

Abstract
We study the enzymatic degradation of an elastic fiber under tension using an anisotropic random-walk model coupled with binding-unbinding reactions that weaken the fiber. The fiber is represented by a chain of elastic springs in series along which enzyme molecules can diffuse. Numerical simulations show that the fiber stiffness decreases exponentially with two distinct regimes. The time constant of the first regime decreases with increasing tension. Using a mean field calculation, we partition the time constant into geometrical, chemical and externally controllable factors, which is corroborated by the simulations. We incorporate the fiber model into a multiscale network model of the extracellular matrix and find that network effects do not mask the exponential decay of stiffness at the fiber level. To test these predictions, we measure the force relaxation of elastin sheets stretched to 20% uniaxial strain in the presence of elastase. The decay of force is exponential and the time constant is proportional to the inverse of enzyme concentration in agreement with model predictions. Furthermore, the fragment mass released into the bath during digestion is linearly related to enzyme concentration that is also borne out in the model. We conclude that in the complex extracellular matrix, feedback between the local rate of fiber digestion and the force the fiber carries acts to attenuate any spatial heterogeneity of digestion such that molecular processes manifest directly at the macroscale. Our findings can help better understand remodeling processes during development or in disease in which enzyme concentrations and/or mechanical forces become abnormal.
Keywords: diffusion, on rate, off rate, cleaving
The extracellular matrix (ECM), the biological structure that supports cells, is composed of elastic fibers such as elastin and collagen. The complex organization of these fibers undergoes a continuous maintenance that requires the catalytic action of enzymes, called proteases (1). In diseases, such as pulmonary emphysema, tissue destruction is thought to be a consequence of the imbalance between protease and antiprotease activity leading to degradation of elastin fibers (2). Biological tissues in vivo are also under tension that may interfere with the enzymatic activity. Indeed, mechanical stretch accelerates the rate of degradation of native ECM during elastase-induced digestion of lung tissue (3, 4), whereas it stabilizes type I collagen against in vitro digestion by collagenases (5, 6).
The elastic and failure properties of single fibers have important biological functions, and these material properties depend on the hierarchical organization of the molecular constituents (7). During digestion, both the molecules and the cross-links in the fiber can be cleaved by enzymes reducing fiber stiffness. Furthermore, following cleavage, an enzyme can unbind, diffuse, bind at a different location and cleave again. This leads to the question: How are the diffusion and binding processes of the enzyme and the subsequent degradation of the fiber affected by the presence of an external mechanical force?
Several different diffusion-reaction models have been used to describe processes at the level of ECM, cell membranes, macromolecules, and DNA (8–13). In addition, the random walk is often used as a diffusion model that takes into account the morphological details of the system. Spring network models also provide a useful framework for analyzing the changes in the mechanical properties of the ECM (14, 15).
Here, we study the decay of stiffness of a single fiber under tension during enzymatic digestion using an anisotropic random-walk model coupled with binding-unbinding reactions. The model takes into account the simultaneous effects of enzyme diffusion, binding and cleaving in the presence of mechanical forces. To compare the results with experiments, we construct a network of fibers as a macroscopic model of the digestion of an ECM tissue under tension. The prediction of the model agrees with experimental data obtained by measuring the decay of tensile force developed by elastin sheets in the presence of enzymes. Furthermore, the amount of fragments leaving the fibers during digestion is analyzed through numerical simulations and compared with experiments. Despite the complexity of the ECM and the diffusion and reaction processes, it is surprising how effectively the single fiber model describes the more complex macroscopic system.
Results and Discussion
We use a modified random walk to mimic anisotropic diffusion of enzyme particles along a fiber and study the digestion of the fiber represented by a chain of springs. Our model consists of a one-dimensional chain of Ns linearly elastic springs in series representing an elastic fiber as in Fig. 1A. The fiber is surrounded by two layers of sites along which particles representing enzymes can diffuse. Periodic boundary conditions are applied in the x direction. Both ends of the chain are subject to a constant force F that mimics tension in the fiber. To simulate enzyme activity along the fiber, we begin with a chain having identical initial spring constants k(t = 0) ≡ k0. The diffusion of enzymes is initiated by releasing a set of particles at random positions in the two layers surrounding the chain. The rules of the random-walk and reaction processes are as follows: (i) pd is the probability for a particle to move right or left parallel to the chain and is associated with diffusion. (ii) pon is the probability for a particle to move up from the bottom layer or down from the top layer. This step represents an enzyme molecule binding to a binding site on the fiber. Only one particle can be bound to a single spring at any time. Note that these probabilities are related by the constraint pon + 2pd = 1. (iii) poff is the probability for a bound particle to move up or down to the top or bottom layer, respectively. This is related to the unbinding step from the fiber. Once a bound particle unbinds, the local spring constant k is reduced by a constant factor γ, k → γk. The reason for this is as follows. The fiber is composed of molecules in parallel and series. If the fiber is a regular array of molecules, cleavage would decrease the number of parallel molecules and the local spring constant would decay as k → k - γ. This would result in a linear decrease in the spring constant during subsequent cleavages. In reality, the molecules overlap and are connected via cross-links in a complex way. Hence, the degradation of the fiber is also a complex process so that simply subtracting γ from k is not appropriate. We therefore model the cleavage process by assuming that the decrease in spring constant is proportional to the actual value, which corresponds to a multiplicative degradation process. This assumes that the process of degradation is far away from the rupture threshold.
Fig. 1.

(A) Schematic diagram of the chain of springs and binding sites used in the model. The binding sites on the springs and the two layers of sites are represented by small and big open circles, respectively. The enzyme particles are shown as filled black circles. The particle at the bottom layer can move up, left, or right whereas the particle at the top can move down, left, or right. The particle on the spring can move only up or down. (B) The binding probability pon defined by Eq. 1 and the diffusion probability pd are a function of the number of visits n at a fixed site. Lines of different styles correspond to Δp = 0.10 (solid lines), Δp = 0.16 (dotted lines), Δp = 0.20 (dashed lines), and Δp = 0.24 (dash-dotted lines). The lines above and below the horizontal dashed line at p = 0.333 correspond to pd and pon, respectively. The vertical dashed line represents the region where the isotropic behavior with p = 0.333 is reached.
The random walk is made anisotropic and time-dependent through the probability pon that depends on the local spring constant k,
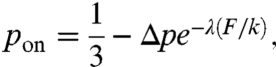 |
[1] |
where Δp is the initial anisotropy and 1/λ is a characteristic length. (Eq. 1) implies the following. Because F is constant, the local stretch increases and more binding sites appear (4, 16), which we represent by an increase in the local pon. This introduces anisotropy in the particle movement and makes the enzyme activity dependent on the local k. The parameter Δp is related to the difficulty of an enzyme to reach a binding site that depends on the surface roughness of the fiber. Fig. 1B shows that at the beginning of the diffusion, the number of times a spring has been visited by particles is n ≈ 1 and initial values of both pon and pd depend on Δp. As the diffusion progresses, pon slowly increases. Around n = 100, pon increases significantly until it approaches the isotropic value of pon = pd = 1/3. When n ≈ 800, the diffusion reaches the isotropic regime in which both diffusion and binding are equally likely. We assume that in this regime, locally the fiber is at its unfolding limit in that the number of binding sites remains constant.
The probability poff is determined by the molecular properties of the specific enzyme and its substrate and is the same for each spring for all times. A particle may remain bound for more than one time step, with probability 1 - poff. We assume that cleavage occurs during unbinding so that k is reduced only when the enzyme unbinds. Thus, k for each spring is a function of time and because it decreases as the spring is repeatedly visited by enzymes, we can write k(t) = γnk0, where n is the total number of visits by time t. The stiffness K(t) of the fiber is calculated as the equivalent stiffness of all Ns springs connected in series,
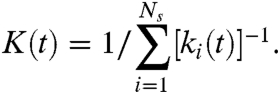 |
[2] |
Next, we study the evolution of K(t) for different parameters. We use a chain composed of Ns = 104 springs while varying the particle numbers (Np = 256, 512, 1024) and the external force F within the interval [0.1,2.5]. The Np and F are related to the experimentally controllable macroscopic parameters of the digestion process. At the microscopic level, we vary poff between [0.1,1.0] whereas Δp is chosen from the interval [0.10,0.24]. Additionally, there are three constant parameters: λ = 0.10, γ = 0.995, and k0 = 1. We simulate the time course of K(t) for t = 2 × 105 time steps. At each time step, we attempt to move all Np particles in the system. We repeat the simulations with 500 different realizations and average K(t) over all runs.
The results for 〈K(t)〉 are plotted on a log-linear scale in Fig. 2. In all cases, 〈K(t)〉 shows two distinct exponentially decreasing regimes with time constants T1 and T2 separated by a cross-over region around t×. For fixed Np, T1 decreases monotonically as F increases. The t× also decreases as a function of F leading to a faster degradation of the fiber. The situation is similar when Np is increased. As the degradation proceeds, the bound particles reach a steady state (see Fig. S1).
Fig. 2.

Log-linear plot of the average stiffness 〈K〉 as a function of diffusion time t for different values of F = 0.1, 0.5, 2.5 and Np = 256, 512 with Δp = 0.20 and poff = 0.5. The black solid line segments at the beginning and end of the simulations represent exponential fits to estimate the value of the time constants T1 and T2, respectively.
To characterize the microscopic properties of the fiber, we calculate the standard deviation 〈σk〉 of all spring constants at a fixed time and average them over all runs in Fig. 3A. Initially, for t ≪ t1, 〈σk〉 increases quickly, which is not influenced by F. When t ≈ 2000, F starts to affect the binding process according to (Eq. 1) and hence 〈σk〉 increases faster for higher F. When t is around t2, 〈σk〉 reaches its maximum followed by a slow decay. The behavior can be confirmed by looking at the spring constant distributions P(k) in Fig. 3B. The width of P(k) has a maximum at t2. For t < t2 and t > t2, P(k) becomes narrower. Also, the peak of P(k) decreases with increasing time.
Fig. 3.

(A) The standard deviation 〈σk〉 of the local spring constants k as a function of time for three values of F = 0.1, 0.5, 2.5 and Np = 512. In panel B, we plot the distribution of spring constants P(k) at time points ti (i = 1, 2, 3) as indicated in panel A for F = 0.1. In both graphs, we use the parameters Δp = 0.20 and poff = 0.5.
A mean field calculation of the rate change in fiber stiffness gives the following result (see Methods and Fig. S2):
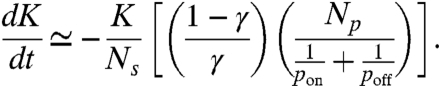 |
[3] |
Assuming that pon does not change during one time step, the solution of this equation is given by K(t) = e-t/T, where
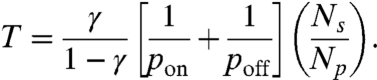 |
[4] |
The first term involves γ, which is related to the average number of molecules in parallel and hence the geometry of the fiber. The second term describes the specific enzyme activity at the microscopic level via the binding and unbinding probabilities. The third term is the inverse of enzyme concentration relative to the binding site density, an external control parameter. Note that the time constant T is symmetric in pon and poff. Thus, if we assume it is the unbinding process that depends on the external force, the results will be identical. Experiments are required to determine if pon, poff or both depend on external force.
The asymptotic limits of (Eq. 4) can be written as:
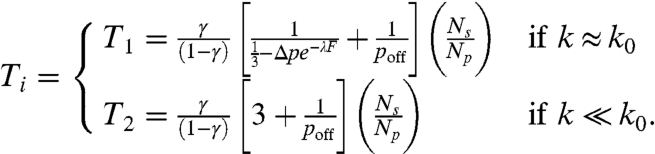 |
[5] |
To compare the numerical simulations with the analytical calculations in the asymptotic limits, we calculate the time constants in Fig. 2 for the two regimes of exponential behavior. The T1 and T2 are estimated from nonoverlapping windows of size Δt = 5,000 as a function of F and poff in the regions K(t) ≈ 1 and K(t) ≪ 1, respectively. The results are compared to the predictions of (Eq. 5) in Fig. 4. Generally, the numerical results confirm the analytical calculations both as a function of F and poff. The T1 decreases with increasing F whereas it diverges when poff becomes less than 1/3. Also, for fixed values of F and poff, T1 decreases with increasing enzyme concentration. Interestingly, F has little effect on T2. This can also be seen in Fig. 2: after the cross-over region, all curves follow the same exponential decay for different F at a fixed value of Np, but decay slower as Np decreases. The difference in T2 between analytical and numerical calculations for low Np is because K, in this limit, takes significantly more time to reach the second regime. We confirm this with additional simulations for Np = 256 for 3 × 105 time steps and find that T2 increases by about 15%, approaching the analytical results. Further simulations also suggest that for a fixed F and poff, T1 increases as Δp increases and T2 remains constant as a function of Δp.
Fig. 4.

Comparison of the mean field calculations and the numerical simulations. Time constants T1 and T2 are plotted as functions of F and poff for Np = 256 (circles), Np = 512 (squares), and Np = 1024 (triangles). The symbols correspond to the numerical simulations and the solid lines are obtained from Eq. 5. In panels A and B, T1 and T2 are shown as a function of F for a constant poff = 0.5. In panels C and D, T1 and T2 are shown as a function of poff for F = 1.0. In all graphs we used Δp = 0.20.
Because the ECM of biological tissues is composed of networks of fibers, we extend our fiber model to a multiscale network that helps understand which phenomena at the higher scale can be deduced from the properties of the fibers and which arise directly from network effects. We construct a random Voronoi network of springs each behaving like a single fiber described above. We take into account that the degradation of a fiber depends on the force it carries because its stiffness decays exponentially with a time constant T that is a function of the force on that fiber. At the macroscopic level, the network is first stretched uniaxially to 20% strain and the equilibrium configuration is determined. Digestion is then initiated by allowing the spring constants to decrease according to the force the springs carry (see Fig. S3). The global force F is computed as the sum of the components of the spring forces at the boundary of the network in the direction of the stretch. Because the network is heterogeneous, the distribution of local forces in the network creates heterogeneity in enzyme activity and hence degradation (see SI Text).
For comparison with experimental data, we normalize F by F0, the force measured before enzyme digestion is initiated (experiments) and before the relaxation process is started (simulations). The force F/F0 as a function of time is shown in Fig. 5A. The force decays exponentially with time constants T that strongly depend on Np. Thus, an important observation is that network effects do not alter the nature of the exponential behavior. This is in agreement with experimental data obtained by measuring the force relaxation in elastin sheets during digestion with elastase (Fig. 5B). Indeed, following a short time period over which the force does not decrease likely due to the diffusion of the enzyme into the tissue, the force shows a region of exponential decay for all three enzyme concentrations. For longer times, fluctuations arise in the experiments because the small forces developed by the tissue are close to the detection limit of the force transducer. These fluctuations are more pronounced for the higher concentrations and likely accompanied by fiber failure not considered in the model. To estimate T from these force curves, we identify the best regions that correspond to an exponential fit, F(t) = F0 exp-t/T, as indicated with straight lines in Fig. 5B. The dependence of T on enzyme concentration obtained from simulations and measured experimentally is displayed in Fig. 5 C and D, respectively. Whereas the magnitudes are not comparable, both results show that T linearly increases with the reciprocal of enzyme concentration. These results are also in agreement with the analytical calculations from Eq. 4.
Fig. 5.

Linear-log plot of the force normalized by the initial force F0 as a function of time for both simulations and experiments. In panel A we show the force calculated by numerical simulations in a network composed of fibers arranged in a Voronoi network. All curves present an exponential decay with a time constant that depends on the number of particles Np. In panel B we show experimental results for the measured force in three elastin sheets. All curves show an exponential decay with a time constant that depends on enzyme concentration. For the numerical results, F0 corresponds to the estimated value obtained from the numerical fit of the data calculated for a single fiber, whereas for the experimental data, F0 is the value of the force measured before addition of enzyme. In panels C and D we plot the time constant T as a function of the inverse of the number of particles Np and enzyme concentration c, respectively. In the simulations, the time constant T is measured in time step and calculated with different initial configurations of the Voronoi network, averaged over 10 runs. The error bars are smaller than the symbol. The experimental time constant T has units of seconds and was obtained from the average over five different experiments for each concentration. The error bars increase as the concentration decreases. In both panels C and D, the solid lines represent the best linear fit to the data with R2 values of 0.999 and 0.998, respectively.
Finally, as a last test of our model, we carry out further simulations to predict the amount of fiber fragments released during enzyme digestion. Because the time dependence of the released mass is difficult to measure, we calculate the fragment release as a function of time and enzyme concentration. Fig. 6 shows that following an initial transient, the fragment mass increases linearly with enzyme concentration at specific time points in excellent agreement with experimentally determined elastin fragment analysis (Inset).
Fig. 6.

The main panel shows the amount of mass M as a function of the number of particles Np. The M represents the sum over all fragments released from the fiber during the digestion process and calculated from numerical simulations on a single fiber. Each solid line corresponds to the increase in M with particle concentration for different fixed time points t during the digestion process. The inset shows the results from experiments: the fragment mass M released into the bath at the end of 60-min digestion with three different elastase concentrations (0.003, 0.006, and 0.012 μg/μL). Each point was obtained from analysis of at least five samples for each concentration. The solid line represents the best linear fit to the data with an R2 value of 0.999.
Conclusions
We have presented a model for the enzymatic digestion of an elastic fiber under tension. We have shown that fiber stiffness decreases exponentially with two regimes separated by a cross-over region. Whereas the first regime has been found experimentally (3), to our knowledge, the second one has not been measured and remains a prediction of the model. Each regime can be associated with an average value of the stiffness along the fiber during enzyme diffusion. In the first regime, the stiffness is dominated by the average local initial stiffness, whereas in the second regime, the stiffness has decreased significantly and almost uniformly throughout the fiber. In the cross-over region, the stiffness is controlled by a wide distribution of local stiffness values. The time constant T1 displays a strong dependence on both F and Np. Our analytical calculations confirm the presence of two regimes and show how the time constants can be partitioned into geometrical, chemical, and externally controllable factors. In the first regime, we expect that the fiber does not reach the failure limit, but after the cross-over, the decrease in stiffness is faster and failure is likely to occur. In future studies, a failure limit should be included in the model with avalanche-like cascades to predict failure stress and strain during digestion.
To predict tissue and organ behavior, we have incorporated the properties of the single fiber into a network creating a functional multiscale model that has become the most useful tool in characterizing the hierarchical organization of ECM structure (7). A surprising finding is that network effects do not dominate the behavior at the macroscopic scale. Instead, the exponential relaxation of the single fiber percolates through scales to dominate the macroscopic behavior. The reason is that the rate of stiffness decline is governed by the force on a fiber. Once the stiffness of a fiber decreases below the average, the force on the fiber decreases, which in turn slows down any further digestion. This negative feedback acts to homogenize spatial heterogeneity in the digestion process within the network (see Fig. S4) and hence macroscopically the average fiber property is observed. The implications are that measurements of the kinetics of digestion in a complex tissue can reveal phenomena at the molecular level.
Finally, the predictions of the model compare well with experimental measurement of the force relaxation in a macroscopic elastin sheet as well as the concentration dependence of released elastin fragments. These results can help better understand developmental growth and maintenance of the ECM as well as the progression of diseases in which enzyme concentrations are high and/or mechanical forces are abnormal such as in pulmonary emphysema or vessel aneurysm.
Methods
Analytical Calculations.
To gain insight into the exponential decay of fiber stiffness, we carry out a simple mean field calculation. The local k at time t depends on the number of times the spring has been visited. We define ti as the time corresponding to the i-th unbinding event along the entire chain. Thus, K at time ti can be written as
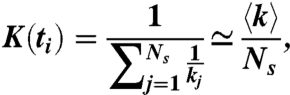 |
[6] |
assuming 〈1/k〉 ≃ 1/〈k〉. Notice that K remains constant for ti ≤ t < ti+1. At time ti+1, an unbinding event occurs at spring m and the corresponding km is reduced to γkm. The new value of K(ti+1) is
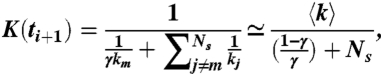 |
[7] |
where we also assume that km ≃ 〈k〉. Thus, from Eqs. 6 and 7, the change ΔK = K(ti+1) - K(ti) in the total stiffness is written as
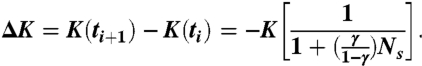 |
[8] |
Next, we consider the average waiting time 〈τ〉 between two unbinding events. During digestion, the number of particles nB that remains bound on the fiber changes, but nB is related to the number of free particles nF because nB + nF = Np. The rate of change of nB is the difference between the average binding rate ponnF and the average unbinding rate poffnB, which can be expressed as dnB/dt = ponnF - poffnB (see SI Text). Assuming that nB has reached a steady state dnB/dt = 0, we obtain nB = ponNp/(pon + poff). Because the unbinding probability is per unit time, 〈τ〉 can now be expressed as
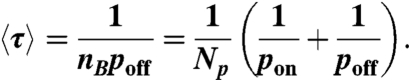 |
[9] |
Finally, we can establish a link between the two processes involved in enzymatic digestion. From Eq. 8, K is reduced by ΔK during the interval Δt = ti+1 - ti. We thus approximate the derivative of K by the discrete change ΔK during the interval Δt ≃ 〈τ〉 as
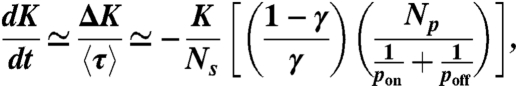 |
[10] |
where we have ignored the 1 in the denominator of Eq. 8. This equation can be solved assuming that pon is approximately constant during one time step.
Numerical Calculation of Fragment Release.
Because the fragments released by the digestion into the bath can be measured experimentally, we carry out simulations to predict how the fragment mass released varies with time and particle concentration Np. We assume that the fiber is composed of many molecules arranged such that enzymes can cleave the molecules and fragments of the fibers are released without separating the fiber into two pieces. To mimic this process, we define a fragment as follows. After a specific spring on the chain has been visited, we examine whether the left or right neighboring spring has been visited the same number of times. The spring that meets this condition constitutes a fragment having unit mass. We continue counting the fragments as the diffusion and reaction processes proceed and the total fragment mass is defined as the sum of all fragments that leave the fiber. We run 500 simulations for each Np and a fixed strain of 20% as in the experiments.
Network Model.
To better understand how the relations derived for the decay in stiffness of a single fiber relate to the decrease in stiffness of an elastin sheet held under fixed strain while being digested by elastase, we consider a two-dimensional (2D) network of fibers held under a fixed strain. To mimic the geometry of the real elastin sheet where the fibers are arranged in a random orientation, we consider a network in which a rectangle representing the elastin sheet is split into Voronoi cells.
We start with a set of points arranged in a regular triangular lattice with line element length L. The position of each of these points is perturbed by adding to its x and y coordinates a random number r that is uniformly distributed between 0 and L. The Voronoi diagram corresponding to this lattice is then computed. When r = 0, the resulting Voronoi will be a perfect hexagonal lattice. As r increases, the resulting network deviates from a regular geometry and becomes a random network. Each line element in this network represents a single fiber made of springs as shown in Fig. 1A.
Initially, all springs in the network are assigned the same stiffness and the entire network is stretched uniaxially in the x direction and the sides corresponding to max(x) and min(x) are fixed in x and y directions. The rest of the network is allowed to move in both x and y directions. The equilibrium configuration of the stretched network is calculated by minimizing the total energy of the network using a steepest descent algorithm. From the single fiber model, we know that the stiffness of each fiber in the network decays exponentially with a time constant T that is a function of the force carried by the fiber, f, the number of enzyme particles, Np, and the rate of decay in stiffness in the absence of force, T0. We obtain this relationship by fitting the T1-F curves in Fig. 4 with low order polynomials, T1 = ϕ(f,Np,T0), which are then used to control the degradation process separately for each fiber in the network. We simulate the decay in stiffness in steps of dt, where at each step every spring constant in the network is decreased as K(t + 1) = K(t)(1 - dt/T1), where T1 varies for each spring depending on the force it carries. The network is then allowed to reach equilibrium before the next time step begins.
Experiments
Tissue Culture.
The ECM constructs containing principally elastin and proteoglycans were obtained as described by Morris and Stone (17). Briefly, the constructs were acquired from neonatal rat aortic smooth muscle cells (NNRSMC) isolated from Sprague–Dawley rats, 1–3 d of age. The NNRSMCs were then grown in culture containing 3.1 mg/mL sodium bicarbonate, 1% sodium pyruvate, 1% penicillin and streptomycin (DV3.7), and 20% fetal bovine serum. The samples were maintained for 6 wk, and the media was changed twice a week. The cells were killed by using 5% sodium azide in Puck’s solution and the constructs were stored at 4 °C. A gelatin solution was then added to the cultures that allowed the constructs to be lifted from the dish intact.
Mechanical Testing.
Tissue mechanics were tested by using a method similar to that described by Black et al. (18). Tissue strips, with dimensions 8 × 5 mm, were attached to metal plates with cyanoacrylate glue. The plates were then attached to steel wires connected to a force transducer (model 403A; Aurora Scientific) and a lever arm (model 300B; Aurora Scientific). The sample bath was filled with 22 mL of phosphate-buffered saline (PBS), and the entire apparatus was placed on a heat stand until the gel melted at 45 °C, leaving only the ECM strip in the stretching system. The PBS was then removed, and the bath was refilled with fresh PBS and maintained at 37 °C. Samples were preconditioned by application of three triangular displacement signals peaking at 10% strain defined as displacement divided by the initial length of the sample. The samples were stretched uniaxially to 20% strain and kept for 5 min for equilibration, then porcine pancreatic elastase was added to the bath. The force was recorded up to 60 min. Five different concentrations of elastase (0.003, 0.006, 0.009, 0.012 and 0.015 μg/μL) were used in each group of five samples. The thickness of the ECM sheets was determined by using a laser scanning confocal microscope (FV-1000; Olympus). The thickness of the sample at 0% strain was approximately 11 ± 2 μm.
Biochemical Analysis.
After mechanical testing was completed on each sample, the contents of the PBS bath were transferred to a 50-mL tube, dried, and stored at 4 °C. The residue within each tube was resolubilized with 2 mL of Milli-Q water and subjected to dialysis (1-kDa molecular weight cut off, GE Healthcare). The amount of soluble elastin in each sample was determined using the Fastin Elastin assay kit (Biocolor). Soluble elastin was precipitated using trichloroacetic acid and HCl, and dye reagent (5,10,15,20-tetraphenyl-21,23-porphine tetrasulfonate) added. The tubes were vortexed to suspend the precipitated elastin with the dye reagent and allowed to incubate on an orbital mixer (150 rpm, 90 min) at room temperature. At the end of the reaction period, the insoluble elastin-dye complex was separated from the soluble dye by centrifugation (10,000 × g, 10 min). A 250-μL quantity of dye dissociation reagent containing guanidine hydrochloride and 1-propanol was added to each tube to release the bound dye into solution. The quantity of dye in solution was determined by measuring absorbance at 513 nm on a SpectraMax 190 microplate reader (Molecular Devices). Replicate samples were averaged and corrected by subtracting the blank average, and elastin content was determined from a standard curve constructed using five concentrations (5–25 μg) of α-elastin. The amount of elastin released into the bath from enzyme digestion of the ECM strip was calculated by multiplying the elastin content of the sample by a ratio of the volume after dialysis to the volume used in the Fastin Elastin assay (500 μL).
Supplementary Material
Acknowledgments.
This work is supported by the Brazilian agency Conselho Nacional de Pesquisas (CNPq) and National Institutes of Health Grants HL090757 and HL088672.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1019188108/-/DCSupplemental.
References
- 1.Ghajar CM, George SC, Putnam AJ. Matrix metalloproteinase control of capillary morphogenesis. Crit Rev Eukaryot Gene Expr. 2008;18:251–78. doi: 10.1615/critreveukargeneexpr.v18.i3.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barnes PJ. Chronic obstructive pulmonary disease. N Engl J Med. 2000;343:269–280. doi: 10.1056/NEJM200007273430407. [DOI] [PubMed] [Google Scholar]
- 3.Jesudason R, Black L, Majumdar A, Stone P, Suki B. Differential effects of static and cyclic stretching during elastase digestion on the mechanical properties of extracellular matrices. J Appl Physiol. 2007;103:803–811. doi: 10.1152/japplphysiol.00057.2007. [DOI] [PubMed] [Google Scholar]
- 4.Jesudason R, et al. Mechanical forces regulate elastase activity and binding site availability in lung elastin. Biophys J. 2010;99:3076–3083. doi: 10.1016/j.bpj.2010.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flynn BP, et al. Mechanical strain stabilizes reconstituted collagen fibrils against enzymatic degradation by mammalian collagenase matrix metalloproteinase 8 (MMP-8) PLoS ONE. 2010;5:e12337. doi: 10.1371/journal.pone.0012337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Camp RJ, et al. Molecular mechanochemistry: Low force switch slows enzymatic cleavage of muman type I collagen monomer. J Am Chem Soc. 2011;133:4073–4078. doi: 10.1021/ja110098b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buehler MJ, Yung YC. Deformation and failure of protein materials in physiologically extreme conditions and disease. Nat Mater. 2009;8:175–188. doi: 10.1038/nmat2387. [DOI] [PubMed] [Google Scholar]
- 8.Abete T, de Candia A, Lairez D, Coniglio A. Percolation model for enzyme gel degradation. Phys Rev Lett. 2004;93:228301. doi: 10.1103/PhysRevLett.93.228301. [DOI] [PubMed] [Google Scholar]
- 9.Dahirel V, Paillusson F, Jardat M, Barbi M, Victor JM. Nonspecific DNA–protein interaction: Why proteins can diffuse along DNA. Phys Rev Lett. 2009;102:228101. doi: 10.1103/PhysRevLett.102.228101. [DOI] [PubMed] [Google Scholar]
- 10.Schmit JD, Kamber E, Kondev J. Lattice model of diffusion-limited bimolecular chemical reactions in confined environments. Phys Rev Lett. 2009;102:218302. doi: 10.1103/PhysRevLett.102.218302. [DOI] [PubMed] [Google Scholar]
- 11.Berry H. Monte Carlo simulations of enzyme reactions in two dimensions: Fractal kinetics and spatial segregation. Biophys J. 2002;83:1891–1901. doi: 10.1016/S0006-3495(02)73953-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Monine MI, Haugh J. Reactions on cell membranes: Comparison of continuum theory and Brownian dynamics simulations. J Chem Phys. 2005;123:074908. doi: 10.1063/1.2000236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gennes PG. Kinetics of diffusion-controlled processes in dense polymer systems. I. Nonentangled regimes. J Chem Phys. 1982;76:3316–3321. [Google Scholar]
- 14.Kantor Y, Webman I. Elastic properties of random percolating systems. Phys Rev Lett. 1984;52:1891–1894. [Google Scholar]
- 15.Bouchiat C, Mézard M. Elasticity model of a supercoiled DNA molecule. Phys Rev Lett. 1998;80:1556–1559. [Google Scholar]
- 16.Klotzsch E, et al. Fibronectin forms the most extensible biological fibers displaying switchable force-exposed cryptic binding sites. Proc Natl Acad Sci USA. 2009;106:18267–18272. doi: 10.1073/pnas.0907518106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morris SM, Stone PJ. Immunocytochemical study of the degradation of elastic fibers in a living extracellular matrix. J Histochem Cytochem. 1995;43:1145–1153. doi: 10.1177/43.11.7560897. [DOI] [PubMed] [Google Scholar]
- 18.Black LD, et al. Effects of elastase on the mechanical and failure properties of engineered elastin-rich matrices. J Appl Physiol. 2005;98:1434–1441. doi: 10.1152/japplphysiol.00921.2004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.