

Abstract
We investigated the changes in gene expression accompanying the development and progression of kidney cancer by use of 31,500-element complementary DNA arrays. We measured expression profiles for paired neoplastic and noncancerous renal epithelium samples from 37 individuals. Using an experimental design optimized for factoring out technological and biological noise, and an adapted statistical test, we found 1738 differentially expressed cDNAs with an expected number of six false positives. Functional annotation of these genes provided views of the changes in the activities of specific biological pathways in renal cancer. Cell adhesion, signal transduction, and nucleotide metabolism were among the biological processes with a large proportion of genes overexpressed in renal cell carcinoma. Down-regulated pathways in the kidney tumor cells included small molecule transport, ion homeostasis, and oxygen and radical metabolism. Our expression profiling data uncovered gene expression changes shared with other epithelial tumors, as well as a unique signature for renal cell carcinoma.
[Expression data for the differentially expressed cDNAs are available as a Web supplement at http://www.dkfz-heidelberg.de/abt0840/whuber/rcc. The array data have been submitted to the GEO data repository under accession no. GSE3.]
Renal cell carcinoma (RCC) is one of the 10 most frequent malignancies in Western societies. Advances in the understanding of the genetics underlying the development of renal epithelial tumors have lead to the recognition of distinctive types of tumors. Genetic alterations play a role in determining both the morphology and the behavior of tumors and underlie the most recent classifications (Kovacs et al. 1997; Störkel et al. 1997). The most common histological subtypes of RCC include clear cell (80%), papillary (∼10%), and chromophobe (<5%) carcinoma. Previous studies have shown that these histological subtypes are genetically and biologically different (Presti et al. 1991; Kovacs et al. 1997). Human RCCs are derived from epithelial cells in the proximal and connecting tubuli. Like many solid tumors, they contain other cell types in addition to carcinoma cells. Especially clear cell RCC is generally well vascularized, and infiltrating immune cells are frequently seen on histological sections.
Many genes and signaling pathways are known to be involved in RCC initiation and progression (Presti et al. 1991; Linehan et al. 1993). Genes potentially involved in kidney cancer include the genes for von Hippel-Lindau (Seizinger et al. 1988; Gnarra et al. 1994), vascular endothelial growth factor (VEGF; Brieger et al. 1999; Takahashi et al. 1999), epidermal growth factor receptor (EGFR; Ishikawa et al. 1990; Moch et al. 1998), transforming growth factor alpha (TGFA; Ishikawa et al. 1990; Lager et al. 1994; Uhlman et al. 1995; Moch et al. 1998), c-myc proto-oncogene (Drabkin et al. 1985; Yao et al. 1988), and vimentin (Moch et al. 1999). However, these molecular markers have not yet gained general use in RCC diagnostics and prognosis. Only tumor stage, determined by tumor extension, regional lymph node involvement, and distant metastases has gained widespread acceptance among pathologists and urologists as an indicator of patient prognosis (Guinan et al. 1997). Moreover, it is likely that many of the genes involved in the initiation and progression of renal cancer are currently unknown. The identification of differentially expressed genes in renal cell carcinoma could lead to the identification of markers for biological phenomena such as invasiveness or metastasis, which would be of significant value for diagnosis, prognosis, and treatment.
The highly parallel analysis of gene expression made possible by the development of cDNA array technology provides a powerful tool for the molecular dissection of cancer. A better understanding of the molecular changes associated with tumor formation and progression could improve the classification of cancer and provide clues to the development of specific therapies for pathogenetically distinct tumor types. The possibility of cancer classification based solely on gene expression monitoring was shown for human acute leukemias (Golub et al. 1999). Gene expression profiling of diffuse large B-cell lymphomas identified two molecularly distinct subtypes with significantly different overall survival (Alizadeh et al. 2000). Moreover, in a variety of solid human tumors and tumor cell lines, variation in gene expression observed by use of this mode of analysis has been correlated to phenotypic characteristics (DeRisi et al. 1996; Alon et al. 1999; Perou et al. 1999, 2000; Bittner et al. 2000; Ross et al. 2000).
We use macroscopically selected samples of RCC and normal corresponding renal tissue. Microscopically, the estimated proportion of non-neoplastic cells in the tumor samples was typically <5%. The choice to use solid tumors, rather than cell lines or microdissected material, was motivated by the fact that it yields important insights into the origin, development, and progression of tumors and immune responses against tumor formation, which would not be available otherwise. Furthermore, it avoids possible artifacts caused by cell line immortalization or RNA amplification technology.
To identify genes that are differentially expressed in different types and stages of epithelial kidney cancer, we analyzed gene expression profiles of primary tumors, metastases, and normal renal tissues. Labeled single-stranded cDNA target was derived from tumor and normal mRNA and hybridized to 31,500-element nylon cDNA arrays. By quantification of the resulting signal from each spot, we obtained a measure for the relative abundance in the tissue samples of mRNA corresponding to each gene. Our study has yielded a well-annotated list of genes that are differentially expressed in renal cell carcinoma. These results should lead toward the identification of kidney tumor-specific marker genes and potential targets for new therapeutic strategies.
RESULTS
Analysis of Gene Expression in Renal Cell Carcinoma
To measure variation in gene expression between renal cell carcinoma and normal renal tissue, we designed a cDNA array carrying a global human cDNA set. We developed an algorithm to select 1 representative clone from each of 41,120 UniGene clusters (Build 17, NCBI). This resulted in 33,792 physically available, noncontaminated I.M.A.G.E. (Lennon et al. 1996) cDNA clones (www.rzpd.de), and the derivation of consensus sequences for each UniGene cluster. Approximately 30% of the clones represented known genes; the remaining 70% were unknown ESTs. Approximately 31,500 cDNA clones from this set were amplified by PCR by use of vector-specific primers and spotted in duplicate to a set of two 22 × 22-cm nylon membranes (Human UniGene 1; www.rzpd.de). An estimated 30% of the UniGene clusters were represented by more than one clone (see Methods), thereby providing internal controls for the reproducibility of gene expression quantitation. We used these cDNA arrays to generate expression profiles from 32 primary RCC samples, matched with normal renal tissue from the same patients, and five liver and pancreas metastases of RCC (Table 1). Radioactively labeled cDNA representations prepared from each patient's tumor and normal messenger RNA sample were hybridized in parallel onto arrays from the same production batch. For the metastases, normal renal tissue from another patient with a primary RCC was used for comparison. Each hybridization was performed twice by use of independently labeled cDNA target from the same mRNA isolation. In total, more than 8,000,000 gene expression measurements were made in 69 malignant and normal samples by use of 50 reusable arrays.
Table 1.
Number of Tumor Samples in Each WHO Stage and Grade
| Stage I | Stage II | Stage III | Stage IV | Metastasis | ||
|---|---|---|---|---|---|---|
| Grade 1 | 8 | 1 | 1 | 10 | ||
| Grade 2 | 4 | 1 | 11 | 5 | 4 | 25 |
| Grade 3 | 2 | 2 | ||||
| Total | 12 | 1 | 14 | 5 | 5 | 37 |
Selection of Differentially Expressed Genes
Despite several reports indicating that tumor (sub)types can be distinguished with DNA microarray data, the methods of data analysis in the presence of considerable technical noise are far from being established (Brazma and Vilo 2000). The identification of differentially expressed genes is biologically important by its own right, as well as an essential step toward classification. Figure 1 shows the histograms of measured tumor/normal ratios for two genes. They indicate systematic differential expression for these genes, but also reflect considerable inter-individual variation, as well as experimental noise. We surveyed the histograms for a large number of genes and ESTs. Ratio-voting criteria are often used to select differentially expressed genes. We used a criterion that marks genes for which 30% of the ratios are >3.5 as up-regulated, and those in which 30% are below a ratio of 1/3.5 as down-regulated. Applied to the data from 31 tumors of stages I, III, or IV, this yielded 230 differentially expressed genes, of which 90 were up-regulated and 140 down-regulated.
Figure 1.

Histograms with tumor-normal ratios (logarithm to base 10) for two genes. (A) fibronectin 1, (B) metallothionein 1G. The bars in the histograms are colored by patient. In most cases, four ratios were measured for each patient. The four subpanels correspond to different tumor stages, stage I, III, and IV primary tumors, and metastases (M). The histograms show systematic trends. (A) Up-regulation, (B) down-regulation, interindividual variations (different-color patches within a histogram), and experimental noise (distribution of same-color patches). The vertical bars indicate ratios of 1/3.5 and 3.5. Histograms for all genes discussed in this article can be found in the Web supplement.
Higher sensitivity and better control over the rate of false positives was obtained through a statistical test that is based on the sign statistic, which we call adapted sign test in the following; it counts for each gene the number of times that its measured intensity in the set of repeated pair-wise comparisons is higher in tumor than in normal tissue. This number is compared with what is expected if the gene is not differentially expressed, and a P value is calculated. In the calculation of the null distribution, correlations between repetitions were taken into account. At an approximate significance level of 2 × 10−4 (≤6 false positive calls expected in the total data set of 31,500 clones), we found 1023 clones up-regulated, and 715 clones down-regulated in tumor tissue (Fig. 2).
Figure 2.

(Top) Histogram of the S-statistic for the set of 31 patients with tumor stages I, III, or IV. (Bottom) Distribution of the S-statistic under the null hypothesis of no differential expression (see Methods). The red bars show the rejection regions at a two-sided significance level of α = 2×10−4.
The two criteria select genes for quite different properties. Whereas the ratio-voting criterion selects genes that are differentially expressed by a large factor in at least a certain fraction of the population, the adapted sign test selects genes that are nearly always differentially expressed by whatever small amount. For our data and with the given parameters, we found the gene selection from ratio voting to be an almost strict subset of the selection from the adapted sign test. It is instructive to consider the power of the tests, for example, to determine how the number of selected genes depends on the number of patient samples profiled, random subsets of the total data set were drawn. For each of these subsets, the size of the selected gene list was calculated. The number of genes identified by the adapted sign test increases continuously with the number of experiments, and does not reach saturation with the present data set size of 37 patients (Fig. 3A). In contrast, the mean number of genes found by the ratio-voting criterion transiently decreases and before reaching a plateau, whereas its variation drops remarkably (Fig. 3B).
Figure 3.

The number of differentially expressed genes identified depends on the data set size. For each sample size between 5 and 36, 32 random subsamples of patients were drawn from the total 37 patients data set. Box plots of the number of selected genes are plotted against the patient sample size. (A) Adapted sign test at significance level 2 × 10−4, (B) ratio-voting criterion with R0 = 3.5 and Φ = 0.3. The horizontal lines in the boxes represent lower and upper quartiles, and median. The lines extending from each end of the box show the extent of the rest of the data. (+) Outliers.
To identify genes that are correlated with different types and stages of renal cell carcinoma, we performed two-sample permutation t-tests for differential expression across different subsets of tumor samples (Table 2). The tests were based on the log ratios between normalized tumor and normal intensities of each patient for the 20,000 clones that did not show consistently low intensities, and were conducted at a significance level of 10−3 (≤20 false positives expected). The comparison of chromophobe versus clear cell RCC yielded the largest set of 123 clones, probably reflecting the different cellular origin, histological characteristics, and cytogenetic background of these 2 tumor types. The class distinction between clear cell carcinoma of stages I–III as opposed to stage IV and metastasis, yielded 44 candidate clones. Even taking into account the expected proportion of false positives, it is likely that we are beginning to identify genes that are involved in tumor progression that could be used in tumor stage diagnosis.
Table 2.
Numbers of Genes ndiff Showing Different Tumor/Normal Ratios between Clinicopathological Subgroups
| Subgroups | ndiff | |
|---|---|---|
| Clear cell (32) | stage I-III (22) vs. IV,M (10) | 43 |
| stage I-IV (27) vs. M (5) | 28 | |
| stage I (9) vs. II-IV,M (23) | 9 | |
| grade 1 (7) vs. 2,3 (25) | 16 | |
| Clear cell (32) vs. chromophobe (4) | 123 |
The genes were identified by permutation t-tests at single-test significance levels of α = 10−3 out of ∼20,000 cDNAs that had no consistently low intensity, leading to an expected number of ≤20 false positives. The “Chromophobe vs. clear cell” distinction shows the most pronounced molecular differences.
Renal Cell Carcinoma Expression Patterns
A comprehensive list of differentially expressed genes in primary RCC was assembled by applying the adapted sign test and the ratio-voting criterion to the data grouped by tumor stages, as well as to the pooled data. The identities of 892 cDNAs have been sequence verified, including all of those referred to here by name. Of these cDNAs, 584 were annotated genes and 308 were ESTs. After excluding different clones representing the same known gene, we found 167 transcripts to be up-regulated and 154 down-regulated in RCC. These genes were classified into the category's biological pathway and cellular component using the terminology proposed by the Gene Ontology Consortium (Ashburner et al. 2000). We based the classification mainly on the functional information available in the GeneCards (http://bio-www.ba.cnr.it:8000/GeneCards/index.html) and Genatlas (http://www.citi2.fr/GENATLAS/welcome.html) databases. Several groups of coexpressed genes provided views of the activities of specific biological pathways (Fig. 4).
Figure 4.

Biological processes involved in RCC. A total of 321 differentially expressed genes were annotated for biological process according to the Gene Ontology proposed by Ashburner et al. (2000). The frequency of up-regulated (light bars) and down-regulated (dark bars) genes in RCC are plotted for the 23 biological processes scored.
A large group of genes involved in cell adhesion was consistently expressed higher in tumor, including fibronectin 1, collagen 4A, and laminin A4 (Fig. 5A). The latter two are major structural components of glomerular basement membranes. Fibronectin is also overexpressed in prostate cancer cells (Sonmez et al. 1995; Suer et al. 1996). Transcripts encoding gene products functioning in different signal transduction pathways, such as thymic hormones (prothymosin α and thymosin β-4), GTP-binding proteins (e.g., guanylate-binding protein 2), kinases (e.g., tyk2), and zinc finger transcriptional regulators (e.g., ZNF76), were more often found to be overexpressed in the tumors. Other biological processes that showed a large proportion of genes overexpressed in renal cell carcinoma were nucleotide and nucleic acid metabolism (encoding gene products involved in mRNA transcription and stability as well as in DNA replication), protein metabolism and modification (mostly ribosomal subunits), cell shape and cell size (several actin-interacting and remodeling proteins; Fig. 5B), and immune response (e.g., MHC molecules). Tumor markers described for renal cell carcinoma that were up-regulated in our data set include vimentin, VEGF, EGF-B, and EGFR.
Figure 5.

Gene expression patterns for genes associated with different biological processes. The colors represent the median tumor-normal ratio for stages I, III, IV, and M patients, respectively. The color scale is shown. More detailed tables and more biological processes are available as on-line supplementary material.
Down-regulated biological pathways in the kidney tumor cells included transport (e.g., renal-specific transport of small molecules; Fig. 5C), ion homeostasis (metallothioneins; Fig. 5D), oxygen and radical metabolism (e.g., glutathione S transferases; Fig. 5E), and electron transport (cytochrome oxidase complex components). Remarkably, the gene for free radical detoxification enzyme superoxide dismutase 2 is strongly overexpressed. Characteristic changes occurred in the carbohydrate metabolism of renal cell carcinoma, confirming and complementing earlier studies (Steinberg et al. 1992). Glycolysis was activated (phosphoglycerate kinase, enolase and phosphofructokinase) and gluconeogenesis was reduced (fructose-1,6-biphosphatase and aldolase B).
Categorizing by cellular compartment showed less pronounced trends for up-regulation or down-regulation. Gene products localized in the extracellular matrix and nucleus were more frequently up-regulated, whereas secreted and mitochondrial proteins were relatively more often down-regulated in tumor tissue (data not shown).
Activation of Cell Communication Pathways
Cell communication seems to be the major target for activation, with many genes involved in cell adhesion and signal transduction being overexpressed (Fig. 4). In carcinomas, including RCC, the interactions of cells with the extracellular matrix are disturbed (Lohi et al. 1998). The interstitial matrix collagens and fibronectin appear to be widely distributed in RCC. As a dynamic component of basement membranes, laminins are important in kidney development and are involved in RCC progression. Two up-regulated metalloproteinase inhibitors, TIMP1 and TIMP2, are involved in embryonal development and in the invasive phenotype of acute myelogenous leukemia (Janowska-Wieczorek et al. 1999). In addition to the extracellular matrix components, the expression of cell surface receptors for extracellular matrix components is disturbed in RCC. We found overexpression of the tumor-associated transmembrane proteins epithelial membrane protein 3, cell differentiation antigen CD68, melanoma adhesion molecule, and GP110. The fibronectin receptor integrin α 5 was overexpressed as well. Extracellular matrix proteins may function in RCC progression by binding and regulating the activity of growth factors, such as transforming growth factor β 1 and basic fibroblast growth factor. Some of the observed changes in signal transduction pathways could reflect cellular responses to these stimuli.
Down-Regulated Pathways and Genes
Kidney-specific pathways appear to be repressed as the tumor cells dedifferentiate, with lower expression of genes involved in small molecule transport, ion homeostasis, and oxygen and radical metabolism (Fig. 4). Down-regulation of specific members of the metallothionein family has been described in RCC (Izawa et al. 1998; Nguyen et al. 2000). In addition, we found genes that are involved in other types of cancer. Mutations of CDKN1C (p57kip2) are associated with sporadic cancers and Beckwith-Wiedemann syndrome suggesting that it is a tumor suppressor candidate (Lee et al. 1995; Matsuoka et al. 1995, 1996; Hatada et al. 1996). Another down-regulated gene involved in cell cycle regulation is GADD45A (growth arrest and DNA-damage-inducible, α), which inhibits the entry of cells into the S phase. The MPP3 protein (membrane protein, palmitoylated 3) is a membrane-associated guanylate kinase involved in coupling the cytoskeleton to the cell membrane, and the human homolog of Drosophila lethal discs large tumor-suppressor protein. Syndecan, a cell surface proteoglycan that links the cytoskeleton to the interstitial matrix is underexpressed in squamous cell carcinoma of the head and neck, whereas melastatin 1 was found down-regulated in a murine melanoma cell line with an aggressive phenotype. The S100 calcium-binding protein A2 gene product may play a role in suppressing tumor cell growth. The latter two genes belong to the biological process of ion homeostasis, which is prominently down-regulated in the RCCs. On the other hand, we found down-regulation of some genes that were described to be up-regulated in other tumor types. The PDZ domain-containing protein (PDZK1) is overexpressed in selected tumors of epithelial origin (Kocher et al. 1999). It is potentially involved in cell proliferation, differentiation, and ion transport. However, in many of the kidney tumors PDZK1 was specifically down-regulated. M1S1 (membrane component, chromosome 1, surface marker 1) is a glycoprotein, identified by monoclonal antibody GA733. This tumor-associated antigen may function as a growth factor receptor and is expressed in normal trophoblast cells, in multistratified epithelia and carcinomas. One of the strongest down-regulated genes was kininogen, which has many physiological functions, including inhibition of cysteine proteases, which could result in extracellular matrix degradation (Muller-Esterl et al. 1985).
Molecular Dissection of Renal Cell Carcinoma
To decide whether clusters of tumor-specific genes were derived from the RCC cells or from other cell types present in the tumor, we compared our expression data with published sets. Ross et al. (2000) found that most cell lines derived from RCCs were characterized by genes whose products are involved in stromal cell functions, such as synthesis and modification of the extracellular matrix. Genes defining this mesenchymal cluster that were also activated in our set of RCCs include melanoma adhesion molecule MUC18, vascular cell adhesion molecule 1, fibronectin 1, caveolin 1, collagen type IV α-1, collagen type V α-2, collagen binding protein 2, lysyl oxidase, and annexin II. Genes characteristically expressed by cell types other than carcinoma cells were detected as well as follows: (1) von Willebrand factor, strongly expressed in endothelial cells, also observed in breast tumors (Perou et al. 2000); (2) markers of macrophage/monocytes, in common with the macrophage cluster expressed in breast tumors (Perou et al. 2000), including CD68 and lysozyme; (3) B lymphocyte-specific genes, for example the B cell activation gene BL34 and the cytokine pre-B cell-enhancing factor; (4) genes involved in antigen processing and presentation, such as MHC class II genes, peptide transporter TAP 1 (RING4), TAP binding protein, and proteasome subunit β type 8 (PSMB8). These results confirm the histological finding of infiltration of the tumors with cells involved in the immune and inflammatory response. Several interferon-induced genes were also specifically expressed in the tumor tissue, including interferon-inducible protein 9–27, interferon α-inducible protein 27 (IFI27), guanylate-binding protein 2 (GBP2), monokine induced by γ interferon (MIG), and interferon-γ-induced protein (IFI 16), PSMB8, and TAP1.
Supplementary Information and Array Data
The array data are reported in the Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) under accession no. GSE3. In addition, the author's Web site (http://www.dkfz-heidelberg.de/abt0840/whuber/rcc) presents a comprehensive collection of the original data and the results, in particular the list of 1738 clones (including the distributions of their estimated fold changes), documentation of the sequence verification, and the expression patterns associated with different biological processes.
DISCUSSION
We have generated a general set of >31,500 human cDNA clones, which represents one of the largest gene sets interrogated in array-based gene expression studies to date. The consensus sequences for all clusters, the cDNA clones, their PCR products, and high-density spotted Nylon cDNA arrays are available at http://www.rzpd.de. Currently, the clone set is being enlarged to 75,000 clones based on UniGene build 90. Here, we have characterized variation in gene expression in a set of surgical specimens of primary and metastasized renal cell carcinoma and normal renal epithelium from 37 different individuals. To eliminate patient-specific variations in gene expression, the primary tumors were directly compared with normal renal tissue from the same patients, and our statistical analysis was optimized for this experimental setup. We present a list of genes that are differentially expressed in neoplasms of renal epithelium compared with normal epithelium (see on-line supplementary information as noted above). Generally, there are two reasons for finding differential expression between primary tissues, different expression levels of genes within the cells, or varying cell type composition. According to histological examination, both the renal cell carcinoma and the normal renal cortex consist to a large majority (estimated at 90%–95%) of epithelial cells. Still, part of the observed differences in gene expression may be influenced by differences in cell type proportion.
The classification according to biological process gives insights into the molecular changes occurring in tumor development and progression (Fig. 5; on-line supplementary information). In addition, we find evidence for a distinction based on gene expression between two different subtypes of RCC, and between different stages of malignancy (Table 2). The largest set of candidate discriminator genes was found in a two-class comparison of clear cell versus chromophobe RCC. These differences in gene expression probably reflect the biological and clinical differences between the histological subtypes. The clear cell carcinoma originates from proximal tubuli that have a mesodermal origin, whereas the less malignant chromophobe carcinoma is derived from connecting ducts with an endodermal origin. Future studies including more chromophobe tumors may elucidate gene expression patterns specific for this difference in embryonal origin.
We used two different criteria to select differentially expressed genes. A ratio-voting criterion selects genes that show a fold change above a threshold in a defined number of experiments (Eisen et al. 1998; Perou et al. 2000). A robust test with a controlled type I error, and power that increases with data set size, is based on the sign statistic. We estimated its null distribution in the presence of correlations. On the basis of data from 37 patients, this resulted in 1738 cDNAs representing differentially expressed genes, with 6 false positives expected. For cells as drastically different as tumor and normal cells, the concept of a well-defined set of truly differentially expressed genes may be evasive. Whereas the ratio-voting criterion may identify the most important candidate genes for robust molecular diagnosis, the more subtle changes in gene expression discovered by the adapted sign test may lead to better insights into the molecular changes involved in cancer development and progression.
The translation of gene expression data to potentially useful targets for molecular diagnosis and treatment depends largely on correct and complete functional annotation. Genes involved in the same biological process often group together in experimental expression clusters (Eisen et al. 1998; Ashburner et al. 2000). In contrast, molecular function and cellular component annotations correlate less well with clustered expression patterns. However, by use of this information, intelligent predictions can be made concerning cancer detection and therapy. Our expression data indicate that RCCs display a mostly mesenchymal expression pattern, in accordance with the mesodermal origin of the tumor cells (Ross et al. 2000). The RCC expression profiling data show both gene expression changes shared with other epithelial tumors, and a unique signature for RCC. The identification and classification of differentially expressed genes is the beginning of a more complete understanding of kidney cancer. An annotated list of the expression data for the categorized genes is presented in the web supplement. On the basis of these results, we have designed a kidney tumor-specific array that will enable higher throughput screening of additional samples and ultimately lead to a clinical classification of RCC on the basis of their expression profiles.
METHODS
Selection of a Global Human Clone Set
To generate a nonredundant human clone set, we post-processed the UniGene clusters (Build 17, NCBI), which represent a large number of human genes. All processing steps are part of the GeneNest software (Haas et al. 2000), which additionally provides an interactive graphical interface to the post-processed UniGene database (http://www.dkfz.de/tbi/services/GeneNest/index). To identify the most reliable, representative clone from each cluster, we analyzed the following criteria (sorted according to their importance): (1) availability of clones at the RZPD; (2) quality of cDNA library of origin; (3) presence of more than one read of the same clone in a cluster, ensuring a higher confidence in the sequence–clone relationship; (4) calculated insert size, selecting for larger inserts; (5) presence of a poly(A) signal. We used a fuzzy logic-based rule system to combine all clone selection criteria to obtain a quality measure of each clone in the entire UniGene set. For each cluster, we selected the clone with the highest quality as representative. To estimate the redundancy of the global clone set, we resequenced >2700 clones of the set and found 12.8% wrongly assigned clones. All of these belonged to clusters that were already represented by another clone. Therefore, the overall redundancy of the clone set was estimated to be 1.44-fold, and the 31,500 clones on the cDNA array represent an estimated 21,875 different transcripts.
The 31,500-Clone Human cDNA Array
From the Human UniGene 1 clone set, cDNA inserts of the clones from the 82 first 384-well microtiter plates were amplified by PCR in a 384-well format (MJ Research) by use of M13 forward (5′-CGTTGTAAAACGACGGCCAGT-3′) and reverse primers (5′-TTTCACACAGGAAACAGCTATGAC-3′). The 31,488 PCR products were transferred in a 4 × 4 pattern onto a set of two 22 × 22-cm Hybond N+ nylon membranes (Amersham Pharmacia Biotech) soaked in 0.4 M NaOH using a Picking-Spotting-Robot (Linear Drives LDT) with 400-μm pins (Genetix). After spotting, the arrays were carefully floated for 2 min on 0.4 M NaOH and 5 × SSC (pH 7.5) successively, air-dried and cross-linked by UV. Every 4 × 4 block contained one spot with PCR product from the bacterial kanamycin resistance gene to serve as a guide spot in automated image analysis, one empty spot to serve as background measurement and DNA from seven clones spotted in duplicate. On each filter, a control plate containing putative housekeeping genes and positive and negative controls was spotted. To assure even quality between subsequent rounds of hybridization, cDNA arrays were prestripped before their first use (Hauser et al. 1998). To check for filter quality, M13 forward oligonucleotide hybridizations were carried out.
Patient Samples
Macroscopically selected samples of the RCC and normal corresponding renal tissue were snap frozen in liquid nitrogen by pathologists within 30 min after dissection. The tumors were staged according to TNM classification (Sobin and Wittekind 1997), graded according to the Fuhrmann grading system (Fuhrmann et al. 1982), and histologically subtyped according to the recommendations of the World Health Organization (Mostofi and Davis 1998). Microscopically, the estimated proportion of non-neoplastic cells in the tumor samples was typically <5%. Total RNA was extracted by use of the standard Trizol method (Life Technologies) and poly(A)+ RNA was selected using Dynabeads according to the manufacturer's recommendations (Dynal).
Hybridization
A total of 500–1000 ng of poly(A)+ RNA was reverse transcribed using 500 ng (dT)18V primer and 50 μCi 33P α-dCTP (Amersham Pharmacia Biotech), 400 μM each of dGTP, dATP, and dTTP, and 200 units of Superscript II reverse transcriptase (Life Technologies). The RNA was removed by hydrolysis, and the first strand cDNA cleaned up by gel filtration on a S-300 spin column (Mobitec). The incorporation rates were usually >30% and similar for reactions carried out simultaneously, yielding ∼10–30 million counts/min. Each prestripped cDNA array was prewetted in 7.5 mL of demineralized water and inserted into a glass tube (24 × 7 cm). An equal volume of prewarmed 2× hybridization solution was added and prehybridization was carried out for 2 h at 65°C in a total volume of 15 mL of 6× SSC, 5× Denhardt's, 0.5% SDS, 50 μg/mL salmon sperm DNA, and 0.5 μg/mL each of freshly denatured Cot-1 DNA (Life Technologies) and (dA)40 oligonucleotide. The first strand cDNA was heat denatured for 5 min at 100°C and divided equally over the two cDNA arrays belonging to one set. For guide spot hybridization, the bacterial kanamycin resistance gene was amplified from a cosmid cloning vector by PCR with the forward primer 5′-AGTGCCGGGGCAGGATCT-3′ and the reverse primer 5′-TCGTGATGGCAGGTTGGG-3′. 5 × 105 cpm of random-primed 33P-labeled kanamycin cDNA was added to each filter. The arrays were hybridized for 20–24 h at 65°C. The wash steps were all performed at 65°C for 10 min in 1× SSC/0.1% SDS, 0.3× SSC/0.1% SDS, 0.3× SSC/0.1% SDS, and 0.1× SSC/0.1% SDS, respectively. Membranes were blotted dry briefly, wrapped in SaranWrap, exposed to PhosphorImager screens for 18–24 h and scanned with the Storm 860 PhosphorImager (Amersham Pharmacia Biotech). The resulting gray level images were partitioned and quantized with Xdigitise version 3.1 (H. Lehrach, MPI for Molecular Genetics, Berlin). After hybridization, membranes were stripped (Hauser et al. 1998) and stored dry at room temperature for 6–8 weeks (approximately two half-lives of 33P) before being rehybridized. Membranes were reused up to six times without significant loss of signal intensities.
Experimental Design, Quality Control, and Normalization
Measurements were repeated in a threefold hierarchical manner. First, clones were spotted in duplicate onto the arrays. Second, each mRNA was labeled and hybridized to two arrays. Third, multiple tissue samples with the same pathological classification were investigated. To obtain a good contrast between the expression levels for tumor and normal tissue, a Latin square design was chosen for the repeated same-patient hybridizations. Hybridizations were done in pairs, with mRNA from tumor and normal tissue of the same patient being prepared at the same time under identical conditions, and hybridized to filters produced during the same spotting run. The intensities were adjusted through affine transformations: Denoting by N the set of background-corrected intensities for normal tissue, and by T the set for tumor tissue, normalization was performed through the transformations N → N + ν, and T → a T + ν (Beißbarth et al. 2000). The multiplicative factor a was determined such that the estimated mode of the distribution of log aT/N, conditional on TN > θ, came to lie at zero. θ was set to the 90%-quantile of TN This robust estimator is based on the assumption that most genes represented on the filters have unchanged expression levels, whereas smaller, but possibly different numbers of genes may be up-regulated and down-regulated. The pseudocount ν was used to regularize the ratio estimation. For the present analysis, we set ν to the 50%-quantile of the pooled intensities (N, aT). The hybridization data reported here are available from the GEO database (http://www.ncbi.nlm.nih.gov/geo/) under the accession numbers GSM81 to GSM422.
Gene Selection Statistics
According to the ratio-voting criterion, a gene is considered differentially expressed if at least 30% of the ratios (aTphr + ν)/(Nphrthinsp;+ ν) above 3.5 or below 1/3.5 is observed. The three indices p, h, and r stand for the three levels of repetition, patient (p), multiple hybridization of the same RNA isolation (h), and duplicate spotting (r). The sign statistic is given by
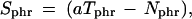 |
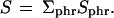 |
Under the null hypothesis of no differential expression, S is approximately normally distributed with mean 0 and a variance that we estimated from the data as follows: The Sph = ΣrSphr can be seen as identically distributed random variables with Sph and Sp'h' uncorrelated for different patients p ≠ p'. There may be correlations between different hybridization rounds of the same patient:
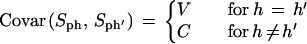 |
The variance of S is E [S2] = N1 V + N2 C, in which N1 is the total number of hybridizations and N2 is the number of pairs of repeated hybridizations from the same sample. V and C were estimated for each gene by the standard unbiased estimators for variance and covariance from the data sample given by the Sph. Global estimates for V and C, with standard deviation reduced by a factor of
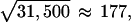 |
were obtained by taking the arithmetic mean over all genes. This assumes equal variances V and covariances C for all genes. The bias caused by the dependence of V and C on the absolute value of S was negligible, restricting the averaging procedure to genes with low absolute values of the S statistic changed the estimated significance level by a factor of at most 1.1.
Two-sample two-sided t-tests for differential expression across different subsets of tumor samples were performed for ∼20,000 cDNAs that did not have consistently low intensity. The t statistic was computed on the mean log ratios Rp (obtained from Rphr by averaging over h and r). Avoiding normality assumptions, the tests were carried out as permutation tests.
Acknowledgments
We thank G. Jakse and R.-H. Ringert for patient samples; J. O'Brien for the first sets of Unigene filters; K. Fellenberg, T. Beißbarth, B. Brors, and A. Frischauf for initial data analysis software and discussion; S. Kirby, M. Peters, and D. Schneider for re-arraying and streaking of contaminated clones; R. Will and R. Wittig for optimizing and performing PCR; S. Wiemann and his team for outstanding sequences; and B. Kornacker and M. Stauch for excellent technical assistance. This work was partly supported by a grant from the German Human Genome Project.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL a.poustka@dkfz-heidelberg.de; FAX 49-6221-423454.
Article published on-line before print: Genome Res., 10.1101/gr.184501.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.184501.
REFERENCES
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- Alon U, Barkai N, Notterman DA, Gish K, Ybarra S, Mack D, Levine AJ. Broad patterns of gene expression revealed by clustering analysis of tumor and normal colon tissues probed by oligonucleotide arrays. Proc Natl Acad Sci. 1999;96:6745–6750. doi: 10.1073/pnas.96.12.6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beißbarth T, Fellenberg K, Brors B, Arribas-Prat R, Boer JM, Hauser NC, Scheideler M, Hoheisel JD, Schütz G, Poustka A, et al. Processing and quality control of DNA array hybridization data. Bioinformatics. 2000;16:1014–1022. doi: 10.1093/bioinformatics/16.11.1014. [DOI] [PubMed] [Google Scholar]
- Bittner M, Meltzer P, Chen Y, Jiang Y, Seftor E, Hendrix M, Radmacher M, Simon R, Yakhini Z, Ben-Dor A, et al. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature. 2000;406:536–540. doi: 10.1038/35020115. [DOI] [PubMed] [Google Scholar]
- Brazma A, Vilo J. Gene expression data analysis. FEBS Lett. 2000;480:17–24. doi: 10.1016/s0014-5793(00)01772-5. [DOI] [PubMed] [Google Scholar]
- Brieger J, Weidt EJ, Schirmacher P, Storkel S, Huber C, Decker HJ. Inverse regulation of vascular endothelial growth factor and VHL tumor suppressor gene in sporadic renal cell carcinomas is correlated with vascular growth: An in vivo study on 29 tumors. J Mol Med. 1999;77:505–510. doi: 10.1007/s001099900022. [DOI] [PubMed] [Google Scholar]
- DeRisi J, Penland L, Brown PO, Bittner ML, Meltzer PS, Ray M, Chen Y, Su YA, Trent JM. Use of a cDNA microarray to analyze gene expression patterns in human cancer. Nat Genet. 1996;14:457–460. doi: 10.1038/ng1296-457. [DOI] [PubMed] [Google Scholar]
- Drabkin HA, Bradley C, Hart I, Bleskan J, Li FP, Patterson D. Translocation of c-myc in the hereditary renal cell carcinoma associated with a t(3;8)(p14.2;q24.13) chromosomal translocation. Proc Natl Acad Sci. 1985;82:6980–6984. doi: 10.1073/pnas.82.20.6980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann SA, Lasky LC, Limas C. Prognostic significance of morphologic parameters in renal cell carcinoma. Am J Surg Pathol. 1982;6:655–663. doi: 10.1097/00000478-198210000-00007. [DOI] [PubMed] [Google Scholar]
- Gnarra JR, Tory K, Weng Y, Schmidt L, Wei MH, Li H, Latif F, Liu S, Chen F, Duh FM, et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat Genet. 1994;7:85–90. doi: 10.1038/ng0594-85. [DOI] [PubMed] [Google Scholar]
- Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, et al. Molecular classification of cancer: Class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- Guinan P, Sobin LH, Algaba F, Badellino F, Kameyama S, MacLennan G, Novick A. TNM staging of renal cell carcinoma: Workgroup No. 3. Union International Contre le Cancer (UICC) and the American Joint Committee on Cancer (AJCC) Cancer. 1997;80:992–993. doi: 10.1002/(sici)1097-0142(19970901)80:5<992::aid-cncr26>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Haas SA, Beißbarth T, Rivals E, Krause A, Vingron M. GeneNest: Automated generation and visualization of gene indices. Trends Genet. 2000;16:521–523. doi: 10.1016/s0168-9525(00)02116-8. [DOI] [PubMed] [Google Scholar]
- Hatada I, Ohashi H, Fukushima Y, Kaneko Y, Inoue M, Komoto Y, Okada A, Ohishi S, Nabetani A, Morisaki H, et al. An imprinted gene p57KIP2 is mutated in Beckwith-Wiedemann syndrome. Nat Genet. 1996;14:171–173. doi: 10.1038/ng1096-171. [DOI] [PubMed] [Google Scholar]
- Hauser N, Vingron M, Scheideler M, Krems B, Hellmuth K, Entian K-D, Hoheisel J. Transcriptional profiling on all open reading frames of Saccaromyces cerevisiae. Yeast. 1998;14:1209–1221. doi: 10.1002/(SICI)1097-0061(19980930)14:13<1209::AID-YEA311>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Ishikawa J, Maeda S, Umezu K, Sugiyama T, Kamidono S. Amplification and overexpression of the epidermal growth factor receptor gene in human renal-cell carcinoma. Int J Cancer. 1990;45:1018–1021. doi: 10.1002/ijc.2910450606. [DOI] [PubMed] [Google Scholar]
- Izawa JI, Moussa M, Cherian MG, Doig G, Chin JL. Metallothionein expression in renal cancer. Urology. 1998;52:767–772. doi: 10.1016/s0090-4295(98)00323-9. [DOI] [PubMed] [Google Scholar]
- Janowska-Wieczorek A, Marquez LA, Matsuzaki A, Hashmi HR, Larratt LM, Boshkov LM, Turner AR, Zhang MC, Edwards DR, Kossakowska AE. Expression of matrix metalloproteinases (MMP-2 and -9) and tissue inhibitors of metalloproteinases (TIMP-1 and -2) in acute myelogenous leukaemia blasts: Comparison with normal bone marrow cells. Br J Haematol. 1999;105:402–411. [PubMed] [Google Scholar]
- Kocher O, Comella N, Gilchrist A, Pal R, Tognazzi K, Brown LF, Knoll JH. PDZK1, a novel PDZ domain-containing protein up-regulated in carcinomas and mapped to chromosome 1q21, interacts with cMOAT (MRP2), the multidrug resistance-associated protein. Lab Invest. 1999;79:1161–1170. [PubMed] [Google Scholar]
- Kovacs G, Akhtar M, Beckwith BJ, Bugert P, Cooper CS, Delahunt B, Eble JN, Fleming S, Ljungberg B, Medeiros LJ, et al. The Heidelberg classification of renal cell tumours. J Pathol. 1997;183:131–133. doi: 10.1002/(SICI)1096-9896(199710)183:2<131::AID-PATH931>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Lager DJ, Slagel DD, Palechek PL. The expression of epidermal growth factor receptor and transforming growth factor alpha in renal cell carcinoma. Mod Pathol. 1994;7:544–548. [PubMed] [Google Scholar]
- Lee MH, Reynisdottir I, Massague J. Cloning of p57KIP2, a cyclin-dependent kinase inhibitor with unique domain structure and tissue distribution. Genes & Dev. 1995;9:639–649. doi: 10.1101/gad.9.6.639. [DOI] [PubMed] [Google Scholar]
- Lennon G, Auffray C, Polymeropoulos M, Soares MB. The I.M.A.G.E. consortium: An integrated molecular analysis of genomes and their expression. Genomics. 1996;33:151–152. doi: 10.1006/geno.1996.0177. [DOI] [PubMed] [Google Scholar]
- Linehan, W.M., Gnarra, J.R., Lerman, M.I., Latif, F., and Zbar, B. 1993. Genetic basis of renal cell cancer. Important Adv. Oncol. 47–70. [PubMed]
- Lohi J, Leivo I, Oivula J, Lehto VP, Virtanen I. Extracellular matrix in renal cell carcinomas. Histol Histopathol. 1998;13:785–796. doi: 10.14670/HH-13.785. [DOI] [PubMed] [Google Scholar]
- Matsuoka S, Edwards MC, Bai C, Parker S, Zhang P, Baldini A, Harper JW, Elledge SJ. p57KIP2, a structurally distinct member of the p21CIP1 Cdk inhibitor family, is a candidate tumor suppressor gene. Genes & Dev. 1995;9:650–662. doi: 10.1101/gad.9.6.650. [DOI] [PubMed] [Google Scholar]
- Matsuoka S, Thompson JS, Edwards MC, Bartletta JM, Grundy P, Kalikin LM, Harper JW, Elledge SJ, Feinberg AP. Imprinting of the gene encoding a human cyclin-dependent kinase inhibitor, p57KIP2, on chromosome 11p15. Proc Natl Acad Sci. 1996;93:3026–3030. doi: 10.1073/pnas.93.7.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moch H, Sauter G, Gasser TC, Bubendorf L, Richter J, Presti JC, Jr, Waldman FM, Mihatsch MJ. EGF-r gene copy number changes in renal cell carcinoma detected by fluorescence in situ hybridization. J Pathol. 1998;184:424–429. doi: 10.1002/(SICI)1096-9896(199804)184:4<424::AID-PATH1223>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Moch H, Schraml P, Bubendorf L, Mirlacher M, Kononen J, Gasser T, Mihatsch MJ, Kallioniemi OP, Sauter G. High-throughput tissue microarray analysis to evaluate genes uncovered by cDNA microarray screening in renal cell carcinoma. Am J Pathol. 1999;154:981–986. doi: 10.1016/S0002-9440(10)65349-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostofi FK, Davis CJ. Histological typing of kidney tumours. International classification of tumours. 2nd edition. Berlin, Heidelberg, New York: World Health Organization. Springer; 1998. [Google Scholar]
- Muller-Esterl W, Fritz H, Kellermann J, Lottspeich F, Machleidt W, Turk V. Genealogy of mammalian cysteine proteinase inhibitors. Common evolutionary origin of stefins, cystatins and kininogens. FEBS Lett. 1985;191:221–226. doi: 10.1016/0014-5793(85)80012-0. [DOI] [PubMed] [Google Scholar]
- Nguyen A, Jing Z, Mahoney PS, Davis R, Sikka SC, Agrawal KC, Abdel-Mageed AB. In vivo gene expression profile analysis of metallothionein in renal cell carcinoma. Cancer Lett. 2000;160:133–140. doi: 10.1016/s0304-3835(00)00534-6. [DOI] [PubMed] [Google Scholar]
- Perou CM, Jeffrey SS, van de Rijn M, Rees CA, Eisen MB, Ross DT, Pergamenschikov A, Williams CF, Zhu SX, Lee JC, et al. Distinctive gene expression patterns in human mammary epithelial cells and breast cancers. Proc Natl Acad Sci. 1999;96:9212–9217. doi: 10.1073/pnas.96.16.9212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- Presti JC, Jr, Rao PH, Chen Q, Reuter VE, Li FP, Fair WR, Jhanwar SC. Histopathological, cytogenetic, and molecular characterization of renal cortical tumors. Cancer Res. 1991;51:1544–1552. [PubMed] [Google Scholar]
- Ross DT, Scherf U, Eisen MB, Perou CM, Rees C, Spellman P, Iyer V, Jeffrey SS, Van de Rijn M, Waltham M, et al. Systematic variation in gene expression patterns in human cancer cell lines. Nat Genet. 2000;24:227–235. doi: 10.1038/73432. [DOI] [PubMed] [Google Scholar]
- Seizinger BR, Rouleau GA, Ozelius LJ, Lane AH, Farmer GE, Lamiell JM, Haines J, Yuen JW, Collins D, Majoor-Krakauer D, et al. Von Hippel-Lindau disease maps to the region of chromosome 3 associated with renal cell carcinoma. Nature. 1988;332:268–269. doi: 10.1038/332268a0. [DOI] [PubMed] [Google Scholar]
- Sobin LH, Wittekind C. TNM classification of malignant tumours. 5th edition. New York: Wiley-Liss; 1997. [Google Scholar]
- Sonmez H, Suer S, Karaarslan I, Baloglu H, Kokoglu E. Tissue fibronectin levels of human prostatic cancer, as a tumor marker. Cancer Biochem Biophys. 1995;15:107–110. [PubMed] [Google Scholar]
- Steinberg P, Storkel S, Oesch F, Thoenes W. Carbohydrate metabolism in human renal clear cell carcinomas. Lab Invest. 1992;67:506–511. [PubMed] [Google Scholar]
- Störkel S, Eble JN, Adlakha K, Amin M, Blute ML, Bostwick DG, Darson M, Delahunt B, Iczkowski K. Classification of renal cell carcinoma: Workgroup No. 1. Union Internationale Contre le Cancer (UICC) and the American Joint Committee on Cancer (AJCC) Cancer. 1997;80:987–989. doi: 10.1002/(sici)1097-0142(19970901)80:5<987::aid-cncr24>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Suer S, Sonmez H, Karaaslan I, Baloglu H, Kokoglu E. Tissue sialic acid and fibronectin levels in human prostatic cancer. Cancer Lett. 1996;99:135–137. doi: 10.1016/0304-3835(95)04084-6. [DOI] [PubMed] [Google Scholar]
- Takahashi A, Sasaki H, Kim SJ, Kakizoe T, Miyao N, Sugimura T, Terada M, Tsukamoto T. Identification of receptor genes in renal cell carcinoma associated with angiogenesis by differential hybridization technique. Biochem Biophys Res Commun. 1999;257:855–859. doi: 10.1006/bbrc.1999.0465. [DOI] [PubMed] [Google Scholar]
- Uhlman DL, Nguyen P, Manivel JC, Zhang G, Hagen K, Fraley E, Aeppli D, Niehans GA. Epidermal growth factor receptor and transforming growth factor alpha expression in papillary and nonpapillary renal cell carcinoma: correlation with metastatic behavior and prognosis. Clin Cancer Res. 1995;1:913–920. [PubMed] [Google Scholar]
- Yao M, Shuin T, Misaki H, Kubota Y. Enhanced expression of c-myc and epidermal growth factor receptor (C- erbB-1) genes in primary human renal cancer. Cancer Res. 1988;48:6753–6757. [PubMed] [Google Scholar]