

Abstract
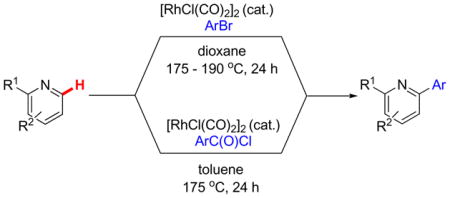
The Rh(I)-catalyzed direct arylation of azines has been developed. Quinolines and 2-substituted pyridines couple with aryl bromides to efficiently afford ortho-arylated azine products using the commercially available and air stable catalyst [RhCl(CO)2]2. Electron-deficient and rich aromatic bromides couple in good yields, and hydroxyl, chloro, fluoro, trifluoromethyl, ether and ketone functionality are compatible with the reaction conditions. Aroyl chlorides also serve as effective azine coupling partners to give ortho-arylation products via a decarbonylation pathway.
Introduction
Methodology for the regioselective arylation of azines is dominated by strategies relying on initial stoichiometric prefunctionalization of the heterocycle, either as the halo or organometallic intermediate, prior to a subsequent arylation step.1 While catalytic direct arylation protocols have been extensively developed for electron-rich heterocycle and arene classes,2 the direct arylation of azines has met with considerably less success despite the enormous importance of azines in drugs.3 Recently, conditions for the catalytic direct arylation of azine N-oxides,4 N-imino-pyridinium ylides,5 and N-phenacyl-pyridinium bromides6 with aryl halides have been developed. These methods represent a major improvement, obviating the need to handle costly and in many instances unstable organometallic azine intermediates; however, they do require preactivation of the azine as well as additional synthetic manipulations to obtain the final arylated azine products. The most direct route to this useful compound class would proceed by catalytic direct arylation of an unactivated azine with readily available electrophilic coupling partners.7, 8 In an initial communication, we documented our efforts in this vein, culminating in a Rh(I)-catalyzed direct arylation of pyridines and quinolines with aryl bromides.9 We now wish to report our studies further exploring the scope of this method, including the discovery that aroyl chlorides are competent electrophilic coupling partners in this direct arylation strategy.10
Results and Discussion
Catalytic Direct Arylation of Azines with Aryl Bromides
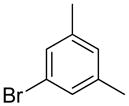
We previously developed a Rh(I)-catalyzed direct arylation of pyridines and quinolines allowing for the efficient arylation of this azine class without prefunctionalization of the heterocycle component.9 The optimized conditions employ the electron-poor catalyst [RhCl(CO)2]2, an aryl bromide as the limiting reagent,11 the appropriate heterocycle starting material (3–6 equiv), and heating in dioxane for 24 h. A more extensive evaluation of heterocycle scope has now established that a variety of substituted pyridines, and to some extent other heterocycles, are effective substrates under these reaction conditions (Table 1). Straight chain (entries 1 and 2), alpha-branched (entry 3), and beta-branched (entry 4) aliphatic substituted pyridines at the C-2 position all undergo efficient cross coupling with 3,5-dimethylbromobenzene. Tetrahydroquinoline also provides the arylated product in good yield (entry 5), as does the more highly substituted 2,4-dimethylpyridine (entry 6). In contrast, when a substituent at the C-2 position is absent arylation does not occur, as observed for the attempted direct arylation of pyridine (entry 7). Consistent with the requirement for C-2 substitution, quinoline also participates in regioselective direct arylation (entry 8), as do chloro- and hydroxy-substituted derivatives (entries 9 and 11, respectively). An appropriately substituted isoquinoline can be regioselectively arylated, albeit in lower yields (entry 12). Further studies exploring the scope in heterocycle identified 2,3-dimethylpyrazine and 4,6-dimethylpyrimidine as competent diazenes for this direct arylation reaction (entries 13 and 14, respectively). We surveyed the use of a variety of 2-heteroatom substituted pyridines (including 2-chloropyridine, 2-aminopyridine and several 2-hydroxypyridine derivatives). In all cases, no arylation product was detected.
Table 1.
Investigation of Scope in Heterocycle
 | ||||
|---|---|---|---|---|
| entry | azine | ArBr | product | yield (%)a |
| 1 | 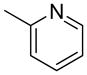 |
 |
1a | 53 |
| 2 | 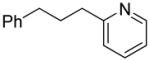 |
“ | 1b | 51 |
| 3 | 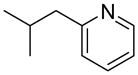 |
“ | 1c | 78 |
| 4 | 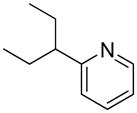 |
“ | 1d | 70 |
| 5 | 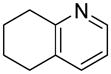 |
“ | 1e | 76 |
| 6 | 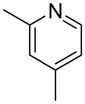 |
“ | 1f | 67 |
| 7 | 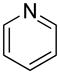 |
“ | 1g | 0 |
| 8 | 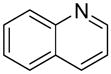 |
“ | 1h | 86b |
| 9 | 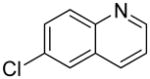 |
“ | 1i | 65b |
| 10 | 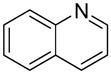 |
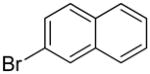 |
1j | 73b |
| 11 | 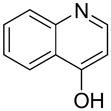 |
“ | 1k | 49c |
| 12 | 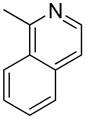 |
“ | 1l | 24b |
| 13 | 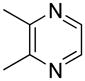 |
“ | 1m | 26b |
| 14 | 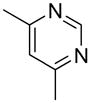 |
“ | 1n | 33b |
Isolated yield of analytically pure product; 0.4 mmol scale in ArBr; 0.8 M absolute concentration in ArBr;
Reaction run at 175 °C and 0.3 M absolute concentration in ArBr;
Reaction run at 165 °C and 0.3 M absolute concentration in ArBr.
The substrate scope in aryl bromide was also evaluated with quinoline as the coupling partner (Table 2). Both electron-rich and electron-poor aryl bromides are accommodated with equal efficiency (compare for example entries 4 and 11). A variety of useful functional groups are tolerated, including aryl and alkyl ethers (entries 3, 4), chloride (entry 8), fluoride (entry 9), and ketone functionality (entry 10). While cross coupling proceeds smoothly with meta substitution on the aryl bromide ring (see for example entry 1), attempted cross coupling with 2-bromotoluene failed to afford product (entry 13), with only unreacted starting material recovered. In the case of 1-bromonaphthalene, the arylated product was obtained in only 9% yield (entry 14).
Table 2.
Investigation of Scope in Aryl Bromide
 | |||
|---|---|---|---|
| entry | ArBr | product | yield (%)a |
| 1 |  |
1h | 86 |
| 2 | 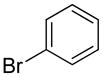 |
1o | 74 |
| 3 | 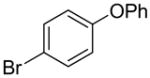 |
1p | 62 |
| 4 | 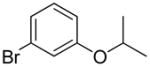 |
1q | 70 |
| 5 | 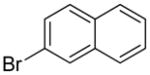 |
1j | 73 |
| 6 | 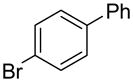 |
1r | 77 |
| 7 | 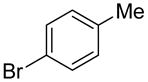 |
1s | 69 |
| 8 | 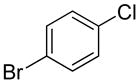 |
1t | 68 |
| 9 | 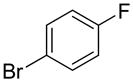 |
1u | 69 |
| 10 | 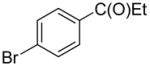 |
1v | 70 |
| 11 | 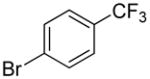 |
1w | 65 |
| 12 | 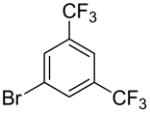 |
1x | 45 |
| 13 | 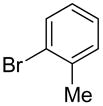 |
1y | 0 |
| 14 | 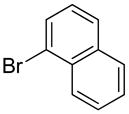 |
1z | 9 |
Isolated yield of analytically pure product; 0.4 mmol scale in ArBr; 0.3 M absolute concentration in ArBr.
Catalytic Direct Arylation of Azines with Aroyl Chlorides
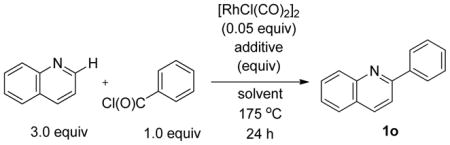
Recent work has documented the use of aroyl chlorides in Rh(I)-catalyzed decarbonylative direct arylation of arenes.10 Owing to the similarity in reaction conditions employed in these systems to our own, we envisioned that this manifold may be amenable to our present system. As such, with the optimized conditions for the direct arylation of quinoline with aryl bromides as a starting point, we evaluated the use of benzoyl chloride as an electrophilic coupling partner (Table 3).12 Under these conditions, an encouraging 32% yield of 2-phenylquinoline was obtained (entry 1). Importantly, we did not observe any formation of ketone, consistent with a decarbonylative manifold for the present reaction. Switching from dioxane to a variety of aromatic solvents proved fruitful, with yields improving under otherwise identical conditions with dichlorobenzene, trifluorotoluene, mesitylene, or toluene as solvent (entries 2–5). We next evaluated the inclusion of inorganic base additives, but consistent with our previous observations for the direct arylation with aryl bromides,9 these additives also proved deleterious when benzoyl chloride is used as the electrophile (entries 6 and 7). The choice of absolute concentration proved important, with the best results being obtained at relatively high concentrations in benzoyl chloride (compare entries 8 and 9). The reactions were typically run at 175 °C, but could be heated to 190 °C with equal efficiency (entry 10). Lastly, the product was obtained in modestly higher yield upon using six equivalents of quinoline under otherwise identical conditions (compare entries 5 and 11).13
Table 3.
Direct Arylation of Quinoline with Benzoyl Chloride
 | ||||
|---|---|---|---|---|
| entry | additive (equiv) | solvent | conc. (M)a | yield (%)b |
| 1 | none | dioxane | 0.3 | 32 |
| 2 | none | dichlorobenzene | 0.3 | 45 |
| 3 | none | trifluorotoluene | 0.3 | 56 |
| 4 | none | mesitylene | 0.3 | 58 |
| 5 | none | toluene | 0.3 | 56 |
| 6 | Na2CO3 (3.0) | toluene | 0.3 | 10 |
| 7 | K3PO4 (3.0) | toluene | 0.3 | 0 |
| 8 | none | toluene | 0.15 | 29 |
| 9 | none | toluene | 0.6 | 64 |
| 10c | none | toluene | 0.3 | 60 |
| 11d | none | toluene | 0.3 | 75 |
Refers to the absolute concentration in benzoyl chloride;
determined by GC analysis with hexamethylbenzene as an internal standard;
reaction run at 190 °C;
6.0 equiv of quinoline employed.
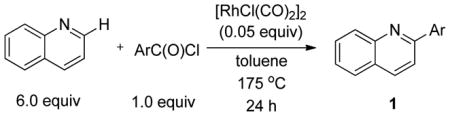
Having identified optimal conditions for the direct arylation of quinoline with benzoyl chloride, we next evaluated the generality of this new protocol with a variety of aroyl chlorides (Table 4). Both benzoyl chloride and 3,5-dimethylbenzoyl chloride provided high isolated yields of the 2-arylquinolines 1h and 1o (entries 1 and 2, respectively). Electron-rich aroyl chlorides coupled efficiently under these conditions, while the use of electron-poor aroyl chlorides proved more challenging (compare entries 3 and 4). Importantly, sterically congested 1-naphthoyl and 1-methylbenzoyl chlorides proved to be competent coupling partners, providing access to the 2-arylquinolines 1z and 1y in good yields (entries 5 and 6, respectively). The successful coupling of 2-substituted aroyl chlorides expands the range of direct arylation products that can be obtained because coupling ortho-substituted aryl bromides was previously found to proceed in very low yields (see Table 2, entries 13 and 14).
Table 4.
Investigation of Scope in Aroyl Chloride
 | |||
|---|---|---|---|
| entry | ArC(O)Cl | product | yield (%)a |
| 1 |  |
1h | 76 |
| 2 | 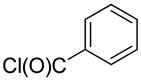 |
1o | 66 |
| 3 | 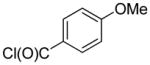 |
1aa | 56 |
| 4 | 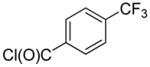 |
1w | 24 |
| 5 | 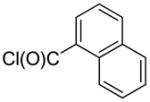 |
1z | 54 |
| 6 | 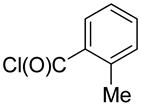 |
1y | 52 |
Isolated yield of analytically pure product; 0.4 mmol scale in ArC(O)Cl; 0.3 M absolute concentration in ArC(O)Cl.
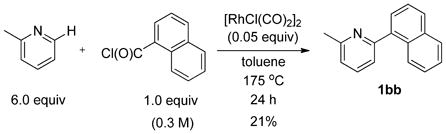
The use of heterocycles other than quinoline was evaluated. For the case of 2-methylpyridine, a 21% yield of 1bb was obtained upon reaction with 1-naphthoyl chloride, indicating that the efficiency of coupling aroyl chlorides is quite sensitive to azine structure (eq 1).14
 |
(1) |
Proposed Mechanism
A plausible mechanism for the Rh(I)-catalyzed direct arylation of azines with aryl bromides is presented in Figure 1. Initial nitrogen coordination of the 2-substituted pyridine to [RhCl(CO)2]2 provides adduct 2. Subsequent ortho C-H activation provides 3, which may tautomerize to N-heterocyclic carbene (NHC) complex 4. We have previously characterized N-heterocyclic carbene (NHC) Rh-complexes for a variety of nitrogen heterocycles15 and others have characterized pyridine- and quinoline-based Os, Ru, and Ir-NHC complexes also prepared via C-H activation.16 Upon solvent or azine coordination and reductive elimination of HX to give 5, oxidative addition of the aryl bromide would then provide 6. Subsequent reductive elimination and exchange with the pyridine starting material would regenerate N-bound Rh complex 2. This catalytic cyclic is also applicable to the direct arylation with aroyl chlorides whereby oxidative addition to 5 would be followed by decarbonylation to generate 6 (Br would be replaced with Cl). Alternative catalytic cycles are possible, for example, oxidative addition of the aryl bromide or aroyl chloride might occur prior to C-H bond activation.17 Mechanistic study will clearly be necessary to discriminate between the different possible catalytic cycles.
Figure 1.

Proposed mechanism for the catalytic direct arylation of azines with aryl bromides.
The proposed mechanism can be used to rationalize the requirement that the pyridine be ortho substituted in order to achieve successful arylation (see entry 7, Table 1). Presumably, unfavorable steric interactions in the N-bound complex of ortho-substituted pyridines increase the equilibrium ratio of the C2-bound rhodium complexes 3 or 4 relative to the N-bound rhodium complex 2. These results are consistent with reports on the preparation and characterization of (2-substituted)-pyridine- and quinoline-based Os and Ir-NHC complexes wherein ortho-substitution was necessary for obtaining the complexes.16 The requirement for ortho-substitution is also consistent with our observations that the Rh-catalyzed intermolecular alkylation of pyridines with alkenes only occurs when the pyridine substrates are substituted at the ortho position.8a
Conclusion
In summary, we have developed a Rh(I)-catalyzed strategy for the direct arylation of azines. A variety of differentially substituted aryl bromides and aroyl chlorides can be used in this protocol, including chloro, fluoro, trifluoromethyl, ether and ketone functionality. The heterocycle is used directly without requiring any prefunctionalization. This strategy represents an expeditious route to an important class of arylated azines and for appropriate substrates should be of broad utility.
Experimental Section
General Procedure for the Catalytic Direct Arylation of Azines with Aryl Bromides
To a 15- or 25-mL sealable flame- or oven-dried Schlenk tube (Kontes No. 218710-0015 or 218710-0025) containing a stir bar was added [RhCl(CO)2]2 (0.0078 g, 0.0200 mmol), the heterocycle (2.400 mmol), the aryl bromide (0.400 mmol), and 1,4-dioxane (reaction solution diluted to a total concentration of 0.30 – 0.80 M). The volume of the heterocycle is not negligible at the concentrations used and must therefore be included in the concentration calculation. The reaction vessel was sealed, removed from the glove box, submerged to the neck in a silicone oil bath, heated at the indicated temperature with stirring for 24 h and then cooled to room temperature. The reaction mixture was transferred to a 60 mL separatory funnel using 3 × 5.0 mL of CH2Cl2 and 2.5 mL of MeOH. The resulting solution was washed with saturated NaHCO3 (1 × 20 mL), the aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The organic extracts were combined, washed with saturated NaCl (1 × 20 mL), dried over Na2SO4, filtered through glass wool and concentrated to dryness under reduced pressure. The crude product mixture was loaded onto a Biotage samplet using a minimal amount of CH2Cl2 (ca. 0.5 mL). This solvent was removed by placing the samplet under reduced pressure in a vacuum dessicator for ca. 15 min, and the samplet was loaded into a Biotage SP1 system. Eluting with an ethyl acetate/hexanes gradient calculated by the instrument from the Rf of the product in a specified ethyl acetate/hexanes mixture provided the desired product.
2-(3,5-Dimethylphenyl)-quinoline (1h)
The reaction was conducted with quinoline (0.3100 g, 2.400 mmol), 1-bromo-3,5-dimethylbenzene (0.0740 g, 0.400 mmol) and 1.0 mL of 1,4-dioxane at 175 °C for 24 h. The crude mixture was purified by flash chromatography using an ethyl acetate/hexanes gradient to provide 0.0800 g, 86% yield of 1h as a clear, colorless oil. 1H NMR (400.13 MHz, CDCl3): δ 8.34 – 8.12 (m, 2 H), 7.91 – 7.67 (m, 5 H), 7.49 – 7.45 (m, 1 H), 7.08 (br s, 1 H), 2.41 (s, 6 H). 13C {1H} NMR (100.61 MHz, CDCl3): δ 157.9, 148.4, 139.8, 138.5, 136.7, 131.2, 129.8, 129.7, 127.6, 127.3, 126.3, 125.6, 119.4, 21.6. HRMS-EI (m/z): calculated for C17H15N: 233.1205; observed: 233.1203.
2-(2-Naphthyl)-4-hydroxyquinoline (1k)
To a 15-mL sealable flame-dried Schlenk tube (Kontes No. 218710-0015) containing a stir bar was added [RhCl(CO)2]2 (0.0038 g, 0.0100 mmol), 4-hydroxyquinoline (0.0436 g, 0.300 mmol), 2-bromonaphthalene (0.0207 g, 0.100 mmol) and 0.33 mL of 1,4-dioxane. The reaction vessel was sealed, removed from the glove box, submerged to the neck in a silicone oil bath, heated at 165 °C for 24 h and then cooled to room temperature. The reaction mixture was transferred to a 60 mL separatory funnel using 3 × 5.0 mL of CH2Cl2 and 2.5 mL of MeOH. The resulting solution was washed with saturated NaHCO3 (1 × 20 mL), and the aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The organic extracts were combined, washed with saturated NaCl (1 × 20 mL), dried over Na2SO4, filtered through glass wool and concentrated to dryness under reduced pressure. The crude product mixture was loaded onto a Biotage samplet using a minimal amount of CH2Cl2 and MeOH (ca. 0.5 mL). This solvent was removed by placing the samplet under reduced pressure in a vacuum dessicator for ca. 15 min, and the samplet was loaded into a Biotage SP1 system. Eluting with a methanol/dichloromethane gradient calculated by the Biotage instrument provided 0.0133 g, 49% yield of 1k as a pale yellow powder. Rf (5% methanol/dichloromethane) 0.45. mp 275 °C (decomposes). 1H NMR (400.13 MHz, DMSO-d6): δ 11.87 (br s, 1 H), 8.48 (s, 1 H), 8.14 – 8.10 (m, 3 H), 8.06 – 8.03 (m, 1 H), 7.96 – 7.94 (m, 1 H), 7.83 – 7.81 (m, 1 H), 7.73 – 7.64 (m, 3 H), 7.38 – 7.35 (m, 1 H), 6.50 (s, 1 H). 13C {1H} NMR (100.61 MHz, DMSO-d6): δ 176.7, 150.3, 141.1, 134.0, 133.0, 132.3, 132.0, 129.1, 129.1, 128.2, 128.0, 127.6, 127.5, 125.4, 125.2, 125.0, 123.8, 119.2, 108.2. HRMS-EI (m/z): calculated for C19H14NO: 272.1070; observed: 272.1072.
1-Methyl-3-(2-naphthyl)-isoquinoline (1l)
The reaction was conducted with 1-methylisoquinoline (0.3432 g, 2.400 mmol), 2-bromonaphthalene (0.0828 g, 0.400 mmol) and 1.00 mL of 1,4-dioxane at 175 °C for 24 h. The crude mixture was purified by flash chromatography using an ethyl acetate/hexanes gradient to provide 0.0236 g, 22% yield of 1l as a white powder. Rf (10% ethyl acetate/hexanes) 0.50. mp 128 – 130 °C. 1H NMR (400.13 MHz, CDCl3): δ 8.72 (s, 1 H), 8.33 – 8.30 (m, 1 H), 8.21 – 8.19 (m, 1 H), 8.12 (s, 1 H), 8.05 – 8.00 (m, 2 H), 7.95 – 7.92 (m, 2 H), 7.76 – 7.72 (m, 1 H), 7.66 – 7.61 (m, 1 H), 7.58 – 7.52 (m, 2 H), 3.14 (s, 3 H). 13C {1H} NMR (100.61 MHz, CDCl3): δ 158.7, 149.8, 137.1, 136.9, 133.7, 133.4, 130.1, 128.7, 128.4, 127.7, 127.7, 126.9, 126.7, 126.2, 126.2, 125.7, 124.8, 115.6, 22.8. HRMS-EI (m/z): calculated for C20H16N: 270.1277; observed: 270.1286.
2-(2-Naphthyl)-5,6-dimethylpyrazine (1m)
The reaction was conducted with 2,3-dimethylpyrazine (0.2594 g, 2.400 mmol), 2-bromonaphthalene (0.0828 g, 0.400 mmol) and 1.10 mL of 1,4-dioxane at 175 °C for 24 h. The crude mixture was purified by flash chromatography using an ethyl acetate/hexanes gradient to provide 0.0153 g, 16% yield of 1m as a pale yellow powder. Rf (10% ethyl acetate/hexanes) 0.20. mp 111 – 112 °C. 1H NMR (400.13 MHz, CDCl3): δ 8.90 (s, 1 H), 8.52 (s, 1 H), 8.18 – 8.16 (m, 1 H), 8.01 – 7.92 (m, 3 H), 7.57 – 7.56 (m, 2 H), 2.70 (s, 3 H), 2.65 (s, 3 H). 13C {1H} NMR (100.61 MHz, CDCl3): δ 151.9, 150.6, 149.3, 138.5, 134.2, 133.7, 133.5, 128.7 (2 carbons), 127.7, 126.7, 126.5, 126.1, 124.1, 22.3, 21.8. HRMS-EI (m/z): calculated for C16H15N2: 235.1230; observed: 235.1233.
2-(2-Naphthyl)-4,6-dimethylpyrimidine (1n)
The reaction was conducted with 4,6-dimethylpyrimidine (0.2594 g, 2.400 mmol), 2-bromonaphthalene (0.0828 g, 0.400 mmol) and 1.10 mL of 1,4-dioxane at 175 °C for 24 h. The crude mixture was purified by flash chromatography using an ethyl acetate/hexanes gradient to provide 0.0205 g, 22% yield of 1n as a white powder. Rf (10% ethyl acetate/hexanes) 0.40. mp 116 – 117 °C. 1H NMR (400.13 MHz, CDCl3): δ 9.02 (s, 1 H), 8.61 – 8.59 (m, 1 H), 8.06 – 7.91 (m, 3 H), 7.57 – 7.54 (m, 2 H), 7.00 (s, 1 H), 2.63 (s, 6 H). 13C {1H} NMR (100.61 MHz, CDCl3): δ 166.9, 135.5, 134.6, 133.4, 129.2, 128.3, 128.1, 127.7, 126.9, 126.1, 125.4, 118.1, 24.3. HRMS-EI (m/z): calculated for C16H15N2: 235.1230; observed: 235.1234.
General Procedure for the Catalytic Direct Arylation of Azines with Aroyl Chlorides
To a 15- or 25-mL sealable flame- or oven-dried Schlenk tube (Kontes No. 218710-0015 or 218710-0025) containing a stir bar was added [RhCl(CO)2]2 (0.0078 g, 0.0200 mmol), the heterocycle (2.400 mmol), the aroyl chloride (0.400 mmol), and toluene (reaction solution diluted to a total concentration of 0.30 M). The volume of the heterocycle is not negligible at the concentrations used and must therefore be included in the concentration calculation. The reaction vessel was sealed, removed from the glove box, submerged to the neck in a silicone oil bath, heated at 175 °C with stirring for 24 h and then cooled to room temperature. The reaction mixture was transferred to a 60 mL separatory funnel using 3 × 5.0 mL of CH2Cl2 and 2.5 mL of MeOH. The resulting solution was washed with saturated NaHCO3 (1 × 20 mL), and the aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The organic extracts were combined, washed with saturated NaCl (1 × 20 mL), dried over Na2SO4, filtered through glass wool and concentrated to dryness under reduced pressure. The crude product mixture was loaded onto a Biotage samplet using a minimal amount of CH2Cl2 (ca. 0.5 mL). This solvent was removed by placing the samplet under reduced pressure in a vacuum dessicator for ca. 15 min, and the samplet was loaded into a Biotage SP1 system. Eluting with an ethyl acetate/hexanes gradient calculated by the instrument from the Rf of the product in a specified ethyl acetate/hexanes mixture provided the desired product.
2-(3,5-Dimethylphenyl)-quinoline (1h)
The reaction was conducted with quinoline (0.3100 g, 2.400 mmol), 3,5-dimethylbenzoyl chloride (0.0674 g, 0.400 mmol) and 1.00 mL of toluene at 175 °C for 24 h. The crude mixture was purified by flash chromatography using an ethyl acetate/hexanes gradient to provide 0.0706 g, 76% yield of 1h as a clear, colorless oil. Analytical data consistent with that previously reported.9
2-Phenyl-quinoline (1o)
The reaction was conducted with quinoline (0.3100 g, 2.400 mmol), benzoyl chloride (0.0562 g, 0.400 mmol) and 1.00 mL of toluene at 175 °C for 24 h. The crude mixture was purified by flash chromatography using an ethyl acetate/hexanes gradient to provide 0.0538 g, 66% yield of 1o as a white powder. Analytical data consistent with that previously reported.9
2-(4-(Trifluoromethyl)phenyl)-quinoline (1w)
The reaction was conducted with quinoline (0.3100 g, 2.400 mmol), 4-(trifluoromethyl)benzoyl chloride (0.0834 g, 0.400 mmol) and 1.00 mL of toluene at 175 °C for 24 h. The crude mixture was purified by flash chromatography using an ethyl acetate/hexanes gradient to provide 0.0321 g, 29% yield of 1w as a white powder. Analytical data consistent with that previously reported.9
2-(1-Methylphenyl)-quinoline (1y)
The reaction was conducted with quinoline (0.3100 g, 2.400 mmol), o-toluoyl chloride (0.0618 g, 0.400 mmol) and 1.00 mL of toluene at 175 °C for 24 h. The crude mixture was purified by flash chromatography using an ethyl acetate/hexanes gradient to provide 0.0455 g, 52% yield of 1y as a white powder. mp 71 – 72 °C. 1H NMR (400.13 MHz, CDCl3): δ 8.27 – 8.25 (m, 1 H), 8.22 – 8.20 (m, 1 H), 7.92 – 7.90 (m, 1 H), 7.81 – 7.77 (m, 1 H), 7.63 – 7.54 (m, 3 H), 7.39 – 7.36 (m, 3 H), 2.47 (s, 3 H). 13C {1H} NMR (100.61 MHz, CDCl3): δ 160.3, 147.9, 140.7, 136.1, 136.0, 130.9, 129.7, 129.7, 129.6, 128.5, 127.5, 126.8, 126.4, 126.1, 122.4, 20.4. HRMS-EI (m/z): calculated for C16H14N: 220.1121; observed: 220.1120.
2-(1-Naphthyl)-quinoline (1z)
The reaction was conducted with quinoline (0.3100 g, 2.400 mmol), 1-naphthoyl chloride (0.0764 g, 0.400 mmol) and 1.00 mL of toluene at 175 °C for 24 h. The crude mixture was purified by flash chromatography using an ethyl acetate/hexanes gradient to provide 0.0553 g, 54% yield of 1z as a pale yellow powder. Rf (10% ethyl acetate/hexanes) 0.30. mp 96 – 98 °C (lit. 96 – 97 °C).18 1H NMR (400.13 MHz, CDCl3): δ 8.35 – 8.33 (m, 1 H), 8.29 – 8.27 (m, 1 H), 8.19 – 8.17 (m, 1 H), 8.02 – 7.96 (m, 3 H), 7.85 – 7.81 (m, 1 H), 7.78 – 7.75 (m, 2 H), 7.67 – 7.63 (m, 2 H), 7.59 – 7.50 (m, 2 H). 13C {1H} NMR (100.61 MHz, CDCl3): δ 159.5, 148.2, 138.8, 136.3, 134.0, 131.3, 129.8, 129.8, 129.1, 128.4, 127.8, 127.6, 127.0, 126.6 (2 carbons), 126.0, 125.7, 125.4, 123.3. HRMS-EI (m/z): calculated for C19H14N: 256.1121; observed: 256.1127.
2-(4-Methoxyphenyl)-quinoline (1aa)
The reaction was conducted with quinoline (0.3100 g, 2.400 mmol), 4-methoxybenzoyl chloride (0.0682 g, 0.400 mmol) and 1.00 mL of toluene at 175 °C for 24 h. The crude mixture was purified by flash chromatography using an ethyl acetate/hexanes gradient to provide 0.0530 g, 56% yield of 1aa as a white powder. Rf (10% ethyl acetate/hexanes) 0.45. mp 123 – 124 °C (lit. 124 – 125 °C).19 1H NMR (400.13 MHz, CDCl3): δ 8.23 – 8.18 (m, 4 H), 7.89 – 7.84 (m, 2 H), 7.78 – 7.73 (m, 1 H), 7.56 – 7.52 (m, 1 H), 7.11 – 7.09 (m, 2 H), 3.93 (s, 3 H). 13C {1H} NMR (100.61 MHz, CDCl3): δ 160.8, 156.9, 148.3, 136.7, 132.3, 129.6, 129.6, 128.9, 127.5, 126.9, 125.9, 118.6, 114.3, 55.4. HRMS-EI (m/z): calculated for C16H14NO: 236.1070; observed: 236.1076.
2-(1-Naphthyl)-6-methyl-pyridine (1bb)
The reaction was conducted with 2-methylpyridine (0.2234 g, 2.400 mmol), 1-naphthoyl chloride (0.0764 g, 0.400 mmol) and 1.00 mL of toluene at 175 °C for 24 h. The crude mixture was purified by flash chromatography using an ethyl acetate/hexanes gradient to provide 0.0183 g, 21% yield of 1bb as a clear, colorless oil. Rf (10% ethyl acetate/hexanes) 0.25. 1H NMR (400.13 MHz, CDCl3): δ 8.10 – 8.08 (m, 1 H), 7.95 – 7.93 (m, 2 H), 7.77 – 7.74 (m, 1 H), 7.64 – 7.48 (m, 4 H), 7.42 – 7.40 (m, 1 H), 7.26 – 7.24 (m, 1 H), 2.72 (s, 3 H). 13C {1H} NMR (100.61 MHz, CDCl3): δ 158.7, 158.3, 138.8, 136.6, 134.0, 131.3, 128.7, 128.3, 127.4, 126.3, 125.8, 125.8, 125.4, 122.1, 121.6, 24.8. HRMS-EI (m/z): calculated for C16H14N: 220.1121; observed: 220.1122.
Supplementary Material
Acknowledgments
This work was supported by NIH Grant GM069559 to J.A.E. and the DOE, Office of Basic Energy Sciences, Chemical Sciences Division, U.S. Department of Energy, under Contract DE-AC03-76SF00098 to R.G.B. A.M.B. was supported by a NRSA postdoctoral fellowship (GM082080).
Footnotes
Supporting Information Available: General experimental information and NMR spectra for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.The use of 2-pyridyl organometallics is particularly troublesome, for leading references on recent advances, see: Metzger A, Bernhardt S, Manolikakes G, Knochel P. Angew Chem Int Ed. 2010;49:4665. doi: 10.1002/anie.201000634.Kim SH, Rieke RD. Tetrahedron. 2010;66:3135.Ackermann L, Potukuchi HK, Kapdi AR, Schulzke C. Chem Eur J. 2010;16:3300. doi: 10.1002/chem.201000032.Knapp DM, Gillis EP, Burke MD. J Am Chem Soc. 2009;131:6961. doi: 10.1021/ja901416p.Deng JZ, Paone DV, Ginnetti AT, Kurihara H, Dreher SD, Weissman SA, Stauffer SR, Burgey CS. Org Lett. 2009;11:345. doi: 10.1021/ol802556f.Billingsley KL, Buchwald SL. Angew Chem Int Ed. 2008;47:4695. doi: 10.1002/anie.200801465.
- 2.For recent reviews on direct arylation of heteroarenes, see: Ackermann L, Vicente R, Kapdi AR. Angew Chem Int Ed. 2009;48:9792. doi: 10.1002/anie.200902996.Bellina F, Rossi R. Tetrahedron. 2009;65:10269.Seregin IV, Gevorgyan V. Chem Soc Rev. 2007;36:1173. doi: 10.1039/b606984n.Alberico D, Scott ME, Lautens M. Chem Rev. 2007;107:174. doi: 10.1021/cr0509760.
- 3.For an analysis of heterocycles used in the preparation of drug candidates, see: Carey JS, Laffan D, Thomson C, Williams MT. Org Biomol Chem. 2006;4:2337. doi: 10.1039/b602413k.
- 4.(a) Campeau LC, Stuart DR, Leclerc JP, Bertrand-Laperle M, Villemure E, Sun HY, Lasserre S, Guimond N, Lecavallier M, Fagnou K. J Am Chem Soc. 2009;131:3291. doi: 10.1021/ja808332k. [DOI] [PubMed] [Google Scholar]; (b) Campeau LS, Rousseaux S, Fagnou K. J Am Chem Soc. 2005;127:18020. doi: 10.1021/ja056800x. [DOI] [PubMed] [Google Scholar]
- 5.Larivee A, Mousseau JJ, Charette AB. J Am Chem Soc. 2008;130:52. doi: 10.1021/ja710073n. [DOI] [PubMed] [Google Scholar]
- 6.Xu J, Cheng G, Su D, Liu Y, Wang X, Hu Y. Chem Eur J. 2009;15:13105. doi: 10.1002/chem.200901399. [DOI] [PubMed] [Google Scholar]
- 7.For a recent example employing organozincs as a nucleophilic arylating reagent for the catalytic direct arylation of azines, see: Tobisu M, Hyodo I, Chatani N. J Am Chem Soc. 2009;131:12070. doi: 10.1021/ja9053509.For a non-regioselective direct arylation of azines with aryl iodides promoted by KOt-Bu, see: Yanagisawa S, Ueda K, Taniguchi T, Itami K. Org Lett. 2008;10:4673. doi: 10.1021/ol8019764.For the direct ortho-arylation of amides of nicotinic and isonicotinic acid, see Wasa M, Worrell BT, Yu JQ. Angew Chem Int Ed. 2010;49:1275. doi: 10.1002/anie.200906104.The Cu-catalyzed direct arylation of pyrimidine and pyridazine with phenyl iodide has been reported, see: Do HQ, Khan RMK, Daugulis O. J Am Chem Soc. 2008;130:15185. doi: 10.1021/ja805688p.
- 8.For the direct alkylation or alkenylation of pyridines, respectively, see: Lewis JC, Bergman RG, Ellman JA. J Am Chem Soc. 2007;129:5332. doi: 10.1021/ja070388z.Nakao Y, Kashihara N, Kanyiva KS, Hiyama T. J Am Chem Soc. 2008;130:16170. doi: 10.1021/ja807258m.Wu J, Cui X, Chen L, Jiang G, Wu Y. J Am Chem Soc. 2009;131:13888. doi: 10.1021/ja902762a.Tsai CC, Shih WC, Fang CH, Li CY, Ong TG, Yap GPA. J Am Chem Soc. 2010;132:11887. doi: 10.1021/ja1061246.Yotphan S, Bergman RG, Ellman JA. Org Lett. 2010;12:2978. doi: 10.1021/ol101002b.Tran LD, Daugulis O. Org Lett. 2010;12:4277. doi: 10.1021/ol101684u.Nakao Y, Yamada Y, Ksahihara N, Hiyama T. J Am Chem Soc. 2010;132:13666. doi: 10.1021/ja106514b.
- 9.Berman AM, Lewis JC, Bergman RG, Ellman JA. J Am Chem Soc. 2008;130:14926. doi: 10.1021/ja8059396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For the use of acid chlorides and acid anhydrides as electrophilic arylating reagents in Rh(I)-catalyzed direct arylation of arenes, see: Zhao X, Yu Z. J Am Chem Soc. 2008;130:8136. doi: 10.1021/ja803154h.Jin W, Yu Z, He W, Ye W, Xiao WJ. Org Lett. 2009;11:1317. doi: 10.1021/ol9000729.
- 11.For a complete list of optimization parameters examined, refer to reference 9.
- 12.The use of benzoic acid anhydride was also evaluated, in this case only trace cross-coupling product was detected.
- 13.Other rhodium complexes served as competent precatalysts for this reaction. Under the conditions described in Table 3, entry 5 the following yields of 1o were obtained with 5 mol% of catalyst: [RhCl(COD)]2, 50%; Rh(acac)(CO)2, 51%; [RhCl(CH2CH2)]2, 37%.
- 14.Similar results were obtained with other substituted pyridines and diazenes; under identical conditions the coupling of 2-(3-pentyl)pyridine with 3,5-dimethylbenzoyl chloride afforded a 16% yield of 2-(3,5-dimethylphenyl)-6-(3-pentyl)pyridine, while the coupling of 2,3-dimethylpyrazine with 3,5-dimethylbenzoyl chloride afforded a 31% assay yield of 2-(3,5-dimethylphenyl)-5,6-dimethylpyrazine.
- 15.(a) Lewis JC, Bergman RG, Ellman JA. Acc Chem Res. 2008;41:1013. doi: 10.1021/ar800042p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wiedemann SH, Lewis JC, Bergman RG, Ellman JA. J Am Chem Soc. 2006;128:2452. doi: 10.1021/ja0576684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Alvarez E, Conejero S, Paneque M, Petronilho A, Poveda ML, Serrano O, Carmona E. J Am Chem Soc. 2006;128:13060. doi: 10.1021/ja0646592. [DOI] [PubMed] [Google Scholar]; (b) Esteruelas MA, Fernandez-Alvarez FJ, Onate E. J Am Chem Soc. 2006;128:13044. doi: 10.1021/ja064979l. [DOI] [PubMed] [Google Scholar]; (c) Buil ML, Esteruelas MA, Garcés K, Oliván M, Oñate E. J Am Chem Soc. 2007;129:10998. doi: 10.1021/ja073673r. [DOI] [PubMed] [Google Scholar]
- 17.For the related Rh-catalyzed alkylation of azines with alkenes C-H activation is followed by alkene insertion suggesting the competence of C-H activation prior to ArBr or aroyl chloride activation. See ref. 8a.
- 18.Yang CH, Tai CC, Huang YT, Sun I. Tetrahedron. 2005;61:4857. [Google Scholar]
- 19.Korn TJ, Schade MA, Cheemala MN, Wirth S, Guevara SA, Cahiez G, Knochel P. Synthesis. 2006;21:3547. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.