

Abstract
Background
Several platforms for the analysis of genome-wide association data are available. However, these platforms focus on the evaluation of the genotype inherited by affected (i.e. case) individuals, whereas for some conditions (e.g. birth defects) the genotype of the mothers of affected individuals may also contribute to risk. For such conditions, it is critical to evaluate associations with both the maternal and the inherited (i.e. case) genotype. When genotype data are available for case-parent triads, a likelihood-based approach using log-linear modeling can be used to assess both the maternal and inherited genotypes. However, available software packages for log-linear analyses are not well suited to the analysis of typical genome-wide association data (e.g. including missing data).
Results
An integrated platform, Maternal and Inherited Analyses for Genome-wide Association Studies (MI-GWAS) for log-linear analyses of maternal and inherited genetic effects in large, genome-wide datasets, is described. MI-GWAS uses SAS and LEM software in combination to appropriately format data, perform the log-linear analyses and summarize the results. This platform was evaluated using existing genome-wide data and was shown to perform accurately and relatively efficiently.
Conclusions
The MI-GWAS platform provides a valuable tool for the analysis of association of a phenotype or condition with maternal and inherited genotypes using genome-wide data from case-parent triads. The source code for this platform is freely available at http://www.sph.uth.tmc.edu/sbrr/mi-gwas.htm.
Background
Candidate gene, and more recently, genome-wide association (GWA) studies have been used to identify associations between several complex diseases and the genotype of affected individuals (i.e. cases) [1-6]. However, for some phenotypes (e.g. birth defects, perinatal outcomes, pediatric cancers), the maternal genotype may also directly contribute to risk, via an effect on the in utero environment [7]. However, despite increasing recognition of the importance of maternal genetic effects in genetic epidemiology studies [8-17], nearly all GWA studies to date have ignored maternal genetic effects [18], which could partially explain why GWA studies have not identified the majority of the genetic contribution to common diseases. Failure to account for maternal genetic effects in previous studies may be due both to lack of appropriate data (i.e. maternal DNA is not collected in traditional case-control studies) and lack of a readily available platform for performing analyses that account for maternal genetic effects using typical GWA data.
The most common study designs used in GWA studies include the case-control and case-parent triad designs. Though the case-control approach has been used more frequently, distinguishing between maternal and inherited genetic effects using this study design requires the addition of samples from mothers of both cases and controls, resulting in increased genotyping costs [7]. Moreover, maternal DNA has not been collected for most existing case-control studies. By contrast, maternal genetic effects can be directly assessed in existing data from case-parent triad GWA studies. However, in the majority of these studies, data have been analyzed using the transmission disequilibrium test (TDT) [19,20], which does not assess maternal genetic effects.
The most commonly used method for assessing maternal and inherited genetic effects using case-parent triad data is a log-linear, likelihood-based approach [21,22]. In addition to evaluating maternal genetic effects, this approach has the advantage (over the TDT) of providing estimates of effect size and, similar to the TDT, does not require the assumption of Hardy-Weinberg equilibrium [21-24]. Further, using this approach, data from incomplete triads can be included in the analysis by use of the expectation-maximization (EM) algorithm [23]. These log-linear analyses can be conducted using standard software (e.g. SAS®, SAS Institute Inc.), but, when data from incomplete triads are included, require programming of the EM, which is cumbersome. The specialized program, LEM [25,26], can also be used to conduct these analyses, and has the advantage over other programs (e.g. SAS) in that it does not require programming of the EM and can be easily programmed to explore a variety of additional types of effects (e.g., gene-gene interaction, gene-environment interaction). However, because LEM requires individual data and program files for each SNP, it is not feasible to conduct a GWA study analysis using LEM as a stand-alone program.
To address the need for an efficient approach for analyzing maternal and inherited genetic effects using GWA data from case-parent triad studies, a novel computational platform was created. This platform uses SAS to prepare the data in an LEM-compatible format, calls LEM to evaluate each SNP, and extracts and summarizes relevant data from the LEM output files. The performance of this platform was evaluated using existing, case-parent triad GWA data.
Implementation
Log-linear Modeling
The theoretical distribution (assuming Mendelian inheritance and mating symmetry) of case-parent triad genotype data (defined by the combination of father, mother, and child genotypes) can be fitted to the observed triad counts by maximum likelihood using the following log-linear model:
The values of E(nF,M,C) correspond to the expected count of each genotype combination (i.e., father, mother, and child genotypes). The γj term stratifies on parental mating type (i.e., each combination of possible parental genotypes), and I is an indicator variable that equals 0, 1, or 2, corresponding to the number of high-risk alleles present in the child's or mother's genotype (C and M respectively). The offset, wF,M,C, accounts for the heterozygous genotype being twice as likely as either homozygous genotype in offspring of double heterozygous matings (assuming Mendelian transmission). Under this model, the risk corresponding to a genotype with one copy of the "high-risk" allele compared to no copies can be estimated by exp(β1) for the inherited genotype and exp(α1) for the maternal genotype, and the risk corresponding to a genotype with two copies compared to no copies can be estimated by exp(β2) for the inherited genotype and exp(α2) for the maternal genotype. The significance of the inherited genetic effect can be evaluated using a two degree of freedom likelihood ratio test (LRT) to compare the log-likelihood of the full model (i.e. Model 1) to that of a reduced model in which β1 = β2 = 0. The null hypothesis for this test is that conditional on parental mating type, the distribution of affected offspring agrees with Mendelian expectation. Similar to the TDT, this LRT provides a test of linkage in the presence of linkage disequilibrium that is not vulnerable to confounding due to population stratification [21,27]. Likewise, the significance of the maternal genetic effect can be evaluated by using the LRT to compare the log-likelihood of the full model to that of a reduced model in which α1 = α2 = 0. The null hypothesis for this test is that reciprocal parental mating types (e.g. Aa × AA and AA × Aa) occur at the same frequency among parents of affected individuals [21-23]. This LRT is vulnerable to a specific form of population stratification that violates the underlying assumption of mating symmetry. Mating asymmetry occurs when reciprocal mating types for a given allele do not occur with equal frequency in the population (e.g. Aa × AA > AA × Aa). The potential for this type of bias can be limited by restricting analyses to matings from the same race and ethnicity [7]. Data from incomplete triads can also be included in these analyses by use of the EM algorithm.
Model 1 is a general (i.e. unrestricted) model, in which no constraints are placed on the relationships between alleles (i.e. no constraints on β1 and β2 or α1 and α2). However, in some circumstances power can be increased by imposing constraints on the general model. For example, a linear constraint can be imposed upon the full model for the genetic effect being tested, leaving the terms for the other genetic effect unconstrained. This allows for a one degree of freedom LRT. The use of a linear constraint has been shown to perform well under a variety of circumstances [7,21-23,28].
LEM Software
The log-linear modeling approach with EM can be implemented using the LEM program, which can be downloaded at: http://spitswww.uvt.nl/~vermunt/#Software. LEM requires individual data and program files for each association test. Thus, for each SNP, it requires one data file and four program files: one for the full model with a linear constraint imposed for inherited effects, one for the full model with a linear constraint imposed for maternal effects, one for the reduced model to test for inherited effects, and one for the reduced model to test for maternal effects. Further, LEM data files must be formatted so that each row contains genotypes for the three members of the triad, whereas it is common for genome-wide genotype data to be outputted as a single file with one individual per row and one allele per column (e.g. PLINK [29] format) (Table 1).
Table 1.
Comparison of PLINK (A) and LEM (B) data format and example data for three hypothetical case-parent triads
| (A) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PLINK Genotype Data for 5 SNPs | |||||||||||||||
| Family Number | Individual ID | Paternal ID | Maternal ID | Sex a | Phenotype b | SNP 1 c | SNP 2 c | SNP 3 c | SNP 4 c | SNP 5 c | |||||
| 1001 | 1001-A | 1001-C | 1001-B | 1 | 1 | 1 | 1 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 2 |
| 1001 | 1001-B | 0 | 0 | 2 | 0 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 1 |
| 1001 | 1001-C | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 0 | 0 |
| 1002 | 1002-A | 1002-C | 1002-B | 2 | 1 | 0 | 0 | 2 | 2 | 1 | 2 | 1 | 1 | 1 | 2 |
| 1002 | 1002-B | 0 | 0 | 2 | 0 | 2 | 2 | 1 | 2 | 0 | 0 | 1 | 1 | 1 | 1 |
| 1002 | 1002-C | 0 | 0 | 1 | 0 | 1 | 1 | 2 | 2 | 1 | 1 | 1 | 2 | 2 | 2 |
| 1003 | 1003-A | 0 | 1003-B | 2 | 1 | 1 | 2 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 1 |
| 1003 | 1003-B | 0 | 0 | 2 | 0 | 1 | 1 | 1 | 2 | 1 | 1 | 0 | 0 | 1 | 1 |
| (B) | |||||||||||||||
| LEM Data File for SNP 1 | |||||||||||||||
| Mother's Genotype d | Father's Genotype d | Case's Genotype d | |||||||||||||
| 1 | 1 | 1 | |||||||||||||
| 3 | 1 | 0 | |||||||||||||
| 1 | 0 | 2 | |||||||||||||
a 1 = male, 2 = female
b 1 = unaffected, 2 = affected
c 0 = allele missing, 1 = allele 1, 2 = allele 2
d 0 = genotype missing, 1 = no high-risk alleles present, 2 = one high-risk alleles present, 3 = two high-risk alleles present
Maternal and Inherited Analyses for Genome-wide Association Studies (MI-GWAS)
SAS version 9.2 was used to develop a platform for the efficient analysis of maternal and inherited genetic effects in GWA data, using LEM to apply the log-linear modeling likelihood-based approach. This platform is called Maternal and Inherited Analyses for Genome-wide Association Studies (MI-GWAS) and is freely available at http://www.sph.uth.tmc.edu/sbrr/mi-gwas.htm.
Briefly, MI-GWAS:
1. Reads data from a single PLINK ped file that contains a separate row of data for each triad member. Each row contains all genotype data for a single individual, and each genotype is coded as two alleles (i.e. two columns of data per genotype). By default, MI-GWAS treats the allele coded as "2" under the Illumina 1/2 allele coding system as the high-risk allele.
2. Converts the PLINK data into one LEM data file per SNP, with a single row of genotype data for each triad, with genotypes in the order of mother, father, and child. These genotypes are coded as 0, 1, or 2, for number of high-risk alleles present (i.e. one column of data per genotype).
3. Creates LEM program files for each SNP - one program file per model.
4. Calls LEM to run the relevant analyses (i.e. full and reduced models) for each SNP.
5. Extracts relevant output (i.e. log-likelihood values, relative risks and corresponding standard errors) from LEM.
6. Calculates relevant LRTs and associated p-values.
7. Computes 95% confidence intervals (CIs) for all relative risks.
8. Merges results with the PLINK marker map file, which includes chromosome number and rs number for each SNP.
9. Sorts results by the LRT p-value, separately for the inherited and maternal genotype, and outputs two corresponding data files.
10. Sorts results by relative risk estimates, separately for the inherited and maternal genotype, and outputs two corresponding data files.
To increase computation efficiency, repeating sections of code (i.e. macros) are used to split these steps into consecutive blocks that process subsets of SNPs (by default 1,000). Each consecutive subset of SNPs is processed separately, and after processing of the last subset is complete, all results are merged. A visual overview of the structure of these steps of the platform is provided in Figure 1.
Figure 1.
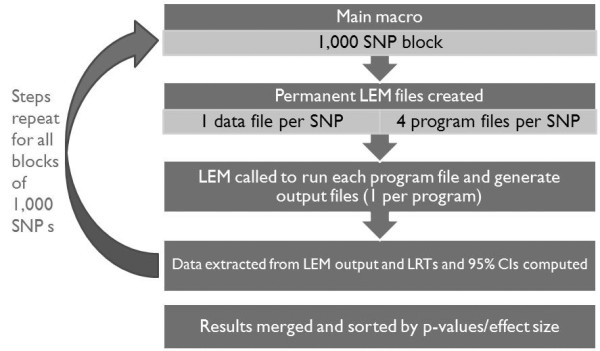
Summary of MI-GWAS platform structure, displaying steps preformed on subsets of 1,000 SNPs at a time.
The use of SAS, a popular statistical software package, allows for flexibility in the MI-GWAS platform with simple programming changes. For example, the user can specify a range of successive SNPs to analyze rather than running all SNPs at once. This allows the user to split the analysis between multiple runs or between multiple computers. The program also includes code that can be modified to allow the user to specify specific individuals, triads, or SNPs to be excluded from the analysis. Further, the log-linear models can be modified to accommodate other family-based association designs (e.g. study designs that incorporate grandparents' genotypes [7]), other relationships between alleles (e.g. dominant inheritance), and to include other effects such as gene-gene and gene-environment interactions and parent of origin effects. By default, a linear constraint is imposed for the effect that is being tested (e.g. inherited genotype) and no constraint is imposed for the other effect (e.g. maternal genotype). The platform can also process imputed data that has been converted to PLINK format. Though by default MI-GWAS processes PLINK-formatted genotype data with Illumina 1/2-based allele coding, the SAS code can be modified to process genotype data that are in other formats.
Evaluation
To validate the MI-GWAS platform and evaluate its performance, it was used to analyze a large, unpublished GWA study dataset derived from case-parent triads ascertained through cases with a conotruncal heart defect. The subject recruitment methods for this study have been previously described [17]. Briefly, case-parent triads were recruited through the Cardiac Center at the Children's Hospital of Philadelphia (CHOP) from 1997-2007 and all participants provided informed consent under a protocol approved by the Institutional Review Boards for the Protection of Human Subjects. Cases had a nonsyndromic, classic conotruncal defect (i.e. tetalogy of Fallot, D-transposition of the great arteries, double outlet right ventricle, truncus arteriosus or interrupted aortic arch) or a related malformation (i.e. perimembranous or posterior malalignment type ventricular septal defect or an isolated aortic arch anomaly). The genotype data were generated from blood or saliva samples using the Illumina HumanHap550 or Human610-Quad BeadChip Platforms at the Center for Applied Genomics at CHOP. Only SNPs present on both platforms were analyzed.
To verify MI-GWAS results for inherited genetic effects using the CHOP dataset, selected SNPs were evaluated for complete triads using both PLINK and MI-GWAS and the chi-square values for the TDT and LRT, respectively, were compared. (The TDT approach under PLINK cannot assess maternal genetic effects and does not incorporate data from incomplete triads into evaluations of inherited genetic effects.) To verify MI-GWAS results for maternal genetic effects using this dataset, selected SNPs were evaluated using both MI-GWAS and LEM run outside of MI-GWAS, and the resulting LRT p-values were compared. In addition, MI-GWAS was used to replicate the findings of a candidate gene study conducted in this study population, using LEM run outside of MI-GWAS [17]. To be consistent with the previous analyses, MI-GWAS was modified to use an unrestricted model of inheritance for both genotypes (i.e. a two degree of freedom LRT), for the latter analysis.
Results
The evaluation dataset included data on 530,350 SNPs from 837 case-parent triads (497 complete triads and 340 triads with one or two members missing). To confirm MI-GWAS results for the test of inherited genetic effect, MI-GWAS was used to analyze all SNPs in the subset of complete triads, and chi-square values for the LRT were compared to TDT chi-square values obtained using PLINK to analyze the same data. The chi-square values for the LRTs of inherited genetic effects generated by MI-GWAS were quite similar to the TDT chi-square values generated using PLINK (Table 2). There were subtle differences in the chi-square values for some of the most significant SNPs, likely due to differences between the two platforms in rounding and/or how numbers are stored (e.g., floating point representation). In addition, identical results were obtained when program and data files generated by MI-GWAS were analyzed using the MI-GWAS platform and LEM run outside of MI-GWAS (data not shown). Finally, the results for maternal and inherited genetic effects from a previously published candidate gene study from the same study population [17] were replicated using MI-GWAS (data not shown).
Table 2.
Comparison of chi-square values from the PLINK TDT and MI-GWAS LRT for inherited genetic effects for a randomly selected set of SNPs on chromosome one and most significant autosomal SNPs.
| SNP | PLINK TDT chi-square value | MI-GWAS log-linear modeling chi-square value | Percent Difference a |
|---|---|---|---|
| Chromosome 1 SNPs | |||
| SNP 1 | 0.02 | 0.02 | 0.00 |
| SNP 2 | 2.22 | 2.22 | 0.00 |
| SNP 3 | 0.91 | 0.91 | 0.00 |
| SNP 4 | 0.09 | 0.09 | 0.00 |
| SNP 5 | 0.24 | 0.24 | 0.00 |
| SNP 6 | 1.58 | 1.58 | 0.00 |
| SNP 7 | 2.97 | 2.98 | 0.00 |
| SNP 8 | 1.22 | 1.22 | 0.00 |
| Most significant autosomal SNPs | |||
| SNP 9 | 22.34 | 24.06 | 0.08 |
| SNP 10 | 22.29 | 22.57 | 0.01 |
| SNP 11 | 21.48 | 21.64 | 0.01 |
| SNP 12 | 20.78 | 21.02 | 0.01 |
| SNP 13 | 19.56 | 19.77 | 0.01 |
a Absolute difference between PLINK chi-square value and MI-GWAS chi-square value divided by PLINK chi-square value
As expected, MI-GWAS running times were somewhat faster on computers with better specifications. For the same 60,000 SNPs, running times ranged from 11 hours 35 minutes to 22 hours 48 minutes for four computers with differing specifications (Table 3).
Table 3.
Running times for the analysis of the same 60,000 SNPs using MI-GWAS on four computers with differing specifications
| Machine specifications | Running Time |
|---|---|
| Intel Core 2 Quad CPU Q9550, 2.83 GHz, 3.21 Gb of RAM | 11 hours 35 minutes |
| Pentium 4 CPU, 3.00 GHz, 2.00 Gb of RAM | 20 hours 52 minutes |
| Pentium D CPU, 3.20 GHz, 1.99 Gb of RAM | 21 hours 24 minutes |
| Intel Xeon CPU E5540, 2.53 GHz, 6.00 Gb of RAM | 22 hours 48 minutes |
Discussion
Computational platforms that can evaluate maternal as well as inherited genetic effects in genome-wide data from case-parent triads have not been previously described. By automating the implementation of such analyses, MI-GWAS provides such a platform. Comparison of results from MI-GWAS and LEM run outside of MI-GWAS, as well as results from MI-GWAS and PLINK, indicate that this platform performs as intended. Further, MI-GWAS performs relatively efficiently. From the observed running times (Table 3), it can be inferred that on a single average modern consumer computer, it may take approximately one week to run a GWA study analysis of ~500,000 SNPs using MI-GWAS. Such an analysis may run overnight if split between around eight average consumer computers.
The MI-GWAS platform has the advantage of using readily available software (i.e. SAS and LEM) and reading a common GWA data input format (i.e. PLINK format). Further, unlike the TDT approach under PLINK, analysis under MI-GWAS uses a log-linear approach that provides estimates of effect size, allows use of data from incomplete triads [21-23], and, most importantly, allows estimation of the significance of maternal effects in addition to inherited effects.
Conclusions
For some conditions, maternal genetic effects may influence the risk of disease in offspring via an effect on the in utero environment. However, maternal genetic effects have not been widely evaluated in GWA studies, at least partially due to lack of a platform designed to analyze maternal genetic effects using GWA data from case-parent triads. The application of the MI-GWAS platform to GWA analyses expands the types of genetic effects that can be evaluated with triad GWA data, which may lead to new insights regarding the etiology of common diseases. Future developments of the MI-GWAS platform will involve improving the efficiency of the platform, and incorporating analyses of additional types of effects (e.g. parental imprinting, interactions).
Availability and Requirements
Project name: MI-GWAS (Maternal and Inherited Analyses for Genome-wide Association Studies)
Project home page: http://www.sph.uth.tmc.edu/sbrr/mi-gwas.htm
Operating system(s): Platform independent
Programming language: SAS®
Other requirements: SAS® release 9.2, LEM release 1.0 (freely available at http://spitswww.uvt.nl/~vermunt/#Software)
Licence: None for MI-GWAS
Any restrictions to use by non-academics: None for MI-GWAS
Authors' contributions
AJA developed and tested the MI-GWAS platform, and wrote the manuscript. LEM conceived the study, provided input on the design and testing of the MI-GWAS platform and helped to draft and edit the manuscript. Both authors read and approved the final manuscript.
Contributor Information
AJ Agopian, Email: aagopian1@yahoo.com.
Laura E Mitchell, Email: laura.e.mitchell@uth.tmc.edu.
Acknowledgements
The authors would like to thank Drs. Elizabeth Goldmuntz and Hakon Hakonarson, as well as their research teams, for providing the evaluation genotype dataset. In addition, they would like to thank Dr. J.C. Huber for helpful comments. This research was supported by grants from the NIH/NHLBI (R01 HL076773 and R21 HL098844).
References
- Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, Green T, Kuballa P, Barmada MM, Datta LW. et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudmundsson J, Sulem P, Manolescu A, Amundadottir LT, Gudbjartsson D, Helgason A, Rafnar T, Bergthorsson JT, Agnarsson BA, Baker A. et al. Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat Genet. 2007;39:631–637. doi: 10.1038/ng1999. [DOI] [PubMed] [Google Scholar]
- Grant SF, Qu HQ, Bradfield JP, Marchand L, Kim CE, Glessner JT, Grabs R, Taback SP, Frackelton EC, Eckert AW. et al. Follow-up analysis of genome-wide association data identifies novel loci for type 1 diabetes. Diabetes. 2009;58:290–295. doi: 10.2337/db08-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakonarson H, Qu HQ, Bradfield JP, Marchand L, Kim CE, Glessner JT, Grabs R, Casalunovo T, Taback SP, Frackelton EC. et al. A novel susceptibility locus for type 1 diabetes on Chr12q13 identified by a genome-wide association study. Diabetes. 2008;57:1143–1146. doi: 10.2337/db07-1305. [DOI] [PubMed] [Google Scholar]
- Easton DF, Pooley KA, Dunning AM, Pharoah PD, Thompson D, Ballinger DG, Struewing JP, Morrison J, Field H, Luben R. et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447:1087–1093. doi: 10.1038/nature05887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell LE, Weinberg CR. Evaluation of offspring and maternal genetic effects on disease risk using a family-based approach: the "pent" design. Am J Epidemiol. 2005;162:676–685. doi: 10.1093/aje/kwi249. [DOI] [PubMed] [Google Scholar]
- Boyles AL, Wilcox AJ, Taylor JA, Shi M, Weinberg CR, Meyer K, Fredriksen A, Ueland PM, Johansen AM, Drevon CA. et al. Oral facial clefts and gene polymorphisms in metabolism of folate/one-carbon and vitamin A: a pathway-wide association study. Genet Epidemiol. 2009;33:247–255. doi: 10.1002/gepi.20376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Put NM, Steegers-Theunissen RP, Frosst P, Trijbels FJ, Eskes TK, van den Heuvel LP, Mariman EC, den Heyer M, Rozen R, Blom HJ. Mutated methylenetetrahydrofolate reductase as a risk factor for spina bifida. Lancet. 1995;346:1070–1071. doi: 10.1016/S0140-6736(95)91743-8. [DOI] [PubMed] [Google Scholar]
- Mitchell LE, Long J, Garbarini J, Paluru P, Goldmuntz E. Variants of folate metabolism genes and risk of left-sided cardiac defects. Birth Defects Res A Clin Mol Teratol. pp. 48–53. [DOI] [PMC free article] [PubMed]
- Reutter H, Birnbaum S, Mende M, Almeida de Assis N, Hoffmann P, Lacava AD, Herms S, Braumann B, Scheer M, Lauster C. et al. Transforming growth factor-beta receptor type 1 (TGFBR1) is not associated with non-syndromic cleft lip with or without cleft palate in patients of Central European descent. Int J Pediatr Otorhinolaryngol. 2009;73(10):1334–8. doi: 10.1016/j.ijporl.2009.06.004. [DOI] [PubMed] [Google Scholar]
- Mills JL, Molloy AM, Parle-McDermott A, Troendle JF, Brody LC, Conley MR, Cox C, Pangilinan F, Orr DJ, Earley M. et al. Folate-related gene polymorphisms as risk factors for cleft lip and cleft palate. Birth Defects Res A Clin Mol Teratol. 2008;82:636–643. doi: 10.1002/bdra.20491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little J, Gilmour M, Mossey PA, Fitzpatrick D, Cardy A, Clayton-Smith J, Hill A, Duthie SJ, Fryer AE, Molloy AM, Scott JM. Folate and clefts of the lip and palate--a U.K.-based case-control study: Part II: Biochemical and genetic analysis. Cleft Palate Craniofac J. 2008;45:428–438. doi: 10.1597/06-151.1. [DOI] [PubMed] [Google Scholar]
- Ehn NL, Cooper ME, Orr K, Shi M, Johnson MK, Caprau D, Dagle J, Steffen K, Johnson K, Marazita ML. et al. Evaluation of fetal and maternal genetic variation in the progesterone receptor gene for contributions to preterm birth. Pediatr Res. 2007;62:630–635. doi: 10.1203/PDR.0b013e3181567bfc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen LE, Hoess K, Mitchell LE, Whitehead AS. Loss of function polymorphisms in NAT1 protect against spina bifida. Hum Genet. 2006;120:52–57. doi: 10.1007/s00439-006-0181-6. [DOI] [PubMed] [Google Scholar]
- Infante-Rivard C, Vermunt JK, Weinberg CR. Excess transmission of the NAD(P)H:quinone oxidoreductase 1 (NQO1) C609T polymorphism in families of children with acute lymphoblastic leukemia. Am J Epidemiol. 2007;165:1248–1254. doi: 10.1093/aje/kwm022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmuntz E, Woyciechowski S, Renstrom D, Lupo PJ, Mitchell LE. Variants of folate metabolism genes and the risk of conotruncal cardiac defects. Circ Cardiovasc Genet. 2008;1:126–132. doi: 10.1161/CIRCGENETICS.108.796342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindorff LA, Junkins HA, Mehta JP, Manolio TA. A Catalog of Published Genome-Wide Association Studies. 2010.
- Spielman RS, Ewens WJ. The TDT and other family-based tests for linkage disequilibrium and association. American Journal of Human Genetics. 1996;59:983–989. [PMC free article] [PubMed] [Google Scholar]
- Benyamin B, Visscher PM, McRae AF. Family-based genome-wide association studies. Pharmacogenomics. 2009;10:181–190. doi: 10.2217/14622416.10.2.181. [DOI] [PubMed] [Google Scholar]
- Weinberg CR, Wilcox AJ, Lie RT. A log-linear approach to case-parent-triad data: assessing effects of disease genes that act either directly or through maternal effects and that may be subject to parental imprinting. Am J Hum Genet. 1998;62:969–978. doi: 10.1086/301802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox AJ, Weinberg CR, Lie RT. Distinguishing the effects of maternal and offspring genes through studies of "case-parent triads". Am J Epidemiol. 1998;148:893–901. doi: 10.1093/oxfordjournals.aje.a009715. [DOI] [PubMed] [Google Scholar]
- Weinberg CR. Allowing for missing parents in genetic studies of case-parent triads. Am J Hum Genet. 1999;64:1186–1193. doi: 10.1086/302337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr JR, Hsu L, Schwartz SM. Assessing maternal genetic associations: a comparison of the log-linear approach to case-parent triad data and a case-control approach. Epidemiology. 2005;16:294–303. doi: 10.1097/01.ede.0000158223.98649.eb. [DOI] [PubMed] [Google Scholar]
- Vermunt JK. LEM: a general program for the analysis of categorical data. Tilberg University; 1997. [Google Scholar]
- van Den Oord EJ, Vermunt JK. Testing for linkage disequilibrium, maternal effects, and imprinting with (In)complete case-parent triads, by use of the computer program LEM. Am J Hum Genet. 2000;66:335–338. doi: 10.1086/302708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielman RS, McGinnis RE, Ewens WJ. Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM) Am J Hum Genet. 1993;52:506–516. [PMC free article] [PubMed] [Google Scholar]
- Starr JR, Hsu L, Schwartz SM. Performance of the log-linear approach to case-parent triad data for assessing maternal genetic associations with offspring disease: type I error, power, and bias. American Journal of Epidemiology. 2005;161:196–204. doi: 10.1093/aje/kwi021. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]