

Abstract
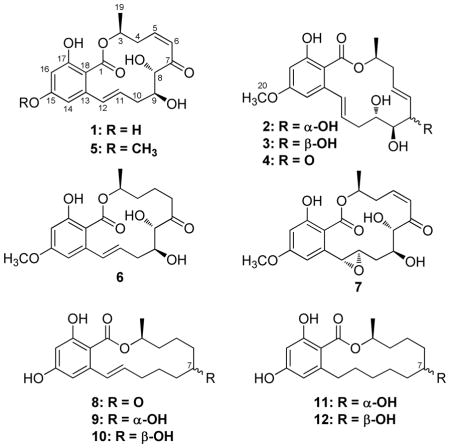
As part of our ongoing investigation of filamentous fungi for anticancer leads, an active fungal extract was identified from the Mycosynthetix library (MSX 63935; related to Phoma sp.). The initial extract exhibited cytotoxic activity against the H460 (human non-small cell lung carcinoma) and SF268 (human astrocytoma) cell lines and was selected for further study. Bioactivity-directed fractionation yielded resorcylic acid lactones (RALs) 1 (a new natural product) and 3 (a new compound) and the known RALs zeaenol (2), 5E-7-oxozeaenol (4), 5Z-7-oxozeaenol (5) and LL-Z1640-1 (6). Reduction of 5E-7-oxozeaenol (4) with sodium borohydride produced 3, which allowed assignment of the absolute configuration of 3. Other known resorcylic acid lactones (7–12) were purchased and assayed in parallel for cytotoxicity with isolated 1–6 to investigate structure-activity relationships in the series. Moreover, the isolated compounds (1–6) were examined for activity in a suite of biological assays, including antibacterial, mitochondria transmembrane potential, and NF-κB. In the latter assay, compounds 1 and 5 displayed sub-micromolar activities that were on par with the positive control, and as such, these compounds may serve as a lead scaffold for future medicinal chemistry studies.
In one component of our collaborative project to identify anticancer leads from diverse natural product study materials,1,2 extracts of filamentous fungi from the Mycosynthetix library, representing over 50,000 accessions, are being investigated systematically. Of the estimated 1.5 million species of fungi in the world, only about 75,000 have been described in the literature,3 and it is likely that an even smaller percentage have been pursued for bioactive secondary metabolites. Yet, many of the pharmaceutical breakthroughs of the 20th century originated in fungal cultures, including the well known examples of antibiotics (i.e. penicillin),4 immunosuppressants (e.g. cyclosporin A),5 and cholesterol lowering agents (e.g. compactin).6 The recently approved natural product analog, fingolimod,7 for the treatment of multiple sclerosis, and a clinical trial on psilocybin for terminally ill cancer patients,8 are but two examples that demonstrate contemporary interest in fungal secondary metabolites. Moreover, recent genomic data supports the potential for fungi to biosynthesize a wealth of compounds.9 In short, and as is often reported in this journal,10 fungi from many different sources continue to be a valuable resource of bioactive secondary metabolites.11
An extract of a filamentous fungus, MSX 63935, isolated from leaf litter collected in Nigeria, displayed promising cytotoxic activity against a human tumor panel.12,13 Previous research on this fungal isolate has examined a variety of biological targets, including assays for methicillin-resistant Staphylococcus aureus (MRSA), tuberculosis (TB), gene regulation, activity in the CNS, and various agricultural pests. However the only positive result was against MRSA and no antibiotic compounds had been isolated.14 Thus, these new cytotoxicity results spawned a bioactivity-directed fractionation study, and a series of resorcylic acid lactones (RALs; 1–6) were isolated and characterized, including a new natural product, 15-O-desmethyl-5Z-7-oxozeaenol (1), and a new compound, 7-epi-zeaenol (3). In conjunction with structurally related analogues purchased from commercial sources, these compounds were evaluated against the human tumor panel, yielding preliminary structure-activity relationship data. The isolated compounds (1–6) were examined also for antibacterial activity, as well as activity in mitochondrial transmembrane potential and NF-κB inhibition assays. The NF-κB inhibition assay revealed promising hits (compounds 1 and 5), both of which had sub-micromolar activities.
Results and Discussion
Through the ongoing exploration of extracts of filamentous fungi for cytotoxicity,1 fungus MSX 63935 showed promising activity (i.e. >96% inhibition of H460 cell growth at 20 μg/mL). The solid phase culture was scaled up to a 2.8 L Fernbach flask, and a 1:1 CHCl3/MeOH extract was generated, which was defatted with a 3:3:4 CH3CN:MeOH:hexanes partition. The resultant organic-soluble material was a white solid, which consisted almost exclusively of compounds 1–6 (see Figure S1 in Supporting Information for an analytical HPLC of this solid). In fact, of all the anticancer leads examined from this library of filamentous fungi over the past three years, MSX 63935 was one of the most proficient producers of small molecules studied to date. This white solid was advanced directly to preparative scale RP-HPLC to isolate all six compounds in >95% purity, as measured by analytical HPLC. Compound 3 was a new compound, 1 was a new natural product, and 2, 4, 5, and 6 were known compounds. Of these, the production of 5 was quite high, in excess of 800 mg from a solid phase culture grown in a 2.8 L Fernbach flask.
The molecular formula for the major compound (5) was C19H22O7, as determined via HRESIMS. The NMR data, in conjunction with the HRMS data and UV maxima (from the PDA detector of the HPLC) at 234, 274, and 314 nm indicated 5 to be the known compound 5Z-7-oxozeaenol (also known as LL-Z1640-2),15,16 first described in 1978 by Ellestad and co-workers.17 The [α]D of 5 was −83, compared to the literature value of −76, indicating the absolute configuration of 5 to be the same as LL-Z1640-2.16 A reference standard was purchased, and the 1H NMR data were identical to the isolated compound (Table S1). The absolute configuration of 5 had been established by synthesis.15,16
The molecular formula for 1 was C18H20O7, as determined via HRESIMS. The 1H NMR spectra of 1 and 5 were very similar (see Tables 1 and S1). The major difference between the NMR data of 1 and 5 was the absence of the methoxy signals at δH/δC 3.80/55.5 in 1 that were present in 5. A new phenol peak was observed at δH 10.39 in 1, further supporting the absence of a methoxy moiety in 1 relative to 5. Compound 1 has been described previously as an intermediate in the synthesis of 5,16 however, it was ascribed the trivial name 15-O-desmethyl-5Z-7-oxozeaenol, since it had not been described from nature previously. The absolute configuration of 1 had been established previously by synthesis.16
Table 1.
NMR Data for Compounds 1 and 3 (700 MHz for 1H, 176 MHz for 13C; chemical shifts in δ, coupling constants in Hz, DMSO-d6)
| Position | 15-O-Desmethyl-5Z-7-oxozeaenol (1) | 7-epi-Zeaenol (3) | ||
|---|---|---|---|---|
| δC | δH mult (J in Hz) | δC | δH mult (J in Hz) | |
| 1 | 170.6 | --- | 169.0 | --- |
| 3 | 73.6 | 5.17, qdd (5.7, 12.4, 2.1) | 71.8 | 5.09, qdd (6.1, 12.4, 2.1) |
| 4a | 36.2 | 2.56, m | 38.6 | 2.35, ddd (15.1, 9.6, 8.9) |
| 4b | 36.2 | 3.19, m | 38.6 | 2.43, m |
| 5 | 142.8 | 6.17, ddd (11.5, 11.5, 2.8) | 127.1 | 5.54, ddd (15.8, 8.2, 4.1) |
| 6 | 126.6 | 6.43, dd (11.5, 2.1) | 133.1 | 5.60, dd (15.8, 6.9) |
| 7 | 200.8 | --- | 74.7 | 4.04, m |
| 8 | 81.6 | 4.39, dd (4.4, 2.1) | 77.6 | 3.44, m |
| 9 | 72.6 | 3.84, m | 72.8 | 3.57, m |
| 10a | 36.0 | 1.98, m | 36.8 | 2.14, m |
| 10b | 36.0 | 2.02, m | 36.8 | 2.52, m |
| 11 | 131.6 | 5.95, ddd (15.3, 10.3, 4.8) | 132.5 | 6.11, ddd (15.8, 7.6, 4.8) |
| 12 | 131.3 | 6.76, d (15.3) | 128.5 | 6.59, d (15.8) |
| 13 | 143.8 | --- | 139.5 | --- |
| 14 | 107.4 | 6.29, d (2.8) | 102.5 | 6.43, d (2.3) |
| 15 | 162.3 | --- | 161.6 | --- |
| 16 | 101.5 | 6.19, d (2.8) | 99.9 | 6.31, d (2.3) |
| 17 | 164.1 | --- | 158.8 | --- |
| 18 | 102.5 | --- | 110.5 | --- |
| 19 | 20.0 | 1.36, d (5.7) | 20.5 | 1.30, d (6.1) |
| 20 | --- | --- | 55.3 | 3.74, s |
| 7-OH | --- | --- | --- | 4.83, br s |
| 8-OH | --- | 4.98, d (4.4) | --- | 4.57, br d (3.9) |
| 9-OH | --- | 5.08, d (5.7) | --- | 4.89, br s |
| 15-OH | --- | 10.39, s | --- | --- |
| 17-OH | --- | 11.82, s | --- | 10.53, s |
The HRESIMS data of compound 2 corresponded to a formula of C19H24O7, indicative of the addition of two protons relative to 5. The 1H NMR spectrum of 2 suggested the identity of 2 as zeaenol, and comparison of the NMR data to literature confirmed this assignment.18 Moreover, the [α]D of 2 was −93, which matched literature data exactly.18 The absolute configuration of 2 had been established previously by X-ray diffraction.18
Compound 3 was isolated as a white powder, and the HRESIMS data also suggested a formula of C19H24O7 as with 2. The 1H NMR data of 3 were similar to 2, however, there were numerous signals that had shifted slightly compared to 2 (see Figures S4 and S5 for 1H and 13C NMR spectra, respectively, of 3). The COSY spectrum of 3 indicated the same carbon skeleton and oxygenation pattern as 2, suggesting that 2 and 3 were diastereomers.
A recent report described a C-7 epimer (C-6′ in their numbering scheme) of aigialomycin B, which differs from 2 by the presence of an epoxide at C-11/C-12 (C-1′/C-2′ in their numbering scheme) instead of an alkene.19 The same report described both C-7 epimers of 5,6-dihydrozeaenol. Hence, given these literature precedents and the 2D-NMR data indicating that 3 and 2 shared an identical skeleton and oxygenation pattern, it was hypothesized that 3 was the C-7 epimer of 2. The reduction of the C-7 ketone of 5E-7-oxozeaenol (4; described below) was performed using sodium borohydride. The major product of the reduction of 4 had the identical retention time and UV spectrum as 3. Moreover, after isolation of the major reduction product by preparative HPLC, the 1H NMR data were identical to that of isolated 3, and thus, the configuration at C-7 for 3 was established as R. That 3 was the major product of the sodium borohydride reduction of 4 was somewhat surprising, as sodium borohydride is known to give mixtures of 1,4- and 1,2-reductions of α,β-unsaturated ketones; only a trace amount of 2 was detected by HPLC in the sodium borohydride reduction of 4. Regardless, these data supported the assignment of 3 as 7-epi-zeaenol.
Compound 4 was isolated as a white powder. HRESIMS data indicated a formula identical to 5 of C19H22O7. NMR data established 4 as the known compound 5E-7-oxozeaenol.18 To the best of our knowledge, the NMR data for 4 have not been reported previously, and thus they are summarized in Table S1. The [α]D value does not appear to have been reported either (see Experimental). Moreover, since the acetonide of 4 has been reported, it was synthesized, and the 1H NMR data were in excellent agreement with literature data.18
Compound 6 was isolated as a white powder. The HRESIMS data indicated a formula identical to 2 and 3 of C19H24O7. The NMR data for 6 matched that of LL-Z1640-1.18 The [α]D of 6 was measured as −78, which compared favorably with the literature value of −81.18 There appears to some confusion in the literature about the absolute configuration of 6, which was determined initially by X-ray diffraction.17 Sugawara et al.18 later reported position C-8 as having an R configuration, whereas the X-ray studies17 indicated C-8 as S; the latter assignment seems to be consistent based on biosynthetic comparisons to the other isolated compounds.
Compounds 1–6, along with other known RALs hypothemycin (7), zearalenone (8), α-zearalenol (9), β-zearalenol (10), α-zearalanol (11), and β-zearalanol (12) were assayed against the three cancer cell lines MCF-7, H460, and SF268. The IC50 results for 1–12 are summarized in Table 2. From these, it was clear that the enone (i.e. compounds 1, 4, 5, and 7), but not exclusively a cis-enone (i.e. note 4), was required for cytotoxic activity. A recent review highlights some of the biology of the resorcylic acid lactones,20 and they are known to inhibit with nanomolar potency the ATPase activity of two critical classes of proteins: the HSP90 chaperone21,22 and a subset of 16 kinases possessing a crucial cysteine residue in their ATPase binding pocket.23 HSP90 client proteins include several oncogenic proteins, such as Src kinase, and HSP90 inhibition results in the dissociation and degradation of these proteins. The activity of the new members of this class of polyketides, described herein, toward these targets remains to be investigated.
Table 2.
Cytotoxicity of Compounds Isolated from MSX 63935 (1–6) and Purchased Standards with Similar Structural Features (7–12) against a Panel of Human Tumor Cell Lines.
| compound | IC50 values (in μM)a |
||||
|---|---|---|---|---|---|
| MCF-7 | H460 | SF268 | HT-29 | MDA-MB-435 | |
| 15-O-desmethyl-5Z-7-oxozeaenol (1) | 10.3 | 4.1 | 8.9 | 11.5 | 15.8 |
| zeaenol (2) | >100 | >100 | >100 | >25 | >25 |
| 7-epi-zeaenol (3) | >100 | >100 | >100 | >25 | >25 |
| 5E-7-oxozeaenol (4) | 4.9 | 1.2 | 5.6 | 4.4 | 5.5 |
| 5Z-7-oxozeaenol (5) | 3.8 | 1.2 | 5.1 | 3.6 | 3.3 |
| LL-Z1640-1 (6) | >50 | >25 | >50 | >50 | >50 |
| hypothemycin (7) | 16.3 | 4.3 | 12.7 | ntc | nt |
| zearalenone (8) | >25 | >25 | >50 | nt | nt |
| α-zearalenol (9) | >25 | >25 | >50 | nt | nt |
| β-zearalenol (10) | >25 | 15.4 | >50 | nt | nt |
| α-zearalanol (11) | >50 | >50 | >50 | nt | nt |
| β-zearalanol (12) | >25 | 9.5 | >25 | nt | nt |
| Camptothecinb | 0.07 | <0.01 | 0.04 | nt | nt |
| Silvestrolb | nt | nt | nt | 0.004 | 0.006 |
Compounds 1–6 were examined for activity in a series of three other assays. With respect to NF-κB inhibitory activity, compound 5 (Table 3) was the most potent, being approximately a halforder of magnitude less potent than the positive control (rocaglamide). Previously, 5 was reported as a potent inhibitor of TAK1, which is a member of the mitogen-activated kinase kinase kinase (MEKK) family that participates in proinflammatory cellular signaling pathways by activating NF-κB.24 As with the cytotoxicity data, the enone moiety seemed to enhance potency, and in this case, the cis relationship (i.e. compounds 1 and 5) was more potent than the trans relationship (i.e. compound 4). In an assay for mitochondrial transmembrane potential (MTP), all compounds displayed IC50 values greater than 10 μM. Since the positive control for this assay, staurosporine, had an IC50 value in the single digit nanomolar range, 1–6 were considered inactive. Finally, compounds 1–6 were examined and found inactive against Escherichia coli, Staphylococcus aureus, Mycobacterium smegmatis, and Bacillus subtilis; MIC values were all > 500 μg/mL.
Table 3.
NF-κB Inhibitory Activity of Compounds Isolated from MSX 63935 (1–6).
| compound | IC50 (μM) |
|---|---|
| 15-O-desmethyl-5Z-7-oxozeaenol (1) | 0.75 |
| zeaenol (2) | >50 |
| 7-epi-zeaenol (3) | >50 |
| 7-oxozeaenol (4) | 11.5 |
| 5Z-7-oxozeaenol (5) | 0.24 |
| LL-Z1640-1 (6) | >50 |
| rocaglamide (positive control) | 0.075 |
In summary, a series of six structurally-related resorcylic acid lactones (1–6) were isolated from MSX 63935. Data in both the cytotoxicity assays and the NF-κB assay suggest that compound 5 was the most promising. Fortuitously, this fungus was a prolific producer of 5, generating over 800 mg per solid phase culture in a 2.8 L Fernbach flask, using standard techniques prior to any growth optimization studies. As such, studies are ongoing to probe the structure-activity relationships further by using 5 as starting materials for medicinal chemistry studies and by modifying the fermentation procedures to generate various analogues.
Experimental Section
General Experimental Procedures
Optical rotations, UV spectra, and IR spectra were obtained on a Rudolph Research Autopol III polarimeter, a Varian Cary 100 Bio UV-vis spectrophotometer, and a Perkin-Elmer Spectrum One with Universal ATR attachment, respectively. NMR experiments were conducted in either DMSO-d6 or CDCl3 with TMS as reference. NMR instrumentation was a Bruker Ultrashield Plus with Avance III console, Topspin software version 2.1, and a QNP style Cryoprobe (operating at 700.13 MHz for 1H, 176.05 MHz for 13C). A JEOL ECA-500 (operating at 500 MHz for 1H, 125 MHz for 13C) was also used for some experiments. HRESIMS was performed on a Waters SYNAPT MS system. HPLC was carried out on Varian Prostar HPLC systems equipped with Prostar 210 pumps and a Prostar 335 photodiode array detector (PDA), with data collected and analyzed using Galaxie Chromatography Workstation software (version 1.9.3.2). For preparative HPLC, a Gemini-NX C18 (5 μm; 250 × 21.2 mm) column was used at a 21.2 mL/min flow rate, while for analytical HPLC, a Gemini-NX C18 (5 μm; 150 × 4.6 mm) column was used with a 1 mL/min flow rate (both from Phenomenex, Inc.). Reference standards of 5Z-7-oxozeaenol (5) and zeaenol (2) were obtained from EMD Chemicals. Zearalenone (8), α-zearalenol (9), β-zearalenol (10), α-zearalanol (11), and β-zearalanol (12) were obtained from Sigma-Aldrich. Hypothemycin (7) was obtained from Enzo Life Sciences. All other reagents and solvents were obtained from Fisher Scientific and were used without further purification.
Producing Organism and Fermentation
Mycosynthetix fungal strain 63935 was isolated in 1992 by Dr. Barry Katz of MYCOsearch from leaf litter collected at the Agricultural Farms, University of Nigeria, Ede. DNA analyses were performed by MIDI Labs, Inc. (Newark, DE), and the D2 variable region of the Large Subunit (LSU) rRNA was sequenced and compared to their database; the closest match could only determine that this fungus was related to Phoma sp.; these data were deposited in Genbank (accession No. JF767207). The culture was stored on a malt extract slant and was transferred periodically. A fresh culture was grown on a similar slant, and a piece was transferred to a medium containing 2% soy peptone, 2% dextrose, and 1% yeast extract (YESD media). Following incubation (7 d) at 22 °C with agitation, the culture was used to inoculate 50 mL of a rice medium, prepared using rice to which was added a vitamin solution and twice the volume of rice with H2O, in a 250 mL Erlenmeyer flask. This was incubated at 22 °C until the culture showed good growth (approximately 14 d). The scale up culture was grown in a 2.8 L Fernbach flask containing 150 g rice and 300 mL H2O and was inoculated using a seed culture grown in YESD medium. This was incubated at 22 °C for 14 d.
Extraction and Isolation
To the large scale solid fermentation was added 500 mL of 1:1 MeOH/CHCl3. The mixture was shaken for 16 h then filtered, and the solvent was evaporated (3.7 g of waxy white solid). The extract was then partitioned between CH3CN/MeOH/hexanes (150/150/200 mL), and the CH3CN/MeOH solubles were separated and the solvent evaporated (1.7 g of white powder, Figure S1, Supporting Information). As such, it was dissolved in 5.5 mL DMSO and purified via seven separate injections by prep-HPLC using a gradient that initiated with 35:65 and increased linearly to 45:55 CH3CN:H2O over 40 min. Compound 1 eluted at ~7.6 min (5.2 mg), 2 at ~9 min (90.7 mg), 3 at ~11.8 min (5.6 mg), 4 at ~14 min (73.1 mg), and 5 and 6 eluted together from 16–20 minutes. The mixture of 5 and 6 was subjected to two further rounds of shave-and-recycle preparative HPLC under the same conditions to yield >95% pure 5 (885.9 mg) and 6 (80.5 mg).
15-O-Desmethyl-5Z-7-oxozeaenol (1)
white solid; [α]D 23 −59 (c 0.07, MeOH); UV (MeOH) λmax (log ε), 232 (4.35), 271 (3.89), 313 (3.62) nm; IR (diamond) νmax 3348, 3146, 2951, 1690, 1646, 1602, 1467, 1350, 1312, 1259, 1216, 1167, 1021, 965 cm−1; 1H NMR (DMSO-d6, 700 MHz), and 13C NMR (DMSO-d6, 175 MHz), see Table 1. HRESIMS m/z 371.1100 [M + Na]+, and 347.1122 [M - H]− (calcd for C18H20O7Na, 371.1107 and C18H19O7, 347.1131, respectively).
Zeaenol (2)
white solid; [α]D 23 −91.9 (c 1.00, MeOH); 1H and 13C NMR data were in good agreement with the literature.18 HRESIMS m/z 387.1413 [M + Na]+ and 363.1442 [M - H]− (calcd for C19H24O7Na, 387.1420, and C19H23O7, 363.1443, respectively).
7-epi-Zeaenol (3)
white solid; [α]D 23 −90.4 (c 0.25, MeOH); UV (MeOH) λmax (log ε), 235 (4.30), 270 (3.94), 312 (3.64) nm; IR (diamond) νmax 3376, 2965, 2930, 2899, 1636, 1602, 1571, 1353, 1312, 1248, 1212, 1157, 1041, 971 cm−1; 1H NMR (DMSO-d6, 700 MHz), and 13C NMR (DMSO-d6, 175 MHz), see Table 1. HRESIMS m/z 387.1410 [M + Na]+ and 363.1446 [M - H]− (calcd for C19H24O7Na, 387.1420, and C19H23O7, 363.1443, respectively).
5E-7-Oxozeaenol (4)
white solid; [α]D 23 = −27.0 (c 0.33, MeOH); 1H and 13C NMR (DMSO-d6, 500 MHz) data for 4, see Table 1. HRESIMS m/z 385.1257 [M + Na]+ and 361.1281 [M - H]− (calcd for C19H22O7Na, 385.1263, and C19H21O7, 361.1287, respectively).
5Z-7-Oxozeaenol (5)
white solid; [α]D 23 = −83.3 (c 0.34, MeOH); 1H and 13C NMR data were in good agreement with the literature.15,16 HRESIMS m/z 385.1252 [M + Na]+ and 361.1278 [M - H]− (calcd for C19H22O7Na, 385.1263, and C19H21O7, 361.1287, respectively).
LL-Z1640-1 (6)
white solid; [α]D 23 = −78.3 (c 0.33, MeOH); 1H and 13C NMR data were in good agreement with the literature.18 HRESIMS m/z 387.1410 [M + Na]+ and 363.1424 [M - H]− (calcd for C19H24O7Na, 387.1420, and C19H23O7, 363.1443, respectively).
Acetonide of 5E-7-oxozeaenol (4a)
To 1.54 mg of 4 was added 100 μL of 2,2-dimethoxypropane and 100 μL of acetone. A trace amount of pyridinium p-toluenesulfonate was added, and the solution was stirred for 2 h at 40°C. H2O (200 μL) was added to the solution, which was then injected directly onto semi-preparative HPLC (ODS-A, 250 × 10 mm i.d., 5 μm; YMC). The elution proceeded at 4 mL/min via a gradient that initiated with 50:50 and increased linearly over 20 min to 0:100 CH3CN:H2O. The acetonide eluted from 14.7–15.3 min and was collected to yield 1.47 mg of 4a (86% yield). The 1H NMR data of 4a were in excellent agreement with literature.18
Reduction of 4
5E-7-Oxozeaenol (4; 4.6 mg) was weighed into a 2 mL reaction vial, and 0.25 mL of both isopropanol and THF were added. The compound dissolved after brief stirring. NaBH4 (10.6 mg) was added with continued stirring. After 30 min, the solution had turned cloudy, and by analytical HPLC the presence of 4 was not discernable. H2O (0.5 mL) was added to this reaction product to dissolve all solids, and this solution was purified in a single step via prep-HPLC using the aforementioned conditions. The major peak was collected between 10–13 min (2.53 mg; 57 % yield) and was confirmed by 1H and 13C NMR to be identical to isolated 3.
Cytotoxicity Assay
The cytotoxicity measurements against the MCF-725 human breast carcinoma (Barbara A. Karmanos Cancer Center), NCI-H46026 human large cell lung carcinoma (HTB-177, American Type Culture Collection (ATCC), and SF-26827 human astrocytoma (NCI Developmental Therapeutics Program) cell lines were performed as described previously.13 Moreover, a second cytotoxicity assay was performed on only the isolated compounds using the HT-2928 human colorectal adenocarcinoma (HTB-38, ATCC) and the MDA-MB-43529 human melanoma (HTB-129, ATCC) cell lines. All cells were propagated in RPMI 1640 medium supplemented with fetal bovine serum (10%), penicillin (100 units/mL), and streptomycin (100 μg/mL). Cells were plated at either 2,500 (HT-29) or 5,000 (MCF-7, MDA-MB-435) cells per well in 96-well microtiter plates and incubated for 16 h in a humidified, 5% CO2 atmosphere at 37 °C. Test substance was then added at the following final concentrations: 25, 5, 1, 0.2 and 0.04 μg/mL. After a 72-hour incubation, an MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3- carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt) assay was performed using a commercially available kit according to the manufacturer’s instructions (CellTiter 96 AQueous One Solution Cell Proliferation Assay, Promega Corp).
Antimicrobial Assay
Antibiotic activity was examined using an agar plate diffusion assay. Overnight cultures of Escherichia coli, Staphylococcus aureus, Mycobacterium smegmatis, and Bacillus subtilis were used to inoculate molten LB media or Middlebrook 7H9 media (Difco) with 1% glycerol, containing 1.5% agar and kept at 50 °C; these were then used to prepare assay plates. Samples (dissolved in 10 μL MeOH) were applied to the surface of the assay dish, and positive controls were treated in a similar manner (penicillin G, novobiocin, and streptomycin; all from Sigma). The bioassay plates were incubated overnight at 37 °C. Biological activity of the standards could be detected to 1 μg/mL (except that penicillin G was active against E. coli at 100 μg/mL only), whereas the test compounds showed no activity at 500 μg/mL and were thus deemed inactive.
NF-κB p65 Assay
An ELISA based NF-κB inhibitory assay was performed as described previously.30 Briefly, HeLa cells (ATCC CCL-2) were treated with various concentrations of test compounds, positive control, or solvent control, and their nuclei extracted using the NE-PER kit (Pierce Biotechnology). The specific binding ability of activated p65 subunits of NF-κB in the nucleus was detected according to the manufacturer’s protocol (EZ-Detect Transcription Factor Assay System ELISA kit, Pierce). Rocaglamide (Enzo Life Sciences International, Inc.,) was used as a positive control (IC50 value of 0.075 μM).
Mitochondria Transmembrane Potential (ΔΨ) Assay
The mitochondrial transmembrane potential assay kit (Cayman Chemical Company) was adapted to detect the ΔΨ using a procedure published previously.31 ΔΨ is used to represent mitochondrial membrane transition events. Briefly, HT-29 cells were treated with various concentrations of the test compounds or positive control. Then, cells were incubated with the lipophilic cationic dye 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzymidazolylcarbocyanide (JC-1; Cayman Chemical Company). After incubation, cells were rinsed with a wash buffer to remove unbound staining reagent. Samples were analyzed by a FLUOstar Optima fluorescence plate reader (BMG Labtech, Inc.) using an excitation wavelength of 485 nm and emission wavelength of 530 nm for JC-1 monomers from apoptotic cells and an excitation wavelength of 560 nm and emission wavelength of 595 nm for JC-1-aggregates from healthy cells. Measurements were performed in triplicate and are representative of at least two independent experiments. For this assay, staurosporine (Cayman Chemical Company, Ann Arbor, MI) was used as a positive control (IC50 value of 2.5 nM).
Supplementary Material
Acknowledgments
This research was supported by P01 CA125066 from the National Cancer Institute/National Institutes of Health, Bethesda, MD, USA. The Golden LEAF Foundation (Rocky Mount, NC) provided partial support to D. J. K. Mycology technical support was provided by Blaise Darveaux and Maurica Lawrence. The authors thank Dr. K. Knagge and M. Su, both of the David H. Murdock Research Institute, Kannapolis, NC, for some NMR and mass spectrometry data and Dr. J. Fuchs (Ohio State University) for helpful suggestions.
Footnotes
Supporting Information Available: 1H NMR spectra for compounds 1–6, 13C NMR spectra for compounds 1 and 3, and an HPLC chromatogram for the crude extract of MSX 63935. This information is available free-of-charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Kinghorn AD, Carache de Blanco EJ, Chai HB, Orjala J, Farnsworth NR, Soejarto DD, Oberlies NH, Wani MC, Kroll DJ, Pearce CJ, Swanson SM, Kramer RA, Rose WC, Fairchild CR, Vite GD, Emanuel S, Jarjoura D, Cope FO. Pure Appl Chem. 2009;81:1051–1063. doi: 10.1351/PAC-CON-08-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Orjala J, Oberlies NH, Pearce CJ, Swanson SM, Kinghorn AD. In: Bioactive Compounds from Natural Sources. 2. Tringali C, editor. Taylor & Francis; London: accepted. [Google Scholar]
- 3.Hawksworth DL, Rossman AY. Phytopathology. 1997;87:888–891. doi: 10.1094/PHYTO.1997.87.9.888. [DOI] [PubMed] [Google Scholar]
- 4.Fleming A. Penicillin, Its Practical Application. Butterworth & co. ltd; London: 1946. p. 380. [Google Scholar]
- 5.Dreyfus M, Harri E, Hofmann H, Pache W, Tscherter H. Eur J Appl Microbiol. 1976;3:125–133. [Google Scholar]
- 6.Brown AG, Smale TC, King TJ, Hasenkamp R, Thompson RH. J Chem Soc, Perkin Trans 1. 1976;11:1165–1170. [PubMed] [Google Scholar]
- 7.Brinkmann V, Billich A, Baumruker T, Heining P, Schmouder R, Francis G, Aradhye S, Burtin P. Nat Rev Drug Discov. 2010;9:883–897. doi: 10.1038/nrd3248. [DOI] [PubMed] [Google Scholar]
- 8.Griffiths R, Richards W, Johnson M, McCann U, Jesse R. J Psychopharmacol. 2008;22:621–632. doi: 10.1177/0269881108094300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Misiek M, Hoffmeister D. Planta Med. 2007;73:103–115. doi: 10.1055/s-2007-967104. [DOI] [PubMed] [Google Scholar]
- 10.The use of the term ‘fungus’ in titles of articles published in the Journal of Natural Products has increased steadily over the last 10 years, from appearing 12 times in 2000 to appearing 31 times in 2009; at the time of manuscript submission, the term was on target to increase to nearly 40 times in 2010.
- 11.Pearce C, Eckard P, Gruen-Wollny I, Hanske FG. In: Natural Product Chemistry for Drug Discovery. Buss AD, Butler MS, editors. The Royal Society of Chemistry; Cambridge: 2010. pp. 215–244. [Google Scholar]
- 12.Alali FQ, El-Elimat T, Li C, Qandil A, Alkofahi A, Tawaha K, Burgess JP, Nakanishi Y, Kroll DJ, Navarro HA, Falkinham JO, III, Wani MC, Oberlies NH. J Nat Prod. 2005;68:173–178. doi: 10.1021/np0496587. [DOI] [PubMed] [Google Scholar]
- 13.Li C, Lee D, Graf TN, Phifer SS, Nakanishi Y, Riswan S, Setyowati FM, Saribi AM, Soejarto DD, Farnsworth NR, Falkinham JO, III, Kroll DJ, Kinghorn AD, Wani MC, Oberlies NH. J Nat Prod. 2009;72:1949–1953. doi: 10.1021/np900572g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.These previous studies were retrieved from the Mycosynthetix database, which covers previous research on these organisms going back over 30 years.
- 15.Tatsuta K, Takano S, Sato T, Nakano S. Chem Lett. 2001:172–173. [Google Scholar]
- 16.Dakas PY, Jogireddy R, Valot G, Barluenga S, Winssinger N. Chemistry. 2009;15:11490–11497. doi: 10.1002/chem.200901373. [DOI] [PubMed] [Google Scholar]
- 17.Ellestad GA, Lovell FM, Perkinson NA, Hargreaves RT, McGahren WJ. J Org Chem. 1978;43:2339–2343. [Google Scholar]
- 18.Sugawara F, Kim KW, Kobayashi K, Uzawa J, Yoshida S, Murofushi N, Takahashi N, Strobel GA. Phytochemistry. 1992;31:1987–1990. [Google Scholar]
- 19.Xu LX, He ZX, Xue JH, Chen XP, Wei XY. J Nat Prod. 2010;73:885–889. doi: 10.1021/np900853n. [DOI] [PubMed] [Google Scholar]
- 20.Winssinger N, Barluenga S. Chem Comm. 2007;22–36 doi: 10.1039/b610344h. [DOI] [PubMed] [Google Scholar]
- 21.Schulte TW, Akinaga S, Soga S, Sullivan W, Stensgard B, Toft D, Neckers LM. Cell Stress Chaperones. 1998;3:100–108. doi: 10.1379/1466-1268(1998)003<0100:arbttn>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharma SV, Agatsuma T, Nakano H. Oncogene. 1998;16:2639–2645. doi: 10.1038/sj.onc.1201790. [DOI] [PubMed] [Google Scholar]
- 23.Schirmer A, Kennedy J, Murli S, Reid R, Santi DV. Proc Natl Acad Sci USA. 2006;103:4234–4239. doi: 10.1073/pnas.0600445103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ninomiya-Tsuji J, Kajino T, Ono K, Ohtomo T, Matsumoto M, Shiina M, Mihara M, Tsuchiya M, Matsumoto K. J Biol Chem. 2003;278:18485–18490. doi: 10.1074/jbc.M207453200. [DOI] [PubMed] [Google Scholar]
- 25.Soule HD, Vazguez J, Long A, Albert S, Brennan M. J Natl Cancer Inst. 1973;51:1409–1416. doi: 10.1093/jnci/51.5.1409. [DOI] [PubMed] [Google Scholar]
- 26.Carney DN, Gazdar AF, Bunn PA, Jr, Guccion JG. Stem Cells. 1982;1:149–164. [PubMed] [Google Scholar]
- 27.Rosenblum ML, Gerosa MA, Wilson CB, Barger GR, Pertuiset BF, de Tribolet N, Dougherty DV. J Neurosurg. 1983;58:170–176. doi: 10.3171/jns.1983.58.2.0170. [DOI] [PubMed] [Google Scholar]
- 28.Fogh J, Trempe G. Human Tumor Cells In Vitro. Plenum; New York: 1975. pp. 115–159. [Google Scholar]
- 29.Rae JM, Creighton CJ, Meck JM, Haddad BR, Johnson MD. Breast Cancer Res Treat. 2007;104:13–19. doi: 10.1007/s10549-006-9392-8. [DOI] [PubMed] [Google Scholar]
- 30.Salim AA, Pawlus AD, Chai HB, Farnsworth NR, Douglas Kinghorn A, Carcache-Blanco EJ. Bioorg Med Chem Lett. 2007;17:109–112. doi: 10.1016/j.bmcl.2006.09.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deng Y, Balunas MJ, Kim JA, Lantvit DD, Chin YW, Chai H, Sugiarso S, Kardono LB, Fong HH, Pezzuto JM, Swanson SM, de Blanco EJ, Kinghorn AD. J Nat Prod. 2009;72:1165–1169. doi: 10.1021/np9001724. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.