

Abstract
We demonstrate for the first time that the pro-inflammatory cytokine interleukin (IL)-18 stimulates rapid and significant proliferation of SMC derived from human saphenous vein (VSMC), but not coronary artery. IL-18 also stimulates VSMC growth. Further investigations revealed that IL-18-induced VSMC proliferation was Wnt Inducible Secreted Protein-1 (WISP1) dependent. In addition to inducing its own expression via phosphotidylinositol 3-kinase/Akt-dependent IKK/NF-κB activation, IL-18 stimulated glycogen synthase kinase 3β phosphorylation and degradation, β-catenin nuclear translocation and stabilization, T-cell factor-lymphoid enhancer binding factor (TCF-LEF) activation, and WISP1 induction. Moreover, WISP1 induced its own expression, and that of survivin and multiple matrix metalloproteinases via β-catenin/TCF-LEF interaction. WISP1 also activated AP-1, but not NF-κB, and induced matrix metalloproteinase (MMP)9 transcription in part via AP-1. Interestingly, WISP1 failed to regulate tissue inhibitors of matrix metalloproteinases (TIMP) expression. These novel findings indicate that IL-18 induces a series of signaling events that result in WISP1-mediated VSMC proliferation, survival and MMP induction that are key components of vein graft stenosis and this may be amplified by IL-18 and WISP1 autoregulation and cross-regulation.
Keywords: Cytokines, interleukin-18, Proliferation, CCN, WISP1, β-catenin
Atherosclerosis is a chronic inflammatory disease. Interleukin (IL)-18 is a proinflammatory cytokine that is associated with the development and progression of atherosclerosis in animal models (Elhage et al., 2003; Li et al., 2008; Maffia et al., 2006; Whitman et al., 2002). Circulating levels of IL-18 are increased in humans with coronary artery disease (CAD), and show a positive correlation with intima-media thickness (Yamagami et al., 2005). Further, IL-18 expression is markedly elevated in atherosclerotic lesions, particularly in unstable plaques (Mallat et al., 2001a). Since IL-18 is a potent inducer of other cytokines and matrix degrading metalloproteinases (MMPs; (Chandrasekar et al., 2006; Reddy et al.), its increased expression may potentiate inflammation, ECM degradation, adverse remodeling, and other related complications. Of note, IL-18 levels are also increased in diabetes and in metabolic syndrome, both major contributing factors for CAD (Hung et al., 2005; Troseid et al., 2009).
IL-18 is synthesized as a pro-form and is cleaved to a mature and biologically active molecule by caspase-1. Both endothelial and smooth muscle cells express IL-18 (Gerdes et al., 2002). IL-18 is also secreted by the infiltrating activated macrophages in atherosclerotic lesions, suggesting that all the cellular components of an inflamed or injured vessel contribute to the increased levels of IL-18 (Gerdes et al., 2002). Secreted mature IL-18 binds to the IL-18 receptor, a complex comprising of a ligand binding α subunit and a signal transducing β subunit. Endothelial cells and SMC express both subunits, and thus are targets of IL-18 autocrine and paracrine effects. Of note, angiotensin II upregulates IL-18Rα in smooth muscle cells in an AP-1-dependent manner (Sahar et al., 2005). Since IL-18 is a potent inducer of AP-1 in SMC (Chandrasekar et al., 2006), and is also an AP-1-responsive gene(Tone et al., 1997), it is possible that IL-18 autoregulation and IL-18Rα overexpression perpetuate inflammatory signals in the vessel wall. Similar to IL-1 receptor antagonist which neutralizes IL-1β, IL-18 binding protein (IL-18BP) binds IL-18 with high affinity and neutralizes its biological activity (Kim et al., 2000), thus making IL-18BP an attractive candidate to diminish IL-18-dependent inflammatory signaling. While the systemic levels of IL-18BP are high in a healthy individual, these levels can be markedly reduced in inflammatory conditions (Mallat et al., 2001b; Mazodier et al., 2005), resulting in unopposed IL-18 pro-inflammatory signaling.
Atherosclerosis is an inflammatory disease characterized by SMC migration and proliferation. IL-18, through the induction of MMPs, also plays a role in extracellular matrix (ECM) degradation and SMC migration. In fact, we have previously shown that IL-18 induces activator protein (AP)-1 and nuclear factor (NF)-κB activation, MMP9 induction and activation, and directional migration of aortic SMC (ASMC; (Chandrasekar et al., 2006)). SMC migration and proliferation are two distinct cellular responses, and both phenomena contribute to transplant vasculopathy, post-angioplasty restenosis, late vein-graft failure, and atherosclerosis. Since similar or distinct signaling pathways contribute to agonist-induced SMC migration and proliferation, and since we already reported that IL-18 stimulates ASMC migration (Chandrasekar et al., 2006), we investigated whether IL-18 also stimulates SMC proliferation, and determined the underlying molecular mechanisms. Although veins are relatively less prone to atherosclerosis, following coronary artery bypass graft surgery, the grafted saphenous veins can develop significant stenosis or occlusion of the vessel due to accelerated SMC proliferation (Mitra et al., 2006; Suma, 1999), suggesting an increased proliferative potential of ‘arterialized’ VSMC. Therefore, we investigated whether IL-18 is mitogenic for SMC, and whether it differentially affects proliferation of SMC derived from saphenous vein as opposed to coronary artery.
The Wnt1-inducible signaling pathway protein-1 (WISP1), a member of the secreted, cysteine-rich CCN (Cyr61-CTGF-Nov; cysteine-rich protein 61, connective tissue growth factor, nephroblastoma-overexpressed gene) family, exerts diverse biological effects, including cell growth and proliferation. We have previously reported that while WISP1 antagonizes TNF-α-induced cardiomyocyte death, it mediates TNF-α-induced cardiac fibroblast proliferation (Venkatachalam et al., 2009), demonstrating pro-survival and pro-mitogenic effects. Since WISP1 has been shown to induce MMPs expression in macrophages (Blom et al., 2009), it is highly likely that WISP1 may also induce MMPs expression in SMC. Thus, IL-18- and WISP1-mediated MMPs may contribute to adverse vessel remodeling, SMC migration and proliferation, and stenosis. Therefore, we also investigated whether IL-18 induces WISP1 expression and whether WISP1 induces MMPs in VSMC.
Our results demonstrate for the first time that IL-18 is a potent and rapid inducer of venous (VSMC), but not arterial (ASMC), SMC proliferation. IL-18 induces its own expression via PI3K/Akt/IKK/NF-κB and that of WISP1 via Akt/GSK3β/β-catenin/TCF-LEF signaling. Further, IL-18 induces VSMC proliferation in part via WISP1, and WISP1 induces its own expression and that of survivin and multiple MMPs via β-catenin/TCF-LEF interaction. WISP1, however, failed to modulate tissue inhibitors of matrix metalloproteinase (TIMP) expression. These results suggest a critical role for IL-18/WISP1 signaling in VSMC proliferation, and identify both as potential therapeutic targets in adverse remodeling, SMC hyperplasia and stenosis of grafted vessels following coronary artery bypass graft surgery.
MATERIALS AND METHODS
Cell culture
Following approval from the Institutional Review Board at the University of Texas Health Science Center in San Antonio and informed consent, discarded saphenous veins of 2–3 cm were obtained from patients undergoing coronary artery bypass graft surgery. Saphenous veins were opened longitudinally, endothelial cells scraped off, and cut into small pieces. The tissues were placed with their luminal surface facing down on a 0.1% gelatin-coated culture flask containing DMEM+20% FCS+antibiotics (100 U/mL penicillin, 50 μg/mL streptomycin), and cultured at 37°C in a humidified atmosphere of 95% air/5% CO2. After 15–20 days, the SMC outgrowths from the explants were trypsinized and replated in tissue culture dishes. Cells were used between passages 3–6. Around 96% of cells were positive for smooth muscle cell α-actin. At 70–80% confluency, the complete media were replaced with respective basal media containing 0.5% bovine serum albumin (conditioning medium). After 48 h incubation, recombinant human (rh) IL-18 or WISP1 was added and cultured for the indicated time periods. Similarly, human coronary artery SMC (ASMC) and culture (Chandrasekar et al., 2006) are described in the Supplementary file.
Cell proliferation
Cells were seeded in triplicate at a concentration of 1×103 cells/well in 200 μl of complete medium in 96-well flat-bottom plates (Costar, Corning, NY, USA). After 24 h incubation, the medium was replaced with conditioning medium, and then incubated for an additional 48 h. The cells were treated with rhIL-18 or rhWISP1 for 48 hrs and pulsed with 0.5 μCi/well [3H]Thymidine (TdR) for the last 16 h of the incubation period. Cells were then harvested on to membranes and the incorporated [3H]TdR was measured using a liquid-scintillation counter (Beckman LS6500; (Chandrasekar et al., 2003)). SMC proliferation was also determined by quantifying the incorporation of the thymidine analogue 5-bromo-2-deoxyuridine(BrdU) into DNA. Cells seeded as above were incubated in complete media for 24 h, followed by 3 days in conditioned media. The cells were then incubated with IL-18 for an additional 72 h. BrdU (10 μM)was present in the last 24 h. BrdU incorporation into DNA was quantified by Cell Proliferation Biotrak ELISA (GE Healthcare) according to the manufacturer’s instructions. Platelet-derived growth factor (PDGF)-BB (10 ng/ml) served as a positive control. Results were expressed as fold increase. Cell proliferation was confirmed by quantifying cell numbers using a Coulter counter.
Cell growth/hypertrophy
The rate of protein synthesis was determined by the incorporation of [3H]leucine as previously described (Chandrasekar et al., 2005; Colston et al., 2007). Quiescent VSMC in 24-well plates were treated or not with IL-18 (1 ng/ml) for 24 h and then incubated with 0.5 μCi [3H]-leucine for an additional 6 h. The radioactivity incorporated into the trichloroacetic acid-precipitable material was determined by liquid scintillation counting.
Adenoviral transduction
SMC were infected at ambient temperature with adenovirusin PBS at the indicated multiplicities of infection (MOI; Supplementary file). After 2h, the adenovirus was replaced with culture mediasupplemented with 0.5% BSA. Assays were carried out 24 h later. The transfection efficiency with the adenoviral (determined using Ad. GFP) and retroviral particles was near 100%, and infection with the viral vectors at indicated MOI and for the duration had no significant effect on SMC shape, adherence, or viability.
Reporter assays
A 4.405-kb hWISP1 promoter-reporter plasmid and its deletion and mutant constructs, 1401nt AKT1 promoter reporter and its mutant construct, 1700nt MMP9 promoter reporter and its deletion (800nt) and mutant constructs, and the 726nt MMP9 promoter are all described in detail in the Supplementary file.
Transcription factor activation
Activation of NF-κB was analyzed by a reporter assay using VSMC transfected with adenoviral NF-κB reporter vector (Ad.NF-κB-Luc, 50 MOI). Similarly, activation of AP-1 was analyzed by a reporter assay using adenoviral transduction of an AP-1 reporter vector (Ad.AP-1-Luc; 50 MOI). Ad-MCS-Luc served as a control. Ad-β-galactosidase (Ad-β-gal; 50 MOI) served as an internal control. β-galactosidase activity in cell extracts was determined using a luminescent β-galactosidase detection kit II (BD Biosciences), and the results are expressed as the ratio of firefly luciferase to β-galactosidase activity measured in relative light units (RLU). TCF-LEF DNA binding activity was analyzed by EMSA using labeled double stranded consensus TCF site (CACTGGGAGCCCTCTCAAAGCCCACACACCCGCCTG) from the WISP1 promoter (AF223404). Specificity of TCF DNA binding activity was verified in competition experiments using unlabeled (cold) consensus and mutant (5′-CACTGGGAGCCCTCTGCTAGCCCACACACCCGCCTG-3′) oligonucleotides. The β-catenin/TCF Optimal Promoter reporter plasmid TOPflash containing 7 TCF-consensus binding sites (Plasmid 12456; Addgene, Cambridge, MA; (Veeman et al., 2003), Addgene, Cambridge, MA). The FOPflash vector (#21-169) containing mutant TCF-binding sites was purchased from Millipore. VSMC were transfected with 2 μg of the indicated vector using Lipofectamine 2000. The firefly luciferase activity was normalized to the Renilla luciferase activity. TCF activation was also analyzed by EMSA.
Analysis of mRNA expression
The expression of IL-18, WISP1 and MMP9 mRNA was analyzed by reverse transcription and real time PCR using Taqman probes. No template controls were also performed for each assay, and samples processed without the reverse transcriptase step served as negative controls. Each cDNA sample was run in triplicate, and the amplification efficiencies of all primer pairs were determined by serial dilutions of input template. Data were analyzed using the 2−ΔΔCt method. Using Actin as the endogenous invariant control gene, all data were normalized and expressed as the fold difference in gene expression in IL-18 or WISP1 treated cells relative to untreated control cells. MMP and tissue inhibitors of metalloproteinases (TIMPs) expression was analyzed as previously described (Chandrasekar et al., 2006) using a qPCR-based array (Extracellular Matrix and Adhesion Molecule PCR array, PAHS-013; SA Biosciences, Frederick, MD) that detects 13 MMPs (MMP 1, 2, 3, 7, 8, 9, 10, 11, 12, 13, 14 [membrane type 1-MMP or MT1-MMP], 15 [MT2-MMP], 16 [MT3-MMP]) and 3 TIMPs (TIMP 1, 2, 3). Data in respective untreated and Ad.GFP infected samples was considered 1, and are shown as fold change.
Analysis of protein expression
Preparation of whole cell homogenates, cytoplasmic, mitochondrial and nuclear protein extraction, immunoblotting, detection of the immunoreactive bands by enhanced chemiluminescence (ECL Plus; GE Healthcare), and their quantification by densitometry were all previously described (Chandrasekar et al., 2006; Reddy et al.; Venkatachalam et al., 2009). α-Tubulin, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), voltage-dependent anion channel (VDAC), and Lamin A/C served as loading and purity controls for whole cell homogenates, cytoplasmic, mitochondrial and nuclear extracts, respectively. For immunoprecipitation, equal amounts of mitochondrial extracts were incubated with specific antibodies attached to agarose beads overnight at 4 °C under slow rotation. After washing 3 times in a buffer containing 50 mm Tris-Cl, 150 mm NaCl, 0.1% Nonidet P-40, the bound proteins were eluted from the beads by boiling in SDS sample buffer for subsequent SDS-PAGE and immunoblotting.
PI3K, Akt, Glycogen synthase kinase (GSK)3β, and IκB kinase (IKK) activity assays
PI3 kinase, Akt, GSK3β and IKK activities were assayed as detailed in the Supplementary file.
Cell death detection
Cell death was analyzed by a photometric enzyme immunoassay(Cell Death Detection ELISAPLUS kit, Roche Applied Science), and is described in the Supplementary file. The nitric oxide donor SNAP served as a positive control.
Statistical analysis
Results are expressed as means ± S.E. For statistical analysis we used analysis of variance followed by an appropriate post hoc multiple comparison test (Tukey method). Data were considered statistically significant at P < 0.05.
RESULTS
IL-18 induces proliferation of venous, but not arterial, SMC
Although veins are regarded as less prone to atherosclerosis, vein grafts show accelerated atherosclerosis and restenosis following coronary artery bypass graft surgery (Mitra et al., 2006; Suma, 1999). Therefore, we compared the effect of IL-18 on SMC derived from both coronary artery (ASMC) and saphenous vein (VSMC). 3H-thymidine and BrdU incorporation were used independently as markers of SMC proliferation. Incubation of quiescent ASMC for 48 h with IL-18 at doses up to 100 ng/ml failed to induce significant proliferation (Fig. 1A). We confirmed this observation using isolates from three different donors. PDGF-BB, used as a positive control, however stimulated robust proliferation. These results were confirmed using BrdU incorporation (data not shown). Since IL-18 failed to modulate ASMC numbers, we next investigated whether IL-18 induces ASMC death. At 10 ng/ml, IL-18 failed to induce any increase in oligonucleosomal fragmented DNA in ASMC cytoplasm (Fig. 1B), whereas SNAP, a nitric oxide donor, induced significant cell death. In contrast to its effects in ASMC, IL-18 induced a marked and significant dose-dependent proliferation of VSMC (Fig. 1C). A significant 2.32-fold increase in VSMC proliferation could be seen with as little as 0.5 ng/ml IL-18, but the response was more robust at 1 ng/ml and higher (Fig. 1C). Confirming the 3H-thymidine incorporation assay (Fig. 1C), addition of IL-18 (1 ng/ml) significantly increased VSMC cell numbers (Fig. 1D; phase contrast microscopy of untreated control and IL-18 treated (1 ng/ml for 48 h) VSMC are shown in Supplementary Figure S1A). Therefore, in all subsequent experiments, IL-18 was used at a concentration of 1 ng/ml. The stimulatory effects of 1 ng/ml IL-18 was blunted when VSMC were incubated with IL-18 neutralizing antibodies or with IL-18BP prior to IL-18 addition (supplementary Figure S1B), thus demonstrating IL-18 specificity. The simultaneous addition of polymyxin B, an endotoxin inhibitor, failed to modulate IL-18 effects, indicating that the response was not mediated by any low levels of endotoxin contaminating the IL-18 preparations (data not shown). Together, these results indicate that at the indicated concentrations and for the duration of treatment, IL-18 is a potent and rapid inducer of VSMC, but not ASMC, proliferation (Fig. 1).
Figure 1.

IL-18 induces VSMC proliferation and hypertrophy via PI3K and Akt
Since IL-18 induced robust proliferation of VSMC (Fig. 1C), we next investigated the underlying molecular mechanisms. Since the PI3K/Akt pathway plays a critical role in cell survival and growth, we investigated whether IL-18 induces PI3K and Akt activation in VSMC. Addition of IL-18 potently induced PI3K activity in quiescent VSMC (Fig. 2A), an effect that could be significantly attenuated by adenoviral transduction of dnPI3Kp85 or by pharmacological inhibition with wortmannin. Further, IL-18 induced time-dependent Akt phosphorylation (Thr308; Fig. 2B) and stimulated its kinase activity (Fig. 2C), effects that were significantly inhibited by Ad.dnAkt1 or Akti-X (Fig. 2D), and adenoviral transduction of dnPI3Kp85 or wortmannin (Fig. 2E). Importantly, infection with Ad.dnPI3Kp85 and Ad.dnAkt1 inhibited IL-18-induced VSMC proliferation (Fig. 2F). Since the PI3K/Akt pathway is also involved in cell growth, we next examined whether IL-18 induces VSMC hypertrophy. Indeed, IL-18 induced VSMC hypertrophy, and its pro-growth effects were blunted following forced expression of dnPI3Kp85 and dnAkt1 by adenoviral transduction (Fig. 2G). These results indicate that IL-18 induces SMC proliferation and hypertrophy, and both of these phenomena are PI3K and Akt dependent (Fig. 2).
Figure 2.

IL-18 autoregulation involves PI3K, Akt, IKK, and NF-κB
IL-18 is an NF-κB responsive gene. Since IL-18 activated PI3K and Akt (Fig. 2), and since IKK is one of the substrates for Akt, and induces IκB phosphorylation and degradation, we next examined whether IL-18 regulates its own expression in VSMC via Akt/IKK/NF-κB pathway. Using an in vitro kinase assay, we found that IL-18 treatment indeed activated IKKβ (Fig. 3A), one of the downstream substrates of Akt, and this activation was also significantly inhibited by Ad.dnAkt1 or Akti-X (Fig. 3A). Overexpression of kdIKKβ also inhibited IL-18 induced IKKβ activity (Fig. 3B). Further, IL-18 activated NF-κB in VSMC, as evidenced by increased NF-κB-dependent reporter gene activation (Fig. 3C) and p65 nuclear translocation (Fig. 3D), effects that were significantly attenuated by Ad.kdIKKβ, Ad.dnp65, and Ad.dnIκB-α. Importantly, IL-18 induced its own expression through a PI3K, Akt, IKK, and NF-κB dependent pathway (Fig. 3E). These results demonstrate that IL-18 induces its own expression in VSMC, and its autoregulation involves PI3K/Akt/IKK/NF-κB (Fig. 3).
Figure 3.

IL-18 induces MMP9 expression in VSMC
MMP9 is an NF-κB-responsive gene (Chandrasekar et al., 2006). Since MMP9 is an important determinant of neointimal thickening in human saphenous vein bypass grafts and is most highly expressed in the highly proliferative neointimal SMC (George et al., 1997), we next investigated whether IL-18 regulated MMP9 expression in VSMC. IL-18 potently induced luciferase activity from a transfected 726nt MMP9 reporter gene construct that contains the NF-κB responsive element, an effect that was markedly inhibited when the core NF-κB DNA-binding sequence was mutated (supplementary Figure 2A). Furthermore, forced expression of dnPI3Kp85, dnAkt, kdIKKβ, and dnp65 significantly blunted both IL-18 induced MMP9 promoter-dependent reporter gene activation (supplementary Figure 2B) and MMP9 mRNA expression (supplementary Figure 2C), indicating that IL-18 induces MMP9 expression in VSMC via PI3K/Akt/IKK-dependent NF-κB activation (supplementary Figure 2).
IL-18 induces Akt-dependent GSK3β phosphorylation
The ubiquitously expressed serine-threonine kinaseGSK3β is an Akt substrate that plays a role in both survival and death in a cell- and stimulus-specific manner. Akt-dependent GSK3β phosphorylation at Ser9 results in its degradation by the ubiquitin-proteasome pathway. IL-18 rapidly induced GSK3β phosphorylation at Ser9 in the VSMC, an effect that was significantly inhibited by the GSK3β inhibitor SB 216763 and LiCl (Fig. 4A). Similarly, pre-treatment with BIO (Meijer et al., 2003), a potent, but reversible, ATP-competitive GSK-3α/β inhibitor attenuated IL-18-mediated GSK3β phosphorylation (data not shown). Further, adenoviral transduction of dnPI3Kp85 and dnAkt attenuated GSK3β phosphorylation (Fig. 4B). Together, these results indicate that IL-18 mediates GSK3β phosphorylation and degradation via PI3K and Akt (Fig. 4).
Figure 4.

IL-18 induces β-catenin-dependent TCF-LEF activation
At steady state, GSK3β is present in an active form in the cytoplasm complexed with β-catenin, axin and adenomatous polyposis coli (APC) protein (Faux et al., 2008; Grimes and Jope, 2001). Following phosphorylation on Ser9, GSK3β is degraded by the proteasome pathway, thus releasing β-catenin. Released β-catenin accumulates in the cytoplasm, translocates to the nucleus, binds TCF-LEF family of transcription factors, and induces gene transcription (Aberle et al., 1997). Therefore, we next examined the effect of IL-18 on β-catenin levels. IL-18 increased the time-dependent accumulation of β-catenin in the cytoplasm (Fig. 5A), followed by its nuclear translocation and stabilization (Fig. 5B), effects that were attenuated by the retroviral transduction of phosphorylation-deficient GSK3β (S9A) (Fig. 5A, 5B). IL-18 also increased TCF-LEF DNA binding activity, an effect that was significantly attenuated by the adenoviral transduction of ICAT, an inhibitor of β-catenin-TCF interaction (Fig. 5C). IL-18 also induced the Top-Flash reporter activity by several fold, as compared to the basal activity of the Fop-Flash reporter, and this effect was similarly inhibited by the forced expression of ICAT (Fig. 5D). Further, forced expression of non-degradable β-catenin (Ad-S37A-β-catenin-HA) enhanced TCF-LEF reporter gene activation (Fig. 5E), indicating that IL-18 induced TCF-LEF activation is β-catenin dependent (Fig. 5).
Figure 5.

IL-18 activates Akt in TCF-LEF-dependent manner
We have demonstrated above that IL-18 stimulates Akt phosphorylation and kinase activity (Fig. 2D, 2E). Since Akt is a TCF-LEF responsive gene (Dihlmann et al., 2005), we next investigated whether IL-18 enhances Akt transcription. VSMC were transfected with either a wild type 1401nt Akt1 promoter-reporter vector, or one in which the four functional TCF-LEF binding elements (TBE) were mutated. Addition of IL-18 markedly induced luciferase activity from the wild type construct (~6 fold greater than from the empty reporter vector; supplementary Figure 3). Mutation of the four TBE reduced this response by ~50% (supplementary Figure 3). These data suggest that IL-18 regulates Akt at multiple levels; increasing Akt gene transcription, phosphorylation and activation. These results indicate a positive amplification in Akt signaling in IL-18-treated VSMC (Fig. S3).
IL-18 induces VSMC proliferation via WISP1
WISP1 is a pro-survival and pro-growth factor. It mediates TNF-α induced cardiac-derived fibroblast proliferation, but inhibits TNF-α induced cardiomyocyte death (Venkatachalam et al., 2009). Since IL-18 induces TCF-LEF activation (Fig. 6C, 6D), and as WISP1 5′cis regulatory region contains multiple TCF-LEF binding sites (Xu et al., 2000), we next investigated whether IL-18 can induce WISP1 expression and stimulate VSMC proliferation via WISP1. Lentiviral transduction of WISP1 shRNA and WISP1 neutralizing antibodies both significantly blunted IL-18-induced VSMC proliferation (Fig. 6A). Further, IL-18 induced time-dependent WISP1 mRNA expression (Fig. 6B), a response that was significantly attenuated by Ad.dnPI3Kp85, Ad.dnAkt, Ad.ICAT, and retroviral GSK3β (S9A) (Fig. 6C), and induced WISP1 protein expression (Fig. 6D). IL-18 also enhanced WISP1 promoter-dependent reporter gene activation, an effect that was significantly blunted when the TBE sites were mutated (Fig. 6E). These results indicate that IL-18 induces WISP1 expression via PI3K/Akt/GSK3β/β-catenin and TCF-LEF, and stimulates VSMC proliferation in part via WISP1 (Fig. 6).
Figure 6.

WISP1 induces its own expression in VSMC
Since WISP1, like IL-18 (Fig. 5A, 5B), can activate β-catenin (Venkatesan et al.), and is also a β-catenin responsive gene (Venkatesan et al.; Xu et al., 2000), we next investigated whether WISP1 induces its own expression in VSMC. We found that addition of WISP1 significantly induced its own mRNA expression in VSMC, and this was markedly attenuated by the forced expression of dnPI3Kp85, dnAkt, GSK3β(S9A), and ICAT (Fig. 7A). Further, WISP1 increased endogenous WISP1 protein in a time dependent manner (Fig. 7B). These results indicate that WISP1 induces its own expression in VSMC in PI3K/Akt/β-catenin/TCF-LEF signaling (Fig. 7).
Figure 7.

WISP1 promotes mitochondrial translocation of β-catenin and its physical association with Bcl-2
Previously we demonstrated that WISP1 is a potential pro-survival factor that induces the expression of anti-apoptotic Bcl-2 in primary cardiomyocytes (Venkatesan et al.). Since β-catenin has been shown to associate with Bcl-2 in leukotriene D4-treated intestinal epithelial cells (Mezhybovska et al., 2006), we next investigated whether WISP1 promotesβ-catenin translocation to mitochondria and association with Bcl-2. WISP1 did promote mitochondrial translocation of β-catenin in a time-dependent manner (supplementary Figure 4A), with significant increases detected as early as 1 h post-treatment. Similarly, WISP1 increased mitochondrial Bcl-2 levels (supplementary Figure 4B). Further, reciprocal immunoprecipitation and immunoblotting revealed that in VSMC mitochondria, WISP1 promotes β-catenin-Bcl-2 physically association (supplementary Figure 4C).
WISP1 induces survivin expression via β-catenin-TCF-LEF signaling
Survivin, a member of the inhibitor of apoptosis protein family, is a TCF-LEF responsive gene, and plays a role in the proliferation of normal hematopoietic cells (Fukuda et al., 2004). We therefore examined whether WISP1 induces survivin expression in VSMC. Indeed WISP1 significantly upregulated survivin expression in VSMC (supplementary Figure 4D), an effect markedly inhibited by the adenoviral transduction of ICAT (supplementary Figure 4E), supporting the role of TCF-LEF in its induction.
WISP1 induces matrix metalloproteinases (MMP), but not tissue inhibitors of MMP (TIMP) expression
Matrix metalloproteinases play critical roles in both SMC migration and proliferation. Since several MMP genes contain one or more TCF-LEF binding sites in their 5′ cis regulatory regions (Yan and Boyd, 2007), and since WISP1-induced survivin expression is blunted by the inhibitor of the β-catenin/TCF binding interaction (ICAT; supplementary Figure 4E), we next investigated whether WISP1 induces MMP expression in VSMC via β-catenin/TCF interaction. Since TIMPs, the MMP inhibitors, also contain TCF-LEF response elements (Yan and Boyd, 2007), we also investigated their regulation. Using a qPCR-based array, we found that WISP1 is a potent inducer of various MMPs in VSMC, including MMP1, 2, 3, 9, and 14, and this response was significantly inhibited by the forced expression of ICAT (supplementary Figure 5). Interestingly, WISP1 failed to regulate TIMPs expression during the 4 h treatment period. These results indicate for the first time that WISP1 induces multiple MMPs in VSMC via β-catenin/TCF-LEF interaction (supplementary Figure 5).
WISP1 induces MMP9 transcription via TCF-LEF
Since MMP9 induction was relatively high following WISP1 treatment (supplementary Figure 5), we next focused on investigating the role of TBE in WISP1-mediated MMP9 transcription. WISP1 significantly increased the activity of a 1.7kb MMP9 promoter reporter construct transfected in to VSMC (Fig. 8A). This effect was attenuated by the deletion of the two distal TBE and a more pronounced inhibition when the two proximal TBE sites were mutated in a 0.8kb deletion construct (Fig. 8A), indicating that WISP1 induces MMP9 transcription in part through TBE. Using the 0.8kb construct, we also found that while forced expression of ICAT by adenoviral transduction inhibited (Fig. 8B), forced expression of phosphorylation-deficient β-catenin (S37A) enhanced MMP9 promoter reporter activity (Fig. 8C), confirming the role of β-catenin-TCF-LEF interaction in MMP9 transcription (Fig. 8).
Figure 8.

WISP1 induces MMP9 transcription in part via AP-1, but not NF-κB
In addition to TCF-LEF, MMP9 is also an AP-1 and NF-κB responsive gene (Chandrasekar et al., 2006; Yan and Boyd, 2007). Since a physical association between TCF-LEF and c-Jun has been previously shown to synergistically regulate MMP transcription and invasion of renal and colonic epithelial cells (Rivat et al., 2003), we next investigated whether AP-1 plays a role in WISP1-mediated MMP9 transcription. At first, we determined the effect of WISP1 on AP-1 activation using a reporter assay. Results in Fig 8D show that WISP1 induces AP-1-dependent reporter gene activation in VSMC, although its effect was more moderate compared to that of the positive control IL-18. Further, adenoviral transduction of dnc-Jun significantly blunted WISP1 induced MMP9 promoter-dependent reporter gene activation (Fig. 8E). Since CTGF, a member of the CCN family has been shown to activate NF-κB incultured murine proximal tubuloepithelial cells (Sanchez-Lopez et al., 2009), and as NF-κB plays a critical role in MMP9 transcription (Chandrasekar et al., 2006), we next investigated whether NF-κB plays a role in WISP1-mediated MMP9 transcription. Our results show that unlike CTGF and IL-18, WISP1 failed to induce NF-κB-dependent reporter gene activation (Fig. 8F). Further, adenoviral transduction of kdIKKβ, dnp65 or dnIκB-α failed to inhibit WISP1-induced MMP9 transcription (Fig. 8G), suggesting that WISP1 induces MMP9 transcription in part via AP-1, but not through NF-κB (Fig. 8).
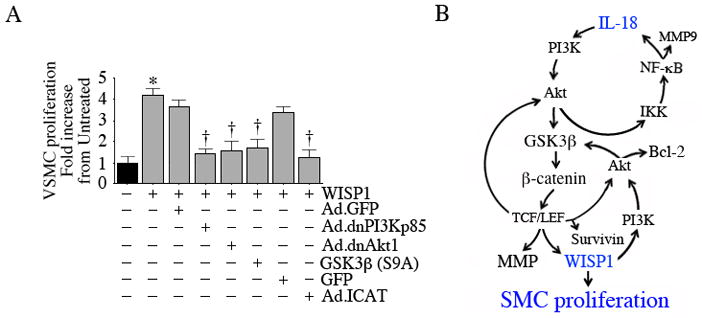
WISP1 induces VSMC proliferation via PI3K/Akt/GSK3ββ-catenin/TCF-LEF
Since WISP1 activates various pro-survival factors (e.g., Bcl-2, surviving; supplementary Figure 4), mediates TNF-α-induced cardiac-derived fibroblast proliferation (Venkatachalam et al., 2009), and since its knockdown blunts IL-18-mediated VSMC proliferation (Fig. 6A), we next investigated whether WISP1 can induce VSMC proliferation. Indeed WISP1 significantly increased VSMC proliferation, an effect that was markedly attenuated by the adenoviral transduction of dnPI3Kp85, dnAkt, GSK3β(S9A), and ICAT (Fig. 9A). Since we demonstrated that WISP1 induces survivin expression (supplementary Figure 4), we next investigated whether survivin modulates WISP1-induced VSMC proliferation. Our unpublished results show that while forced expression of mutant survivin moderately attenuated WISP1-induced VSMC proliferation, overexpression of wild type survivin was additive. These results indicate that WISP1 is a potent inducer of VSMC proliferation (Fig. 9).
Figure 9.
DISCUSSION
Our results demonstrate for the first time that IL-18 is a potent and rapid inducer of saphenous vein SMC proliferation, and that this response is partly mediated by WISP1. IL-18 also induces VSMC hypertrophy. IL-18 induces its own expression via PI3K/Akt/IKK/NF-κB and β-catenin/TCF-LEF signaling, and that of WISP1 via GSK3β/β-catenin/TCF-LEF. Further, WISP1 induces the prosurvival factors survivin and Bcl-2, and promotes their physical interaction in mitochondria. Both IL-18 and WISP1 induce multiple matrix metalloproteinases, but not their tissue inhibitors (TIMPs), via β-catenin/TCF-LEF interaction. These results suggest a critical role for IL-18/WISP1 signaling in SMC hyperplasia and stenosis in vein grafts following coronary artery bypass graft surgery.
Although relatively less prone to atherosclerosis in situ (Mitra et al., 2006; Suma, 1999), saphenous veins used as arterial grafts show accelerated atherosclerosis and stenosis, suggesting that arterialization increases their susceptibility to neointimal hyperplasia and reduced patency. Here we show that IL-18 is a more potent mitogen for venous SMC than for coronary artery SMC. The mitogenic effect of IL-18 on VSMC was detectable as early as 2 days following addition, and was markedly inhibited by IL-18 neutralizing antibodies or IL-18 binding protein. Furthermore, IL-18 is an NF-κB responsive gene (Tone et al., 1997), as well as an inducer of NF-κB activation. In the VSMC, we found that IL-18 induced its own expression through PI3K/Akt/IKK-dependent NF-κB activation. Thus in vivo, following its initial induction, IL-18 may sustain its own expression by autoregulation, and thereby further enhance SMC hyperplasia in the graft.
We also show for the first time that IL-18 activates PI3K/Akt-dependent glycogen synthase kinase (GSK)3β phosphorylation and degradation, β-catenin nuclear translocation and stabilization, and TCF-LEF DNA binding and gene activation. Recently it was reported that GSK3β may act as a selective negative regulator of NF-κB activity (Vines et al., 2006). Here we report similar observations, and show that while nuclear levels of NF-κB were considerably increased in the IL-18-treated VSMC, GSK3β levels were significantly reduced. β-catenin is also known to negatively regulate NF-κB, however in a cell type-specific manner. In cancer cell lines for example, β-catenin was shown to physically interact with NF-κB and reduce its DNA binding activity (Deng et al., 2002). Further, both IKKα and IKKβ can phosphorylateβ-catenin, resulting in its degradation (Lamberti et al., 2001) and the NF-κB component RelA may suppressβ-catenin/TCF dependent transcription (Masui et al., 2002). However, in vascular SMC, β-catenin was shown to mediate IκB degradation and thus activate NF-κB (Wang et al., 2004). Furthermore, we found that adenoviral transduction of non-degradable β-catenin decreased total IκB levels in IL-18-treated VSMC in a time dependent manner (data not shown). Thus IL-18 induced GSK3β degradation and the subsequent release of β-catenin might activate NF-κB and induce κB-dependent pro-mitogenic gene expression in VSMC. Thus IL-18 may sustain its expression in vascular SMC by inducing and maintaining NF-κB activity through more than one mechanism.
IL-18 induced VSMC proliferation in part through WISP1. Both WISP1 neutralizing antibodies and retroviral shRNA-mediated knockdown significantly attenuated IL-18-mediated VSMC proliferation. Addition of WISP1 to VSMC induced both its own expression as well as that of survivin and Bcl-2. Survivin exerts anti-apoptotic and pro-growth effects in a cell type and stimulus-specific manner. It also plays a role in vein graft hyperplasia (Wang et al., 2005). It inhibits both intrinsic and extrinsic pro-apoptotic signaling pathways. By binding to pro-caspase-9, survivin prevents its activation by cytochrome c, thus blocking the intrinsic apoptotic pathway (Kasof and Gomes, 2001). Further, by inhibiting caspases 3, 6, and 7, survivin blocks the extrinsic apoptotic pathway. We believe that survivin might play a similar role in WISP1-treated VSMC. Consistent with our earlier results in cardiomyocytes (Venkatesan et al.), we found that WISP1 induces Bcl-2 expression and translocation to mitochondria. Similar to survivin, Bcl-2 blocks cytochrome c release from mitochondria (Yang et al., 1997), thus preventing intrinsic apoptotic signaling. WISP1 also promoted mitochondrial translocation of β-catenin and its physical association with Bcl-2. Similar results have been reported in leukotriene D4 (LTD4) treated intestinal epithelial cells (Mezhybovska et al., 2006). Interestingly, in those cells, Bcl-2 overexpression increased basal and LTD4 induced TCF-LEF-dependent reporter gene activation via mechanisms yet to be determined (Mezhybovska et al., 2006). Together, these results demonstrate that by inducing survivin and Bcl-2 expression, WISP1 exerts pro-survival and pro-growth effects in VSMC. These results however, contrast with those with Nov3, another CCN family member, which has been shown to suppress SMC proliferation and migration, and neointimal hyperplasia (Shimoyama et al.). Similarly, CTGF which is known to activate NF-κB incultured murine proximal tubuloepithelial cells (Sanchez-Lopez et al., 2009), has been shown to induce human aortic smooth muscle cell death, but at higher doses and after longer incubations (Hishikawa et al., 2000), indicating that the effects of WISP1 are not shared with other members of the CCN family. Therefore targeting WISP1 specfically may attenuate intimal hyperplasia in vivo.
Using an array-based technology, we also report for the first time that WISP1 significantly upregulates the expression of multiple matrix metalloproteinases in VSMC. Adenoviral transduction of ICAT (inhibitor of the β-catenin/TCF binding interaction) significantly blunted the expression levels of MMP 1, 2, 3, 9, and 14, supporting a role for β-catenin/TCF-LEF interaction in their transcription. It is interesting to note that while all these MMPs, with the exception of MMP12, contain one or more putative TCF-LEF binding sites (Yan and Boyd, 2007), MMP12 contains at least 4 putative AP-1 binding sites. Since WISP1 induces AP-1-dependent reporter gene activation, it is possible that MMP12 was induced via AP-1 activation. It is surprising however that WISP1 failed to significantly modulate MMP7, 8, and 13 in VSMC, as these MMPs also contain either AP-1 (MMP13) or TCF-LEF and AP-1 binding sites (MMP7 and 8). These results suggest that regulation of MMPs is complex, stimulus-specific, cell-type dependent, and probably time-dependent, and thus needs further investigation. Another potentially interesting observation is that WISP1 failed to significantly modulate TIMPs expression in VSMC, despite the presence of AP-1 binding sites in TIMPs 1 and 2 promoter regions (Yan and Boyd, 2007). The array does not contain TIMP4. Since TIMP4 contains AP-1 and multiple TCF-LEF binding site, it is possible that WISP1 may induce its expression in VSMC. Here we investigated the expression levels of MMPs and TIMPs at 4 h after WISP1 treatment. It is possible that, similar to the effect of IL-18 on cardiomyocytes (Reddy et al.), WISP1 may upregulate TIMPs expression in VSMC at later time periods, and our future studies will investigate this possibility. Importantly, these results indicate that by favoring early induction of MMPs, WISP1 may play a role in adverse remodeling, SMC hyperplasia and stenosis in saphenous vein grafts in vivo.
Similar to WISP1 and consistent with our results with ASMC, IL-18 induced MMP9 expression in the VSMC. Both IL-18 and WISP1 induced GSK3β phosphorylation and degradation, β-catenin nuclear translocation and TCF-LEF activation. While AP-1 plays a role in WISP-1-induced MMP9 transcription, NF-κB appears not to be involved, suggesting a level of crosstalk between β-catenin and members of the AP-1, but not NF-κB, families of proteins. In fact, a physical interaction between β-catenin and c-Jun and c-Fos has been previously demonstrated (Toualbi et al., 2007). Using GST pull-down assays, these authors showed an interaction between the armadillo repeat domain of β-catenin with the DNA-binding domain of c-Jun, and of the C-terminal domain of β-catenin with the N-terminal domain of c-Fos. They also reported that overexpression of AP-1 activates the transcription of two β-catenin target genes, cyclin D1 and c-myc, by a mechanism independent of the AP-1 site, but fully dependent on the TCF-binding site (Toualbi et al., 2007). It is possible therefore that WISP1 induced AP-1 and β-catenin may synergistically induce MMPs expression in VSMC.
Our results demonstrate that IL-18 induces both proliferation and hypertrophy of VSMC. Since SMC undergo dedifferentiation from a contractile to a synthetic phenotype prior to migration and proliferation, and since proliferation and hypertrophy of SMC occur in parallel as reported here, we do not rule out the possibility that the cells undergoing proliferation and hypertrophy may be from different lineages, occur at different time intervals and may result from activation of distinct signal transduction pathways. However, our results demonstrate that IL-18 induces proliferation and hypertrophy of VSMC via activation of PI3K and Akt. Our future studies will investigate the downstream signaling pathways involved in IL-18-induced VSMC hypertrophy.
Our principle findings are that; 1) IL-18 stimulates proliferation of venous but not arterial SMC through a mechanism that partly involves WISP1. 2) In VMSC, IL-18 induces its own expression and that of WISP1, and WISP1 also induces its own expression. Their persistent expression may contribute to VSMC proliferation in vivo. 3) In addition to inducing Akt activation, IL-18 also stimulates AKT gene transcription. Since Akt is a pro-survival factor, its increased expression and activation may contribute to VSMC survival and proliferation in vivo. 4) Both IL-18 and WISP1 induce MMPs expression through β-catenin and AP-1 activation. While IL-18 also activates NF-κB, WISP1 fails to modulate its activity. Importantly, WISP1 induces MMP, but not TIMP expression in VSMC. Enhanced MMP expression results in ECM degradation, SMC migration and proliferation. 5) WISP1 induces the anti-apoptotic Bcl-2 and TCF-LEF-responsive survivin expression in VSMC. Survivin has been shown to induce SMC hyperplasia (42). 6) Since WISP1 activates AP-1, and since IL-18 is an AP-1 responsive gene, it is plausible that WISP1 may induce IL-18 expression in VSMC. In conclusion, our studies indicate that both IL-18 and WISP1 activate similar and multiple cell survival pathways in VSMC, and their coexpression and regulation might contribute to intimal hyperplasia, stenosis, and graft failure following coronary artery bypass graft surgery in vivo.
Supplementary Material
Acknowledgments
Funded by: Biomedical Laboratory Research and Development Service of the VA Office of Research and Development, Award number 1I01BX000246 NIH-NHLBI grant HL-86787, HL-70241 and HL-80682
Abbreviation: The following abbreviation were used
- ASMC
aortic smooth muscle cells
- AP-1
activator protein-1
- Bcl-2
B cell leukemia-2
- CCN
Cyr61/CTGF/Nov
- Cyr61
cysteine-rich protein 61
- CTGF
connective tissue growth factor
- GSK
glycogen synthase kinase
- MOI
multiplicity of infection
- PI3K
phosphoinositide 3-kinase
- ICAT
inhibitor of the β-catenin/TCF binding interaction
- WISP
Wnt-1 inducible signaling pathway protein
- WNT
wingless-type MMTV integration site family
- TCF-LEF
T-cell factor-lymphoid enhancer binding factor
- dn
dominant negative
- kd
kinase deficient
- ECM
extracellular matrix
- EMSA
electrophoretic mobility shift assay
- GFP
green fluorescent protein
- IκB
inhibitory κB
- IKK
IκB kinase
- MMP
matrix metalloproteinase
- TIMP
tissue inhibitor of MMP
- NF-κB
nuclear factor κB
- PDGF
platelet-derived growth factor
- RT-qPCR
reverse transcriptase quantitative polymerase chain reaction
- shRNA
short hairpin RNA
- TNF-α
tumor necrosis factor-α
- Nov
nephroblastoma-overexpressed gene
- VSMC
saphenous vein smooth muscle cells
Footnotes
The contents of this report do not represent the views of the Department of Veterans Affairs or the United States Government.
References
- Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997;16(13):3797–3804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom AB, Brockbank SM, van Lent PL, van Beuningen HM, Geurts J, Takahashi N, van der Kraan PM, van de Loo FA, Schreurs BW, Clements K, Newham P, van den Berg WB. Involvement of the Wnt signaling pathway in experimental and human osteoarthritis: prominent role of Wnt-induced signaling protein 1. Arthritis Rheum. 2009;60(2):501–512. doi: 10.1002/art.24247. [DOI] [PubMed] [Google Scholar]
- Chandrasekar B, Mummidi S, Claycomb WC, Mestril R, Nemer M. Interleukin-18 is a pro-hypertrophic cytokine that acts through a phosphatidylinositol 3-kinase-phosphoinositide-dependent kinase-1-Akt-GATA4 signaling pathway in cardiomyocytes. J Biol Chem. 2005;280(6):4553–4567. doi: 10.1074/jbc.M411787200. [DOI] [PubMed] [Google Scholar]
- Chandrasekar B, Mummidi S, Mahimainathan L, Patel DN, Bailey SR, Imam SZ, Greene WC, Valente AJ. Interleukin-18-induced human coronary artery smooth muscle cell migration is dependent on NF-kappaB-and AP-1-mediated matrix metalloproteinase-9 expression and is inhibited by atorvastatin. J Biol Chem. 2006;281(22):15099–15109. doi: 10.1074/jbc.M600200200. [DOI] [PubMed] [Google Scholar]
- Chandrasekar B, Mummidi S, Perla RP, Bysani S, Dulin NO, Liu F, Melby PC. Fractalkine (CX3CL1) stimulated by nuclear factor kappaB (NF-kappaB)-dependent inflammatory signals induces aortic smooth muscle cell proliferation through an autocrine pathway. Biochem J. 2003;373(Pt 2):547–558. doi: 10.1042/BJ20030207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colston JT, de la Rosa SD, Koehler M, Gonzales K, Mestril R, Freeman GL, Bailey SR, Chandrasekar B. Wnt-induced secreted protein-1 is a prohypertrophic and profibrotic growth factor. Am J Physiol Heart Circ Physiol. 2007;293(3):H1839–1846. doi: 10.1152/ajpheart.00428.2007. [DOI] [PubMed] [Google Scholar]
- Deng J, Miller SA, Wang HY, Xia W, Wen Y, Zhou BP, Li Y, Lin SY, Hung MC. beta-catenin interacts with and inhibits NF-kappa B in human colon and breast cancer. Cancer Cell. 2002;2(4):323–334. doi: 10.1016/s1535-6108(02)00154-x. [DOI] [PubMed] [Google Scholar]
- Dihlmann S, Kloor M, Fallsehr C, von Knebel Doeberitz M. Regulation of AKT1 expression by beta-catenin/Tcf/Lef signaling in colorectal cancer cells. Carcinogenesis. 2005;26(9):1503–1512. doi: 10.1093/carcin/bgi120. [DOI] [PubMed] [Google Scholar]
- Elhage R, Jawien J, Rudling M, Ljunggren HG, Takeda K, Akira S, Bayard F, Hansson GK. Reduced atherosclerosis in interleukin-18 deficient apolipoprotein E-knockout mice. Cardiovasc Res. 2003;59(1):234–240. doi: 10.1016/s0008-6363(03)00343-2. [DOI] [PubMed] [Google Scholar]
- Faux MC, Coates JL, Catimel B, Cody S, Clayton AH, Layton MJ, Burgess AW. Recruitment of adenomatous polyposis coli and beta-catenin to axin-puncta. Oncogene. 2008;27(44):5808–5820. doi: 10.1038/onc.2008.205. [DOI] [PubMed] [Google Scholar]
- Fukuda S, Mantel CR, Pelus LM. Survivin regulates hematopoietic progenitor cell proliferation through p21WAF1/Cip1-dependent and -independent pathways. Blood. 2004;103(1):120–127. doi: 10.1182/blood-2003-05-1756. [DOI] [PubMed] [Google Scholar]
- George SJ, Zaltsman AB, Newby AC. Surgical preparative injury and neointima formation increase MMP-9 expression and MMP-2 activation in human saphenous vein. Cardiovasc Res. 1997;33(2):447–459. doi: 10.1016/s0008-6363(96)00211-8. [DOI] [PubMed] [Google Scholar]
- Gerdes N, Sukhova GK, Libby P, Reynolds RS, Young JL, Schonbeck U. Expression of interleukin (IL)-18 and functional IL-18 receptor on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for atherogenesis. J Exp Med. 2002;195(2):245–257. doi: 10.1084/jem.20011022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol. 2001;65(4):391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- Hishikawa K, Nakaki T, Fujii T. Connective tissue growth factor induces apoptosis via caspase 3 in cultured human aortic smooth muscle cells. Eur J Pharmacol. 2000;392(1–2):19–22. doi: 10.1016/s0014-2999(00)00115-1. [DOI] [PubMed] [Google Scholar]
- Hung J, McQuillan BM, Chapman CM, Thompson PL, Beilby JP. Elevated interleukin-18 levels are associated with the metabolic syndrome independent of obesity and insulin resistance. Arterioscler Thromb Vasc Biol. 2005;25(6):1268–1273. doi: 10.1161/01.ATV.0000163843.70369.12. [DOI] [PubMed] [Google Scholar]
- Kasof GM, Gomes BC. Livin, a novel inhibitor of apoptosis protein family member. J Biol Chem. 2001;276(5):3238–3246. doi: 10.1074/jbc.M003670200. [DOI] [PubMed] [Google Scholar]
- Kim SH, Eisenstein M, Reznikov L, Fantuzzi G, Novick D, Rubinstein M, Dinarello CA. Structural requirements of six naturally occurring isoforms of the IL-18 binding protein to inhibit IL-18. Proc Natl Acad Sci U S A. 2000;97(3):1190–1195. doi: 10.1073/pnas.97.3.1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamberti C, Lin KM, Yamamoto Y, Verma U, Verma IM, Byers S, Gaynor RB. Regulation of beta-catenin function by the IkappaB kinases. J Biol Chem. 2001;276(45):42276–42286. doi: 10.1074/jbc.M104227200. [DOI] [PubMed] [Google Scholar]
- Li JM, Eslami MH, Rohrer MJ, Dargon P, Joris I, Hendricks G, Baker S, Cutler BS. Interleukin 18 binding protein (IL18-BP) inhibits neointimal hyperplasia after balloon injury in an atherosclerotic rabbit model. J Vasc Surg. 2008;47(5):1048–1057. doi: 10.1016/j.jvs.2007.12.005. [DOI] [PubMed] [Google Scholar]
- Maffia P, Grassia G, Di Meglio P, Carnuccio R, Berrino L, Garside P, Ianaro A, Ialenti A. Neutralization of interleukin-18 inhibits neointimal formation in a rat model of vascular injury. Circulation. 2006;114(5):430–437. doi: 10.1161/CIRCULATIONAHA.105.602714. [DOI] [PubMed] [Google Scholar]
- Mallat Z, Corbaz A, Scoazec A, Besnard S, Leseche G, Chvatchko Y, Tedgui A. Expression of interleukin-18 in human atherosclerotic plaques and relation to plaque instability. Circulation. 2001a;104(14):1598–1603. doi: 10.1161/hc3901.096721. [DOI] [PubMed] [Google Scholar]
- Mallat Z, Corbaz A, Scoazec A, Graber P, Alouani S, Esposito B, Humbert Y, Chvatchko Y, Tedgui A. Interleukin-18/interleukin-18 binding protein signaling modulates atherosclerotic lesion development and stability. Circ Res. 2001b;89(7):E41–45. doi: 10.1161/hh1901.098735. [DOI] [PubMed] [Google Scholar]
- Masui O, Ueda Y, Tsumura A, Koyanagi M, Hijikata M, Shimotohno K. RelA suppresses the Wnt/beta-catenin pathway without exerting trans-acting transcriptional ability. Int J Mol Med. 2002;9(5):489–493. [PubMed] [Google Scholar]
- Mazodier K, Marin V, Novick D, Farnarier C, Robitail S, Schleinitz N, Veit V, Paul P, Rubinstein M, Dinarello CA, Harle JR, Kaplanski G. Severe imbalance of IL-18/IL-18BP in patients with secondary hemophagocytic syndrome. Blood. 2005;106(10):3483–3489. doi: 10.1182/blood-2005-05-1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer L, Skaltsounis AL, Magiatis P, Polychronopoulos P, Knockaert M, Leost M, Ryan XP, Vonica CA, Brivanlou A, Dajani R, Crovace C, Tarricone C, Musacchio A, Roe SM, Pearl L, Greengard P. GSK-3-selective inhibitors derived from Tyrian purple indirubins. Chem Biol. 2003;10(12):1255–1266. doi: 10.1016/j.chembiol.2003.11.010. [DOI] [PubMed] [Google Scholar]
- Mezhybovska M, Wikstrom K, Ohd JF, Sjolander A. The inflammatory mediator leukotriene D4 induces beta-catenin signaling and its association with antiapoptotic Bcl-2 in intestinal epithelial cells. J Biol Chem. 2006;281(10):6776–6784. doi: 10.1074/jbc.M509999200. [DOI] [PubMed] [Google Scholar]
- Mitra AK, Gangahar DM, Agrawal DK. Cellular, molecular and immunological mechanisms in the pathophysiology of vein graft intimal hyperplasia. Immunol Cell Biol. 2006;84(2):115–124. doi: 10.1111/j.1440-1711.2005.01407.x. [DOI] [PubMed] [Google Scholar]
- Reddy VS, Prabhu SD, Mummidi S, Valente AJ, Venkatesan B, Shanmugam P, Delafontaine P, Chandrasekar B. Interleukin-18 induces EMMPRIN expression in primary cardiomyocytes via JNK/Sp1 signalingand MMP-9 in part via EMMPRIN and through AP-1 and NF-kappaB activation. Am J Physiol Heart Circ Physiol. 299(4):H1242–1254. doi: 10.1152/ajpheart.00451.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivat C, Le Floch N, Sabbah M, Teyrol I, Redeuilh G, Bruyneel E, Mareel M, Matrisian LM, Crawford HC, Gespach C, Attoub S. Synergistic cooperation between the AP-1 and LEF-1 transcription factors in activation of the matrilysin promoter by the src oncogene: implications in cellular invasion. FASEB J. 2003;17(12):1721–1723. doi: 10.1096/fj.03-0132fje. [DOI] [PubMed] [Google Scholar]
- Sahar S, Dwarakanath RS, Reddy MA, Lanting L, Todorov I, Natarajan R. Angiotensin II enhances interleukin-18 mediated inflammatory gene expression in vascular smooth muscle cells: a novel cross-talk in the pathogenesis of atherosclerosis. Circ Res. 2005;96(10):1064–1071. doi: 10.1161/01.RES.0000168210.10358.f4. [DOI] [PubMed] [Google Scholar]
- Sanchez-Lopez E, Rayego S, Rodrigues-Diez R, Rodriguez JS, Rodriguez-Vita J, Carvajal G, Aroeira LS, Selgas R, Mezzano SA, Ortiz A, Egido J, Ruiz-Ortega M. CTGF promotes inflammatory cell infiltration of the renal interstitium by activating NF-kappaB. J Am Soc Nephrol. 2009;20(7):1513–1526. doi: 10.1681/ASN.2008090999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoyama T, Hiraoka S, Takemoto M, Koshizaka M, Tokuyama H, Tokuyama T, Watanabe A, Fujimoto M, Kawamura H, Sato S, Tsurutani Y, Saito Y, Perbal B, Koseki H, Yokote K. CCN3 inhibits neointimal hyperplasia through modulation of smooth muscle cell growth and migration. Arterioscler Thromb Vasc Biol. 30(4):675–682. doi: 10.1161/ATVBAHA.110.203356. [DOI] [PubMed] [Google Scholar]
- Suma H. Arterial grafts in coronary bypass surgery. Ann Thorac Cardiovasc Surg. 1999;5(3):141–145. [PubMed] [Google Scholar]
- Tone M, Thompson SA, Tone Y, Fairchild PJ, Waldmann H. Regulation of IL-18 (IFN-gamma-inducing factor) gene expression. J Immunol. 1997;159(12):6156–6163. [PubMed] [Google Scholar]
- Toualbi K, Guller MC, Mauriz JL, Labalette C, Buendia MA, Mauviel A, Bernuau D. Physical and functional cooperation between AP-1 and beta-catenin for the regulation of TCF-dependent genes. Oncogene. 2007;26(24):3492–3502. doi: 10.1038/sj.onc.1210133. [DOI] [PubMed] [Google Scholar]
- Troseid M, Seljeflot I, Hjerkinn EM, Arnesen H. Interleukin-18 is a strong predictor of cardiovascular events in elderly men with the metabolic syndrome: synergistic effect of inflammation and hyperglycemia. Diabetes Care. 2009;32(3):486–492. doi: 10.2337/dc08-1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeman MT, Slusarski DC, Kaykas A, Louie SH, Moon RT. Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Curr Biol. 2003;13(8):680–685. doi: 10.1016/s0960-9822(03)00240-9. [DOI] [PubMed] [Google Scholar]
- Venkatachalam K, Venkatesan B, Valente AJ, Melby PC, Nandish S, Reusch JE, Clark RA, Chandrasekar B. WISP1, a pro-mitogenic, pro-survival factor, mediates tumor necrosis factor-alpha (TNF-alpha)-stimulated cardiac fibroblast proliferation but inhibits TNF-alpha-induced cardiomyocyte death. J Biol Chem. 2009;284(21):14414–14427. doi: 10.1074/jbc.M809757200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesan B, Prabhu SD, Venkatachalam K, Mummidi S, Valente AJ, Clark RA, Delafontaine P, Chandrasekar B. WNT1-inducible signaling pathway protein-1 activates diverse cell survival pathways and blocks doxorubicin-induced cardiomyocyte death. Cell Signal. 22(5):809–820. doi: 10.1016/j.cellsig.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vines A, Cahoon S, Goldberg I, Saxena U, Pillarisetti S. Novel anti-inflammatory role for glycogen synthase kinase-3beta in the inhibition of tumor necrosis factor-alpha-and interleukin-1beta-induced inflammatory gene expression. J Biol Chem. 2006;281(25):16985–16990. doi: 10.1074/jbc.M602446200. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Sui XX, Simosa HF, Jain MK, Altieri DC, Conte MS. Regulation of vein graft hyperplasia by survivin, an inhibitor of apoptosis protein. Arterioscler Thromb Vasc Biol. 2005;25(10):2081–2087. doi: 10.1161/01.ATV.0000183885.66153.8a. [DOI] [PubMed] [Google Scholar]
- Wang X, Adhikari N, Li Q, Guan Z, Hall JL. The role of [beta]-transducin repeat-containing protein ([beta]-TrCP) in the regulation of NF-[kappa]B in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2004;24(1):85–90. doi: 10.1161/01.ATV.0000104012.40720.c4. [DOI] [PubMed] [Google Scholar]
- Whitman SC, Ravisankar P, Daugherty A. Interleukin-18 enhances atherosclerosis in apolipoprotein E(−/−) mice through release of interferon-gamma. Circ Res. 2002;90(2):E34–38. doi: 10.1161/hh0202.105292. [DOI] [PubMed] [Google Scholar]
- Xu L, Corcoran RB, Welsh JW, Pennica D, Levine AJ. WISP-1 is a Wnt-1-and beta-catenin-responsive oncogene. Genes Dev. 2000;14(5):585–595. [PMC free article] [PubMed] [Google Scholar]
- Yamagami H, Kitagawa K, Hoshi T, Furukado S, Hougaku H, Nagai Y, Hori M. Associations of serum IL-18 levels with carotid intima-media thickness. Arterioscler Thromb Vasc Biol. 2005;25(7):1458–1462. doi: 10.1161/01.ATV.0000168417.52486.56. [DOI] [PubMed] [Google Scholar]
- Yan C, Boyd DD. Regulation of matrix metalloproteinase gene expression. J Cell Physiol. 2007;211(1):19–26. doi: 10.1002/jcp.20948. [DOI] [PubMed] [Google Scholar]
- Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275(5303):1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.