

Summary
Bacterial lipid homeostasis plays an important role for the adaptation to changing environments and under conditions of antimicrobial treatment. The tRNA-dependent aminoacylation of the phospholipid phosphatidylglycerol catalyzed by aminoacyl-phosphatidylglycerol synthases was shown to render various organisms less susceptible to antibacterial agents. Therefore, this type of enzyme might provide a new target to potentiate the efficacy of existing antimicrobials. This study makes use of the Pseudomonas aeruginosa alanyl-phosphatidylglycerol synthase to identify the minimal core domain of this transmembrane protein which is capable of alanyl-phosphatidylglycerol biosynthesis. Using this catalytic fragment we established a reliable activity assay which was used to study the enzymatic mechanism by analyzing an overall of 33 mutant proteins in vitro. Substrate recognition was analyzed by using aminoacylated microhelices as analogs of the natural tRNA substrate. The enzyme even tolerated mutated versions of this minimal substrate which indicates that neither the intact tRNA, nor the individual sequence of the acceptor stem is a determinant for substrate recognition. Furthermore, the analysis of derivatives of phosphatidylglycerol indicated that the polar headgroup of the phospholipid is specifically recognized by the enzyme, whereas modification of an individual fatty acid or even the deletion of a single fatty acid did not abolish A-PG synthesis.
Keywords: Pseudomonas aeruginosa, A-PGS, alanyl-phosphatidylglycerol synthase, tRNA recognition, aminoacyl phospholipid, substrate specificity
Introduction
Bacterial membranes function as a natural barrier upon environmental changes. Therefore, continuous adaptation is needed in order to cope with compounds that are potentially harmful for the integrity of the membrane. For example cationic antimicrobial peptides which can be found in all living species as part of the intrinsic defences of most organisms have been shown to directly interact with the negatively charged membrane as an antibacterial target. A comparable mode of interaction was also suggested for various cationic antibiotics. One major bacterial strategy in response to such cationic compounds is the aminoacylation of the phospholipid phosphatidylglycerol (PG) thereby reducing the overall net negative charge of the membrane. Aminoacyl-PGs are either zwitter-ionic phospholipids or alternatively they possess a positive net charge, dependent on the utilized amino acid (Roy & Ibba, 2008, MacFarlane, 1962, Houtsmuller & van Deenen, 1963, Fischer & Leopold, 1999, Sohlenkamp et al., 2007, Klein et al., 2009). Besides this, it is believed, that the modification of PG additionally results in an overall alteration of the biophysical properties of the membrane (Roy, 2009, Sacre et al., 1977). According to this, formation of lysyl-phosphatidylglycerol (L-PG) under low pH conditions in Staphylococcus aureus and Enterococcus faecalis (formerly known as Streptococcus faecalis) was already described in the 60ties (Houtsmuller & van Deenen, 1965). Furthermore, in 1962 MacFarlane has found alanyl-phosphatidylglycerol (A-PG) in Clostridium welchii (today known as C. perfringens) (MacFarlane, 1962). In E. faecalis A-PG and L-PG were found simultaneously. Moreover, aminoacylation of PG with arginine, glycine and ornithine, respectively, has been described (Houtsmuller & van Deenen, 1963, Gould & Lennarz, 1967, Kocun, 1970). In 2001 the enzyme responsible for the formation of L-PG in Gram-positive S. aureus was found during studies of the bacterial immune escape mechanisms (Peschel et al., 2001). An mprF (mprF – ‘multiple peptide resistance Factor’) mutant strain of S. aureus was found incapable of L-PG synthesis and was thereby rendered sensitive to cationic defensins when compared with the wild type strain. Studies with the orthologous mprF gene (lmo1695) from Gram-positive Listeria monocytogenes revealed an analogous resistance mechanism (Thedieck et al., 2006). Comparison of the mprF mutant with the corresponding S. aureus wild type strain did not reveal any differences of bacterial growth (Peschel et al., 2001, Klein et al., 2009). Related proteome analyses were indicative of only minor differences when the S. aureus mprF mutant was compared to the parental strain (Sievers et al., 2010).
For the opportunistic Gram-negative pathogen Pseudomonas aeruginosa it was shown that A-PG synthesis catalyzed by alanyl-phosphatidylglycerol synthase (A-PGS) mediates the resistance against CrCl3 and the antibacterial compounds protamine sulphate, cefsulodin and sodium lactate (Klein et al., 2009). Furthermore, it was shown that the A-PG content of the bacterial membrane is drastically increased in response to acidic growth conditions (Klein et al., 2009). Most organisms only contain a single aminoacyl-PG synthase (aa-PGS) gene. However, in the Gram-positive bacterium C. perfringens SM101 two homologous genes were identified, one coding for a lysyl-phosphatidylglycerol synthase (L-PGS) and an additional one coding for an A-PGS. The formation of A-PG and L-PG was shown to be tRNA-dependent with Ala-tRNAAla and Lys-tRNALys as substrate, respectively (Staubitz et al., 2004, Klein et al., 2009, Roy & Ibba, 2008). Under in vitro conditions for the Enterococcus faecium aa-PGS a relaxed specificity for lysine, arginine and alanine was shown, whereas for the orthologous Bacillus subtilis protein a primary specificity for lysine, in parallel with a relaxed specificity allowing for A-PG synthesis was observed under in vitro conditions (Roy & Ibba, 2009). In a recent publication we could clearly rule out such an extended specificity for the A-PGS enzyme from P. aeruginosa (Klein et al., 2009). However, formation of both isomers of A-PG (2’ and 3’A-PG) was indicated. To date it is not clear whether lysylation of PG, alanylation of PG but also the modification with glycine and arginine have a comparable effect on the physiology of the organism. However, for C. perfringens formation of A-PG and L-PG was speculated as a mechanism for fine-tuning of the biophysical properties of the bacterial membrane (Roy & Ibba, 2008).
Based on computational analysis for almost all aa-PGS enzymes a two domain architecture consisting of an N-terminal transmembrane domain and an additional C-terminal domain was proposed using TMPred (Hofmann & Stoffel, 1993). The N-terminal domains from various organisms are highly variable in size (approximately 228 (C. perfringens A-PGS) to 542 (P. aeruginosa A-PGS) amino acid residues) and share only a sequence identity of approximately 15 %. In contrast to this the C-terminal domains of aa-PGS can be found highly conserved with a sequence identity of approximately 30 %. Based on these theoretical findings it was concluded, that the C-terminal domain might be essential for aminoacyl-PG synthesis.
Only recently Ernst et al. (2009) described the analysis of a truncated L-PGS in vivo. Deletion of 8 of the initial transmembrane helices out of the postulated 14 transmembrane helices comprising the N-terminal transmembrane domain still resulted in L-PGS activity for the enzyme from S. aureus (Ernst et al., 2009). With in vitro experiments for B. subtilis L-PGS and the C. perfringens A-PGS it was shown that a mutant protein with a truncation of the complete hydrophobic N-terminal domain still allows for enzymatic activity (Roy & Ibba, 2009). However, these mutant proteins did not sustain detectable lipid modification under in vivo conditions (Roy & Ibba, 2009).
Based on the amino acid sequence identity observed for the C-terminal domain it was concluded, that the key amino acid residues responsible for A-PG or L-PG catalysis are conserved among all aa-PGS enzymes. Two types of amino acid residues with potential relevance for A-PGS catalysis have been described in the literature: mutations in the mprF genes identified from clinical isolates of S. aureus, which were correlated to an increased daptomycin resistance (Kosowska-Shick et al., 2009, Friedman et al., 2006, Julian et al., 2007). These mutations have been found located in the N-terminal transmembrane domain at positions without a high degree of sequence conservation. It was proposed, that the potential flippase activity located on this domain might be affected (Ernst et al., 2009, Ernst & Peschel, 2011). Furthermore, in a mutagenesis study for the L-PGS from S. aureus 6 conserved amino acid residues of the C-terminal domain with relevance for L-PG synthesis have been identified (Ernst et al., 2009). However these analyses did not provide insight into the related enzymatic mechanism.
The inhibition of aa-PGS is a promising strategy to render pathogenic bacteria more susceptible to antibacterial agents. These compounds might include the existing library of nowadays antibiotics, but also the arsenal of naturally occurring antimicrobial agents of the human host. In order to find a rationale for the development of efficient inhibitor molecules the understanding of the enzymatic mechanism is of prime importance.
This study is focused on the investigation of the catalytic mechanism for the A-PG formation in P. aeruginosa. For this purpose we establish a minimal A-PGS catalytic fragment as a versatile tool for the elucidation of the catalytic mechanism in vitro. This robust test system clearly reflects the in vivo properties of A-PGS catalysis and allows for the accurate differentiation of enzymatic activities. Based on a site-directed mutagenesis study comprising an overall of 33 mutant proteins in combination with chemical modification experiments we propose a catalytic mechanism for A-PGS catalysis. Furthermore, we elucidate potential determinants of the PG substrate and for the tRNA co-substrate. Besides this, we analyze the topology of the C-terminus of the membrane protein which is relevant for A-PGS activity.
Results and Discussion
A-PGS543-881 produced in E. coli is enzymatically active
In a previous report it was shown that the A-PGS dependent lipid modification in P. aeruginosa employs the activated amino acid substrate Ala-tRNAAla. For this transesterification it was shown that A-PGS is a transmembrane protein which is located in the inner membrane, whereas the reaction product A-PG was shown to be localized in the inner and the outer membrane (Klein et al., 2009). The routine analysis of A-PGS catalysis is strongly hampered in vitro due to the requirement for a solubilization step in the presence of detergent molecules for the native protein. Therefore, a ‘soluble’ domain providing full transesterification activity would be beneficial for the biochemical characterization of A-PGS catalysis and all future experiments to identify potential A-PGS inhibitors. Computational analyses using the prediction program TMPred (Hofmann & Stoffel, 1993) strongly indicated the presence of an N-terminal domain composed of 14 transmembrane helices followed by a C-terminal more hydrophilic domain (residues 543-881) (compare Fig. 4).
FIG. 4. Schematic representation of the domain architecture and the catalytic mechanism of P. aeruginosa A-PGS.
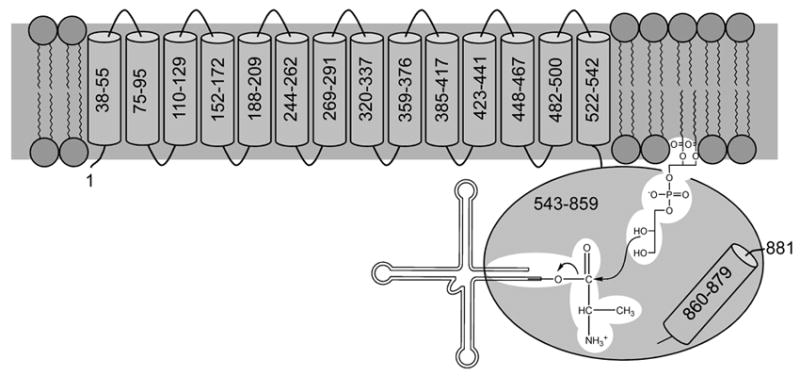
The N-terminal transmembrane domain containing 14 transmembrane helices is responsible for the anchoring of A-PGS in the inner bacterial membrane. Besides this, this part of the protein might harbor a potential flippase activity as recently described for S. aureus MprF (Ernst et al., 2009, Ernst & Peschel, 2011). The cytoplasmatic, C-terminal domain (residues 543-881, P. aeruginosa numbering) contains all elements responsible for A-PGS catalysis. However, deletion of the predicted C-terminal hydrophobic helix (residues 860 – 879) abolishes A-PGS activity. Localization of the C-terminus of A-PGS on the cytoplasmic site of the inner membrane was indicated by immunogold labelling experiments, therefore it is concluded that these residues are important constituents of the catalytic hydrophilic domain. The proposed catalytic reaction mechanism of A-PGS includes the direct transesterification of the alanyl-moiety of Ala-tRNAAla onto the 2’ hydroxyl group (or alternatively onto the 3’ hydroxyl group) of the lipid substrate. The 2’ hydroxyl group nucleophilically attacking the α-carbonyl group of Ala-tRNAAla is indicated. All elements of the PG and of the Ala-tRNAAla substrate with relevance for the A-PGS substrate recognition are highlighted in white.
To elucidate the potential role of the C-terminal domain a standard expression system was constructed (pGEX-6P-1/PA0920Δaa1-542) which allowed for the recombinant production of A-PGS residues 543-881 N-terminally fused to glutathione S-transferase (GST). E. coli BL 21 (λDE3) cells heterologously producing GST-A-PGS543-881 were harvested 3 h after protein induction and the total lipids of the E. coli host were extracted and subsequently separated using established 2D-TLC techniques. This procedure allows for the identification of the de novo biosynthesis of A-PG in E. coli under in vivo conditions due to the absence of any A-PGS related genes in the host (Fig. 1A, panel c). The employed in vivo assay resulted in the accumulation of approximately 10 % A-PG for the A-PGS543-881 variant when compared to the total lipid content (Fig. 1A, panel a). This value is higher than the observed 5 % for the overproduction of wild type A-PGS in E. coli TOP10 (Fig 1A, panel b) (Klein et al., 2009), which can be ascribed to the increased overproduction level for the truncated protein variant. This initial experiment is indicative for the efficient conversion of the E. coli specific PG substrate by the GST-A-PGS543-881 fusion protein under in vivo conditions.
FIG. 1. In vivo activity of A-PGS variants (A) and purification of recombinant A-PGS variants (B).
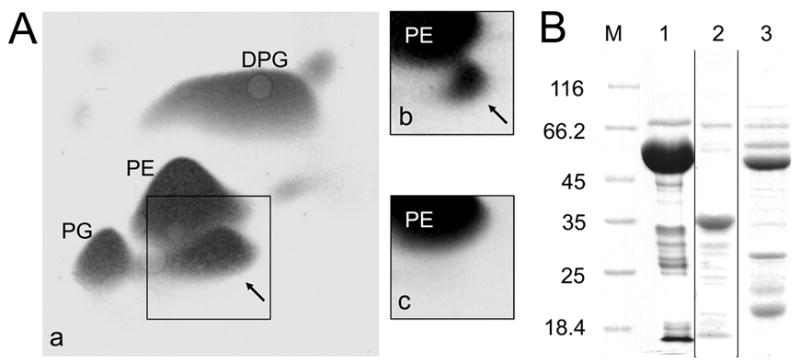
A. Separation of polar membrane lipids by 2D-TLC and detection by 5 % (w/v) molybdatophosphoric acid staining is shown. Lipids were extracted from E. coli cells overproducing GST-A-PGS543-881 (panel a), His6-A-PGS (panel b) and solely GST (panel c) as a negative control. The lipids phosphatidylethanolamine (PE), phosphatidylglycerol (PG), and diphosphatidylglycerol (DPG) are indicated. Alanyl-phosphatidylglycerol (A-PG) is highlighted by black arrows.
B. SDS-PAGE analysis of purified GST-A-PGS543-881, A-PGS543-881 and GST-A-PGS543-855N proteins. Indicated proteins were produced recombinantly in E. coli BL21 (λDE3), purified chromatographically and subsequently analyzed by 12 % SDS-PAGE and Coomassie Brilliant Blue staining. Lane M, molecular mass marker; relative molecular masses (* 1000) are indicated. Lane 1, purified GST-A-PGS543-881; lane 2, A-PGS543-881; lane 3, purified GST-A-PGS543-855N.
In a comparable in vivo experiment for the related S. aureus MprF protein (L-PGS) it was recently shown that 6 of the transmembrane helices are required for maintaining a functional enzyme (Ernst et al., 2009). When all transmembrane helices of MprF1 from C. perfringens or from MprF of B. subtilis were deleted residual enzymatic activity was obtained under in vitro conditions (Roy & Ibba, 2009). However, no detectable lipid modification was observed with the employed in vivo assay, which might indicate that slight, fragment specific disarrangements of the overall structure under the employed assay conditions might be responsible for the loss of enzymatic activity. These results indicate that the newly established A-PGS543-881 variant protein from P. aeruginosa is an appropriate enzyme to investigate the enzymatic mechanism of A-PGS or A-PGS related systems.
Purification and characterization of A-PGS543-881
For the subsequent analysis of A-PGS543-881 under in vitro conditions the truncated protein was purified to apparent homogeneity in the absence of detergent molecules and subjected to a biochemical characterization.
After binding of the GST-A-PGS543-881 fusion protein to a Glutathione Sepharose 4FF column, A-PGS543-881 was either eluted intact with 10 mM glutathione (calculated molecular weight 65’130 Da) (Fig. 1B, lane 1) or liberated from the resin by PreScission™ protease treatment as indicated by a dominant band with a relative molecular weight of approximately 38’000 in SDS-PAGE analyses (calculated molecular weight 38’720 Da) (Fig. 1B, lane 2). The obtained protein was subsequently concentrated by ultrafiltration and subjected to an analytical gel permeation chromatography which indicated a native molecular mass of 34’000 (data not shown). This result is indicative for a monomeric soluble A-PGS543-881 protein. The integrity of the 12 N-terminal amino acid residues of the truncated protein was confirmed by N-terminal protein sequencing. In UV/Vis absorption spectroscopic or fluorescence spectroscopic experiments no evidence for a chromogenic cofactor was obtained.
Assays for A-PGS543-881 activity
In vitro assay
The in vitro activity assay using purified GST-A-PGS543-881 made use of an ATP-regenerating system, 2 mM ATP, 20 μM of 14C labelled L-alanine and 500 μl of a crude cellular extract from E. coli cells overproducing P. aeruginosa alanyl-tRNA synthetase (AlaRS) (E. coli Rosetta (DE3) pLysS carrying pET28b(+)PA0903) to provide the substrate molecules PG and Ala-tRNAAla. Subsequently the polar lipids were extracted and characterized by 2D-TLC and autoradiography. In Fig. 2A a typical in vitro standard assay for GST-A-PGS543-881 is shown. Control experiments in the absence of GST-A-PGS543-881 (Fig. 2A, panel b) or with RNase A pre-treatment (Fig. 2A, panel c) did not result in any detectable amounts of A-PG. However, incubation with 20 μM of GST-A-PGS543-881 resulted in the formation of high amounts of A-PG (Fig. 2A, panel a). For the quantification of the enzymatic activity of A-PGS543-881 [1-14C] L-alanine incorporation into the lipid fraction was analyzed after lipid extraction and scintillation counting. In the presence of 10 μM GST-A-PGS543-881, this type of experiment resulted in a linear increase of the A-PG formation over a time scale of 10 min (data not shown). A specific activity of 27 pmol · mg-1 · min-1 was determined which is comparable to the measured value of 23 pmol · mg-1 · min-1 for the full length A-PGS protein.
FIG. 2. In vitro analysis of A-PG formation (A), modified in vitro analysis of A-PGS catalysis using different tRNA species (B) and localization analysis of the C-terminus of A-PGS (C).
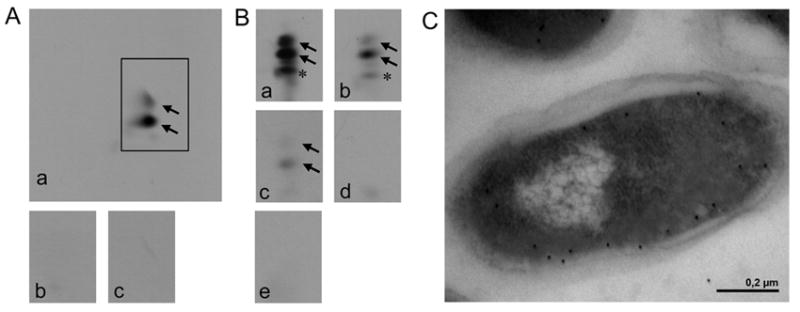
A. The activity of purified GST-A-PGS543-881 was analyzed by 2D-TLC of 14C-labelled phospholipids, A-PG formation is visualized by autoradiography. In vitro assay for GST-A-PGS543-881: a crude cellular extract from E. coli Rosetta (DE3) pLysS cells overproducing AlaRS enables the synthesis of Ala-tRNAAla (panel a); control reactions in the absence of A-PGS or after RNase A pre-treatment (panel b and c) are shown.
B. Modified in vitro assay for GST-A-PGS543-881 in the presence of recombinantly purified AlaRS enables the analysis of different tRNA substrates: a tRNA mixture from E. coli (panel a), purified tRNAAla1 or tRNAAla2 from P. aeruginosa which was synthesized by in vitro transcription (panel b or c). Control reactions in the absence of A-PGS or in the presence of RNase A (100 μg/ml) are shown (panel d or e). Formation of 2’ and 3’A-PG is highlighted by black arrows. * This additional lipid spot is observed due to the heterogeneity of the employed egg yolk lecithin lipid fraction. It is proposed that this is a result of the A-PGS dependent aminoacylation of a partly hydrolyzed PG molecule. Experiments using different sources of PG did not result in any compound migrating onto this specific position of the 2D-TLC.
C. The subcellular localization of a myc epitope C-terminally fused to the A-PGS was immune electron microscopically detected. Immunogold labelling was performed in the presence of a myc-specific antibody. Gold particles, indicative for the localization of the myc epitope, almost exclusively decorate the cytosolic part of the inner membrane.
Modified in vitro assay
To analyze A-PGS substrate recognition a modified in vitro activity assay was required. This experimental approach was also used in combination with tRNAAla which was previously synthesized by in vitro transcription. This technique results in a tRNA substrate molecule without any posttranscriptional modifications.
The modified A-PGS in vitro assay mixture contained purified PG and recombinantly purified AlaRS in the presence of a commercially available mixture of tRNAs from E. coli, or alternatively in the presence of purified tRNAAla1 or tRNAAla2 from P. aeruginosa which were produced by in vitro transcription (Sampson & Uhlenbeck, 1988). The lipid content was extracted and 14C labelled compounds were analyzed by autoradiography after 2D-TLC. To quantify the A-PG formation in chemical modification experiments, in experiments with in vitro transcribed tRNAs or in the presence of microhelices, the polar lipids were analyzed by scintillation counting after lipid extraction from the assay mixture.
In Fig. 2B (panel a) a typical modified in vitro GST-A-PGS543-881 assay using a tRNA mixture from E. coli as substrate is shown. Clearly A-PG synthesis was observed. This was also the case for experiments containing in vitro transcribed tRNAAla1 or tRNAAla2 from P. aeruginosa which do not carry any posttranscriptional modifications (Fig. 2B, panel b and c). Control experiments in the absence of GST-A-PGS543-881 or after RNase A pre-treatment did not result in any detectable A-PG synthesis (Fig. 2B, panel d and e). These experiments clearly indicate that the specific sequence variations of the tRNAs from E. coli do not abolish A-PG synthesis. Furthermore, we conclude that A-PGS substrate recognition is not dependent on posttranscriptional modifications. The established in vitro activity assay and the modified in vitro activity assay (also when using an in vitro transcribed tRNA substrate) in combination with the A-PGS543-881 variant protein is an appropriate system for the elucidation of the fundamental biochemical features of A-PGS catalysis.
A-PGS catalysis is not dependent on a covalent intermediate of serine, cysteine or histidine
The analysis of the enzymatic mechanism of A-PGS (or of orthologous enzymes) is of central importance for all subsequent efforts for the identification of efficient inhibitor molecules. For this purpose we employed our newly established in vitro and also the modified in vitro activity assay in combination with classical chemical modification experiments and in combination with an extended mutagenesis study. Presence of an activated nucleophilic side chain is an important feature of enzymes performing covalent catalysis (Moser et al., 1999, Perozich et al., 1999, Iqbal et al., 2009, Scheiner & Lipscomb, 1975). In many cases compounds like PMSF, DIPFP (inhibiting serine residues) allow for the direct chemical modification of such active site nucleophiles resulting in the covalent inactivation of the enzyme. When the A-PGS543-881 from P. aeruginosa was treated with these two chemical modification reagents at concentrations up to 1 mM PMSF or DIPFP no significant loss of A-PGS activity was observed (compare Table S1). In parallel mass spectroscopic (MS) experiments, no derivatized A-PGS protein was detectable, whereas the employed positive controls clearly resulted in the covalent derivatization of the serine nucleophile of trypsin when using PMSF (increase of 154.2 Da) and DIPFP (increase of 163.8 Da) under the employed assay conditions.
To substantiate these observations highly conserved amino acid residues with potential relevance for the catalytic mechanism of A-PGS were identified in an amino acid sequence alignment composed of A-PGS and L-PGS proteins from various organisms (supplemental Fig. S1). An overall of 41 amino acid residues were found 100 % conserved in all available A-PGS or L-PGS sequences of the database. To elucidate the basis of A-PG catalysis we initiated an extended mutagenesis study focussing on all conserved polar amino acid residues of the alignment (D579, K580, Y635, K654, E657, E658, S709, D710, E720, S724, Y732, S763, D765, R768, M778, Y831, R837, K840, K842; P. aeruginosa numbering).
However, the employed sequence alignment did not reveal a highly conserved cysteinyl or histidinyl residue. Therefore, involvement of a possible thiol ester intermediate or of a covalent histidinyl-intermediate was ruled out. To provide good insight into the functional role of the 19 identified polar residues, all of these amino acid positions were mutagenized into a residue with a comparable size (conservative exchange) or alternatively into a residue with differing size or charge characteristics. The initial characterization of all 33 mutant enzymes was performed in an E. coli in vivo activity assay analogously as described in a recent publication (Ernst et al., 2009). This type of analysis only revealed mutant proteins showing activities similar to the wild type enzyme (K654Q, K654S, E657Q, E658D, E658Q, S709A, S709N, D710A, D710N, E720Q, S763A, S763N, M778A, R837S, R837Q, K842S, K842Q), mutants with decreased activities (D579A, D579N, E657D, S724A, Y732A) and with no detectable A-PGS activity (K580S, K580Q, Y635A, S724N, D765A, D765N, R768S, R768Q, Y831A, K840S, K840Q) (Table 1, compare Fig. S2 for classification). However a precise quantification of the relative A-PGS activity was hampered due to the accumulation of comparable amounts of A-PG in the bacterial membrane of E. coli despite greatly varying enzymatic activities (time course 3 h).
Table 1. Enzymatic activities of A-PGS543-881 variants under in vivo and in vitro conditions.
Standard in vivo and in vitro A-PGS543-881 assays were performed as described under “Experimental Procedures”. The specific activity observed in the in vitro activity assay for wild type A-PGS543-881 was set as 100 % and all other values of mutant proteins were related to this value (detection limit: 0.5 %). A-PG formation in the in vivo assay was classified as: + + +, activity comparable to wild type enzyme; +, decreased activity when compared to wild type enzyme; -, no detectable A-PGS activity. The observed activity in the in vivo assay for wild type A-PGS543-881 and mutants K654S (+ + +), Y732A (+) and D765N (-) can be found visualized in supplemental figure S2.
| mutation | In vivo activity | Relative in vitro activity ± standard deviation [%] |
|---|---|---|
| Wild type A-PGS543-881 | + + + | 100 ± 0 |
| D579A | + | 3 ± 1 |
| D579N | + | 5 ± 2 |
| K580S | - | 0 |
| K580Q | - | 0 |
| Y635A | - | 0 |
| K654S | + + + | 46 ± 2 |
| K654Q | + + + | 65 ± 1 |
| E657D | + | 4 ± 1 |
| E657Q | + + + | 21 ± 4 |
| E658D | + + + | 28 ± 7 |
| E658Q | + + + | 19 ± 3 |
| S709A | + + + | 5 ± 1 |
| S709N | + + + | 17 ± 3 |
| D710A | + + + | 26 ± 3 |
| D710N | + + + | 30 ± 3 |
| E720Q | + + + | 10 ± 2 |
| S724A | + | 7 ± 2 |
| S724N | - | 0 |
| Y732A | + | 3 ± 1 |
| S763A | + + + | 23 ± 5 |
| S763N | + + + | 11 ± 1 |
| D765A | - | 0 |
| D765N | - | 0 |
| R768S | - | 0 |
| R768Q | - | 0 |
| M778A | + + + | 50 ± 2 |
| Y831A | - | 0 |
| R837S | + + + | 65 ± 8 |
| R837Q | + + + | 100 ± 8 |
| K840S | - | 0 |
| K840Q | - | 0 |
| K842S | + + + | 50 ± 2 |
| K842Q | + + + | 100 ± 3 |
By contrast, the newly established in vitro activity assay allowed for the accurate determination of the specific activity of mutant proteins under initial-rate conditions. Therefore, lipid samples of the in vitro assay taken at various time points were analyzed by scintillation counting. All activities indicated in Table 1 are averaged on the basis of three independent experiments.
Numerous mutagenesis studies for enzymes employing covalent catalysis resulted in a complete loss of enzymatic activity when the active site nucleophile residue was mutagenized (Moser et al., 1999, Carter & Wells, 1988, Corey & Craik, 1992). However mutagenesis of residues serine 709 and 763 into asparagine residues resulted in residual A-PGS activities of 17 ± 3 % and 11 ± 1 % when compared to the wild type enzyme. Also when those two residues were altered into alanine residual activities of 5 ± 1 % and 23 ± 5 % were observed which rules out a potential role as an active site nucleophile. In contrast to this, mutant protein S724N did not sustain any A-PG formation. But the analysis of the more ‘conservative mutant’ S724A clearly indicated a residual activity of 7 ± 2 %. This observation is not compatible with the involvement of a S724 nucleophile in the active site of A-PGS.
The significantly reduced activities of the employed mutant proteins might indicate that residues S709, S724 and S763 play an important role in A-PGS substrate recognition. Steric effects and/or the reduced ability for the formation of salt bridges might be responsible for the impaired activities of the related mutant proteins.
The results of the chemical modification experiments and the results of the mutagenesis study do not give evidence for a covalent seryl, cysteinyl or histidinyl intermediate of P. aeruginosa A-PGS catalysis. Therefore, a direct transesterification mechanism was proposed.
Mutation of aspartate and glutamate residues
Aspartate and glutamate residues are good candidates as potential acid-base catalysts of the A-PGS dependent transesterification. Besides this, also covalent aspartate and glutamate reaction intermediates can be found in the literature for enzymes like tRNA guanine transglycosylase and for CoA-transferases (Xie et al., 2003, Berthold et al., 2008). However, to date there is no description of a transesterification process which employs a covalent aspartate or glutamate intermediate (anhydride intermediate).
Mutation experiments for residues aspartate 579, 710 and 765 revealed an important role of these residues for A-PGS catalysis as indicated by residual activities of 3 ± 1 %, 26 ± 3 %, and 0 % (D579A, D710A and D765A) and 5 ± 2 %, 30 ± 3 % and 0 % (D579N, D710N and D765N). Furthermore mutation of residues glutamate 657, 658 and 720 resulted in systematically decreased A-PGS activities of 4 ± 1 % or 21 ± 4 % (E657D, E657Q), 28 ± 7 % or 19 ± 3 % (E658D, E658Q) and of 10 ± 2 % (E720Q). Obtained values for this set of mutant enzymes argue for the involvement of these glutamate and aspartate residues in A-PGS catalysis. Steric effects or the absence of polar interactions upon substrate binding might be responsible for the reduced activities. Both mutant proteins of aspartate 765 did not sustain any detectable activity which might indicate a key role (for example in acid-base catalysis) in A-PG formation. Obtained results are clearly confirmed in a related study for S. aureus MprF in which residues D546/579, E624/657, E658/720 and D731/765 (S. aureus/P. aeruginosa numbering) have been mutated into alanine residues (Ernst et al., 2009).
Mutation of lysine and arginine residues
Chemical modification experiments in the presence of HPG (modification of arginine residues) at concentrations of up to 10 mM did not indicate the direct involvement of an arginine residue in substrate recognition (compare Table S1). Protein samples of this modification experiment were also analyzed by tandem MS.
For the further characterization of the functional role of the highly conserved residues K580, K654, R768, R837, K840 and K842 an overall of twelve mutant proteins were analyzed in the newly established in vitro activity assay. The conservative mutation of residues lysine 842 and 654 into glutamine resulted in activities of 100 ± 3 % and 65 ± 1 %, whereas mutation into a serine residue still allowed for residual activities of 50 ± 2 % and 46 ± 2 %. Mutation of residue arginine 837 into glutamine and serine resulted in activities of 100 ± 8 % and 65 ± 8 %, respectively.
Residues arginine 837 and lysine 842 might be responsible for polar interactions which can be substituted by using a glutaminyl residue at the respective positions of A-PGS.
However, when residues K580, R768 and K840 were mutated into serine or alternatively into glutamine residues, no A-PGS activity was detectable. These results are clearly confirmed in a related study for the S. aureus MprF in which residues K547/580, R734/768 and K806/840 were mutated into alanine (S. aureus/P. aeruginosa numbering) (Ernst et al., 2009).
From these results a direct role for A-PGS catalysis (for example in acid-base catalysis) was concluded.
HPG treatment of the native protein clearly resulted in the covalent modification of up to three arginine residues of the A-PGS543-881 protein as indicated by an increase of 395.5 Da (theoretical molecular weight of unmodified protein = 38719.2 Da). This experiment clearly indicated that three residues (out of the overall 33 arginine residues) have been derivatized without any influence on the enzymatic activity of this protein fragment.
For both mutant proteins R837S and R837Q no reduced A-PGS activity was detected. However, mutagenesis of arginine 768 into serine or glutamine completely abolished A-PG formation. This residue might be buried in the employed modification experiment and is possibly involved in inter-protein salt bridge formation.
Mutation of methionine and tyrosine residues
When highly conserved tyrosine residues were mutagenized into alanine no residual activity was observed for mutant proteins Y635A and Y831A whereas mutant Y732A only showed a residual activity of 3 ± 1 %. These results are indicative for an important role of these three residues. However, due to the partial hydrophobic character of the tyrosyl side chain it is unclear whether these residues are directly involved in A-PGS catalysis.
When methionine 778 was altered into an alanine a reduced activity of 50 ± 2 % was observed. The partial loss of non-polar interactions might be responsible for the reduced activity of mutant M778A.
A-PGS catalysis is not dependent on metal ions
To further elucidate the principles of A-PGS catalysis involvement of metal ions was analyzed. For this purpose the potential inhibitory effect of the chelating agents EDTA and 1,10-phenanthroline were verified. When the modified in vitro activity assay (using a tRNA Mix from E. coli) was performed in the presence of up to 20 mM EDTA or up to 5 mM 1,10-phenanthroline no significant inactivation was observed (Table S1). However in the presence of 10 mM and 20 mM 1,10-phenanthroline a residual activity of approximately 35 % and 20 %, respectively, was determined. Since these experiments could not clearly rule out the involvement of a “tightly bound” metal ion the purified protein GST-A-PGS543-881 was concentrated (10.5 mg/ml – 161 μM; specific activity 27 pmol · mg-1 · min-1) and subjected to inductively coupled plasma - mass spectrometry (ICP-MS) experiments.
This type of analysis did not reveal any ferrous, manganese, nickel, copper, or zinc ions above the background level of the employed buffer system (compare Table S2). The purified protein fraction only revealed substoichiometric amounts of magnesium (19 μM). In the context of the dramatically higher chelator concentrations of the employed inhibition experiments (10 mM - 20 mM) metal-dependent A-PGS catalysis was ruled out.
The postulated C-terminal helix of A-PGS is essential for A-PG synthesis
Based on theoretical analysis for the aa-PGS from B. subtilis and C. perfringens an additional C-terminal transmembrane helix was proposed (Roy & Ibba, 2009). Computational analysis of the overall domain architecture of P. aeruginosa A-PGS also revealed a potential transmembrane helix (amino acid residues 860 to 879) located at the C-terminus of the membrane protein. To elucidate the functional role of this putative transmembrane segment of A-PGS the C-terminally truncated variant proteins A-PGS1-855N and A-PGS543-855N were analyzed under in vivo and in vitro conditions, respectively. In the employed in vivo activity assay the A-PGS1-855N mutant did not sustain any detectable A-PGS activity. This was also the case when the purified mutant protein A-PGS543-855N (compare Fig. 1B, lane 3) was analyzed in the in vitro assay. From these results we conclude that amino acid residues 856 – 881 play a crucial role for the activity of P. aeruginosa A-PGS.
Localization of the C-terminus of A-PGS
To investigate the presence of the postulated C-terminal transmembrane helix we determined the subcellular localization of the C-terminus of A-PGS by immunogold labelling of ultrathin plastic-embedded cell sections. For this purpose a myc epitope C-terminally fused to wild type A-PGS was immune electron microscopically detected, with a myc-specific antibody. In Fig. 2C gold particles, indicative for the localization of the myc epitope, almost exclusively decorate the cytosolic part of the inner membrane. This experiment confirms the results of our previous biochemical analysis which indicated the localization of A-PGS in the inner bacterial membrane (Klein et al., 2009). Furthermore this new methodology revealed a homogeneous distribution of A-PGS in the bacterial membrane. However, the cytoplasmic orientation of the C-terminus clearly is in disagreement with the computational prediction program TMPred (Hofmann & Stoffel, 1993). Based on our labelling experiments we conclude that the postulated C-terminal helix is not a transmembrane helix that penetrates the bacterial membrane. This part of the molecule might be a hydrophobic protein segment which is part of the ‘soluble’ catalytic domain. These results were also confirmed by our localization analysis of constructs A-PGS543-855N and A-PGS543-881. After the separation of the membrane fraction from the cytosolic fraction both variant proteins were exclusively detected in the soluble protein fraction (data not shown). Besides this, our gel permeation experiments revealed a monomeric, monomodal protein fragment A-PGS543-881 which is inconsistent with a highly hydrophobic transmembrane segment.
The C-terminal localization and the requirement of residues 856 – 881 for A-PGS activity might indicate that this C-terminal part of the molecule is directly involved in substrate recognition and/or catalysis. Furthermore, we conclude that P. aeruginosa A-PGS does not possess a C-terminal transmembrane helix.
Specificity of PG substrate recognition
Phospholipids from E. coli are preferentially esterified with palmitic acid, hexadec-9-enoic acid and octadec-11-enoic acid, whereas phospholipids from P. aeruginosa are predominantly condensed with palmitic acid and methylene octadecanoic acid (Cronan & Rock, 1996, Hancock & Meadow, 1969). The employed in vivo activity assay might indicate that minor modifications of the fatty acid side chains of PG are tolerated by the A-PGS system. However, there are several investigations for phospholipid biosynthetic enzymes showing a high degree of fatty acid specificity (Henneberry et al., 2002, MacDonald et al., 1988).
To determine the specificity of A-PGS substrate recognition several derivatives of PG (Fig. 3A) were analyzed in the modified in vitro activity assay (using synthetic lipid variants). The most abundant fatty acids of the employed standard PG from egg yolk lecithin (Sigma Aldrich) are oleic acid (18:1; (9Z)-octadec-9-enoic acid), linoleic acid (18:2; cis, cis-9,12-octadecadienoic acid) and palmitic acid (16:0; hexadecanoic acid) (Fredriksson et al., 2006) (compare Fig. 3A). In parallel control reactions in the absence of the phospholipid were employed.
FIG. 3. Analysis of A-PGS substrate recognition by using derivatives of PG (A) and variants of tRNAAla (B).
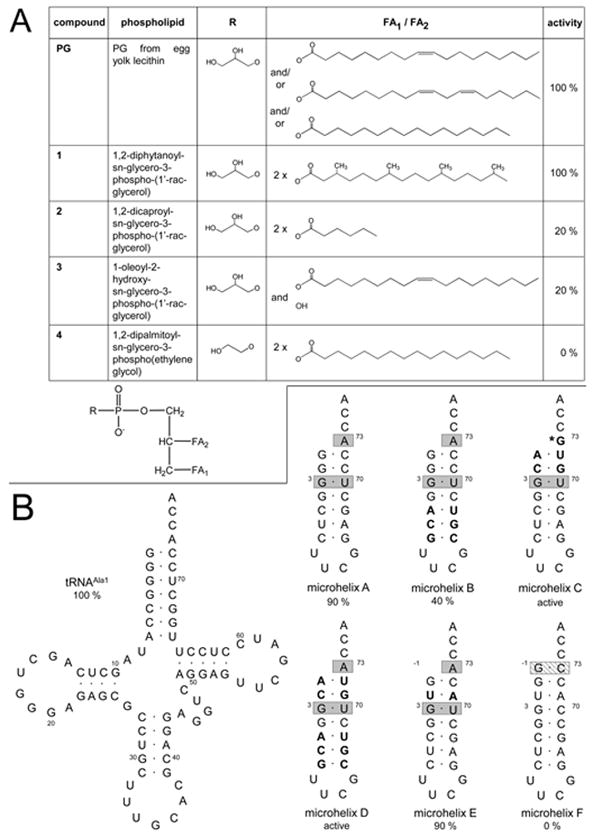
A. Structures of the employed analogs of PG are indicated. R represents the polar headgroup of the phospholipid, FA1 and FA2 represents the individual fatty acid residues. The relative activities of the individual compounds in the modified in vitro assay are indicated, standard deviations are ± 10 %.
B. Comparison of the tRNA sequences of the employed microhelices with the wild type tRNAAla1 sequence from P. aeruginosa. Bases that are essential determinants for E. coli AlaRS or for E. coli HisRS are highlighted with gray boxes and with a striped box respectively. All bases that are modified when compared to microhelix A are highlighted in bold font. Numbering of microhelices is based on full-length E. coli tRNAAla. * Efficient aminoacylation despite the mutated ‘discriminator base’ A73G has been shown earlier (Hou & Schimmel, 1988).
To analyze the influence of the overall structure of the individual fatty acid, we made use of a PG molecule carrying two saturated C16 fatty acids, each modified by fourfold methylation (compound 1). With this substrate no reduction of the specific activity was observed, which might indicate, that the integrity of the overall structure of the hydrophobic side chain (methylation or presence of double bonds) is not relevant for A-PGS substrate recognition. When we made use of a PG derivative containing two C6 fatty acids (instead of the usual C16 or C18 compounds) (compound 2) a residual activity of 20 % was determined which again demonstrates that the overall integrity of the fatty acid side chain is not a prerequisite for A-PGS catalysis. One might speculate that the reduced activity indicates the hampering of the direct interaction with the fatty acid and/or the glycerol phosphate moiety of this compound. This conclusion was corroborated by using a PG derivative carrying solely one C18 fatty acid (compound 3). This truncated substrate molecule still resulted in a residual activity of 20 %. During A-PG catalysis the 2’ hydroxyl group or alternatively the 3’ hydroxyl group of the glycerophosphate moiety of PG nucleophilically attacks the carboxylate function of Ala-tRNAAla. To study the functional role of the polar headgroup of the phospholipid for A-PGS substrate recognition we also analyzed a truncated substrate carrying an ethylene glycol phosphate instead of the glycerol phosphate (compound 4). This substrate analog did not result in any detectable A-PGS activity. From these experiments we conclude that the polar headgroup of A-PG is an important determinant for A-PGS substrate recognition.
Substrate specificity for Ala-tRNAAla
In a previous investigation for the A-PGS from P. aeruginosa we could clearly show that A-PGS catalysis in vivo and also in vitro is highly specific for the transesterification of the alanyl-moiety from Ala-tRNAAla (Klein et al., 2009). Important experiments with relevance for the understanding of the substrate specificity of A-PGS have been already performed more than 40 years ago, when Gould and co-workers tested several modified Ala-tRNA derivatives as substrate for A-PGS from the Gram-positive bacterium C. perfringens (Gould et al., 1968). All substrate analogs carrying modified amino acid moieties (N-acetylalanyl-tRNAAla, lactyl-tRNAAla and phenylalanyl-tRNAAla) did not sustain any A-PGS activity which clearly indicates that this part of the aminoacylated tRNA substrate is recognized with a high degree of specificity. Furthermore, it was shown, that also the tRNAAla part of the molecule is relevant for substrate recognition, since alanyl-tRNACys was not tolerated as an A-PGS substrate (Gould et al., 1968). However, Roy and Ibba (2008) were able to show that the overall integrity of tRNAAla is not a prerequisite for C. perfringens A-PGS catalysis (Roy & Ibba, 2008). In the present study we make use of several microhelices (Fig. 3B) to mimic the minimal part of the tRNAAla acceptor stem to elucidate the main determinants of the P. aeruginosa A-PGS catalysis. Specific P. aeruginosa tRNAAla1 was synthesized by in vitro transcription and aminoacylated with radioactively labelled alanine. Purified aminoacylated tRNAAla1 or microhelices were subjected to the modified A-PGS in vitro assay. The relative activity of A-PGS543-881 for in vitro transcribed tRNAAla1 (Fig. 3B) was set as 100 % and all other values were related to that.
When microhelix A (Nagan et al., 1999) was used as a substrate analog a relative activity of 90 % was observed. This result clearly indicates that no D-loop, anticodon-loop, variable loop or TΨC-loop is required for A-PGS substrate recognition. The acceptor stem of this microhelix carries only five of the terminal base pairings (G1-C72, G2-C71, G3-U70, G4-C69, C5-G68) which can be found in the natural substrate. Accordingly all potential determinants of this minimal substrate must be located on this part of the molecule. Therefore, microhelix B was analogously analyzed to characterize the potential role of base pair C5-G68. The enclosed mutation C5A-G68U resulted in a residual activity of only 40 %, which clearly indicates that this position is highly relevant for A-PGS catalysis. However, microhelix E also resulted in 90 % activity as observed for reference microhelix A, which clearly indicates that the U2-A71 base pair is not an important determinant for this enzyme. Bases G3, U70 and A73 are tRNAAla identity elements for recognition by the AlaRS, these positions are essential for the efficient aminoacylation by AlaRS. Microhelix C or microhelix D carrying additional mutations at position G1-C72 (among other mutated positions) were only inefficiently aminoacylated in the employed assay. Therefore, a potential role as identity elements for A-PGS catalysis was analyzed in a coupled AlaRS/A-PGS assay. This type of analysis clearly indicated that mutation of position G1-C72 does not impede A-PG formation. These two experiments clearly rule out these two positions as tRNAAla identity element for A-PGS catalysis.
Very early investigations with chemically modified ‘alanyl moieties’ are indicative for a highly specific recognition of the alanyl part of the substrate (Gould et al., 1968). To substantiate this idea for the A-PGS from P. aeruginosa we aminoacylated microhelix F with histidine. Comparison of microhelix F with microhelix E (which is an efficient substrate for A-PG synthesis) indicates only three variations of individual bases (G-1, C70, C73). This artificial substrate molecule did not result in any detectable A-PGS activity, suggesting that the alanyl-moiety is an important determinant for A-PGS substrate recognition. However, for this experiment an influence from the three base variations of microhelix F can not be clearly ruled out.
From the results of the present study we propose that substrate recognition of the membrane localized A-PGS enzyme only includes the acceptor stem of the soluble substrate molecule. One might speculate that five of the terminal base pairings and especially the C5-G68 base pair are required to direct the alanyl-moiety of Ala-tRNAAla into the active site of the enzyme.
The enzymatic mechanism of A-PGS: An initial proposal
In this study we continued our characterization of the P. aeruginosa A-PGS enzyme. The present work is based on our soluble purified enzyme derived from a suitable N-terminal truncation (of 542 amino acids) that possesses close to wild type activity.
A-PGS catalysis at the water-lipid interface requires accurate substrate recognition for the phospholipid PG and concurrently for the cytosolic co-substrate Ala-tRNAAla. Despite the presence of a large hydrophobic N-terminal transmembrane domain all elements for the catalytic function are localized in the C-terminal hydrophilic domain. This part of the enzyme is competent for the transesterification of the alanyl-moiety. Thereby, the 2’ or 3’ hydroxyl group of the lipid substrate might nucleophilically attack the α-carbonyl group of Ala-tRNAAla which is functioning as an activated alanyl-ester substrate. Concurrently the huge tRNAAla moiety acts as an efficient ‘leaving group’. We propose that this process does not involve a covalent enzyme localized intermediate and proceeds without the participation of any metal-ions.
Based on our expanded mutagenesis study we conclude that amino acids K580, D765, R768 and K840 are key residues for the formation of A-PG.
The polar headgroup of PG is specifically recognized by the enzyme, whereas the overall integrity of both fatty acid residues is not a prerequisite for A-PGS catalysis. Out of the huge tRNA co-substrate, only the presence of the five terminal base pairings of the acceptor stem is required for catalysis. These aspects of A-PGS catalysis can be found summarized in the schematic representation shown in Fig. 4. The minor specificity which was observed for the recognition of the C5-G68 base pair of the minimal substrate highlights the dominant role of the alanyl-moiety for A-PGS substrate recognition. According to these observations it becomes clear that “Crick’s adaptor molecule” is mainly used as an activated amino acid substrate in A-PGS catalysis. This “abuse of the tRNA molecule” in phospholipid biosynthesis might be a relict of an ancient RNA world.
Further work is needed to corroborate the proposed enzymatic mechanism. These efforts should be focused on the structural investigation of A-PGS.
Experimental Procedures
Plasmid construction
The gene sequence encoding the amino acid residues 543 to 881 of A-PGS from P. aeruginosa PAO1 was amplified using vector pBAD-His-A/PA0920 (Klein et al., 2009) by PCR with primers 5pGPaBamf and 6pGPaXhor (primer sequences are given in supplemental Table S3). The resulting PCR fragment was digested and cloned into the BamHI/XhoI site of pGEX-6P-1 (GE Healthcare, Freiburg, Germany) to yield pGEX-6P-1/PA0920Δaa1-542 (strains and plasmids are given in supplemental Table S4). Open reading frame PA0903 encoding the AlaRS from P. aeruginosa was amplified from genomic DNA with primers 27alaSNdeIfw and 28alaSBclIrv. The PCR fragment was digested with NdeI and BclI and cloned into the NdeI/BamHI site of pET28b(+) (Novagen, Darmstadt, Germany) to yield pET28b(+)PA0903.
Mutagenesis and truncation of P. aeruginosa A-PGS
Vectors pGEX-6P-1/A-PGS543-855N and pBAD-His-A/PA09201-855N were constructed by site-directed mutagenesis with the QuikChange™ kit (Stratagene, La Jolla, CA) according to the manufacturer’s instruction using pGEX-6P-1/PA0920Δaa1-542 and pBAD-His-A/PA0920, respectively, as templates with primers QCA856NSTOPfw and QCA856NSTOPrv. Amino acid substitutions in A-PGS543-881 were introduced using the QuikChange™ kit in combination with primers 7 to 72.
Preparation and purification of RNA transcripts
The two isoacceptor tRNAAla genes (PA4280.3, PA3133.2) from P. aeruginosa were cloned using oligonucleotides 73 - 76 into pUC18 or pUC119 vector that allowed for in vitro T7 RNA polymerase run-off transcription after plasmid cleavage with BstNI (Sampson & Uhlenbeck, 1988). The tRNAs were purified either via 12 % polyacrylamide gel electrophoresis in the presence of 8 M urea according to (Pande et al., 1991) or via MonoQ chromatography according to (Jahn et al., 1991).
Production and purification of GST-A-PGS543-881 and GST-A-PGS543-855N in E. coli
Glutathione S-transferase (GST)-tagged A-PGS543-881 or A-PGS543-855N was overproduced in E. coli BL21 (λDE3) (Stratagene) employing pGEX-6P-1/PA0920Δaa1-542 and pGEX-6P-1/A-PGS543-855N, respectively. Cells were cultivated at 37 °C in 500 ml LB medium with 100 μg/ml ampicillin. Protein production was induced with 50 μM isopropyl-1-thio-β-D-galactopyranoside (IPTG) at 0.5 OD578. Cells were further cultivated at 37 °C for 3 h (A-PGS543-881) or at 17 °C for 5 h (A-PGS543-855N), harvested by centrifugation, suspended in buffer A (phosphate buffered saline (PBS) (137 mM NaCl; 2.7 mM KCl; 10 mM Na2HPO4; 2 mM KH2PO4; pH 7.4) with 2 mM DTT), and disrupted by a single passage through a French Press® (Thermo Fisher Scientific, Waltham, MA) at 19’200 p.s.i.. After ultracentrifugation (1 h, 110’000 * g, 4 °C) the resulting supernatant was loaded onto Glutathione Sepharose 4FF (GE Healthcare) equilibrated with buffer A. After extensive washing with 10 column volumes of buffer A, followed by 10 column volumes of buffer B (50 mM Tris-HCl; 2 mM DTT; pH 8.0), the protein was eluted in buffer B containing 10 mM glutathione or was liberated from the column by PreScission™ Protease (GE Healthcare) treatment for 16 h.
Production and purification of His6-tagged HisRS from E. coli and AlaRS from P. aeruginosa and E. coli
For purification of His6-tagged AlaRS from P. aeruginosa, E. coli Rosetta (DE3) pLysS carrying pET28b(+)PA0903 was cultivated at 37 °C in 500 ml LB medium with 34 μg/ml chloramphenicol and 30 μg/ml kanamycin. Protein production was induced with 200 μM IPTG at 0.5 OD578. Cells were further cultivated at 30 °C for 4 h, harvested by centrifugation and suspended in buffer C (35 mM Tris-HCl; 300 mM NaCl; 40 mM MgCl2; 10 % (w/v) glycerol; pH 7.2) and disrupted by French Press® as described above to yield the crude cellular extract. After ultracentrifugation the supernatant was subjected to a nickel-loaded Chelating Sepharose 4FF (GE Healthcare) and purified according to the manufacturer’s instructions with 500 mM imidazole in buffer C. His6-tagged E. coli AlaRS and histidyl-tRNA synthetase (HisRS) were overproduced using the E. coli ASKA library clone JW2667 and JW2498 (Kitagawa et al., 2005), respectively. Cultures were grown at 37 °C in LB medium with 30 μg/ml chloramphenicol, recombinant protein was produced by autoinduction as described (Studier, 2005). Cells were harvested by centrifugation and suspended in buffer D (50 mM Tris-HCl; 200 mM NaCl; 20 mM MgCl2; 1 mM β-mercaptoethanol; pH 7.0), and broken by sonication. After ultracentrifugation the supernatant was applied to Ni-NTA (Qiagen, Hilden, Germany) metal affinity resin and purified according to the manufacturer’s instructions with 1 M imidazole in buffer D. The eluted enzyme was supplemented with 10 % (v/v) of glycerol and stored at −20 °C. SDS–PAGE of purified proteins followed by staining with Coomassie blue revealed greater than 95 % purity.
tRNA aminoacylation reactions
Purified, synthetic microhelices were purchased from Integrated DNA Technologies (Coralville, IA). tRNA transcripts and microhelices were denatured at 80 °C and 60 °C for 2 min, respectively, followed by addition of 10 mM MgCl2 and immediate cooling on ice to facilitate RNA folding. AlaRS aminoacylation reactions were performed at room temperature according to (Swairjo et al., 2004) using 20 μM of [1-14C] L-alanine (51 μCi μmol-1, Moravek Biochemicals, Brea, CA), 10 μM of tRNA (tRNA Mix from E. coli strain W (Sigma Aldrich, St. Louis, MO) or in vitro transcribed tRNAAla) or microhelices, respectively, and 1 μM of AlaRS. All experiments to elucidate tRNA substrate recognition were performed in the presence of 4.75 μM of [2, 3-3H] L-alanine (52 μCi μmol-1, GE Healthcare).
HisRS aminoacylation was carried out at 37 °C according to (Connolly et al., 2004) using 21.7 μM [U-14C] L-histidine (322 μCi μmol-1, GE Healthcare), 10 μM of tRNA microhelix and 1 μM of HisRS.
Extraction and analysis of polar membrane lipids
Bacteria or membrane fragments were harvested by centrifugation for 5 or 30 min, respectively, at 11’000 * g. The resulting sediment was used for a modified method of Bligh and Dyer (1959) extraction of polar membrane lipids as previously described (Klein et al., 2009). Lipids were either analyzed by 2D-TLC or liquid scintillation analysis.
Determination of in vivo A-PGS activity
For the determination of the in vivo A-PGS activity 5 ml of an E. coli cell culture overproducing wild type or variant A-PGS protein were harvested by centrifugation. Then lipids were extracted and separated by 2D-TLC.
Determination of in vitro A-PGS activity
The in vitro A-PGS activity assay makes use of an E. coli crude cellular extract to provide the substrate molecules PG and Ala-tRNAAla. Therefore, 500 μl of a crude cellular extract from E. coli Rosetta (DE3) pLysS carrying pET28b(+)PA0903 was supplemented with 2 - 20 μM of purified GST-A-PGS543-881 or GST-A-PGS543-855N, 2 mM ATP, an ATP-regenerating system (consisting of 18 mM creatine phosphate and 25 U of creatine phosphokinase) and 20 μM of [1-14C] L-alanine in a total volume of 700 μl at 37 °C under vigorous shaking (1’000 rpm). Samples of 100 μl volume were heat inactivated (5 min at 60 °C) and subjected to lipid extraction. All subsequent assays for the determination of A-PGS543-881 mutant activities were standardized by analyzing the individual proteins at a concentration of 10 μM for 1 h at 37 °C. The activities for wild type GST-A-PGS543-881 obtained were set as 100 %; all other values of mutant GST-A-PGS543-881 were related to this. Experiments were performed in triplicate and were completed by control experiments in which GST-A-PGS543-881 was omitted or in which the crude cellular extract was pre-treated with 100 μg/ml RNase A for 25 min. The deduced detection limit of the assay was 0.5 %.
Specific activity of A-PGS543-881 and full length A-PGS was determined as described previously (Klein et al., 2009).
Modification of the in vitro A-PGS activity assay
A modified in vitro A-PGS activity assay was performed using purified aminoacylated tRNA or alternatively the tRNA substrate was aminoacylated in a coupled AlaRS/A-PGS experiment. For the modified in vitro assay with aminoacylated tRNA, 0.2 – 5 μM of purified GST-A-PGS543-881 and 2 – 20 nM aminoacylated tRNA was added to a commercially available PG fraction [2 mg/ml PG from egg yolk lecithin (Sigma Aldrich, St. Louis, MO)] supplemented with 1.76 mg/ml Triton X-100 in 50 mM Tris-HCl, pH 8.0. For the coupled AlaRS/A-PGS in vitro assay the crude cellular extract of the above mentioned in vitro assay was substituted with 5 μM of purified AlaRS, 10 - 15 μM tRNA, and 2.7 mM PG in aminoacylation buffer according to (Swairjo et al., 2004). Samples of 100 μl volume were inactivated by the addition of 375 μl MeOH/chloroform (2:1, v/v) and subsequently subjected to lipid extraction.
Chemical modification of A-PGS543-881
To characterize potential amino acid residues with relevance for the A-PGS reaction mechanism, 10 pmol of A-PGS543-881 were chemically modified with reagents showing a high degree of specificity for individual amino acids. For this purpose, the affinity purification of GST-A-PGS543-881 was performed in the absence of DTT. After extensive washing with PBS the target protein was liberated from the column by PreScission™ Protease treatment. The obtained eluate fraction (~ 60 μM) was incubated in the presence of phenylmethylsulfonyl fluoride (PMSF, Roth, Karlsruhe, Germany) (0.1, 1 mM) and diisopropylfluorophosphate (DIPFP, Pierce, Bonn, Germany) (0.1, 1 mM) at 4 °C for 45 min respectively; or alternatively in the presence of p-hydroxyphenylglyoxal (HPG, Pierce, Bonn, Germany) (0.5, 1, 5, 10 mM) at 20 °C in the dark for 45 min. These protein fractions were excessively dialysed against PBS and subjected to in vitro assays in the presence of 10 mM ATP (ATP-regenerating system omitted) or to modified in vitro assays with aminoacylated tRNAAla. To study the influence of metal chelating agents both types of activity assays were also incubated in the presence of ethylenediaminetetraacetic acid (EDTA) (3, 10, 20 mM) or 1,10-phenanthroline (1, 3, 5, 10, 20 mM), respectively.
Mass spectroscopy of chemically modified A-PGS proteins
All chemical modification experiments using A-PGS543-881 were concurrently analyzed by mass spectroscopy. For this purpose the native and derivatized proteins after desalting were subjected to nanospray mass spectroscopy on a QTOF 2 instrument and the resulting spectra deconvoluted using the MaxEnt. I software package (Waters, Manchester, UK).
This type of experiment allowed the direct detection and approximate quantification of modifications by the observation of characteristic mass increments compared to the unmodified protein. These experiments were validated by analogous modification of trypsin from bovine pancreas (Sigma Aldrich) as positive control for modification with PMSF and DIPFP to confirm the efficiency of the employed agents under the employed assay conditions.
Analysis of A-PGS substrate recognition by using artificial PG derivatives
Artificial lipids were employed to identify essential determinants of the PG substrate. For this purpose the coupled AlaRS/A-PGS modified in vitro assay was performed in the presence of PG derivatives (Fig. 3A) (Avanti Polar Lipids, Inc., Alabaster, AL) at a concentration of 2 mg/ml.
Recognition of the tRNA substrate
To determine the tRNA identity elements of A-PGS catalysis P. aeruginosa wild type in vitro transcribed tRNAAla1 and tRNA microhelices (microhelix A, B, E and F) (Fig. 3B) were aminoacylated and subjected to a modified in vitro assay containing 5 μM of GST-A-PGS543-881. Experiments were performed in triplicate; standard deviations are ± 10 %. The obtained A-PGS activity using in vitro transcribed tRNAAla1 was set as 100 %. The employed AlaRS failed to efficiently aminoacylate tRNA microhelices C and D; therefore a coupled AlaRS/A-PGS modified in vitro assay was performed. Each microhelix (20 μM) was pre-incubated at 37 °C for 30 min in the standard AlaRS aminoacylation reaction buffer (500 μl). A-PGS catalysis was then initiated by the addition of 500 μl reaction buffer containing 5.4 mM of PG and 10 μM GST-A-PGS543-881. Experiments were performed in duplicate.
Immunogold-labelling
E. coli TOP10 pBADmyc-His-A/PA0920 was cultivated as described (Klein et al., 2009). Bacteria were fixed with 1 % formaldehyde in the growth medium at 4 °C. After washing with cacodylate buffer (0.1 M cacodylate; 0.01 M CaCl2; 0.01 M MgCl2; 0.09 M saccharose; pH 6.9) containing 10 mM glycine samples were dehydrated with a graded series of aceton (10, 30, 50, 70, 90, 100 %) and embedded in LRW resin. Ultrathin sections were cut with a diamond knife and collected onto butvar coated nickel grids (300 mesh). Sections were incubated with anti-myc epitope mouse monoclonal IgG1 (Invitrogen, Karlsruhe, Germany) (1:50 dilution of the stock solution containing 1 mg/ml IgG1 antibodies) for 16 h at 4 °C. After washing with PBS bound antibodies were made visible with goat anti-mouse IgG antibodies coupled to 10 nm gold-nanoparticles (1:200 dilution of the stock solution, incubation for 1 h at room temperature). After washing with PBS containing 1 % Tween 20, an additional washing with PBS and distilled water was performed. Then air-dried sections were counterstained with 4 % aqueous uranyl acetate for 3 min before examination in a Zeiss transmission electron microscope EM910 at an acceleration voltage of 80 kV and calibrated magnifications. Images were recorded digitally with a Slow-Scan CCD-Camera (ProScan, 1024×1024, Scheuring, Germany) with ITEM-Software (Olympus Soft Imaging Solutions, Münster, Germany). Images were corrected for brightness and contrast applying Adobe photoshop CS 3.
Determination of native molecular mass
Analytical gel permeation chromatography was performed as described elsewhere (Moser et al., 1999).
Determination of protein concentration
The concentration of purified protein was determined using the Bradford reagent (Sigma Aldrich, St. Louis, MO) according to the manufacturer’s instructions with bovine serum albumin as standard.
UV-visible light absorption spectroscopy and fluorescence measurements
UV-visible light spectra of purified recombinant A-PGS543-881 were recorded from 260 – 900 nm using a V-550 spectrometer (Jasco, Groß Umstadt, Germany). Fluorescence spectra via LS50B-luminescence spectrometer (PerkinElmer, Boston, MA) were monitored to detect possible fluorescent cofactors. Therefore, purified A-PGS543-881 was excited from 250 - 450 nm. Fluorescence emission maxima were detected from 250 – 800 nm.
Inductively coupled plasma - mass spectrometry
The fusion protein GST-A-PGS543-881 was affinity purified and subsequently concentrated to 10.5 mg/ml (161 μM). In a parallel procedure the sole GST protein was overproduced from vector pGEX-6P-1 (GE Healthcare), purified and concentrated to 4 mg/ml (154 μM). The obtained GST protein fraction was employed to determine the background level of the purification strategy.
To determine protein-bound metal ions (Cu2+, Fe2+, Mg2+, Mn2+, Ni2+, Zn2+) inductively coupled plasma - mass spectrometry (ICP-MS) was performed (CURRENTA Bayer-Analytics, Leverkusen, Germany). All experiments were performed in duplicate.
N-terminal amino acid sequence determination
Automated Edman degradation was used to confirm the identity of purified proteins.
Supplementary Material
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (grant MO 1749/1-1) and from the National Institute of General Medical Sciences (to D.S.). I.U.H. is a Postdoctoral Fellow of the Deutsche Forschungsgemeinschaft. Technical assistance of Ina Schleicher for the electron microscopic work is acknowledged. Special thanks go to our mentor Dieter Jahn for his continuous support.
References
- Berthold CL, Toyota CG, Richards NG, Lindqvist Y. Reinvestigation of the catalytic mechanism of formyl-CoA transferase, a class III CoA-transferase. J Biol Chem. 2008;283:6519–6529. doi: 10.1074/jbc.M709353200. [DOI] [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Carter P, Wells JA. Dissecting the catalytic triad of a serine protease. Nature. 1988;332:564–568. doi: 10.1038/332564a0. [DOI] [PubMed] [Google Scholar]
- Connolly SA, Rosen AE, Musier-Forsyth K, Francklyn CS. G-1:C73 recognition by an arginine cluster in the active site of Escherichia coli histidyl-tRNA synthetase. Biochemistry. 2004;43:962–969. doi: 10.1021/bi035708f. [DOI] [PubMed] [Google Scholar]
- Corey DR, Craik CS. An investigation into the minimum requirements for peptide hydrolysis by mutation of the catalytic triad of trypsin. J Am Chem Soc. 1992;114:1784–1790. [Google Scholar]
- Cronan JE, Rock CO. Escherichia coli and Salmonella thyphimurium. American Society for Microbiology Press; Washington, DC: 1996. Biosynthesis of membrane lipids. [Google Scholar]
- Ernst CM, Peschel A. Broad-spectrum antimicrobial peptide resistance by MprF-mediated aminoacylation and flipping of phospholipids. Mol Microbiol. 2011;80:290–299. doi: 10.1111/j.1365-2958.2011.07576.x. [DOI] [PubMed] [Google Scholar]
- Ernst CM, Staubitz P, Mishra NN, Yang SJ, Hornig G, Kalbacher H, Bayer AS, Kraus D, Peschel A. The bacterial defensin resistance protein MprF consists of separable domains for lipid lysinylation and antimicrobial peptide repulsion. PLoS Pathog. 2009;5:e1000660. doi: 10.1371/journal.ppat.1000660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer W, Leopold K. Polar lipids of four Listeria species containing L-lysylcardiolipin, a novel lipid structure, and other unique phospholipids. Int J Syst Bacteriol. 1999;49:653–662. doi: 10.1099/00207713-49-2-653. [DOI] [PubMed] [Google Scholar]
- Fredriksson S, Elwinger K, Pickova J. Fatty acid and carotenoid composition of egg yolk as an effect of microalgae addition to feed formula for laying hens. Food Chemistry. 2006;99:530–537. [Google Scholar]
- Friedman L, Alder JD, Silverman JA. Genetic changes that correlate with reduced susceptibility to daptomycin in Staphylococcus aureus. Antimicrob Agents Chemother. 2006;50:2137–2145. doi: 10.1128/AAC.00039-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould RM, Lennarz WJ. Biosynthesis of aminoacyl derivatives of phosphatidylglycerol. Biochem Biophys Res Commun. 1967;26:512–515. doi: 10.1016/0006-291x(67)90578-5. [DOI] [PubMed] [Google Scholar]
- Gould RM, Thornton MP, Liepkalns V, Lennarz WJ. Participation of aminoacyl transfer ribonucleic acid in aminoacyl phosphatidylglycerol synthesis. II. Specificity of alanyl phosphatidylglycerol synthetase. J Biol Chem. 1968;243:3096–3104. [PubMed] [Google Scholar]
- Hancock IC, Meadow PM. The extractable lipids of Pseudomonas aeruginosa. Biochim Biophys Acta. 1969;187:366–379. doi: 10.1016/0005-2760(69)90010-1. [DOI] [PubMed] [Google Scholar]
- Henneberry AL, Wright MM, McMaster CR. The major sites of cellular phospholipid synthesis and molecular determinants of Fatty Acid and lipid head group specificity. Mol Biol Cell. 2002;13:3148–3161. doi: 10.1091/mbc.01-11-0540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann K, Stoffel W. TMBASE - A database of membrane spanning protein segments. Biol Chem Hoppe-Seyler. 1993;374:166. [Google Scholar]
- Hou YM, Schimmel P. A simple structural feature is a major determinant of the identity of a transfer RNA. Nature. 1988;333:140–145. doi: 10.1038/333140a0. [DOI] [PubMed] [Google Scholar]
- Houtsmuller UM, van Deenen L. Identification of a bacterial phospholipid as an O-ornithine ester of phosphatidyl glycerol. Biochim Biophys Acta. 1963;70:211–213. doi: 10.1016/0006-3002(63)90743-1. [DOI] [PubMed] [Google Scholar]
- Houtsmuller UM, van Deenen LL. On the amino acid esters of phosphatidyl glycerol from bacteria. Biochim Biophys Acta. 1965;106:564–576. doi: 10.1016/0005-2760(65)90072-x. [DOI] [PubMed] [Google Scholar]
- Iqbal A, Clifton IJ, Bagonis M, Kershaw NJ, Domene C, Claridge TD, Wharton CW, Schofield CJ. Anatomy of a simple acyl intermediate in enzyme catalysis: combined biophysical and modeling studies on ornithine acetyl transferase. J Am Chem Soc. 2009;131:749–757. doi: 10.1021/ja807215u. [DOI] [PubMed] [Google Scholar]
- Jahn MJ, Jahn D, Kumar AM, Söll D. Mono Q chromatography permits recycling of DNA template and purification of RNA transcripts after T7 RNA polymerase reaction. Nucleic Acids Res. 1991;19:2786. doi: 10.1093/nar/19.10.2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julian K, Kosowska-Shick K, Whitener C, Roos M, Labischinski H, Rubio A, Parent L, Ednie L, Koeth L, Bogdanovich T, Appelbaum PC. Characterization of a daptomycin-nonsusceptible vancomycin-intermediate Staphylococcus aureus strain in a patient with endocarditis. Antimicrob Agents Chemother. 2007;51:3445–3448. doi: 10.1128/AAC.00559-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E, Toyonaga H, Mori H. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 2005;12:291–299. doi: 10.1093/dnares/dsi012. [DOI] [PubMed] [Google Scholar]
- Klein S, Lorenzo C, Hoffmann S, Walther JM, Storbeck S, Piekarski T, Tindall BJ, Wray V, Nimtz M, Moser J. Adaptation of Pseudomonas aeruginosa to various conditions includes tRNA-dependent formation of alanyl-phosphatidylglycerol. Mol Microbiol. 2009;71:551–565. doi: 10.1111/j.1365-2958.2008.06562.x. [DOI] [PubMed] [Google Scholar]
- Kocun FJ. Amino acid containing phospholipids as major components of the phospholipids of Streptococcus faecalis 10C1. Biochim Biophys Acta. 1970;202:277–282. doi: 10.1016/0005-2760(70)90189-x. [DOI] [PubMed] [Google Scholar]
- Kosowska-Shick K, Clark C, Pankuch GA, McGhee P, Dewasse B, Beachel L, Appelbaum PC. Activity of telavancin against staphylococci and enterococci determined by MIC and resistance selection studies. Antimicrob Agents Chemother. 2009;53:4217–4224. doi: 10.1128/AAC.00742-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald ML, Mack KF, Williams BW, King WC, Glomset JA. A membrane-bound diacylglycerol kinase that selectively phosphorylates arachidonoyl-diacylglycerol. Distinction from cytosolic diacylglycerol kinase and comparison with the membrane-bound enzyme from Escherichia coli. J Biol Chem. 1988;263:1584–1592. [PubMed] [Google Scholar]
- MacFarlane MG. Characterization of Lipoamino-Acids as O-Amino-Acid Esters of Phosphatidyl-Glycerol. Nature. 1962;196:136–138. [Google Scholar]
- Moser J, Lorenz S, Hubschwerlen C, Rompf A, Jahn D. Methanopyrus kandleri glutamyl-tRNA reductase. J Biol Chem. 1999;274:30679–30685. doi: 10.1074/jbc.274.43.30679. [DOI] [PubMed] [Google Scholar]
- Nagan MC, Kerimo SS, Musier-Forsyth K, Cramer CJ. Wild-Type RNA MicrohelixAla and 3:70 Variants: Molecular Dynamics Analysis of Local Helical Structure and Tightly Bound Water. J Am Chem Soc. 1999;121:7310–7317. [Google Scholar]
- Pande S, Jahn D, Söll D. Histidine tRNA guanylyltransferase from Saccharomyces cerevisiae. I. Purification and physical properties. J Biol Chem. 1991;266:22826–22831. [PubMed] [Google Scholar]
- Perozich J, Nicholas H, Wang BC, Lindahl R, Hempel J. Relationships within the aldehyde dehydrogenase extended family. Protein Sci. 1999;8:137–146. doi: 10.1110/ps.8.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschel A, Jack RW, Otto M, Collins LV, Staubitz P, Nicholson G, Kalbacher H, Nieuwenhuizen WF, Jung G, Tarkowski A, van Kessel KP, van Strijp JA. Staphylococcus aureus resistance to human defensins and evasion of neutrophil killing via the novel virulence factor MprF is based on modification of membrane lipids with l-lysine. J Exp Med. 2001;193:1067–1076. doi: 10.1084/jem.193.9.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy H. Tuning the properties of the bacterial membrane with aminoacylated phosphatidylglycerol. IUBMB Life. 2009;61:940–953. doi: 10.1002/iub.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy H, Ibba M. RNA-dependent lipid remodeling by bacterial multiple peptide resistance factors. Proc Natl Acad Sci U S A. 2008;105:4667–4672. doi: 10.1073/pnas.0800006105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy H, Ibba M. Broad range amino acid specificity of RNA-dependent lipid remodeling by multiple peptide resistance factors. J Biol Chem. 2009;284:29677–29683. doi: 10.1074/jbc.M109.046367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacre MM, El Mashak EM, Tocanne JF. A monolayer (π,ΔV) study of the ionic properties of alanylphosphatidylglycerol: effects of pH and ions. Chem Phys Lipids. 1977;20:305–318. doi: 10.1016/0009-3084(77)90071-8. [DOI] [PubMed] [Google Scholar]
- Sampson JR, Uhlenbeck OC. Biochemical and physical characterization of an unmodified yeast phenylalanine transfer RNA transcribed in vitro. Proc Natl Acad Sci U S A. 1988;85:1033–1037. doi: 10.1073/pnas.85.4.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiner S, Lipscomb WN. Molecular orbital studies of enzyme activity: Catalytic mechanism of serine proteinases. Proc Nat Acad Sci USA. 1975;73:432–436. doi: 10.1073/pnas.73.2.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers S, Ernst CM, Geiger T, Hecker M, Wolz C, Becher D, Peschel A. Changing the phospholipid composition of Staphylococcus aureus causes distinct changes in membrane proteome and membrane-sensory regulators. Proteomics. 2010;10:1685–1693. doi: 10.1002/pmic.200900772. [DOI] [PubMed] [Google Scholar]
- Sohlenkamp C, Galindo-Lagunas KA, Guan Z, Vinuesa P, Robinson S, Thomas-Oates J, Raetz CR, Geiger O. The lipid lysyl-phosphatidylglycerol is present in membranes of Rhizobium tropici CIAT899 and confers increased resistance to polymyxin B under acidic growth conditions. Mol Plant Microbe Interact. 2007;20:1421–1430. doi: 10.1094/MPMI-20-11-1421. [DOI] [PubMed] [Google Scholar]
- Staubitz P, Neumann H, Schneider T, Wiedemann I, Peschel A. MprF-mediated biosynthesis of lysylphosphatidylglycerol, an important determinant in staphylococcal defensin resistance. FEMS Microbiol Lett. 2004;231:67–71. doi: 10.1016/S0378-1097(03)00921-2. [DOI] [PubMed] [Google Scholar]
- Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Swairjo MA, Otero FJ, Yang XL, Lovato MA, Skene RJ, McRee DE, Ribas de Pouplana L, Schimmel P. Alanyl-tRNA synthetase crystal structure and design for acceptor-stem recognition. Mol Cell. 2004;13:829–841. doi: 10.1016/s1097-2765(04)00126-1. [DOI] [PubMed] [Google Scholar]
- Thedieck K, Hain T, Mohamed W, Tindall BJ, Nimtz M, Chakraborty T, Wehland J, Jänsch L. The MprF protein is required for lysinylation of phospholipids in listerial membranes and confers resistance to cationic antimicrobial peptides (CAMPs) on Listeria monocytogenes. Mol Microbiol. 2006;62:1325–1339. doi: 10.1111/j.1365-2958.2006.05452.x. [DOI] [PubMed] [Google Scholar]
- Xie W, Liu X, Huang RH. Chemical trapping and crystal structure of a catalytic tRNA guanine transglycosylase covalent intermediate. Nat Struct Biol. 2003;10:781–788. doi: 10.1038/nsb976. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.