
D. Traumatic brain injury
Traumatic brain injury (TBI) is the leading cause of death among young people in developed countries (162). While the primary injury is due to impact from an external source, secondary damage can be evident after the initial incident (162). ROS, glutamate excitotoxicity, mitochondrial dysfunction, and cytokines are all induced by TBI (34, 256). Increases in free radical scavengers such as superoxide dismutase, catalase, GPX (105), and neurotrophic factors such as nerve growth factor (NGF) might act as physiological compensatory actions to prevent excessive brain damage. In rats, intraventricular infusion of nerve growth factor serves to increase catalase and GPX activities by 1 day post-trauma and continues until day three (105, 191). Studies have pointed to a biphasic regulation of hippocampal nerve growth factor and GPX, suggesting a more complex temporal regulation of the redox cycle in TBI patients (64). When compared to adult animals, young mice (21 days old) do not demonstrate increased GPX, even though baseline GPX levels are the same, suggesting an impaired redox response to TBI (76). Overexpression of GPX in mice prevents TBI-induced decreases in energy-coupling mitochondrial dysregulation, whereas GPX knock out mice show an enhanced sensitivity in mitochondrial dysfunction to TBI compared to wild types (304). During mitochondrial respiration experiments, brain mitochondrial dysfunction was reversed by adding the calcium chelator, egtazic acid mitochondria isolated from GPX knock out and wild-type animals (304), implying an essential role for Ca2+ in TBI-induced mitochondrial dysfunction.
Decreases in mitochondrial and cytosolic GSH contribute to dysregulation of mitochondrial electron transfer after TBI in rats. However, NAC pretreatment 5 min preinjury or up to 1 h postinjury does replete GSH to normal levels (303). Depletion of intracellular GSH with BSO exacerbates injury in brain endothelial cells and preincubation with GSH reduces injury as measured by lactate dehydrogenase levels (98). NAC treatment 15 min after TBI increases GSH, superoxide dismutase, and GPX activity and decreases TBI-induced increases in malondialdehyde (119). Further evidence for the beneficial effects of GSH comes from Zn2+-induced cell death studies. Free Zn2+ is released from synapses in excess during TBI, is colocalized with glutamate, and contributes to the brain injury. Zn acetate is able to inactivate glutathione reductase via an NADPH-dependant mechanism and increase GSSG/GSH ratios in astrocytes (22). In addition, Zn acetate treatment increases intracellular ROS. This mechanism is distinct from that of carmustine, a glutathione reductase inhibitor. Carmustine did not alter GSSG/GSH ratios or generate ROS (22), suggesting that Zn may be a primary contributor to astrocyte toxicity (22). Further, preincubation of primary cortical neurons with GSH partially blocked toxicity and completely blocked Zn2+-induced primary astrocyte toxicities (43). This is especially important since astrocytes are responsible for clearing extracellular glutamate through the glutamate transporters, GLT-1 and GLAST. Astrocyte cell death is evident under severe hypoxia, and high glutamate levels can downregulate GLT-1 and GLAST expression (152). This would impair the ability of astrocytes to clear excessive glutamate. Excess glutamate release can lead to neuronal excitotoxicity as a consequence of an influx of Ca2+ through NMDA receptors and microdialysis studies have shown that TBI elevates glutamate in preclinical (121) and clinical samples (34). In fact, glutamate levels can be correlated with injury severity, which is brain region specific (121). There is lower expression of the antiapoptotic gene Bcl-2 for as long as 12 months after TBI in the rat hippocampus, an area crucial in memory formation (189). Mice overexpressing GPX show an acute decrease in nitrotyrosine and an increased number of hippocampal neurons and improved spatial memory 3 months post-TBI (277). However, in young mice (postnatal day 21) GPX overexpression is unable to prevent TBI-induced deficits in hippocampal neuronal proliferation and survival later in life (214), indicating that the age at which TBI occurs is important for functional recovery. In a comprehensive time-dependent antioxidant and hippocampal synaptic protein study, 24 h postinjury TBI caused a reduction in GSH and GSH/GSSG ratio, an increase in GSSG, decreases in GPX activity and glutathione reductase activity, decreases in Cu/Zn-SOD and Mn-SOD activity, and an increase in levels of 4-hydroxynonenal, acrolein, protein carbonyl content, and 3-nitrotyrosine (11). Further, there were time-dependent decreases in synapsin-1, PSD-95, and SAP-97 in the hippocampus as well as increases in FJB, neuronal death, staining in the dentate gyrus, CA3, and CA1 hippocampal subfields (11). Although these variables are numerous, at least one central component of the damage control revolves around GSH. One of the major thiol sensitive transcription factors is nuclear factor erythroid 2-related factor 2 (Nrf2). Nrf2 knock out mice have higher levels of inflammatory cytokines such as TNF-alpha, IL-1beta, and IL-6 and more pronounced brain edema and neurological deficits compared to wild types (135). In addition, they have lower levels of GSTA1 and NADPH:quinone oxidoreductase (135). As may be expected, mild repetitive TBI in rats can cause increases in ROS that lead to long lasting and sustained alterations in the redox system (267). This may have important implications for recurring concussions in humans where permanent redox system dysregulation may occur.
As measured by microtubule-associated protein light chain 3 II (LC3 II) expression patterns, TBI is also accompanied by increased autophagy (148). Given that ROS is an essential factor in both autophagy and TBI, treatment with the antioxidant gamma-glutamylcysteinyl ethyl ester (GCEE) partially reduced LC3 II expression and improved behavioral outcomes (148). In another study, after experimentally induced TBI, intraperitoneal GCEE treatments decreased oxidized protein carbonyls and 3-nitrotyrosine levels (226). The fact that this treatment can be administered postinjury may have implications for clinical treatment strategies. Perhaps less expected, oral administration of GSNO in adult male rats reduces blood–brain barrier (BBB) injury and macrophage activation after TBI (142). In addition, GSNO treatment reverses TBI-induced decreases of BBB integrity proteins, ZO-1 and occludin and improves behavioral outcomes (142).
In clinical studies, total antioxidant reserve, including ascorbate and GSH, is decreased in the cerebrospinal fluid (CSF) of infants or children after TBI (18). Other studies found that hypothermia treatment after TBI in infants and children preserved the antioxidant reserve in CSF, with GSH levels inversely correlated with temperature control (17). Perhaps this suggests that hypothermia can be used as a therapeutic approach to maintain redox status in TBI patients. Taken together, treatment strategies to increase GSH (pharmacologically), GPX activity, and hypothermia may ameliorate the side effects of TBI-induced damage.
XII. Neurological Diseases and Redox Pathways
Altered GSH levels, changes in redox status, and oxidative stress underlie many neurodegenerative diseases such as Parkinson's disease (PD), Alzheimer's disease (AD), Huntington's disease (HD), and Friedreich's ataxia (FRDA) (92, 168, 169). For example, low GSH levels were thought to be the result of PD, but subsequent data provide evidence that changes in GSH levels precede the onset of PD or HD symptomology (254) underscoring the importance of GSH and redox regulation in these conditions. After nitric oxide generation a recent study provided evidence for S-glutathionylation of neuronal and astrocyte proteins (Fig. 12) (308). Further, this led to an increase in GSNO and GSSG with a shift in the redox potential toward a more oxidized state. Specific decreases in neuronal GSNO reductase and GSSG reductase were liabilities to recovery with the consequence that neurons were more susceptible than astrocytes to further NO exposure. This could be one reason for the selective degeneration of neurons in the different disease states.
FIG. 12.

Glutathione synthesis in neurons and astrocytes. Both neurons and astrocytes are able to synthesize glutathione through γ-GCS and glutathione synthetase, whereas only astrocytes are able to export GSH. Extracellular GSH can be broken down and the constitutive amino acids (glutamate, cysteine, and glycine) taken up by neurons for subsequent GSH synthesis. In addition, astrocytes are able to exchange glutamate for cystine through the cystine-glutamate exchange of system xc-, which allows cystine to be used for GSH synthesis. Further, it has been shown that GSH can act as a neurotransmitter through binding to NMDA, AMPA, or even GSH receptors. (To see this illustration in color the reader is referred to the web version of this article at www.liebertonline.com/ars).
A. Parkinson's disease
PD is characterized by selective loss of dopaminergic neurons in the pars compacta of the substantia nigra and by the presence of Lewy bodies. The loss of dopamine neurons results in a decrease in striatal dopamine that leads to difficulty in movement coordination and execution. In addition, ubiquitin and α-synuclein containing Lewy bodies are considered hallmarks of the disease (74) and it has been suggested that oxidative damage or misfolded proteins can initiate the formation of Lewy bodies that ultimately cause dopamine neuronal degeneration (242). Pharmacotherapeutic treatment for PD has focused on increasing dopamine levels with the dopamine precursor, L-3,4-dihydroxyphenylalanine (L-DOPA), or combination therapies with a decarboxylase inhibitor such as carbidopa or benserazide. In addition, prolonging the half-life of levodopa with cathechol-O-methyltransferase inhibitors or inhibiting the enzymatic breakdown of dopamine with monoamine oxidase type B inhibitors are strategies to provide symptomatic relief of the debilitating symptoms (269). However, current treatments do not prevent the inevitable degeneration of the dopaminergic system and more research is needed on the cause of the dopaminergic cell loss. One current hypothesis is that oxidative stress can cause damage that may lead to misfolded proteins preceding Lewy body formation and subsequent dopaminergic neurodegeneration (59, 97). This is further strengthened by studies showing increased levels of damaged proteins in the brains of PD patients (97) and impairments in the ubiquitin proteasomal system and autophagy lysosomal pathways (234). More specifically, nitrated tyrosine residues in α-synuclein have been observed in Lewy bodies in the brains of patients with PD, suggesting impairment of the antioxidant defense (97).
S-nitrosylation and S-glutathionylation can impact mitochondrial complex I biology (19, 187). For instance, the mitochondrial thiol transferase, Grx2, catalyzes protein S-glutathionylation of complex I in the presence of GSSG and results in inhibition of complex I activity (19). Mitochondrial complex I is especially sensitive to changes in redox state (38, 187) and complex I inhibition elicits selective neuronal loss, which contributes to behavioral effects that are characteristic of PD. The neurotoxin and mitochondrial complex I inhibitor, 1-methyl-1,2,3,6-tetrahydropyridine (MPTP), causes selective damage to dopaminergic neurons of the nigrostriatal pathway (62). More specifically, the damage is caused by the active metabolite of MPTP, MPP+, which increases oxidative stress, the opening of the mitochondrial permeability transition pore, the release of cytochrome c, and caspase activation (83, 285). After dopamine depletion, there is an early detectable decrease in total GSH levels and subsequent mitochondrial complex I impairment, followed by striatal dopaminergic cell loss and neurodegeneration (254). Another complex I inhibitor, rotenone, is used in vitro and in vivo as an experimental model for PD. Rotenone decreased the GSH redox potential, and the antioxidant and GSH precursor, NAC, reversed the rotenone-induced change in the redox potential (103). The authors used carotid body chemoreceptor cells as a model to determine the impact of mitochondrial respiratory chain blockers and a mitochondrial uncoupler on ROS production by measuring GSH, GSSG, and the redox potential. The respiratory chain blockers utilized were rotenone, 3-nitropropionic acid (3-NP), antimycin A, sodium azide, and the uncoupler 2,4-dinitrophenol. A rotenone concentration of 1 μM was able to significantly lower the redox potential with 2 mM NAC reversing the rotenone-induced decrease in the oxidative status. In addition, rotenone treatment elicited a dose-dependent activation response in chemoreceptor cells as measured by 3H catecholamine release.
Inducible downregulation of γ-glutamyl cysteine ligase (the rate-limiting enzyme in de novo glutathione synthesis) in catecholaminergic neurons in transgenic mice leads to reversible thiol-oxidation-dependent mitochondrial complex I inhibition (40). This was followed by nigrostriatal neurodegeneration in an age-related manner (40). In contrast, treatment with γ-glutamylcysteine ethyl ester, a GSH precursor, provided in vitro and in vivo protection against MPTP-induced PD symptoms (41). There are inherent difficulties in treating PD patients with GSH or restoring GSH levels with antioxidants. GSH does not effectively cross the blood–brain barrier, meaning that de novo synthesis of GSH in the CNS is the primary source. Cysteine administration is also not an effective means of elevating GSH in the CNS in vivo. NAC does not cross the blood–brain barrier (175), but is able to increase GSH levels through supply of cysteine after being metabolized (287). A disadvantage is that cysteine can interact with glutamate receptors, which may lead to excitotoxicity (198, 216), whereas NAC has been reported to be an oxidative stressor at high concentrations in vivo (114) and intrastriatal infusion of NAC caused neuronal toxicity (183). In addition to cysteine toxicity, nucleophilic addition of cysteine with dopamine metabolites such as dihydroxyphenylacetate (dopac) leads to formation of a cysteinylcathechol, 5-S-cysteinyl-3,4-dihydroxyphenylacetate. 5-S-cysteinyl-3,4-dihydroxyphenylacetate is a dopaminergic neurotoxin and inhibits complex I activity (181), which ultimately may contribute to PD. While GSH ethyl ester does provide protection against striatal damage (41), central delivery and the toxicity of the ethanol formed after ethyl ester cleavage (312) limits the use of this GSH precursor to ameliorate PD symptoms. As a whole, impaired GSH levels may be the first in a range of antioxidant and redox system impairments that ultimately contribute to the PD symptomology and future treatment strategies could beneficially focus on GSH modulation in the CNS.
Nitric oxide GSH derivatives such as S-nitrosoglutathione (GSNO) could protect substantia nigra dopaminergic neurons from oxidative stress in vivo (223). Increased lipid peroxidation in vivo was caused by intranigral infusion of either ferrous citrate (4.2 and 10 nmol caused an 8- and 13-fold increase) or peroxynitrate (33.6 nmol caused only a 40% increase). GSNO and GSH dose-dependently blocked the formation of hydroxyl radicals of both compounds. In addition, photodegraded GSNO and GSSG (100 μM) had no effect on iron-induced or peroxynitrate-induced lipid peroxidation (223). GSNO partly mediates cytoprotective effects through S-nitrosylation-induced inhibition of proteolysis and cytotoxicity caused by caspases and HIV-1 protease (42). This protective effect seems tissue and cell specific, since stimulation of HEK293 cells with GSNO caused S-nitrosylation of thioredoxin and subsequently activation of ASK1 (264). More recent studies confirm that GSNO is a mediator of P-SSG in neurons and further characterization of target proteins may lead to a better understanding of the protective effects of GSNO (308).
GGT, a biomarker for liver dysfunction and alcohol consumption, is highly enriched in those brain endothelial cells that constitute the blood–brain barrier and is responsible for metabolizing extracellular glutathione into the GSH precursor amino acid, glutamate, the dipeptide gamma-glutamylamino acid, and/or cysteinylglycine. These amino acids and dipeptides can subsequently be used for synthesis of reduced GSH in the intracellular environment (296). Astrocytes can transport cysteinylglycine (71), and both neurons (72) and glia (71) are able to utilize this dipeptide for GSH biosynthesis (Fig. 12). Serum contains GGT (163) and some metabolism of GSH could occur, but it is unclear if cysteinylglycine can be transported across the blood–brain barrier. However, blood–brain barrier GGT and amino dipeptidases can still metabolize cysteinylglycine into cysteine, and this can subsequently be transported across the blood–brain barrier and used for GSH synthesis.
Recent studies suggest that GGT may be involved in the generation of ROS (69, 75). In the presence of iron, GSH can induce lipid peroxidation and NFκB activation. These effects are GGT dependent, since they were prevented by the GGT transition state inhibitor, serine-borate complex (69). In contrast, upregulation of GGT is a compensatory reaction to GSH depletion and mitochondrial complex I inhibition, possibly representing an adaptive response to maintain or increase intracellular GSH levels (39).
Interestingly, GSTP is the only member expressed in dopaminergic substantia nigra neurons (32, 257). A recent study has shown that inhibition of GSTP by RNA interference and/or pharmacological inhibition of GSTP increases sensitivity to MPTP-induced PD, whereas GSTP1P2−/− mice show an increased sensitivity to MPTP (257). Further, overexpression of three GSTP variants, GSTP1B, 1C, and 1D, provided protection against rotenone-induced neuron loss (251), whereas another study revealed a significant association between the GSTP1B allele and sporadic PD (284). These data demonstrate the importance of GSTP and the role of genetic polymorphisms in PD etiology, and while catalysis of S-glutathionylation through GSTP may play an role in PD, Grx has been shown to have a significant impact on redox regulation in PD. The loss of DJ-1 protein levels, a gene linked to early onset PD, is evident after Grx1 downregulation, but not depletion of GSH (237). In addition, the death-associated protein Daxx translocates from the nucleus and causes cell death, whereas overexpression of DJ-1 prevents the Daxx-induced cell death. As a translational observation, L-DOPA as a first-line treatment for PD causes a decrease in Grx activity without a change in enzyme levels in dopaminergic SHSY5Y cells (236). Further, inhibition of Grx leads to neuronal apoptosis, which may be mediated by reactive quinones after L-DOPA oxidation (13, 236). Reactive quinone adducts, which are also evident in the substantia nigra of patients with PD (261), may contribute to the side effect profile of L-DOPA in the treatment of PD.
B. Alzheimer's disease
AD is the most common form of dementia and an estimated 24.3 million are affected worldwide (195). AD can be classified into sporadic and familial subtypes with sporadic AD being the most common (195). It is characterized by impairment of cognitive functions, impacting daily activities and eventually resulting in death. The disease starts anatomically from the entorhinal cortex and spreads via the hippocampus to other cortical areas. The presence of senile plaques (SP) and neurofibrillary tangles has been regarded as a histopathological hallmark of AD (209) and is further characterized by synaptic loss in brain areas that are responsible for learning and memory (67, 195, 199). A major component of senile plaques is amyloid beta-peptide (AB), a 40–42 amino acid peptide that is cleaved from amyloid precursor protein (APP) by beta- and gamma-secretases (282). AB is a cytotoxic peptide that exists as soluble and insoluble forms that can induce neuronal death, impair mitochondrial function, and increase ROS (14, 100, 206, 306). There is substantial evidence for the role of oxidative stress and mitochondrial impairment in AD (131, 225) and targeting mitochondria has been proposed as a therapeutic mechanism (225). On a genomic level there is an increase in mitochondrial cytochrome c oxidase II and IV mRNA ratios in the hippocampus, inferior parietal lobule, and cerebellum, which may contribute to the dysregulation in oxidative metabolism of patients with AD (8). Recent evidence for the regulation of APP by microRNAs (miRNAs) has sparked interest as potential treatment strategies for AD (116). miRNAs are small, noncoding RNAs highly enriched in the brain that are involved in mRNA translational regulation; for review see ref. (188). More specifically, miRNAs of the miR-20a family are endogenous regulators of APP (116) and overexpression of hsa-miR-106a and hsa-miR520c repress APP translation (205). In contrast, decreases in miR-106b (116), miR-107 (293), and in the miR cluster, miR-29a/b-1 (117), were found in patients with AD. The decrease in miR-107 and niR-29a/b-1 coincided with an increase in beta-site APP-cleaving enzyme 1 (BACE1) mRNA (117, 293). While it is known that BACE1 is responsible for AB generation (282), it is clear that further research into miRNAs may point to novel targets for neurodegenerative diseases such as AD.
Utilizing proteomic methods, it has been found in clinical studies that the most pronounced mitochondrial protein expression changes occur in the early stages of AD (161). By labeling mitochondrial proteins with isotope coded affinity tags, it was determined that pathways responsible for ATP utilization were preferentially activated (161). Additional proteomic studies pointed to an increase in oxidized proteins and proteins damaged through oxidation by ROS (31). Oxidative damage to proteins can be measured by an increase in carbonyl content (6, 7), which in AD is associated with a reduction in GSH and an increase in GSSG levels leading to an altered redox balance (29). These oxidatively modified proteins include creatine kinase, glutamine synthase, ubiquitin carboxy-terminal hydrolase L-1 (31), dihydropyrimidinase-related protein 2, and alpha-enolase (31). The change in redox could lead to S-glutathionylation of proteins. In fact, there is an increase in p53 S-glutathionylation in the inferior parietal lobule (66) and elevated serum levels of GGT (309) in patients with AD. These data have been corroborated by preclinical studies that have shown a reduction in GSH, an increase in GSSG and a decrease in the GSH/GSSG ratio in the brains of aged rats (314). More specifically, these changes were evident in the cortex, striatum, midbrain, and cerebellum of both male and female rats. In addition, there was a concomitant decrease in γ-GCS and increases in GGT, GST, and glutathione peroxidase. Males also showed an increase in glutathione reductase in all brain areas and in females an increase in the cortex and midbrain only (314). Treatment of neuronal cell cultures with AB resulted in oxidation of 14-3-3 and glyceraldehyde-3-phosphate dehydrogenase, which is prevented by pretreatment with gamma-glutamylcysteine ethyl ester (24). Other studies have found a protective effect for GSNO on AB-induced neurotoxicity in primary rat neuronal cultures, which involved activation of cGMP-dependent protein kinase, PI3K, and ERK (137). S-glutathionylation via GSTP has only recently emerged and as such the mechanism and impact of GST-mediated redox regulation has not been established. We do know, however, that polymorphisms that lead to alterations in the catalytic activity of GSTP are causally linked with neurodegenerative disorders. While preclinical data on GSTP is scarce (see above), clinical studies found that AD patients who were carriers of the GSTP1*C allelic variant, but not GSTM1, had a faster decline in cognitive functions (259). A similar study found an increase in the V allele of the GSTP1 polymorphism in AD, whereas the GSTT1 polymorphism was higher in controls (213). In addition, postmortem brains of patients with AD show an increase in Grx, which is oxidized by AB in vitro, and subsequent translocation of Daxx and apoptosis (9). The increase in Grx could be a compensatory mechanism of the human brain since, Grx overexpression prevented AB-induced toxicity. Taken together, the data from both clinical and preclinical studies underscore to the importance of redox regulation in AD.
C. Huntington's disease
Another neurodegenerative disorder affected directly or indirectly by mitochondrial dysfunction is HD. HD is caused by a pathological elongation of the CAG repeat in exon 1 of the gene encoding the Huntington protein. The mutated gene leads to a stretch of glutamine (Q) residues near the Huntington N-terminal domain, with more than 39 CAG/Q repeats almost always leading to HD [for review see ref. (16)]. There is accumulating evidence that the pathological polyglutamine repeats might contribute directly or indirectly to mitochondrial dysfunction and oxidative stress or vice versa (81, 87, 288). Huntington overexpressing, transfected neuronal cells showed an increase in ROS levels and cytochrome c release. The polyglutamine-induced cell death was reduced by overexpression of heat shock protein 27 and inhibition of cytochrome c release (302). Further, both NAC and GSH provided protection against cell death (302), indicating a role for redox regulation in HD.
Animal models for HD include the 150/150Q mutant Huntington knock-in mice and the R6 transgenic mouse model. The R6 transgenic mouse model consists of the R6/1 and R6/2 lines, with the R6/2 being the better characterized of the two for the study of HD (154). Generally, symptom progression is delayed, whereas life expectancy is longer for the R6/1 versus the R6/2 strain. Given the importance of oxidative stress and mitochondrial dysfunction, it is therefore not surprising that there is interest in redox regulation in R6/2 mice. While there is no difference in cellular GSH between R6/2 and wild-type mice, the transgenic mice do show an increase in mitochondrial GSH in the cortex and striatum (45). In addition, R6/2 mice show a decrease in glutamate-cysteine ligase activity in the cortex and a decrease in glucose-6-phosphate dehydrogenase (G6PD) activity in the striatum (45). The decrease in the enzymatic activity may point to impairment in GSH utilization, whereas the increase in GSH may reflect a compensatory reaction to the oxidative stress. Another study in R6/2 mice found an increase in forebrain GSH, whereas there were increases in Grx, GSH reductase, and glutamate-cysteine ligase mRNA levels (84). Daily treatment for 4 weeks with 112 mg/kg/day cystamine reversed the mRNA changes in GSH reductase and glutamate-cysteine ligase to control levels (84). In the related R6/1 transgenic strain, there is an increase in striatal lipid peroxidation (207) and in ROS production, whereas striatal glutathione peroxidase activity remained unchanged (208). These data suggest that alterations in the redox status may contribute to the observed HD phenotype of the transgenic mice.
In addition to transgenic mouse models, treatment with 3-NP results in a HD phenotype and represents a recognized pharmacological model for HD [for review see ref. (26)]. More specifically, 3-NP treatment causes alterations in cognitive deficits, mitochondrial dysfunction, and redox status. The primary mechanism underlying 3-NP-induced striatal toxicity is inhibition of respiratory chain complex II activity (158) and succinate dehydrogenase (SDH) activity, which is reduced in patients with HD (27, 106). In rats treated with 3-NP there was an increase in protein carbonyl groups, lipid peroxidation, and total nitrite (nitrite+nitrate) levels in striatal synaptosomes, a reduction in GSH, catalase, GSH peroxidase, and SDH activity in the striatum (278). Transcranial magnetic field stimulation (TMS) at 60 Hz and 0.7 mT for 4 days partially reversed the 3-NP-induced changes in protein carbonyl levels, total nitrite levels, lipid peroxidation products, GSH, GSH peroxidase, catalase, and SDH activity (278). It has been shown that the nitric oxide carrier, GSNO, decreases 3-NP-induced neurotoxicity in fetal rat striatal cultures and increases cell survival. This GSNO-induced protective mechanism was more potent than treatment with the antioxidant, NAC (138). Given the fact that GSNO treatment provided protection against iron-induced hydroxyl radical- and peroxynitrate formation in substantia nigra dopaminergic neurons in vivo (223), it is not surprising that GSNO partially protected neurons from 3-NP. GSNO could be providing protection not only by inhibiting 3-NP-induced hydroxyl or peroxynitrate formation (290), but also inhibiting caspase activity (23, 180). Additional evidence for the involvement of nitric oxide in 3-NP-induced neurotoxicity comes from experiments that have shown efficacy for the NO inhibitor, l-NAME, in combination with the immunosuppressant FK506, in reversing cognitive deficits and GSH decreases in vivo (147).
In vivo clinical studies have found potential peripheral biomarkers in the plasma of patients with HD. Plasma malondialdehyde was elevated and showed a correlation with disease severity (35). In addition, there was a reduction in erythrocyte Cu/Zn-superoxide dismutase and glutathione peroxidase activity (35). Further in vivo evidence for increased oxidative stress in patients with HD includes higher plasma lipid peroxidation and lower GSH levels (145). Younger asymptomatic HD gene carriers had a similar profile, which suggests that oxidative stress precedes the onset of HD symptoms. These preclinical and clinical data suggest that redox regulation may be a valuable pharmacotherapeutic tool for the treatment of HD.
D. Friedreich's ataxia
The most common form of hereditary ataxias is FRDA, which is caused by a pathological expansion of the GAA triplet repeat in the first intron of the frataxin protein (215). The length of the repeats is positively correlated with the severity of the disease and results in the loss of the mitochondrial protein, frataxin. This protein plays an essential role in the binding of free iron and low levels are thought to lead to mitochondrial dysfunction, accumulation of free iron, and subsequent generation of ROS (93). In a knockout mouse model of frataxin, there is time-dependent intramitochondrial iron accumulation, a deficiency in mitochondrial complex I–III activity and aconitase activity (215). While studies on S-glutathionylation in FRDA are limited, one did find an increase in protein S-glutathionylation in fibroblasts of patients with FRDA (204). In addition, it was found that actin was S-glutathionylated probably through an increase in ROS due to iron overload (204). FRDA fibroblasts showed a significant disarrangement of F-actin, which was reversed by treatment with GSH. This suggests that the cellular abnormalities may have been caused by oxidative stress. In a recent study, the spinal cord of patients with FRDA exhibited an increase in tyrosinated tubulin and phosphorylated neurofilaments, which colocalized with increased protein S-glutathionylation (260). Taken together, these studies suggest that abnormal cellular S-glutathionylation and oxidative stress may play a role in FRDA, implying that antioxidants may therefore be potential therapeutic interventions. In a small clinical study, patients with FRDA responded to treatment with idebenone, a short chain analog of coenzyme Q10. Larger meta-analysis found that idebenone treatment was more effective than placebo in halting and reversing the hypertrophic cardiomyopathy associated with FRDA, but did not improve neurological symptoms (141, 275). In contrast to PD genetic studies that found differences in GSTP polymorphisms, similar studies in patients with FRDA found no such differences. However, FRDA patients did show an increase in the general specific activity of GST in erythrocytes (82), suggesting the importance of redox regulation in FRDA.
E. Amylotrophic lateral sclerosis
Although there are limited studies in the importance of redox conditions to the etiology of ALS, one recent report (88) suggests that activation of Cu, Zn-superoxide dismutase (SOD1) may involve several post-translational modifications, including copper and zinc binding, as well as formation of an intramolecular disulfide bond. There is a metal chaperone for SOD1, CCS, which is involved in intracellular copper loading in SOD1 (55). It appears that CCS may have dual roles. Not only does it deliver copper to SOD1 under stringent copper limiting conditions, but it also facilitates the conversion of the disulfide-reduced immature SOD1 into the active disulfide-containing enzyme. Thus, two new functions may be attributed to CCS, oxygen-dependent sulfhydryl oxidase, and disulfide isomerase-like activities. The CCS activation of SOD1 seems to be dependent upon cellular exposure to conditions of oxidative stress. The structure and protein aggregation status of SOD1 are quite dependent upon cellular redox balance, which links it to the neurodegenerative disease amyotrophic lateral sclerosis (ALS). Recent studies showed that S-glutathionylation of SOD1 is pertinent to ALS (298). SOD1 isolated from human erythrocytes was shown to be S-glutathionylated at Cys 111 and promotes SOD1 monomer formation, a prerequisite step in SOD1 aggregation.
XIII. Conclusions
GSH homeostasis is an evolutionarily well-conserved process for controlling intracellular redox conditions. The versatility of GSH comes from the nucleophilicity of its cysteine residue, allowing covalent modifications of a variety of cell proteins that have cysteine residues located in basic regions. The addition of a bulky tripeptide and a net negative charge through the glutamic acid residue can influence the structure/function of the protein. There is value to the concept that S-glutathionylation, as a post-translational modification, may have consequences every bit as critical as phosphorylation, perhaps providing a contiguous relationship between sulfur and phosphorus biochemistry. Aberrations in redox homeostasis frequently manifest through alterations in GSH and associated pathways have been conditionally linked with the etiology and progression of a number of human diseases of aging exemplified by cancer and neurological dementias. Interpretation of some of the published material does not always allow for the separation of cause:effect relationships with respect to the involvement of oxidative stress, protein modifications, and their involvement in or consequences to these pathologies. Nevertheless, there is reason to be optimistic that ongoing and continued attention to the variety of components of the S-glutathionylation cycle may help to identify possible biomarkers for diagnosis, progression, and response to therapies in these diseases.
Abbreviations Used
- 2D SDS-PAGE
two-dimensional sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- 3-NP
3-nitropropionic acid
- AB
amyloid beta-peptide
- AD
Alzheimer's disease
- APP
amyloid precursor protein
- ASK1
apoptosis signaling kinase 1
- ATF6
activating transcription factor 6
- Bcl-2
B-cell lymphoma 2
- cAMP
cyclic adenosine monophosphate
- CG
cysteinyl-glycine
- CHOP
C/EBP homologous protein
- EGF
epidermal growth factor
- ER
endoplasmic reticulum
- ERK
extracellular signal-regulated kinase
- Ero1
ER oxidase 1
- FRDA
Friedreich's ataxia
- γ-GCS
γ-glutamylcysteine synthase
- GAP
GTPase-activating protein
- GCL
glutamate cysteine ligase
- GGT
gamma-glutamyltransferase
- Grx
glutaredoxin
- GSH
reduced glutathione
- GSNO
S-nitrosoglutathione
- GSSG
oxidized glutathione
- GST
glutathione-S-transferase
- HD
Huntington's disease
- IKK
I kappa B kinase
- JNK
c-Jun N-terminal kinase
- L-DOPA
L-3,4-dihydroxyphenylalanine
- MALDI-TOF
matrix-assisted laser desorption/ionization time-of-flight
- MAPK
mitogen-activated protein kinase
- Mdm2
murine double minute 2
- MEKK1
MAPK/ERK kinase kinase 1
- miRNAs
microRNAs
- MPTP
1-methyl-1,2,3,6-tetrahydropyridine
- MRP
multidrug resistant protein
- NAC
N-acetyl-L-cysteine
- NFκB
nuclear factor kappa B
- PABA/NO
(O2-{2,4-dinitro-5-[4-(N-methylamino)benzoyloxy]phenyl}1-(N,N-dimethylamino)diazen-1-ium-1,2-diolate)
- PD
Parkinson's disease
- PDI
protein disulfide isomerase
- PDK
phosphoinosityl-dependent kinase
- PERK
pancreatic ER kinase
- PI3K
phosphoinositide 3 kinase
- PKA
protein kinase A
- PKC
protein kinase C
- PP2A
protein phosphatase 2A
- Prx
peroxiredoxin
- PTEN
phosphatase and tensin homolog deleted from chromosome 10
- PTP
protein tyrosine phosphatase
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- RyR
ryanodine receptor
- SDH
succinate dehydrogenase
- SERCA
sarcoplasmic endoplasmic reticulum calcium ATPase
- SHP
Src-homology protein tyrosine phospphatase
- SP
senile plaques
- TNFα
tumor necrosis factor alpha
- TRAF2
TNF receptor-associated factor 2
- UPR
unfolded protein response
Footnotes
Reviewing Editors: Adam Benham, Isabella Dalle-Donne, Christopher H. Lillig, John Mieyal, Peep Palumaa, Han-A Park, Fiorella Piemonte, Pierre-Marie Girard, and Nanduri Prabhakar
References
- 1.Adachi T. Pimentel DR. Heibeck T. Hou X. Lee YJ. Jiang B. Ido Y. Cohen RA. S-glutathiolation of Ras mediates redox-sensitive signaling by angiotensin II in vascular smooth muscle cells. J Biol Chem. 2004;279:29857–29862. doi: 10.1074/jbc.M313320200. [DOI] [PubMed] [Google Scholar]
- 2.Adachi T. Weisbrod RM. Pimentel DR. Ying J. Sharov VS. Schoneich C. Cohen RA. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med. 2004;10:1200–1207. doi: 10.1038/nm1119. [DOI] [PubMed] [Google Scholar]
- 3.Adler V. Yin Z. Fuchs SY. Benezra M. Rosario L. Tew KD. Pincus MR. Sardana M. Henderson CJ. Wolf CR. Davis RJ. Ronai Z. Regulation of JNK signaling by GSTp. EMBO J. 1999;18:1321–1334. doi: 10.1093/emboj/18.5.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Agazie YM. Hayman MJ. Molecular mechanism for a role of SHP2 in epidermal growth factor receptor signaling. Mol Cell Biol. 2003;23:7875–7886. doi: 10.1128/MCB.23.21.7875-7886.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahsan MK. Lekli I. Ray D. Yodoi J. Das DK. Redox regulation of cell survival by the thioredoxin superfamily: an implication of redox gene therapy in the heart. Antioxid Redox Signal. 2009;11:2741–2758. doi: 10.1089/ars.2009.2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aksenov M. Aksenova M. Butterfield DA. Markesbery WR. Oxidative modification of creatine kinase BB in Alzheimer's disease brain. J Neurochem. 2000;74:2520–2527. doi: 10.1046/j.1471-4159.2000.0742520.x. [DOI] [PubMed] [Google Scholar]
- 7.Aksenov MY. Aksenova MV. Butterfield DA. Geddes JW. Markesbery WR. Protein oxidation in the brain in Alzheimer's disease. Neuroscience. 2001;103:373–383. doi: 10.1016/s0306-4522(00)00580-7. [DOI] [PubMed] [Google Scholar]
- 8.Aksenov MY. Tucker HM. Nair P. Aksenova MV. Butterfield DA. Estus S. Markesbery WR. The expression of several mitochondrial and nuclear genes encoding the subunits of electron transport chain enzyme complexes, cytochrome c oxidase, and NADH dehydrogenase, in different brain regions in Alzheimer's disease. Neurochem Res. 1999;24:767–774. doi: 10.1023/a:1020783614031. [DOI] [PubMed] [Google Scholar]
- 9.Akterin S. Cowburn RF. Miranda-Vizuete A. Jimenez A. Bogdanovic N. Winblad B. Cedazo-Minguez A. Involvement of glutaredoxin-1 and thioredoxin-1 in beta-amyloid toxicity and Alzheimer's disease. Cell Death Differ. 2006;13:1454–1465. doi: 10.1038/sj.cdd.4401818. [DOI] [PubMed] [Google Scholar]
- 10.Anathy V. Aesif SW. Guala AS. Havermans M. Reynaert NL. Ho YS. Budd RC. Janssen-Heininger YM. Redox amplification of apoptosis by caspase-dependent cleavage of glutaredoxin 1 and S-glutathionylation of Fas. J Cell Biol. 2009;184:241–252. doi: 10.1083/jcb.200807019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ansari MA. Roberts KN. Scheff SW. Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury. Free Radic Biol Med. 2008;45:443–452. doi: 10.1016/j.freeradbiomed.2008.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aracena-Parks P. Goonasekera SA. Gilman CP. Dirksen RT. Hidalgo C. Hamilton SL. Identification of cysteines involved in S-nitrosylation, S-glutathionylation, and oxidation to disulfides in ryanodine receptor type 1. J Biol Chem. 2006;281:40354–40368. doi: 10.1074/jbc.M600876200. [DOI] [PubMed] [Google Scholar]
- 13.Asanuma M. Miyazaki I. Diaz-Corrales FJ. Ogawa N. Quinone formation as dopaminergic neuron-specific oxidative stress in the pathogenesis of sporadic Parkinson's disease and neurotoxin-induced parkinsonism. Acta Med Okayama. 2004;58:221–233. doi: 10.18926/AMO/32105. [DOI] [PubMed] [Google Scholar]
- 14.Atamna H. Amino acids variations in Amyloid-beta peptides, mitochondrial dysfunction, and new therapies for Alzheimer's disease. J Bioenerg Biomembr. 2009;41:457–464. doi: 10.1007/s10863-009-9246-2. [DOI] [PubMed] [Google Scholar]
- 15.Barrett WC. DeGnore JP. Konig S. Fales HM. Keng YF. Zhang ZY. Yim MB. Chock PB. Regulation of PTP1B via glutathionylation of the active site cysteine 215. Biochemistry. 1999;38:6699–6705. doi: 10.1021/bi990240v. [DOI] [PubMed] [Google Scholar]
- 16.Bauer PO. Nukina N. The pathogenic mechanisms of polyglutamine diseases and current therapeutic strategies. J Neurochem. 2009;110:1737–1765. doi: 10.1111/j.1471-4159.2009.06302.x. [DOI] [PubMed] [Google Scholar]
- 17.Bayir H. Adelson PD. Wisniewski SR. Shore P. Lai Y. Brown D. Janesko-Feldman KL. Kagan VE. Kochanek PM. Therapeutic hypothermia preserves antioxidant defenses after severe traumatic brain injury in infants and children. Crit Care Med. 2009;37:689–695. doi: 10.1097/CCM.0b013e318194abf2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bayir H. Kagan VE. Tyurina YY. Tyurin V. Ruppel RA. Adelson PD. Graham SH. Janesko K. Clark RS. Kochanek PM. Assessment of antioxidant reserves and oxidative stress in cerebrospinal fluid after severe traumatic brain injury in infants and children. Pediatr Res. 2002;51:571–578. doi: 10.1203/00006450-200205000-00005. [DOI] [PubMed] [Google Scholar]
- 19.Beer SM. Taylor ER. Brown SE. Dahm CC. Costa NJ. Runswick MJ. Murphy MP. Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins: implications for mitochondrial redox regulation and antioxidant DEFENSE. J Biol Chem. 2004;279:47939–47951. doi: 10.1074/jbc.M408011200. [DOI] [PubMed] [Google Scholar]
- 20.Bini L. Magi B. Marzocchi B. Arcuri F. Tripodi S. Cintorino M. Sanchez JC. Frutiger S. Hughes G. Pallini V. Hochstrasser DF. Tosi P. Protein expression profiles in human breast ductal carcinoma and histologically normal tissue. Electrophoresis. 1997;18:2832–2841. doi: 10.1002/elps.1150181519. [DOI] [PubMed] [Google Scholar]
- 21.Birkenmeier G. Stegemann C. Hoffmann R. Gunther R. Huse K. Birkemeyer C. Posttranslational modification of human glyoxalase 1 indicates redox-dependent regulation. PLoS One. 5:e10399. doi: 10.1371/journal.pone.0010399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bishop GM. Dringen R. Robinson SR. Zinc stimulates the production of toxic reactive oxygen species (ROS) and inhibits glutathione reductase in astrocytes. Free Radic Biol Med. 2007;42:1222–1230. doi: 10.1016/j.freeradbiomed.2007.01.022. [DOI] [PubMed] [Google Scholar]
- 23.Bizat N. Hermel JM. Boyer F. Jacquard C. Creminon C. Ouary S. Escartin C. Hantraye P. Kajewski S. Brouillet E. Calpain is a major cell death effector in selective striatal degeneration induced in vivo by 3-nitropropionate: implications for Huntington's disease. J Neurosci. 2003;23:5020–5030. doi: 10.1523/JNEUROSCI.23-12-05020.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boyd-Kimball D. Sultana R. Poon HF. Mohmmad-Abdul H. Lynn BC. Klein JB. Butterfield DA. Gamma-glutamylcysteine ethyl ester protection of proteins from Abeta(1–42)-mediated oxidative stress in neuronal cell culture: a proteomics approach. J Neurosci Res. 2005;79:707–713. doi: 10.1002/jnr.20393. [DOI] [PubMed] [Google Scholar]
- 25.Brewer JW. Diehl JA. PERK mediates cell-cycle exit during the mammalian unfolded protein response. Proc Natl Acad Sci U S A. 2000;97:12625–12630. doi: 10.1073/pnas.220247197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brouillet E. Jacquard C. Bizat N. Blum D. 3-Nitropropionic acid: a mitochondrial toxin to uncover physiopathological mechanisms underlying striatal degeneration in Huntington's disease. J Neurochem. 2005;95:1521–1540. doi: 10.1111/j.1471-4159.2005.03515.x. [DOI] [PubMed] [Google Scholar]
- 27.Browne SE. Bowling AC. MacGarvey U. Baik MJ. Berger SC. Muqit MM. Bird ED. Beal MF. Oxidative damage and metabolic dysfunction in Huntington's disease: selective vulnerability of the basal ganglia. Ann Neurol. 1997;41:646–653. doi: 10.1002/ana.410410514. [DOI] [PubMed] [Google Scholar]
- 28.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 29.Calabrese V. Sultana R. Scapagnini G. Guagliano E. Sapienza M. Bella R. Kanski J. Pennisi G. Mancuso C. Stella AM. Butterfield DA. Nitrosative stress, cellular stress response, and thiol homeostasis in patients with Alzheimer's disease. Antioxid Redox Signal. 2006;8:1975–1986. doi: 10.1089/ars.2006.8.1975. [DOI] [PubMed] [Google Scholar]
- 30.Casagrande S. Bonetto V. Fratelli M. Gianazza E. Eberini I. Massignan T. Salmona M. Chang G. Holmgren A. Ghezzi P. Glutathionylation of human thioredoxin: a possible crosstalk between the glutathione and thioredoxin systems. Proc Natl Acad Sci U S A. 2002;99:9745–9749. doi: 10.1073/pnas.152168599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Castegna A. Aksenov M. Thongboonkerd V. Klein JB. Pierce WM. Booze R. Markesbery WR. Butterfield DA. Proteomic identification of oxidatively modified proteins in Alzheimer's disease brain. Part II: dihydropyrimidinase-related protein 2, alpha-enolase and heat shock cognate 71. J Neurochem. 2002;82:1524–1532. doi: 10.1046/j.1471-4159.2002.01103.x. [DOI] [PubMed] [Google Scholar]
- 32.Castro-Caldas M. Neves Carvalho A. Peixeiro I. Rodrigues E. Lechner MC. Gama MJ. GSTpi expression in MPTP-induced dopaminergic neurodegeneration of C57BL/6 mouse midbrain and striatum. J Mol Neurosci. 2009;38:114–127. doi: 10.1007/s12031-008-9141-z. [DOI] [PubMed] [Google Scholar]
- 33.Chakrabarty S. Huang S. Modulation of chemosensitivity in human colon carcinoma cells by downregulating protein kinase C alpha expression. J Exp Ther Oncol. 1996;1:218–221. [PubMed] [Google Scholar]
- 34.Chamoun R. Suki D. Gopinath SP. Goodman JC. Robertson C. Role of extracellular glutamate measured by cerebral microdialysis in severe traumatic brain injury. J Neurosurg. 2010;113:564–570. doi: 10.3171/2009.12.JNS09689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen CM. Wu YR. Cheng ML. Liu JL. Lee YM. Lee PW. Soong BW. Chiu DT. Increased oxidative damage and mitochondrial abnormalities in the peripheral blood of Huntington's disease patients. Biochem Biophys Res Commun. 2007;359:335–340. doi: 10.1016/j.bbrc.2007.05.093. [DOI] [PubMed] [Google Scholar]
- 36.Chen J. Chen CL. Rawale S. Chen CA. Zweier JL. Kaumaya PT. Chen YR. Peptide-based antibodies against glutathione-binding domains suppress superoxide production mediated by mitochondrial complex I. J Biol Chem. 285:3168–3180. doi: 10.1074/jbc.M109.056846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheng G. Ikeda Y. Iuchi Y. Fujii J. Detection of S-glutathionylated proteins by glutathione S-transferase overlay. Arch Biochem Biophys. 2005;435:42–49. doi: 10.1016/j.abb.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 38.Chinta SJ. Andersen JK. Reversible inhibition of mitochondrial complex I activity following chronic dopaminergic glutathione depletion in vitro: implications for Parkinson's disease. Free Radic Biol Med. 2006;41:1442–1448. doi: 10.1016/j.freeradbiomed.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 39.Chinta SJ. Kumar JM. Zhang H. Forman HJ. Andersen JK. Up-regulation of gamma-glutamyl transpeptidase activity following glutathione depletion has a compensatory rather than an inhibitory effect on mitochondrial complex I activity: implications for Parkinson's disease. Free Radic Biol Med. 2006;40:1557–1563. doi: 10.1016/j.freeradbiomed.2005.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chinta SJ. Kumar MJ. Hsu M. Rajagopalan S. Kaur D. Rane A. Nicholls DG. Choi J. Andersen JK. Inducible alterations of glutathione levels in adult dopaminergic midbrain neurons result in nigrostriatal degeneration. J Neurosci. 2007;27:13997–14006. doi: 10.1523/JNEUROSCI.3885-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chinta SJ. Rajagopalan S. Butterfield DA. Andersen JK. In vitro and in vivo neuroprotection by gamma-glutamylcysteine ethyl ester against MPTP: relevance to the role of glutathione in Parkinson's disease. Neurosci Lett. 2006;402:137–141. doi: 10.1016/j.neulet.2006.03.056. [DOI] [PubMed] [Google Scholar]
- 42.Chiueh CC. Andoh T. Lai AR. Lai E. Krishna G. Neuroprotective strategies in Parkinson's disease: protection against progressive nigral damage induced by free radicals. Neurotox Res. 2000;2:293–310. doi: 10.1007/BF03033799. [DOI] [PubMed] [Google Scholar]
- 43.Cho IH. Im JY. Kim D. Kim KS. Lee JK. Han PL. Protective effects of extracellular glutathione against Zn2+-induced cell death in vitro and in vivo. J Neurosci Res. 2003;74:736–743. doi: 10.1002/jnr.10794. [DOI] [PubMed] [Google Scholar]
- 44.Chong ZZ. Maiese K. The Src homology 2 domain tyrosine phosphatases SHP-1 and SHP-2: diversified control of cell growth, inflammation, and injury. Histol Histopathol. 2007;22:1251–1267. doi: 10.14670/hh-22.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choo YS. Mao Z. Johnson GV. Lesort M. Increased glutathione levels in cortical and striatal mitochondria of the R6/2 Huntington's disease mouse model. Neurosci Lett. 2005;386:63–68. doi: 10.1016/j.neulet.2005.05.065. [DOI] [PubMed] [Google Scholar]
- 46.Christofk HR. Vander Heiden MG. Harris MH. Ramanathan A. Gerszten RE. Wei R. Fleming MD. Schreiber SL. Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- 47.Chu F. Ward NE. O'Brian CA. PKC isozyme S-cysteinylation by cystine stimulates the pro-apoptotic isozyme PKC delta and inactivates the oncogenic isozyme PKC epsilon. Carcinogenesis. 2003;24:317–325. doi: 10.1093/carcin/24.2.317. [DOI] [PubMed] [Google Scholar]
- 48.Chu F. Ward NE. O'Brian CA. Potent inactivation of representative members of each PKC isozyme subfamily and PKD via S-thiolation by the tumor-promotion/progression antagonist glutathione but not by its precursor cysteine. Carcinogenesis. 2001;22:1221–1229. doi: 10.1093/carcin/22.8.1221. [DOI] [PubMed] [Google Scholar]
- 49.Ciuchi E. Odetti P. Prando R. Relationship between glutathione and sorbitol concentrations in erythrocytes from diabetic patients. Metabolism. 1996;45:611–613. doi: 10.1016/s0026-0495(96)90032-3. [DOI] [PubMed] [Google Scholar]
- 50.Clement MV. Stamenkovic I. Superoxide anion is a natural inhibitor of FAS-mediated cell death. EMBO J. 1996;15:216–225. [PMC free article] [PubMed] [Google Scholar]
- 51.Conklin DJ. Haberzettl P. Prough RA. Bhatnagar A. Glutathione-S-transferase P protects against endothelial dysfunction induced by exposure to tobacco smoke. Am J Physiol Heart Circ Physiol. 2009;296:H1586–H1597. doi: 10.1152/ajpheart.00867.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Corti A. Paolicchi A. Franzini M. Dominici S. Casini AF. Pompella A. The S-thiolating activity of membrane gamma-glutamyltransferase: formation of cysteinyl-glycine mixed disulfides with cellular proteins and in the cell microenvironment. Antioxid Redox Signal. 2005;7:911–918. doi: 10.1089/ars.2005.7.911. [DOI] [PubMed] [Google Scholar]
- 53.Cross JV. Templeton DJ. Oxidative stress inhibits MEKK1 by site-specific glutathionylation in the ATP-binding domain. Biochem J. 2004;381:675–683. doi: 10.1042/BJ20040591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cruz CM. Rinna A. Forman HJ. Ventura AL. Persechini PM. Ojcius DM. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem. 2007;282:2871–2879. doi: 10.1074/jbc.M608083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Culotta VC. Klomp LW. Strain J. Casareno RL. Krems B. Gitlin JD. The copper chaperone for superoxide dismutase. J Biol Chem. 1997;272:23469–23472. doi: 10.1074/jbc.272.38.23469. [DOI] [PubMed] [Google Scholar]
- 56.Curtis JM. Grimsrud PA. Wright WS. Xu X. Foncea RE. Graham DW. Brestoff JR. Wiczer BM. Ilkayeva O. Cianflone K. Muoio DE. Arriaga EA. Bernlohr DA. Down regulation of adipose glutathione S-transferase leads to increased protein carbonylation, oxidative stress and mitochondrial dysfunction. Diabetes. 2010;59:1132–1142. doi: 10.2337/db09-1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dalle-Donne I. Giustarini D. Rossi R. Colombo R. Milzani A. Reversible S-glutathionylation of Cys 374 regulates actin filament formation by inducing structural changes in the actin molecule. Free Radic Biol Med. 2003;34:23–32. doi: 10.1016/s0891-5849(02)01182-6. [DOI] [PubMed] [Google Scholar]
- 58.Dalle-Donne I. Rossi R. Giustarini D. Colombo R. Milzani A. Actin S-glutathionylation: evidence against a thiol-disulphide exchange mechanism. Free Radic Biol Med. 2003;35:1185–1193. doi: 10.1016/s0891-5849(03)00504-5. [DOI] [PubMed] [Google Scholar]
- 59.Danielson SR. Andersen JK. Oxidative and nitrative protein modifications in Parkinson's disease. Free Radic Biol Med. 2008;44:1787–1794. doi: 10.1016/j.freeradbiomed.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Darmaun D. Smith SD. Sweeten S. Hartman BK. Welch S. Mauras N. Poorly controlled type 1 diabetes is associated with altered glutathione homeostasis in adolescents: apparent resistance to N-acetylcysteine supplementation. Pediatr Diabetes. 2008;9:577–582. doi: 10.1111/j.1399-5448.2008.00436.x. [DOI] [PubMed] [Google Scholar]
- 61.Darmaun D. Smith SD. Sweeten S. Sager BK. Welch S. Mauras N. Evidence for accelerated rates of glutathione utilization and glutathione depletion in adolescents with poorly controlled type 1 diabetes. Diabetes. 2005;54:190–196. doi: 10.2337/diabetes.54.1.190. [DOI] [PubMed] [Google Scholar]
- 62.Dauer W. Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 63.de Hemptinne V. Rondas D. Vandekerckhove J. Vancompernolle K. Tumour necrosis factor induces phosphorylation primarily of the nitric-oxide-responsive form of glyoxalase I. Biochem J. 2007;407:121–128. doi: 10.1042/BJ20070379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.DeKosky ST. Taffe KM. Abrahamson EE. Dixon CE. Kochanek PM. Ikonomovic MD. Time course analysis of hippocampal nerve growth factor and antioxidant enzyme activity following lateral controlled cortical impact brain injury in the rat. J Neurotrauma. 2004;21:491–500. doi: 10.1089/089771504774129838. [DOI] [PubMed] [Google Scholar]
- 65.Demasi M. Shringarpure R. Davies KJ. Glutathiolation of the proteasome is enhanced by proteolytic inhibitors. Arch Biochem Biophys. 2001;389:254–263. doi: 10.1006/abbi.2001.2332. [DOI] [PubMed] [Google Scholar]
- 66.Di Domenico F. Cenini G. Sultana R. Perluigi M. Uberti D. Memo M. Butterfield DA. Glutathionylation of the pro-apoptotic protein p53 in Alzheimer's disease brain: implications for AD pathogenesis. Neurochem Res. 2009;34:727–733. doi: 10.1007/s11064-009-9924-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dickerson BC. Sperling RA. Large-scale functional brain network abnormalities in Alzheimer's disease: insights from functional neuroimaging. Behav Neurol. 2009;21:63–75. doi: 10.3233/BEN-2009-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dinoto L. Deture MA. Purich DL. Structural insights into Alzheimer filament assembly pathways based on site-directed mutagenesis and S-glutathionylation of three-repeat neuronal Tau protein. Microsc Res Tech. 2005;67:156–163. doi: 10.1002/jemt.20195. [DOI] [PubMed] [Google Scholar]
- 69.Djavaheri-Mergny M. Accaoui MJ. Rouillard D. Wietzerbin J. Gamma-glutamyl transpeptidase activity mediates NF-kappaB activation through lipid peroxidation in human leukemia U937 cells. Mol Cell Biochem. 2002;232:103–111. doi: 10.1023/a:1014834315936. [DOI] [PubMed] [Google Scholar]
- 70.Dominici S. Valentini M. Maellaro E. Del Bello B. Paolicchi A. Lorenzini E. Tongiani R. Comporti M. Pompella A. Redox modulation of cell surface protein thiols in U937 lymphoma cells: the role of gamma-glutamyl transpeptidase-dependent H2O2 production and S-thiolation. Free Radic Biol Med. 1999;27:623–635. doi: 10.1016/s0891-5849(99)00111-2. [DOI] [PubMed] [Google Scholar]
- 71.Dringen R. Kranich O. Loschmann PA. Hamprecht B. Use of dipeptides for the synthesis of glutathione by astroglia-rich primary cultures. J Neurochem. 1997;69:868–874. doi: 10.1046/j.1471-4159.1997.69020868.x. [DOI] [PubMed] [Google Scholar]
- 72.Dringen R. Pfeiffer B. Hamprecht B. Synthesis of the antioxidant glutathione in neurons: supply by astrocytes of CysGly as precursor for neuronal glutathione. J Neurosci. 1999;19:562–569. doi: 10.1523/JNEUROSCI.19-02-00562.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dunstone MA. Dai W. Whisstock JC. Rossjohn J. Pike RN. Feil SC. Le Bonniec BF. Parker MW. Bottomley SP. Cleaved antitrypsin polymers at atomic resolution. Protein Sci. 2000;9:417–420. doi: 10.1110/ps.9.2.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Engelender S. Ubiquitination of alpha-synuclein and autophagy in Parkinson's disease. Autophagy. 2008;4:372–374. doi: 10.4161/auto.5604. [DOI] [PubMed] [Google Scholar]
- 75.Enoiu M. Aberkane H. Salazar JF. Leroy P. Groffen J. Siest G. Wellman M. Evidence for the pro-oxidant effect of gamma-glutamyltranspeptidase-related enzyme. Free Radic Biol Med. 2000;29:825–833. doi: 10.1016/s0891-5849(00)00370-1. [DOI] [PubMed] [Google Scholar]
- 76.Fan P. Yamauchi T. Noble LJ. Ferriero DM. Age-dependent differences in glutathione peroxidase activity after traumatic brain injury. J Neurotrauma. 2003;20:437–445. doi: 10.1089/089771503765355513. [DOI] [PubMed] [Google Scholar]
- 77.Feldman DE. Chauhan V. Koong AC. The unfolded protein response: a novel component of the hypoxic stress response in tumors. Mol Cancer Res. 2005;3:597–605. doi: 10.1158/1541-7786.MCR-05-0221. [DOI] [PubMed] [Google Scholar]
- 78.Fiaschi T. Cozzi G. Raugei G. Formigli L. Ramponi G. Chiarugi P. Redox regulation of beta-actin during integrin-mediated cell adhesion. J Biol Chem. 2006;281:22983–22991. doi: 10.1074/jbc.M603040200. [DOI] [PubMed] [Google Scholar]
- 79.Findlay VJ. Tapiero H. Townsend DM. Sulfiredoxin: a potential therapeutic agent? Biomed Pharmacother. 2005;59:374–379. doi: 10.1016/j.biopha.2005.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Findlay VJ. Townsend DM. Morris TE. Fraser JP. He L. Tew KD. A novel role for human sulfiredoxin in the reversal of glutathionylation. Cancer Res. 2006;66:6800–6806. doi: 10.1158/0008-5472.CAN-06-0484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Firdaus WJ. Wyttenbach A. Diaz-Latoud C. Currie RW. Arrigo AP. Analysis of oxidative events induced by expanded polyglutamine huntingtin exon 1 that are differentially restored by expression of heat shock proteins or treatment with an antioxidant. FEBS J. 2006;273:3076–3093. doi: 10.1111/j.1742-4658.2006.05318.x. [DOI] [PubMed] [Google Scholar]
- 82.Fisher PB. Goldstein NI. Bonner DP. Mechlinski W. Bryson V. Schaffner CP. Toxicity of amphotericin B and its methyl ester toward normal and tumor cell lines. Cancer Res. 1975;35:1996–1999. [PubMed] [Google Scholar]
- 83.Fiskum G. Starkov A. Polster BM. Chinopoulos C. Mitochondrial mechanisms of neural cell death and neuroprotective interventions in Parkinson's disease. Ann N Y Acad Sci. 2003;991:111–119. doi: 10.1111/j.1749-6632.2003.tb07469.x. [DOI] [PubMed] [Google Scholar]
- 84.Fox JH. Barber DS. Singh B. Zucker B. Swindell MK. Norflus F. Buzescu R. Chopra R. Ferrante RJ. Kazantsev A. Hersch SM. Cystamine increases L-cysteine levels in Huntington's disease transgenic mouse brain and in a PC12 model of polyglutamine aggregation. J Neurochem. 2004;91:413–422. doi: 10.1111/j.1471-4159.2004.02726.x. [DOI] [PubMed] [Google Scholar]
- 85.Franco R. Cidlowski JA. Apoptosis and glutathione: beyond an antioxidant. Cell Death Differ. 2009;16:1303–1314. doi: 10.1038/cdd.2009.107. [DOI] [PubMed] [Google Scholar]
- 86.Fratelli M. Demol H. Puype M. Casagrande S. Eberini I. Salmona M. Bonetto V. Mengozzi M. Duffieux F. Miclet E. Bachi A. Vandekerckhove J. Gianazza E. Ghezzi P. Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc Natl Acad Sci U S A. 2002;99:3505–3510. doi: 10.1073/pnas.052592699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fukui H. Moraes CT. Extended polyglutamine repeats trigger a feedback loop involving the mitochondrial complex III, the proteasome and huntingtin aggregates. Hum Mol Genet. 2007;16:783–797. doi: 10.1093/hmg/ddm023. [DOI] [PubMed] [Google Scholar]
- 88.Furukawa Y. O'Halloran TV. Posttranslational modifications in Cu,Zn-superoxide dismutase and mutations associated with amyotrophic lateral sclerosis. Antioxid Redox Signal. 2006;8:847–867. doi: 10.1089/ars.2006.8.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gallogly MM. Shelton MD. Qanungo S. Pai HV. Starke DW. Hoppel CL. Lesnefsky EJ. Mieyal JJ. Glutaredoxin regulates apoptosis in cardiomyocytes via NFkappaB targets Bcl-2 and Bcl-xL: implications for cardiac aging. Antioxid Redox Signal. 2010;12:1339–1353. doi: 10.1089/ars.2009.2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gallogly MM. Starke DW. Leonberg AK. Ospina SM. Mieyal JJ. Kinetic and mechanistic characterization and versatile catalytic properties of mammalian glutaredoxin 2: implications for intracellular roles. Biochemistry. 2008;47:11144–11157. doi: 10.1021/bi800966v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ganji SH. Qin S. Zhang L. Kamanna VS. Kashyap ML. Niacin inhibits vascular oxidative stress, redox-sensitive genes, and monocyte adhesion to human aortic endothelial cells. Atherosclerosis. 2009;202:68–75. doi: 10.1016/j.atherosclerosis.2008.04.044. [DOI] [PubMed] [Google Scholar]
- 92.Gardner TW. Eller AW. Friberg TR. Reduction of severe macular edema in eyes with poor vision after panretinal photocoagulation for proliferative diabetic retinopathy. Graefes Arch Clin Exp Ophthalmol. 1991;229:323–328. doi: 10.1007/BF00170689. [DOI] [PubMed] [Google Scholar]
- 93.Gatchel JR. Zoghbi HY. Diseases of unstable repeat expansion: mechanisms and common principles. Nat Rev Genet. 2005;6:743–755. doi: 10.1038/nrg1691. [DOI] [PubMed] [Google Scholar]
- 94.Gate L. Majumdar RS. Lunk A. Tew KD. Increased myeloproliferation in glutathione S-transferase pi-deficient mice is associated with a deregulation of JNK and Janus kinase/STAT pathways. J Biol Chem. 2004;279:8608–8616. doi: 10.1074/jbc.M308613200. [DOI] [PubMed] [Google Scholar]
- 95.Gerits N. Kostenko S. Shiryaev A. Johannessen M. Moens U. Relations between the mitogen-activated protein kinase and the cAMP-dependent protein kinase pathways: comradeship and hostility. Cell Signal. 2008;20:1592–1607. doi: 10.1016/j.cellsig.2008.02.022. [DOI] [PubMed] [Google Scholar]
- 96.Ghezzi P. Romines B. Fratelli M. Eberini I. Gianazza E. Casagrande S. Laragione T. Mengozzi M. Herzenberg LA. Protein glutathionylation: coupling and uncoupling of glutathione to protein thiol groups in lymphocytes under oxidative stress and HIV infection. Mol Immunol. 2002;38:773–780. doi: 10.1016/s0161-5890(01)00114-6. [DOI] [PubMed] [Google Scholar]
- 97.Giasson BI. Duda JE. Murray IV. Chen Q. Souza JM. Hurtig HI. Ischiropoulos H. Trojanowski JQ. Lee VM. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science. 2000;290:985–989. doi: 10.1126/science.290.5493.985. [DOI] [PubMed] [Google Scholar]
- 98.Gidday JM. Beetsch JW. Park TS. Endogenous glutathione protects cerebral endothelial cells from traumatic injury. J Neurotrauma. 1999;16:27–36. doi: 10.1089/neu.1999.16.27. [DOI] [PubMed] [Google Scholar]
- 99.Giles GI. The redox regulation of thiol dependent signaling pathways in cancer. Curr Pharm Des. 2006;12:4427–4443. doi: 10.2174/138161206779010549. [DOI] [PubMed] [Google Scholar]
- 100.Giovanna C. Cecchi C. Pensalfini A. Bonini SA. Ferrari-Toninelli G. Liguri G. Memo M. Uberti D. Generation of reactive oxygen species by beta amyloid fibrils and oligomers involves different intra/extracellular pathways. Amino Acids. 2010;38:1101–1106. doi: 10.1007/s00726-009-0319-7. [DOI] [PubMed] [Google Scholar]
- 101.Giustarini D. Milzani A. Aldini G. Carini M. Rossi R. Dalle-Donne I. S-nitrosation versus S-glutathionylation of protein sulfhydryl groups by S-nitrosoglutathione. Antioxid Redox Signal. 2005;7:930–939. doi: 10.1089/ars.2005.7.930. [DOI] [PubMed] [Google Scholar]
- 102.Givertz MM. Colucci WS. New targets for heart-failure therapy: endothelin, inflammatory cytokines, and oxidative stress. Lancet. 1998;352(Suppl 1):SI34–SI38. doi: 10.1016/s0140-6736(98)90017-4. [DOI] [PubMed] [Google Scholar]
- 103.Gomez-Nino A. Agapito MT. Obeso A. Gonzalez C. Effects of mitochondrial poisons on glutathione redox potential and carotid body chemoreceptor activity. Respir Physiol Neurobiol. 2009;165:104–111. doi: 10.1016/j.resp.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 104.Gopalakrishna R. Jaken S. Protein kinase C signaling and oxidative stress. Free Radic Biol Med. 2000;28:1349–1361. doi: 10.1016/s0891-5849(00)00221-5. [DOI] [PubMed] [Google Scholar]
- 105.Goss JR. Taffe KM. Kochanek PM. DeKosky ST. The antioxidant enzymes glutathione peroxidase and catalase increase following traumatic brain injury in the rat. Exp Neurol. 1997;146:291–294. doi: 10.1006/exnr.1997.6515. [DOI] [PubMed] [Google Scholar]
- 106.Gu M. Gash MT. Mann VM. Javoy-Agid F. Cooper JM. Schapira AH. Mitochondrial defect in Huntington's disease caudate nucleus. Ann Neurol. 1996;39:385–389. doi: 10.1002/ana.410390317. [DOI] [PubMed] [Google Scholar]
- 107.Gumireddy K. Sun F. Klein-Szanto AJ. Gibbins JM. Gimotty PA. Saunders AJ. Schultz PG. Huang Q. In vivo selection for metastasis promoting genes in the mouse. Proc Natl Acad Sci U S A. 2007;104:6696–6701. doi: 10.1073/pnas.0701145104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Haendeler J. Thioredoxin-1 and posttranslational modifications. Antioxid Redox Signal. 2006;8:1723–1728. doi: 10.1089/ars.2006.8.1723. [DOI] [PubMed] [Google Scholar]
- 109.Hamnell-Pamment Y. Lind C. Palmberg C. Bergman T. Cotgreave IA. Determination of site-specificity of S-glutathionylated cellular proteins. Biochem Biophys Res Commun. 2005;332:362–369. doi: 10.1016/j.bbrc.2005.04.130. [DOI] [PubMed] [Google Scholar]
- 110.Hampton MB. Orrenius S. Dual regulation of caspase activity by hydrogen peroxide: implications for apoptosis. FEBS Lett. 1997;414:552–556. doi: 10.1016/s0014-5793(97)01068-5. [DOI] [PubMed] [Google Scholar]
- 111.Han D. Hanawa N. Saberi B. Kaplowitz N. Mechanisms of liver injury. III. Role of glutathione redox status in liver injury. Am J Physiol Gastrointest Liver Physiol. 2006;291:G1–G7. doi: 10.1152/ajpgi.00001.2006. [DOI] [PubMed] [Google Scholar]
- 112.Hanigan MH. Frierson HF., Jr. Immunohistochemical detection of gamma-glutamyl transpeptidase in normal human tissue. J Histochem Cytochem. 1996;44:1101–1108. doi: 10.1177/44.10.8813074. [DOI] [PubMed] [Google Scholar]
- 113.Hansen RE. Roth D. Winther JR. Quantifying the global cellular thiol-disulfide status. Proc Natl Acad Sci U S A. 2009;106:422–427. doi: 10.1073/pnas.0812149106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Harvey BH. Joubert C. du Preez JL. Berk M. Effect of chronic N-acetyl cysteine administration on oxidative status in the presence and absence of induced oxidative stress in rat striatum. Neurochem Res. 2008;33:508–517. doi: 10.1007/s11064-007-9466-y. [DOI] [PubMed] [Google Scholar]
- 115.Hatahet F. Ruddock LW. Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid Redox Signal. 2009;11:2807–2850. doi: 10.1089/ars.2009.2466. [DOI] [PubMed] [Google Scholar]
- 116.Hebert SS. Horre K. Nicolai L. Bergmans B. Papadopoulou AS. Delacourte A. De Strooper B. MicroRNA regulation of Alzheimer's amyloid precursor protein expression. Neurobiol Dis. 2009;33:422–428. doi: 10.1016/j.nbd.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 117.Hebert SS. Horre K. Nicolai L. Papadopoulou AS. Mandemakers W. Silahtaroglu AN. Kauppinen S. Delacourte A. De Strooper B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer's disease correlates with increased BACE1/beta-secretase expression. Proc Natl Acad Sci U S A. 2008;105:6415–6420. doi: 10.1073/pnas.0710263105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hetz C. Bernasconi P. Fisher J. Lee AH. Bassik MC. Antonsson B. Brandt GS. Iwakoshi NN. Schinzel A. Glimcher LH. Korsmeyer SJ. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science. 2006;312:572–576. doi: 10.1126/science.1123480. [DOI] [PubMed] [Google Scholar]
- 119.Hicdonmez T. Kanter M. Tiryaki M. Parsak T. Cobanoglu S. Neuroprotective effects of N-acetylcysteine on experimental closed head trauma in rats. Neurochem Res. 2006;31:473–481. doi: 10.1007/s11064-006-9040-z. [DOI] [PubMed] [Google Scholar]
- 120.Hidalgo C. Sanchez G. Barrientos G. Aracena-Parks P. A transverse tubule NADPH oxidase activity stimulates calcium release from isolated triads via ryanodine receptor type 1 S-glutathionylation. J Biol Chem. 2006;281:26473–26482. doi: 10.1074/jbc.M600451200. [DOI] [PubMed] [Google Scholar]
- 121.Hinzman J. Thomas T. Burmeister JJ. Quintero J. Huettl P. Pomerleau F. Gerhardt G. Lifshitz J. Diffuse brain injury elevates tonic glutamate levels and potassium-evoked glutamate release in discrete brain regions at two days post-injury: an enzyme-based microelectrode array study. J Neurotrauma. 2010;27:889–899. doi: 10.1089/neu.2009.1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hiratsuka S. Watanabe A. Aburatani H. Maru Y. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol. 2006;8:1369–1375. doi: 10.1038/ncb1507. [DOI] [PubMed] [Google Scholar]
- 123.Hirose M. Hayano T. Shirai H. Nakamura H. Kikuchi M. Isolation of anti-glutathione antibodies from a phage display library. Protein Eng. 1998;11:243–248. doi: 10.1093/protein/11.3.243. [DOI] [PubMed] [Google Scholar]
- 124.Hoffmann P. Woon J. Rowley KG. Karschimkus C. Nelson CL. Dragicevic G. O'Neal D. Wilson A. Croft KD. Mori TA. Kemp BE. Best JD. Jenkins AJ. Glutathionyl haemoglobin is not increased in diabetes nor related to glycaemia, complications, dyslipidaemia, inflammation or other measures of oxidative stress. Diabetes Res Clin Pract. 2008;80:e1–e3. doi: 10.1016/j.diabres.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 125.Hopkins FG. On an Autoxidisable Constituent of the Cell. Biochem J. 1921;15:286–305. doi: 10.1042/bj0150286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Huang KP. Huang FL. Glutathionylation of proteins by glutathione disulfide S-oxide. Biochem Pharmacol. 2002;64:1049–1056. doi: 10.1016/s0006-2952(02)01175-9. [DOI] [PubMed] [Google Scholar]
- 127.Huang Z. Pinto JT. Deng H. Richie JP., Jr. Inhibition of caspase-3 activity and activation by protein glutathionylation. Biochem Pharmacol. 2008;75:2234–2244. doi: 10.1016/j.bcp.2008.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hudemann C. Lonn ME. Godoy JR. Zahedi Avval F. Capani F. Holmgren A. Lillig CH. Identification, expression pattern, and characterization of mouse glutaredoxin 2 isoforms. Antioxid Redox Signal. 2009;11:1–14. doi: 10.1089/ars.2008.2068. [DOI] [PubMed] [Google Scholar]
- 129.Humphries KM. Juliano C. Taylor SS. Regulation of cAMP-dependent protein kinase activity by glutathionylation. J Biol Chem. 2002;277:43505–43511. doi: 10.1074/jbc.M207088200. [DOI] [PubMed] [Google Scholar]
- 130.Hutchens S. Manevich Y. He L. Tew KD. Townsend DM. Cellular resistance to a nitric oxide releasing glutathione S-transferase P-activated prodrug, PABA/NO. Invest New Drugs. doi: 10.1007/s10637-010-9407-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Iijima-Ando K. Hearn SA. Shenton C. Gatt A. Zhao L. Iijima K. Mitochondrial mislocalization underlies Abeta42-induced neuronal dysfunction in a Drosophila model of Alzheimer's disease. PLoS One. 2009;4:e8310. doi: 10.1371/journal.pone.0008310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Irani K. Xia Y. Zweier JL. Sollott SJ. Der CJ. Fearon ER. Sundaresan M. Finkel T. Goldschmidt-Clermont PJ. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science. 1997;275:1649–1652. doi: 10.1126/science.275.5306.1649. [DOI] [PubMed] [Google Scholar]
- 133.Jaffrey SR. Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. Sci STKE. 2001;2001:pl1. doi: 10.1126/stke.2001.86.pl1. [DOI] [PubMed] [Google Scholar]
- 134.Jha N. Jurma O. Lalli G. Liu Y. Pettus EH. Greenamyre JT. Liu RM. Forman HJ. Andersen JK. Glutathione depletion in PC12 results in selective inhibition of mitochondrial complex I activity. Implications for Parkinson's disease. J Biol Chem. 2000;275:26096–26101. doi: 10.1074/jbc.M000120200. [DOI] [PubMed] [Google Scholar]
- 135.Jin W. Wang H. Yan W. Zhu L. Hu Z. Ding Y. Tang K. Role of Nrf2 in protection against traumatic brain injury in mice. J Neurotrauma. 2009;26:131–139. doi: 10.1089/neu.2008.0655. [DOI] [PubMed] [Google Scholar]
- 136.Jinushi M. Vanneman M. Munshi NC. Tai YT. Prabhala RH. Ritz J. Neuberg D. Anderson KC. Carrasco DR. Dranoff G. MHC class I chain-related protein A antibodies and shedding are associated with the progression of multiple myeloma. Proc Natl Acad Sci U S A. 2008;105:1285–1290. doi: 10.1073/pnas.0711293105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ju TC. Chen SD. Liu CC. Yang DI. Protective effects of S-nitrosoglutathione against amyloid beta-peptide neurotoxicity. Free Radic Biol Med. 2005;38:938–949. doi: 10.1016/j.freeradbiomed.2004.12.019. [DOI] [PubMed] [Google Scholar]
- 138.Ju TC. Yang YT. Yang DI. Protective effects of S-nitrosoglutathione against neurotoxicity of 3-nitropropionic acid in rat. Neurosci Lett. 2004;362:226–231. doi: 10.1016/j.neulet.2004.03.028. [DOI] [PubMed] [Google Scholar]
- 139.Kang PT. Yun J. Kaumaya PP. Chen YR. Design and use of peptide-based antibodies decreasing superoxide production by mitochondrial complex I and complex II. Biopolymers. 2010 doi: 10.1002/bip.21457. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Karin M. Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3:221–227. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 141.Kearney M. Orrell RW. Fahey M. Pandolfo M. Antioxidants and other pharmacological treatments for Friedreich ataxia. Cochrane Database Syst Rev. 2009;4:CD007791. doi: 10.1002/14651858.CD007791.pub2. [DOI] [PubMed] [Google Scholar]
- 142.Khan M. Im YB. Shunmugavel A. Gilg AG. Dhindsa RK. Singh AK. Singh I. Administration of S-nitrosoglutathione after traumatic brain injury protects the neurovascular unit and reduces secondary injury in a rat model of controlled cortical impact. J Neuroinflammation. 2009;6:32. doi: 10.1186/1742-2094-6-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kim YJ. Lee WS. Ip C. Chae HZ. Park EM. Park YM. Prx1 suppresses radiation-induced c-Jun NH2-terminal kinase signaling in lung cancer cells through interaction with the glutathione S-transferase Pi/c-Jun NH2-terminal kinase complex. Cancer Res. 2006;66:7136–7142. doi: 10.1158/0008-5472.CAN-05-4446. [DOI] [PubMed] [Google Scholar]
- 144.Klatt P. Molina EP. Lamas S. Nitric oxide inhibits c-Jun DNA binding by specifically targeted S-glutathionylation. J Biol Chem. 1999;274:15857–15864. doi: 10.1074/jbc.274.22.15857. [DOI] [PubMed] [Google Scholar]
- 145.Klepac N. Relja M. Klepac R. Hecimovic S. Babic T. Trkulja V. Oxidative stress parameters in plasma of Huntington's disease patients, asymptomatic Huntington's disease gene carriers and healthy subjects: a cross-sectional study. J Neurol. 2007;254:1676–1683. doi: 10.1007/s00415-007-0611-y. [DOI] [PubMed] [Google Scholar]
- 146.Krautwald S. Buscher D. Kummer V. Buder S. Baccarini M. Involvement of the protein tyrosine phosphatase SHP-1 in Ras-mediated activation of the mitogen-activated protein kinase pathway. Mol Cell Biol. 1996;16:5955–5963. doi: 10.1128/mcb.16.11.5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kumar P. Kumar A. Neuroprotective effect of cyclosporine and FK506 against 3-nitropropionic acid induced cognitive dysfunction and glutathione redox in rat: possible role of nitric oxide. Neurosci Res. 2009;63:302–314. doi: 10.1016/j.neures.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 148.Lai Y. Hickey RW. Chen Y. Bayir H. Sullivan ML. Chu CT. Kochanek PM. Dixon CE. Jenkins LW. Graham SH. Watkins SC. Clark RS. Autophagy is increased after traumatic brain injury in mice and is partially inhibited by the antioxidant gamma-glutamylcysteinyl ethyl ester. J Cereb Blood Flow Metab. 2008;28:540–550. doi: 10.1038/sj.jcbfm.9600551. [DOI] [PubMed] [Google Scholar]
- 149.Landino LM. Brown CM. Edson CA. Gilbert LJ. Grega-Larson N. Wirth AJ. Lane KC. Fluorescein-labeled glutathione to study protein S-glutathionylation. Anal Biochem. 2010;402:102–104. doi: 10.1016/j.ab.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Law RH. Zhang Q. McGowan S. Buckle AM. Silverman GA. Wong W. Rosado CJ. Langendorf CG. Pike RN. Bird PI. Whisstock JC. An overview of the serpin superfamily. Genome Biol. 2006;7:216. doi: 10.1186/gb-2006-7-5-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lee E. Jeong J. Kim SE. Song EJ. Kang SW. Lee KJ. Multiple functions of Nm23-H1 are regulated by oxido-reduction system. PLoS One. 2009;4:e7949. doi: 10.1371/journal.pone.0007949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lehmann C. Bette S. Engele J. High extracellular glutamate modulates expression of glutamate transporters and glutamine synthetase in cultured astrocytes. Brain Res. 2009;1297:1–8. doi: 10.1016/j.brainres.2009.08.070. [DOI] [PubMed] [Google Scholar]
- 153.Levesque JP. Liu F. Simmons PJ. Betsuyaku T. Senior RM. Pham C. Link DC. Characterization of hematopoietic progenitor mobilization in protease-deficient mice. Blood. 2004;104:65–72. doi: 10.1182/blood-2003-05-1589. [DOI] [PubMed] [Google Scholar]
- 154.Li JY. Popovic N. Brundin P. The use of the R6 transgenic mouse models of Huntington's disease in attempts to develop novel therapeutic strategies. NeuroRx. 2005;2:447–464. doi: 10.1602/neurorx.2.3.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Liao BC. Hsieh CW. Lin YC. Wung BS. The glutaredoxin/glutathione system modulates NF-{kappa}B activity by glutathionylation of p65 in cinnamaldehyde-treated endothelial cells. Toxicol Sci. 2010;116:151–163. doi: 10.1093/toxsci/kfq098. [DOI] [PubMed] [Google Scholar]
- 156.Lim SY. Raftery M. Cai H. Hsu K. Yan WX. Hseih HL. Watts RN. Richardson D. Thomas S. Perry M. Geczy CL. S-nitrosylated S100A8: novel anti-inflammatory properties. J Immunol. 2008;181:5627–5636. doi: 10.4049/jimmunol.181.8.5627. [DOI] [PubMed] [Google Scholar]
- 157.Lind C. Gerdes R. Hamnell Y. Schuppe-Koistinen I. von Lowenhielm HB. Holmgren A. Cotgreave IA. Identification of S-glutathionylated cellular proteins during oxidative stress and constitutive metabolism by affinity purification and proteomic analysis. Arch Biochem Biophys. 2002;406:229–240. doi: 10.1016/s0003-9861(02)00468-x. [DOI] [PubMed] [Google Scholar]
- 158.Liot G. Bossy B. Lubitz S. Kushnareva Y. Sejbuk N. Bossy-Wetzel E. Complex II inhibition by 3-NP causes mitochondrial fragmentation and neuronal cell death via an NMDA- and ROS-dependent pathway. Cell Death Differ. 2009;16:899–909. doi: 10.1038/cdd.2009.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Loh K. Deng H. Fukushima A. Cai X. Boivin B. Galic S. Bruce C. Shields BJ. Skiba B. Ooms LM. Stepto N. Wu B. Mitchell CA. Tonks NK. Watt MJ. Febbraio MA. Crack PJ. Andrikopoulos S. Tiganis T. Reactive oxygen species enhance insulin sensitivity. Cell Metab. 2009;10:260–272. doi: 10.1016/j.cmet.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lomas DA. Evans DL. Finch JT. Carrell RW. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature. 1992;357:605–607. doi: 10.1038/357605a0. [DOI] [PubMed] [Google Scholar]
- 161.Lynn BC. Wang J. Markesbery WR. Lovell MA. Quantitative changes in the mitochondrial proteome from subjects with mild cognitive impairment, early stage, and late stage Alzheimer's disease. J Alzheimers Dis. 2010;19:325–339. doi: 10.3233/JAD-2010-1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Maas AI. Stocchetti N. Bullock R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008;7:728–741. doi: 10.1016/S1474-4422(08)70164-9. [DOI] [PubMed] [Google Scholar]
- 163.Magnani M. Novelli G. Palloni R. Human plasma glutathione oxidation in normal and pathological conditions. Clin Physiol Biochem. 1984;2:287–290. [PubMed] [Google Scholar]
- 164.Malik G. Nagy N. Ho YS. Maulik N. Das DK. Role of glutaredoxin-1 in cardioprotection: an insight with Glrx1 transgenic and knockout animals. J Mol Cell Cardiol. 2008;44:261–269. doi: 10.1016/j.yjmcc.2007.08.022. [DOI] [PubMed] [Google Scholar]
- 165.Manevich Y. Feinstein SI. Fisher AB. Activation of the antioxidant enzyme 1-CYS peroxiredoxin requires glutathionylation mediated by heterodimerization with pi GST. Proc Natl Acad Sci U S A. 2004;101:3780–3785. doi: 10.1073/pnas.0400181101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Manevich Y. Townsend DM. Hutchens S. Tew KD. Diazeniumdiolate mediated nitrosative stress alters nitric oxide homeostasis through intracellular calcium and S-glutathionylation of nitric oxide synthetase. PLoS One. 2010;5:e14151. doi: 10.1371/journal.pone.0014151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Mannervik B. Danielson UH. Glutathione transferases—structure and catalytic activity. CRC Crit Rev Biochem. 1988;23:283–337. doi: 10.3109/10409238809088226. [DOI] [PubMed] [Google Scholar]
- 168.Martin HL. Teismann P. Glutathione—a review on its role and significance in Parkinson's disease. FASEB J. 2009;23:3263–3272. doi: 10.1096/fj.08-125443. [DOI] [PubMed] [Google Scholar]
- 169.Mason JK. Recording HIV status on police computers. BMJ. 1992;304:995–996. doi: 10.1136/bmj.304.6833.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Matozaki T. Murata Y. Saito Y. Okazawa H. Ohnishi H. Protein tyrosine phosphatase SHP-2: a proto-oncogene product that promotes Ras activation. Cancer Sci. 2009;100:1786–1793. doi: 10.1111/j.1349-7006.2009.01257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Matsuda M. Masutani H. Nakamura H. Miyajima S. Yamauchi A. Yonehara S. Uchida A. Irimajiri K. Horiuchi A. Yodoi J. Protective activity of adult T cell leukemia-derived factor (ADF) against tumor necrosis factor-dependent cytotoxicity on U937 cells. J Immunol. 1991;147:3837–3841. [PubMed] [Google Scholar]
- 172.McClung JP. Roneker CA. Mu W. Lisk DJ. Langlais P. Liu F. Lei XG. Development of insulin resistance and obesity in mice overexpressing cellular glutathione peroxidase. Proc Natl Acad Sci U S A. 2004;101:8852–8857. doi: 10.1073/pnas.0308096101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.McIlwain CC. Townsend DM. Tew KD. Glutathione S-transferase polymorphisms: cancer incidence and therapy. Oncogene. 2006;25:1639–1648. doi: 10.1038/sj.onc.1209373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.McKiernan E. O'Brien K. Grebenchtchikov N. Geurts-Moespot A. Sieuwerts AM. Martens JW. Magdolen V. Evoy D. McDermott E. Crown J. Sweep FC. Duffy MJ. Protein kinase Cdelta expression in breast cancer as measured by real-time PCR, western blotting and ELISA. Br J Cancer. 2008;99:1644–1650. doi: 10.1038/sj.bjc.6604728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.McLellan LI. Lewis AD. Hall DJ. Ansell JD. Wolf CR. Uptake and distribution of N-acetylcysteine in mice: tissue-specific effects on glutathione concentrations. Carcinogenesis. 1995;16:2099–2106. doi: 10.1093/carcin/16.9.2099. [DOI] [PubMed] [Google Scholar]
- 176.Meek DW. Hupp TR. The regulation of MDM2 by multisite phosphorylation-opportunities for molecular-based intervention to target tumours? Semin Cancer Biol. 2010;20:19–28. doi: 10.1016/j.semcancer.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 177.Mieyal JJ. Gallogly MM. Qanungo S. Sabens EA. Shelton MD. Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid Redox Signal. 2008;10:1941–1988. doi: 10.1089/ars.2008.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Mieyal JJ. Starke DW. Gravina SA. Dothey C. Chung JS. Thioltransferase in human red blood cells: purification and properties. Biochemistry. 1991;30:6088–6097. doi: 10.1021/bi00239a002. [DOI] [PubMed] [Google Scholar]
- 179.Mohr S. Hallak H. de Boitte A. Lapetina EG. Brune B. Nitric oxide-induced S-glutathionylation and inactivation of glyceraldehyde-3-phosphate dehydrogenase. J Biol Chem. 1999;274:9427–9430. doi: 10.1074/jbc.274.14.9427. [DOI] [PubMed] [Google Scholar]
- 180.Mohr S. Zech B. Lapetina EG. Brune B. Inhibition of caspase-3 by S-nitrosation and oxidation caused by nitric oxide. Biochem Biophys Res Commun. 1997;238:387–391. doi: 10.1006/bbrc.1997.7304. [DOI] [PubMed] [Google Scholar]
- 181.Montine TJ. Picklo MJ. Amarnath V. Whetsell WO., Jr. Graham DG. Neurotoxicity of endogenous cysteinylcatechols. Exp Neurol. 1997;148:26–33. doi: 10.1006/exnr.1997.6662. [DOI] [PubMed] [Google Scholar]
- 182.Monzon FA. Ogino S. Hammond ME. Halling KC. Bloom KJ. Nikiforova MN. The role of KRAS mutation testing in the management of patients with metastatic colorectal cancer. Arch Pathol Lab Med. 2009;133:1600–1606. doi: 10.5858/133.10.1600. [DOI] [PubMed] [Google Scholar]
- 183.Munoz AM. Rey P. Soto-Otero R. Guerra MJ. Labandeira-Garcia JL. Systemic administration of N-acetylcysteine protects dopaminergic neurons against 6-hydroxydopamine-induced degeneration. J Neurosci Res. 2004;76:551–562. doi: 10.1002/jnr.20107. [DOI] [PubMed] [Google Scholar]
- 184.Murakami K. Kondo T. Ohtsuka Y. Fujiwara Y. Shimada M. Kawakami Y. Impairment of glutathione metabolism in erythrocytes from patients with diabetes mellitus. Metabolism. 1989;38:753–758. doi: 10.1016/0026-0495(89)90061-9. [DOI] [PubMed] [Google Scholar]
- 185.Murata H. Ihara Y. Nakamura H. Yodoi J. Sumikawa K. Kondo T. Glutaredoxin exerts an antiapoptotic effect by regulating the redox state of Akt. J Biol Chem. 2003;278:50226–50233. doi: 10.1074/jbc.M310171200. [DOI] [PubMed] [Google Scholar]
- 186.Naito C. Niwa T. Analysis of glutathionyl hemoglobin levels in diabetic patients by electrospray ionization liquid chromatography-mass spectrometry: effect of vitamin E administration. J Chromatogr B Biomed Sci Appl. 2000;746:91–94. doi: 10.1016/s0378-4347(00)00121-3. [DOI] [PubMed] [Google Scholar]
- 187.Naoi M. Maruyama W. Yi H. Inaba K. Akao Y. Shamoto-Nagai M. Mitochondria in neurodegenerative disorders: regulation of the redox state and death signaling leading to neuronal death and survival. J Neural Transm. 2009;116:1371–1381. doi: 10.1007/s00702-009-0309-7. [DOI] [PubMed] [Google Scholar]
- 188.Nelson PT. Wang WX. Rajeev BW. MicroRNAs (miRNAs) in neurodegenerative diseases. Brain Pathol. 2008;18:130–138. doi: 10.1111/j.1750-3639.2007.00120.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Neves G. Cooke SF. Bliss TV. Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Nat Rev Neurosci. 2008;9:65–75. doi: 10.1038/nrn2303. [DOI] [PubMed] [Google Scholar]
- 190.Nigam R. Anderson DJ. Lee SF. Bennett BM. Isoform-specific biotransformation of glyceryl trinitrate by rat aortic glutathione S-transferases. J Pharmacol Exp Ther. 1996;279:1527–1534. [PubMed] [Google Scholar]
- 191.Nistico G. Ciriolo MR. Fiskin K. Iannone M. De Martino A. Rotilio G. NGF restores decrease in catalase and increases glutathione peroxidase activity in the brain of aged rats. Neurosci Lett. 1991;130:117–119. doi: 10.1016/0304-3940(91)90241-k. [DOI] [PubMed] [Google Scholar]
- 192.Niture SK. Velu CS. Bailey NI. Srivenugopal KS. S-thiolation mimicry: quantitative and kinetic analysis of redox status of protein cysteines by glutathione-affinity chromatography. Arch Biochem Biophys. 2005;444:174–184. doi: 10.1016/j.abb.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 193.Niwa T. Naito C. Mawjood AH. Imai K. Increased glutathionyl hemoglobin in diabetes mellitus and hyperlipidemia demonstrated by liquid chromatography/electrospray ionization-mass spectrometry. Clin Chem. 2000;46:82–88. [PubMed] [Google Scholar]
- 194.Noguera-Mazon V. Lemoine J. Walker O. Rouhier N. Salvador A. Jacquot JP. Lancelin JM. Krimm I. Glutathionylation induces the dissociation of 1-Cys D-peroxiredoxin non-covalent homodimer. J Biol Chem. 2006;281:31736–31742. doi: 10.1074/jbc.M602188200. [DOI] [PubMed] [Google Scholar]
- 195.Nussbaum RL. Ellis CE. Alzheimer's disease and Parkinson's disease. N Engl J Med. 2003;348:1356–1364. doi: 10.1056/NEJM2003ra020003. [DOI] [PubMed] [Google Scholar]
- 196.Nwose EU. Jelinek HF. Richards RS. Kerr PG. Erythrocyte oxidative stress in clinical management of diabetes and its cardiovascular complications. Br J Biomed Sci. 2007;64:35–43. doi: 10.1080/09674845.2007.11732754. [DOI] [PubMed] [Google Scholar]
- 197.Obin M. Shang F. Gong X. Handelman G. Blumberg J. Taylor A. Redox regulation of ubiquitin-conjugating enzymes: mechanistic insights using the thiol-specific oxidant diamide. FASEB J. 1998;12:561–569. doi: 10.1096/fasebj.12.7.561. [DOI] [PubMed] [Google Scholar]
- 198.Olney JW. Zorumski C. Price MT. Labruyere J. L-cysteine, a bicarbonate-sensitive endogenous excitotoxin. Science. 1990;248:596–599. doi: 10.1126/science.2185543. [DOI] [PubMed] [Google Scholar]
- 199.Ondrejcak T. Klyubin I. Hu NW. Barry AE. Cullen WK. Rowan MJ. Alzheimer's disease amyloid beta-protein and synaptic function. Neuromolecular Med. 2010;12:13–26. doi: 10.1007/s12017-009-8091-0. [DOI] [PubMed] [Google Scholar]
- 200.Opie LH. Commerford PJ. Gersh BJ. Pfeffer MA. Controversies in ventricular remodelling. Lancet. 2006;367:356–367. doi: 10.1016/S0140-6736(06)68074-4. [DOI] [PubMed] [Google Scholar]
- 201.Pan S. Berk BC. Glutathiolation regulates tumor necrosis factor-alpha-induced caspase-3 cleavage and apoptosis: key role for glutaredoxin in the death pathway. Circ Res. 2007;100:213–219. doi: 10.1161/01.RES.0000256089.30318.20. [DOI] [PubMed] [Google Scholar]
- 202.Paolicchi A. Dominici S. Pieri L. Maellaro E. Pompella A. Glutathione catabolism as a signaling mechanism. Biochem Pharmacol. 2002;64:1027–1035. doi: 10.1016/s0006-2952(02)01173-5. [DOI] [PubMed] [Google Scholar]
- 203.Park JW. Mieyal JJ. Rhee SG. Chock PB. Deglutathionylation of 2-Cys peroxiredoxin is specifically catalyzed by sulfiredoxin. J Biol Chem. 2009;284:23364–23374. doi: 10.1074/jbc.M109.021394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Pastore A. Tozzi G. Gaeta LM. Bertini E. Serafini V. Di Cesare S. Bonetto V. Casoni F. Carrozzo R. Federici G. Piemonte F. Actin glutathionylation increases in fibroblasts of patients with Friedreich's ataxia: a potential role in the pathogenesis of the disease. J Biol Chem. 2003;278:42588–42595. doi: 10.1074/jbc.M301872200. [DOI] [PubMed] [Google Scholar]
- 205.Patel N. Hoang D. Miller N. Ansaloni S. Huang Q. Rogers JT. Lee JC. Saunders AJ. MicroRNAs can regulate human APP levels. Mol Neurodegener. 2008;3:10. doi: 10.1186/1750-1326-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Pavlov PF. Petersen CH. Glaser E. Ankarcrona M. Mitochondrial accumulation of APP and Abeta: significance for Alzheimer disease pathogenesis. J Cell Mol Med. 2009;13:4137–4145. doi: 10.1111/j.1582-4934.2009.00892.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Perez-Severiano F. Rios C. Segovia J. Striatal oxidative damage parallels the expression of a neurological phenotype in mice transgenic for the mutation of Huntington's disease. Brain Res. 2000;862:234–237. doi: 10.1016/s0006-8993(00)02082-5. [DOI] [PubMed] [Google Scholar]
- 208.Perez-Severiano F. Santamaria A. Pedraza-Chaverri J. Medina-Campos ON. Rios C. Segovia J. Increased formation of reactive oxygen species, but no changes in glutathione peroxidase activity, in striata of mice transgenic for the Huntington's disease mutation. Neurochem Res. 2004;29:729–733. doi: 10.1023/b:nere.0000018843.83770.4b. [DOI] [PubMed] [Google Scholar]
- 209.Perrin RJ. Fagan AM. Holtzman DM. Multimodal techniques for diagnosis and prognosis of Alzheimer's disease. Nature. 2009;461:916–922. doi: 10.1038/nature08538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Peskin AV. Winterbourn CC. Taurine chloramine is more selective than hypochlorous acid at targeting critical cysteines and inactivating creatine kinase and glyceraldehyde-3-phosphate dehydrogenase. Free Radic Biol Med. 2006;40:45–53. doi: 10.1016/j.freeradbiomed.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 211.Pham FH. Sugden PH. Clerk A. Regulation of protein kinase B and 4E-BP1 by oxidative stress in cardiac myocytes. Circ Res. 2000;86:1252–1258. doi: 10.1161/01.res.86.12.1252. [DOI] [PubMed] [Google Scholar]
- 212.Pineda-Molina E. Klatt P. Vazquez J. Marina A. Garcia de Lacoba M. Perez-Sala D. Lamas S. Glutathionylation of the p50 subunit of NF-kappaB: a mechanism for redox-induced inhibition of DNA binding. Biochemistry. 2001;40:14134–14142. doi: 10.1021/bi011459o. [DOI] [PubMed] [Google Scholar]
- 213.Pinhel MA. Nakazone MA. Cacao JC. Piteri RC. Dantas RT. Godoy MF. Godoy MR. Tognola WA. Conforti-Froes ND. Souza D. Glutathione S-transferase variants increase susceptibility for late-onset Alzheimer's disease: association study and relationship with apolipoprotein E epsilon4 allele. Clin Chem Lab Med. 2008;46:439–445. doi: 10.1515/CCLM.2008.102. [DOI] [PubMed] [Google Scholar]
- 214.Potts MB. Rola R. Claus CP. Ferriero DM. Fike JR. Noble-Haeusslein LJ. Glutathione peroxidase overexpression does not rescue impaired neurogenesis in the injured immature brain. J Neurosci Res. 2009;87:1848–1857. doi: 10.1002/jnr.21996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Puccio H. Simon D. Cossee M. Criqui-Filipe P. Tiziano F. Melki J. Hindelang C. Matyas R. Rustin P. Koenig M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat Genet. 2001;27:181–186. doi: 10.1038/84818. [DOI] [PubMed] [Google Scholar]
- 216.Puka-Sundvall M. Eriksson P. Nilsson M. Sandberg M. Lehmann A. Neurotoxicity of cysteine: interaction with glutamate. Brain Res. 1995;705:65–70. doi: 10.1016/0006-8993(95)01139-0. [DOI] [PubMed] [Google Scholar]
- 217.Qanungo S. Starke DW. Pai HV. Mieyal JJ. Nieminen AL. Glutathione supplementation potentiates hypoxic apoptosis by S-glutathionylation of p65-NFkappaB. J Biol Chem. 2007;282:18427–18436. doi: 10.1074/jbc.M610934200. [DOI] [PubMed] [Google Scholar]
- 218.Qanungo S. Wang M. Nieminen AL. N-Acetyl-L-cysteine enhances apoptosis through inhibition of nuclear factor-kappaB in hypoxic murine embryonic fibroblasts. J Biol Chem. 2004;279:50455–50464. doi: 10.1074/jbc.M406749200. [DOI] [PubMed] [Google Scholar]
- 219.Rafii S. Lyden D. S100 chemokines mediate bookmarking of premetastatic niches. Nat Cell Biol. 2006;8:1321–1323. doi: 10.1038/ncb1206-1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Ralat LA. Manevich Y. Fisher AB. Colman RF. Direct evidence for the formation of a complex between 1-cysteine peroxiredoxin and glutathione S-transferase pi with activity changes in both enzymes. Biochemistry. 2006;45:360–372. doi: 10.1021/bi0520737. [DOI] [PubMed] [Google Scholar]
- 221.Ranganathan S. Walsh ES. Godwin AK. Tew KD. Cloning and characterization of human colon glyoxalase-I. J Biol Chem. 1993;268:5661–5667. [PubMed] [Google Scholar]
- 222.Rao RK. Clayton LW. Regulation of protein phosphatase 2A by hydrogen peroxide and glutathionylation. Biochem Biophys Res Commun. 2002;293:610–616. doi: 10.1016/S0006-291X(02)00268-1. [DOI] [PubMed] [Google Scholar]
- 223.Rauhala P. Lin AM. Chiueh CC. Neuroprotection by S-nitrosoglutathione of brain dopamine neurons from oxidative stress. FASEB J. 1998;12:165–173. doi: 10.1096/fasebj.12.2.165. [DOI] [PubMed] [Google Scholar]
- 224.Ravi K. Brennan LA. Levic S. Ross PA. Black SM. S-nitrosylation of endothelial nitric oxide synthase is associated with monomerization and decreased enzyme activity. Proc Natl Acad Sci U S A. 2004;101:2619–2624. doi: 10.1073/pnas.0300464101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Reddy PH. Role of mitochondria in neurodegenerative diseases: mitochondria as a therapeutic target in Alzheimer's disease. CNS Spectr. 2009;14:8–13. doi: 10.1017/s1092852900024901. ; discussion 16–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Reed TT. Owen J. Pierce WM. Sebastian A. Sullivan PG. Butterfield DA. Proteomic identification of nitrated brain proteins in traumatic brain-injured rats treated postinjury with gamma-glutamylcysteine ethyl ester: insights into the role of elevation of glutathione as a potential therapeutic strategy for traumatic brain injury. J Neurosci Res. 2009;87:408–417. doi: 10.1002/jnr.21872. [DOI] [PubMed] [Google Scholar]
- 227.Regazzoni L. Panusa A. Yeum KJ. Carini M. Aldini G. Hemoglobin glutathionylation can occur through cysteine sulfenic acid intermediate: electrospray ionization LTQ-Orbitrap hybrid mass spectrometry studies. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:3456–3461. doi: 10.1016/j.jchromb.2009.05.020. [DOI] [PubMed] [Google Scholar]
- 228.Reynaert NL. Ckless K. Guala AS. Wouters EF. van der Vliet A. Janssen-Heininger YM. In situ detection of S-glutathionylated proteins following glutaredoxin-1 catalyzed cysteine derivatization. Biochim Biophys Acta. 2006;1760:380–387. doi: 10.1016/j.bbagen.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 229.Reynaert NL. van der Vliet A. Guala AS. McGovern T. Hristova M. Pantano C. Heintz NH. Heim J. Ho YS. Matthews DE. Wouters EF. Janssen-Heininger YM. Dynamic redox control of NF-kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc Natl Acad Sci U S A. 2006;103:13086–13091. doi: 10.1073/pnas.0603290103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Rinna A. Torres M. Forman HJ. Stimulation of the alveolar macrophage respiratory burst by ADP causes selective glutathionylation of protein tyrosine phosphatase 1B. Free Radic Biol Med. 2006;41:86–91. doi: 10.1016/j.freeradbiomed.2006.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Rokutan K. Thomas JA. Johnston RB., Jr. Phagocytosis and stimulation of the respiratory burst by phorbol diester initiate S-thiolation of specific proteins in macrophages. J Immunol. 1991;147:260–264. [PubMed] [Google Scholar]
- 232.Rosengard AM. Krutzsch HC. Shearn A. Biggs JR. Barker E. Margulies IM. King CR. Liotta LA. Steeg PS. Reduced Nm23/Awd protein in tumour metastasis and aberrant Drosophila development. Nature. 1989;342:177–180. doi: 10.1038/342177a0. [DOI] [PubMed] [Google Scholar]
- 233.Rossi R. Giustarini D. Milzani A. Dalle-Donne I. Membrane skeletal protein S-glutathionylation and hemolysis in human red blood cells. Blood Cells Mol Dis. 2006;37:180–187. doi: 10.1016/j.bcmd.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 234.Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443:780–786. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- 235.Ruscoe JE. Rosario LA. Wang T. Gate L. Arifoglu P. Wolf CR. Henderson CJ. Ronai Z. Tew KD. Pharmacologic or genetic manipulation of glutathione S-transferase P1–1 (GSTpi) influences cell proliferation pathways. J Pharmacol Exp Ther. 2001;298:339–345. [PubMed] [Google Scholar]
- 236.Sabens EA. Distler AM. Mieyal JJ. Levodopa deactivates enzymes that regulate thiol-disulfide homeostasis and promotes neuronal cell death: implications for therapy of Parkinson's disease. Biochemistry. 2010;49:2715–2724. doi: 10.1021/bi9018658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Saeed U. Ray A. Valli RK. Kumar AM. Ravindranath V. DJ-1 loss by glutaredoxin but not glutathione depletion triggers Daxx translocation and cell death. Antioxid Redox Signal. 2010;13:127–144. doi: 10.1089/ars.2009.2832. [DOI] [PubMed] [Google Scholar]
- 238.Saitoh M. Nishitoh H. Fujii M. Takeda K. Tobiume K. Sawada Y. Kawabata M. Miyazono K. Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Salama I. Malone PS. Mihaimeed F. Jones JL. A review of the S100 proteins in cancer. Eur J Surg Oncol. 2008;34:357–364. doi: 10.1016/j.ejso.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 240.Sampathkumar R. Balasubramanyam M. Sudarslal S. Rema M. Mohan V. Balaram P. Increased glutathionylated hemoglobin (HbSSG) in type 2 diabetes subjects with microangiopathy. Clin Biochem. 2005;38:892–899. doi: 10.1016/j.clinbiochem.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 241.Sandler AB. Molecular targeted agents in non-small-cell lung cancer. Clin Lung Cancer 5 Suppl. 2003;1:S22–S28. [PubMed] [Google Scholar]
- 242.Savitt JM. Dawson VL. Dawson TM. Diagnosis and treatment of Parkinson disease: molecules to medicine. J Clin Invest. 2006;116:1744–1754. doi: 10.1172/JCI29178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Schrauwen P. Hesselink MK. Oxidative capacity, lipotoxicity, and mitochondrial damage in type 2 diabetes. Diabetes. 2004;53:1412–1417. doi: 10.2337/diabetes.53.6.1412. [DOI] [PubMed] [Google Scholar]
- 244.Schuppe-Koistinen I. Gerdes R. Moldeus P. Cotgreave IA. Studies on the reversibility of protein S-thiolation in human endothelial cells. Arch Biochem Biophys. 1994;315:226–234. doi: 10.1006/abbi.1994.1494. [DOI] [PubMed] [Google Scholar]
- 245.Seo YH. Carroll KS. Profiling protein thiol oxidation in tumor cells using sulfenic acid-specific antibodies. Proc Natl Acad Sci U S A. 2009;106:16163–16168. doi: 10.1073/pnas.0903015106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Sevier CS. Qu H. Heldman N. Gross E. Fass D. Kaiser CA. Modulation of cellular disulfide-bond formation and the ER redox environment by feedback regulation of Ero1. Cell. 2007;129:333–344. doi: 10.1016/j.cell.2007.02.039. [DOI] [PubMed] [Google Scholar]
- 247.Sharma R. Brown D. Awasthi S. Yang Y. Sharma A. Patrick B. Saini MK. Singh SP. Zimniak P. Singh SV. Awasthi YC. Transfection with 4-hydroxynonenal-metabolizing glutathione S-transferase isozymes leads to phenotypic transformation and immortalization of adherent cells. Eur J Biochem. 2004;271:1690–1701. doi: 10.1111/j.1432-1033.2004.04067.x. [DOI] [PubMed] [Google Scholar]
- 248.Shelton MD. Distler AM. Kern TS. Mieyal JJ. Glutaredoxin regulates autocrine and paracrine proinflammatory responses in retinal glial (muller) cells. J Biol Chem. 2009;284:4760–4766. doi: 10.1074/jbc.M805464200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Shelton MD. Kern TS. Mieyal JJ. Glutaredoxin regulates nuclear factor kappa-B and intercellular adhesion molecule in Muller cells: model of diabetic retinopathy. J Biol Chem. 2007;282:12467–12474. doi: 10.1074/jbc.M610863200. [DOI] [PubMed] [Google Scholar]
- 250.Shelton MD. Mieyal JJ. Regulation by reversible S-glutathionylation: molecular targets implicated in inflammatory diseases. Mol Cells. 2008;25:332–346. [PMC free article] [PubMed] [Google Scholar]
- 251.Shi M. Bradner J. Bammler TK. Eaton DL. Zhang J. Ye Z. Wilson AM. Montine TJ. Pan C. Identification of glutathione S-transferase pi as a protein involved in Parkinson disease progression. Am J Pathol. 2009;175:54–65. doi: 10.2353/ajpath.2009.081019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Shin BK. Wang H. Yim AM. Le Naour F. Brichory F. Jang JH. Zhao R. Puravs E. Tra J. Michael CW. Misek DE. Hanash SM. Global profiling of the cell surface proteome of cancer cells uncovers an abundance of proteins with chaperone function. J Biol Chem. 2003;278:7607–7616. doi: 10.1074/jbc.M210455200. [DOI] [PubMed] [Google Scholar]
- 253.Shiomi T. Tsutsui H. Matsusaka H. Murakami K. Hayashidani S. Ikeuchi M. Wen J. Kubota T. Utsumi H. Takeshita A. Overexpression of glutathione peroxidase prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation. 2004;109:544–549. doi: 10.1161/01.CIR.0000109701.77059.E9. [DOI] [PubMed] [Google Scholar]
- 254.Sian J. Dexter DT. Lees AJ. Daniel S. Agid Y. Javoy-Agid F. Jenner P. Marsden CD. Alterations in glutathione levels in Parkinson's disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol. 1994;36:348–355. doi: 10.1002/ana.410360305. [DOI] [PubMed] [Google Scholar]
- 255.Siscovick DS. Sotoodehnia N. Rea TD. Raghunathan TE. Jouven X. Lemaitre RN. Type 2 diabetes mellitus and the risk of sudden cardiac arrest in the community. Rev Endocr Metab Disord. 2010;11:53–59. doi: 10.1007/s11154-010-9133-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Slemmer JE. Shacka JJ. Sweeney MI. Weber JT. Antioxidants and free radical scavengers for the treatment of stroke, traumatic brain injury and aging. Curr Med Chem. 2008;15:404–414. doi: 10.2174/092986708783497337. [DOI] [PubMed] [Google Scholar]
- 257.Smeyne M. Boyd J. Raviie Shepherd K. Jiao Y. Pond BB. Hatler M. Wolf R. Henderson C. Smeyne RJ. GSTpi expression mediates dopaminergic neuron sensitivity in experimental parkinsonism. Proc Natl Acad Sci U S A. 2007;104:1977–1982. doi: 10.1073/pnas.0610978104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Smith SP. Shaw GS. A novel calcium-sensitive switch revealed by the structure of human S100B in the calcium-bound form. Structure. 1998;6:211–222. doi: 10.1016/s0969-2126(98)00022-7. [DOI] [PubMed] [Google Scholar]
- 259.Spalletta G. Bernardini S. Bellincampi L. Federici G. Trequattrini A. Ciappi F. Bria P. Caltagirone C. Bossu P. Glutathione S-transferase P1 and T1 gene polymorphisms predict longitudinal course and age at onset of Alzheimer disease. Am J Geriatr Psychiatry. 2007;15:879–887. doi: 10.1097/JGP.0b013e3180547076. [DOI] [PubMed] [Google Scholar]
- 260.Sparaco M. Gaeta LM. Santorelli FM. Passarelli C. Tozzi G. Bertini E. Simonati A. Scaravilli F. Taroni F. Duyckaerts C. Feleppa M. Piemonte F. Friedreich's ataxia: oxidative stress and cytoskeletal abnormalities. J Neurol Sci. 2009;287:111–118. doi: 10.1016/j.jns.2009.08.052. [DOI] [PubMed] [Google Scholar]
- 261.Spencer JP. Jenner A. Butler J. Aruoma OI. Dexter DT. Jenner P. Halliwell B. Evaluation of the pro-oxidant and antioxidant actions of L-DOPA and dopamine in vitro: implications for Parkinson's disease. Free Radic Res. 1996;24:95–105. doi: 10.3109/10715769609088005. [DOI] [PubMed] [Google Scholar]
- 262.Stark AA. Zeiger E. Pagano DA. Glutathione metabolism by gamma-glutamyltranspeptidase leads to lipid peroxidation: characterization of the system and relevance to hepatocarcinogenesis. Carcinogenesis. 1993;14:183–189. doi: 10.1093/carcin/14.2.183. [DOI] [PubMed] [Google Scholar]
- 263.Sullivan DM. Wehr NB. Fergusson MM. Levine RL. Finkel T. Identification of oxidant-sensitive proteins: TNF-alpha induces protein glutathiolation. Biochemistry. 2000;39:11121–11128. doi: 10.1021/bi0007674. [DOI] [PubMed] [Google Scholar]
- 264.Sumbayev VV. S-nitrosylation of thioredoxin mediates activation of apoptosis signal-regulating kinase 1. Arch Biochem Biophys. 2003;415:133–136. doi: 10.1016/s0003-9861(03)00199-1. [DOI] [PubMed] [Google Scholar]
- 265.Tagliabue M. Pinach S. Di Bisceglie C. Brocato L. Cassader M. Bertagna A. Manieri C. Pescarmona GP. Glutathione levels in patients with erectile dysfunction, with or without diabetes mellitus. Int J Androl. 2005;28:156–162. doi: 10.1111/j.1365-2605.2005.00528.x. [DOI] [PubMed] [Google Scholar]
- 266.Tao L. English AM. Protein S-glutathiolation triggered by decomposed S-nitrosoglutathione. Biochemistry. 2004;43:4028–4038. doi: 10.1021/bi035924o. [DOI] [PubMed] [Google Scholar]
- 267.Tavazzi B. Vagnozzi R. Signoretti S. Amorini AM. Belli A. Cimatti M. Delfini R. Di Pietro V. Finocchiaro A. Lazzarino G. Temporal window of metabolic brain vulnerability to concussions: oxidative and nitrosative stresses—part II. Neurosurgery. 2007;61:390–395. doi: 10.1227/01.neu.0000255525.34956.3f. ; discussion 395–396. [DOI] [PubMed] [Google Scholar]
- 268.Tew KD. Glutathione-associated enzymes in anticancer drug resistance. Cancer Res. 1994;54:4313–4320. [PubMed] [Google Scholar]
- 269.Thomas B. Parkinson's disease: from molecular pathways in disease to therapeutic approaches. Antioxid Redox Signal. 2009;11:2077–2082. doi: 10.1089/ars.2009.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Tian G. Xiang S. Noiva R. Lennarz WJ. Schindelin H. The crystal structure of yeast protein disulfide isomerase suggests cooperativity between its active sites. Cell. 2006;124:61–73. doi: 10.1016/j.cell.2005.10.044. [DOI] [PubMed] [Google Scholar]
- 271.Townsend DM. S-glutathionylation: indicator of cell stress and regulator of the unfolded protein response. Mol Interv. 2007;7:313–324. doi: 10.1124/mi.7.6.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Townsend DM. Findlay VJ. Fazilev F. Ogle M. Fraser J. Saavedra JE. Ji X. Keefer LK. Tew KD. A glutathione S-transferase pi-activated prodrug causes kinase activation concurrent with S-glutathionylation of proteins. Mol Pharmacol. 2006;69:501–508. doi: 10.1124/mol.105.018523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Townsend DM. Manevich Y. He L. Hutchens S. Pazoles CJ. Tew KD. Novel role for glutathione S-transferase pi. Regulator of protein S-glutathionylation following oxidative and nitrosative stress. J Biol Chem. 2009;284:436–445. doi: 10.1074/jbc.M805586200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Townsend DM. Manevich Y. He L. Xiong Y. Bowers RR., Jr. Hutchens S. Tew KD. Nitrosative stress-induced S-glutathionylation of protein disulfide isomerase leads to activation of the unfolded protein response. Cancer Res. 2009;69:7626–7634. doi: 10.1158/0008-5472.CAN-09-0493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Trujillo-Martin MM. Serrano-Aguilar P. Monton-Alvarez F. Carrillo-Fumero R. Effectiveness and safety of treatments for degenerative ataxias: a systematic review. Mov Disord. 2009;24:1111–1124. doi: 10.1002/mds.22564. [DOI] [PubMed] [Google Scholar]
- 276.Tsuchida S. Maki T. Sato K. Purification and characterization of glutathione transferases with an activity toward nitroglycerin from human aorta and heart. Multiplicity of the human class Mu forms. J Biol Chem. 1990;265:7150–7157. [PubMed] [Google Scholar]
- 277.Tsuru-Aoyagi K. Potts MB. Trivedi A. Pfankuch T. Raber J. Wendland M. Claus CP. Koh SE. Ferriero D. Noble-Haeusslein LJ. Glutathione peroxidase activity modulates recovery in the injured immature brain. Ann Neurol. 2009;65:540–549. doi: 10.1002/ana.21600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Tunez I. Drucker-Colin R. Jimena I. Medina FJ. Munoz Mdel C. Pena J. Montilla P. Transcranial magnetic stimulation attenuates cell loss and oxidative damage in the striatum induced in the 3-nitropropionic model of Huntington's disease. J Neurochem. 2006;97:619–630. doi: 10.1111/j.1471-4159.2006.03724.x. [DOI] [PubMed] [Google Scholar]
- 279.Tyagi SC. Reversible inhibition of neutrophil elastase by thiol-modified alpha-1 protease inhibitor. J Biol Chem. 1991;266:5279–5285. [PubMed] [Google Scholar]
- 280.Uehara T. Nakamura T. Yao D. Shi ZQ. Gu Z. Ma Y. Masliah E. Nomura Y. Lipton SA. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006;441:513–517. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- 281.van Pel M. van Os R. Velders GA. Hagoort H. Heegaard PM. Lindley IJ. Willemze R. Fibbe WE. Serpina1 is a potent inhibitor of IL-8-induced hematopoietic stem cell mobilization. Proc Natl Acad Sci U S A. 2006;103:1469–1474. doi: 10.1073/pnas.0510192103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Vassar R. Kovacs DM. Yan R. Wong PC. The beta-secretase enzyme BACE in health and Alzheimer's disease: regulation, cell biology, function, and therapeutic potential. J Neurosci. 2009;29:12787–12794. doi: 10.1523/JNEUROSCI.3657-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Velu CS. Niture SK. Doneanu CE. Pattabiraman N. Srivenugopal KS. Human p53 is inhibited by glutathionylation of cysteines present in the proximal DNA-binding domain during oxidative stress. Biochemistry. 2007;46:7765–7780. doi: 10.1021/bi700425y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Vilar R. Coelho H. Rodrigues E. Gama MJ. Rivera I. Taioli E. Lechner MC. Association of A313 G polymorphism (GSTP1*B) in the glutathione-S-transferase P1 gene with sporadic Parkinson's disease. Eur J Neurol. 2007;14:156–161. doi: 10.1111/j.1468-1331.2006.01590.x. [DOI] [PubMed] [Google Scholar]
- 285.Viswanath V. Wu Y. Boonplueang R. Chen S. Stevenson FF. Yantiri F. Yang L. Beal MF. Andersen JK. Caspase-9 activation results in downstream caspase-8 activation and bid cleavage in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson's disease. J Neurosci. 2001;21:9519–9528. doi: 10.1523/JNEUROSCI.21-24-09519.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Vousden KH. Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–283. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- 287.Wade LA. Brady HM. Cysteine and cystine transport at the blood-brain barrier. J Neurochem. 1981;37:730–734. doi: 10.1111/j.1471-4159.1982.tb12548.x. [DOI] [PubMed] [Google Scholar]
- 288.Wang H. Lim PJ. Karbowski M. Monteiro MJ. Effects of overexpression of huntingtin proteins on mitochondrial integrity. Hum Mol Genet. 2009;18:737–752. doi: 10.1093/hmg/ddn404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Wang J. Boja ES. Tan W. Tekle E. Fales HM. English S. Mieyal JJ. Chock PB. Reversible glutathionylation regulates actin polymerization in A431 cells. J Biol Chem. 2001;276:47763–47766. doi: 10.1074/jbc.C100415200. [DOI] [PubMed] [Google Scholar]
- 290.Wang J. Green PS. Simpkins JW. Estradiol protects against ATP depletion, mitochondrial membrane potential decline and the generation of reactive oxygen species induced by 3-nitroproprionic acid in SK-N-SH human neuroblastoma cells. J Neurochem. 2001;77:804–811. doi: 10.1046/j.1471-4159.2001.00271.x. [DOI] [PubMed] [Google Scholar]
- 291.Wang J. Tekle E. Oubrahim H. Mieyal JJ. Stadtman ER. Chock PB. Stable and controllable RNA interference: investigating the physiological function of glutathionylated actin. Proc Natl Acad Sci U S A. 2003;100:5103–5106. doi: 10.1073/pnas.0931345100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Wang T. Arifoglu P. Ronai Z. Tew KD. Glutathione S-transferase P1–1 (GSTP1–1) inhibits c-Jun N-terminal kinase (JNK1) signaling through interaction with the C terminus. J Biol Chem. 2001;276:20999–21003. doi: 10.1074/jbc.M101355200. [DOI] [PubMed] [Google Scholar]
- 293.Wang WX. Rajeev BW. Stromberg AJ. Ren N. Tang G. Huang Q. Rigoutsos I. Nelson PT. The expression of microRNA miR-107 decreases early in Alzheimer's disease and may accelerate disease progression through regulation of beta-site amyloid precursor protein-cleaving enzyme 1. J Neurosci. 2008;28:1213–1223. doi: 10.1523/JNEUROSCI.5065-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Wang XZ. Ron D. Stress-induced phosphorylation and activation of the transcription factor CHOP (GADD153) by p38 MAP Kinase. Science. 1996;272:1347–1349. doi: 10.1126/science.272.5266.1347. [DOI] [PubMed] [Google Scholar]
- 295.Ward NE. Stewart JR. Ioannides CG. O'Brian CA. Oxidant-induced S-glutathiolation inactivates protein kinase C-alpha (PKC-alpha): a potential mechanism of PKC isozyme regulation. Biochemistry. 2000;39:10319–10329. doi: 10.1021/bi000781g. [DOI] [PubMed] [Google Scholar]
- 296.Whitfield JB. Gamma glutamyl transferase. Crit Rev Clin Lab Sci. 2001;38:263–355. doi: 10.1080/20014091084227. [DOI] [PubMed] [Google Scholar]
- 297.Whiting PH. Kalansooriya A. Holbrook I. Haddad F. Jennings PE. The relationship between chronic glycaemic control and oxidative stress in type 2 diabetes mellitus. Br J Biomed Sci. 2008;65:71–74. doi: 10.1080/09674845.2008.11732800. [DOI] [PubMed] [Google Scholar]
- 298.Wilcox KC. Zhou L. Jordon JK. Huang Y. Yu Y. Redler RL. Chen X. Caplow M. Dokholyan NV. Modifications of superoxide dismutase (SOD1) in human erythrocytes: a possible role in amyotrophic lateral sclerosis. J Biol Chem. 2009;284:13940–13947. doi: 10.1074/jbc.M809687200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Winkler IG. Hendy J. Coughlin P. Horvath A. Levesque JP. Serine protease inhibitors serpina1 and serpina3 are down-regulated in bone marrow during hematopoietic progenitor mobilization. J Exp Med. 2005;201:1077–1088. doi: 10.1084/jem.20042299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Wouters MA. Fan SW. Haworth NL. Disulfides as redox switches: from molecular mechanisms to functional significance. Antioxid Redox Signal. 2010;12:53–91. doi: 10.1089/ars.2009.2510. [DOI] [PubMed] [Google Scholar]
- 301.Wu Y. Fan Y. Xue B. Luo L. Shen J. Zhang S. Jiang Y. Yin Z. Human glutathione S-transferase P1–1 interacts with TRAF2 and regulates TRAF2-ASK1 signals. Oncogene. 2006;25:5787–5800. doi: 10.1038/sj.onc.1209576. [DOI] [PubMed] [Google Scholar]
- 302.Wyttenbach A. Sauvageot O. Carmichael J. Diaz-Latoud C. Arrigo AP. Rubinsztein DC. Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin. Hum Mol Genet. 2002;11:1137–1151. doi: 10.1093/hmg/11.9.1137. [DOI] [PubMed] [Google Scholar]
- 303.Xiong Y. Peterson PL. Lee CP. Effect of N-acetylcysteine on mitochondrial function following traumatic brain injury in rats. J Neurotrauma. 1999;16:1067–1082. doi: 10.1089/neu.1999.16.1067. [DOI] [PubMed] [Google Scholar]
- 304.Xiong Y. Shie FS. Zhang J. Lee CP. Ho YS. The protective role of cellular glutathione peroxidase against trauma-induced mitochondrial dysfunction in the mouse brain. J Stroke Cerebrovasc Dis. 2004;13:129–137. doi: 10.1016/j.jstrokecerebrovasdis.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 305.Xu X. Stambrook PJ. Two murine GSTpi genes are arranged in tandem and are differentially expressed. J Biol Chem. 1994;269:30268–30273. [PubMed] [Google Scholar]
- 306.Yang TT. Hsu CT. Kuo YM. Cell-derived soluble oligomers of human amyloid-beta peptides disturb cellular homeostasis and induce apoptosis in primary hippocampal neurons. J Neural Transm. 2009;116:1561–1569. doi: 10.1007/s00702-009-0311-0. [DOI] [PubMed] [Google Scholar]
- 307.Yao D. Brownlee M. Hyperglycemia-induced reactive oxygen species increase expression of the receptor for advanced glycation end products (RAGE) and RAGE ligands. Diabetes. 2010;59:249–255. doi: 10.2337/db09-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Yap LP. Garcia JV. Han DS. Cadenas E. Role of nitric oxide-mediated glutathionylation in neuronal function. Potential regulation of energy utilization. Biochem J. 2010;428:85–93. doi: 10.1042/BJ20100164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Yavuz BB. Yavuz B. Halil M. Cankurtaran M. Ulger Z. Cankurtaran ES. Aytemir K. Ariogul S. Serum elevated gamma glutamyltransferase levels may be a marker for oxidative stress in Alzheimer's disease. Int Psychogeriatr. 2008;20:815–823. doi: 10.1017/S1041610208006790. [DOI] [PubMed] [Google Scholar]
- 310.Yin Z. Ivanov VN. Habelhah H. Tew K. Ronai Z. Glutathione S-transferase p elicits protection against H2O2-induced cell death via coordinated regulation of stress kinases. Cancer Res. 2000;60:4053–4057. [PubMed] [Google Scholar]
- 311.Yu CX. Li S. Whorton AR. Redox regulation of PTEN by S-nitrosothiols. Mol Pharmacol. 2005;68:847–854. doi: 10.1124/mol.104.010504. [DOI] [PubMed] [Google Scholar]
- 312.Zeevalk GD. Manzino L. Sonsalla PK. Bernard LP. Characterization of intracellular elevation of glutathione (GSH) with glutathione monoethyl ester and GSH in brain and neuronal cultures: relevance to Parkinson's disease. Exp Neurol. 2007;203:512–520. doi: 10.1016/j.expneurol.2006.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Zhou SG. Wang P. Pi RB. Gao J. Fu JJ. Fang J. Qin J. Zhang HJ. Li RF. Chen SR. Tang FT. Liu PQ. Reduced expression of GSTM2 and increased oxidative stress in spontaneously hypertensive rat. Mol Cell Biochem. 2008;309:99–107. doi: 10.1007/s11010-007-9647-7. [DOI] [PubMed] [Google Scholar]
- 314.Zhu Y. Carvey PM. Ling Z. Age-related changes in glutathione and glutathione-related enzymes in rat brain. Brain Res. 2006;1090:35–44. doi: 10.1016/j.brainres.2006.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Zhukova L. Zhukov I. Bal W. Wyslouch-Cieszynska A. Redox modifications of the C-terminal cysteine residue cause structural changes in S100A1 and S100B proteins. Biochim Biophys Acta. 2004;1742:191–201. doi: 10.1016/j.bbamcr.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 316.Zou Z. Anisowicz A. Hendrix MJ. Thor A. Neveu M. Sheng S. Rafidi K. Seftor E. Sager R. Maspin, a serpin with tumor-suppressing activity in human mammary epithelial cells. Science. 1994;263:526–529. doi: 10.1126/science.8290962. [DOI] [PubMed] [Google Scholar]