

Abstract
The polar Felkin-Anh, Cornforth, and Cram-chelation models predict that the addition of organometallic reagents to silyl–protected α–hydroxy ketones proceeds via a non-chelation pathway to give anti-diol addition products. This prediction has held true for the vast majority of additions reported in the literature and few methods for chelation-controlled additions of organometallic reagents to silyl–protected α–hydroxy ketones have been introduced. Herein, we present a general and highly diastereoselective method for the addition of dialkylzincs and (E)-di-, (E)-tri- and (Z)-disubstituted vinylzinc reagents to α-silyloxy ketones using alkyl zinc halide Lewis acids, RZnX, to give chelation-controlled products (dr ≥18:1). The compatibility of organozinc reagents with other functional groups makes this method potentially very useful in complex molecule synthesis.
1. Introduction
The prevailing strategy in complex molecule synthesis is to use existing substrate stereochemistry to direct the introduction of new stereogenic centers.1–4 This approach has been particularly useful in the stereoselective generation of diol motifs by addition of organometallic nucleophiles to chiral protected α- and β-hydroxy aldehydes and ketones. The paradigm to predict the stereochemical outcomes of these additions is based on the polar Felkin–Anh, Cornforth, and Cram-chelation models.5–18 The key factor in the additions to α– and β–hydroxy aldehydes and ketones is the size of the protecting group.19 Small protecting groups, such as MOM and Bn, promote carbonyl addition via a chelation-controlled pathway (Figure 1).7,20 Conversely, sterically encumbering silyl protecting groups favor a non-chelation pathway, giving rise to 1,2-anti diol addition products.5,7,21 There are very few exceptions to this paradigm.22–28 Thus, when the desired protecting group for the global protecting group strategy does not provide the requisite stereochemistry in the diastereoselective addition, chemists have relied on the use of enantioenriched stoichiometric auxiliaries, optically active stoichiometric additives,5,29–37 or chiral catalysts38–42 to override substrate control.
Figure 1.

Polar Felkin-Anh, Cornforth, and Cram-chelation models for additions to protected α-hydroxy aldehydes and ketones. Recent studies have highlighted the importance of the Cornforth model over the polar Felkin-Anh model.13,14
Based on our previous studies,43–45 we recently developed the first general protocol to achieve chelation–controlled additions of organometallic reagents to α–silyloxy aldehydes utilizing alkyl zinc halide Lewis acids.46 Dialkylzinc and functionalized dialkylzinc reagents add to α-silyloxy aldehydes with excellent selectivity for the chelation-controlled addition product (Scheme 1A). Moreover, we have shown that a variety of vinylzinc reagents undergo addition to α-silyloxy aldehydes to generate (E)-di-, (E)-tri-, and (Z)-disubstituted allylic alcohols with high diastereoselectivity, strongly favoring chelation–controlled products (Scheme 1B and C).
Scheme 1.

Diastereoselective Addition of Dialkylzinc and Vinylzinc Reagents to α–Silyloxy Aldehydes
There are limited examples of methods to promote highly diastereoselective chelation-controlled additions of organometallic reagents to α-silyloxy ketones. The products of these additions contain vicinal secondary and tertiary carbinols, a motif that is found in natural products such as fostriecin, among others.47–53 To fill the gap in the methodology, we set out to develop a versatile method for the chelation-controlled addition of organometallic reagents to α-silyloxy ketones. We perceived three potential problems. First, it is well known that ketones are significantly less reactive than aldehydes. Second, compounding this reactivity issue, organozinc reagents are very mild alkyl donors and either do not react or react very slowly with ketones in the absence of Lewis acids. Finally, although we have presented evidence that EtZnCl can reversibly chelate to α-silyloxy ketones (1H NMR),46 alkyl zinc halides are relatively weak Lewis acids and it was not clear if they would activate ketones sufficiently to promote addition of dialkylzinc reagents. The low nucleophilicity of dialkylzinc reagents combined with the mild Lewis acidity of RZnX can result in deprotonation of the ketone substrate and generation of enolates. The resulting enolates react with a second equivalent of substrate to give rise to aldol side products. Despite these challenges, we report herein a highly diastereoselective method for the chelation-controlled addition of a variety of organozinc reagents to α–silyloxy ketones.
2. Experimental Section
General Methods
All reactions were performed under a nitrogen atmosphere using oven-dried glassware and standard Schlenk or vacuum line techniques. The progress of all reactions was monitored by thin-layer chromatography. Toluene and dichloromethane were dried through alumina columns. Racemic ketone derivatives were prepared by literature method; nucleophilic addition of Grignard or organolithium reagents to trans-2-methyl-2-butenal was followed by ozonolysis and protection. Enantiopure ketones were prepared from the corresponding enantiopure madelic acid or methyl lactate according to liturature procedure.54,55 Alkyl zinc halides were prepared by literature methods.56,57 All chemicals were obtained from Acros, Sigma-Aldrich, or GFS Chemicals unless otherwise described. The 1H NMR and 13C{1H} NMR spectra were obtained using a Brüker AM-500 Fourier transform NMR spectrometer at 500 and 125 MHz, respectively. Chemical shifts are reported in units of parts per million (ppm) downfield from tetramethylsilane and all coupling constants are reported in hertz. The infrared spectra were obtained using a Perkin-Elmer 1600 series spectrometer. Thin-layer chromatography was carried out on Whatman pre-coated silica gel 60 F-254 plates and visualized by ultra-violet light or by staining with ceric ammonium molybdate or 2,4-dinitrophenylhydrazine stain. Silica gel (230–400 mesh, Silicycle) was used for air-flashed chromatography. Analysis of diastereomeric ratios was performed by gas chromatography using a Hewlett-Packard 6890 or Agilent Technologies 7890A GC with a Beta-Dex or Chirasil-Dex Column or by 1H NMR of the crude reaction products. Analysis of enantiomeric excess was performed using a Hewlett-Packard 1100 series HPLC and Chiralcel OD-H or Chiralpak AD-H column. High-resolution mass spectra were measured using a Waters 2695 Separations Module (1S0RR23444). Relative stereochemistry was determined by comparison to literature values or comparison to corresponding Grignard or organolithium addition products. Vinyllithium29 reagents were made from the corresponding vinyliodides58 according to literature procedure.
Cautionary Note
Diorganozinc reagents and tert-butyllithium are pyrophoric. Extreme caution should be used in their handling and proper laboratory attire is highly recommended.
General Dialkylzinc Procedure. General Procedure A
A dry 10 mL Schlenk flask, which was evacuated and backfilled with N2 three times, was charged with the alkyl zinc chloride (1.75 mmol, neat solid), dialkylzinc (0.75 mmol), and toluene (1 mL). The flask was then cooled to −15 °C followed by dropwise addition of ketone solution (0.5 mmol in 0.5 mL toluene). The reaction was monitored by TLC until completion. The reaction mixture was quenched with saturated aq. NH4Cl (2 mL) followed by addition of Et2O (5 mL). The organic layer was separated and the aqueous solution extracted with Et2O (3 × 10 mL). The combined organic layers were successively washed with brine, dried over MgSO4, and filtered. The filtrate was concentrated in vacuo and purified by column chromatography on silica gel.
General Dialkylzinc Addition Procedure for Butyl-substituted Ketones. General Procedure B
A dry 10 mL Schlenk flask, which was evacuated and backfilled with N2 three times, was charged with the solid alkyl zinc chloride (2 mmol), dialkylzinc (2 mmol), and toluene (1 mL). The ketone solution (0.5 mmol in 0.5 mL toluene) was then added dropwise followed by heating to 50 °C. The reaction was monitored by TLC until completion (12–24 h). The reaction mixture was then quenched with saturated aq. NH4Cl (2 mL) followed by addition of Et2O (5 mL). The organic layer was separated and the aqueous solution extracted with Et2O (3 × 10 mL). The combined organic layers were successively washed with brine, dried over MgSO4, and filtered. The filtrate was concentrated in vacuo and purified by column chromatography on silica gel
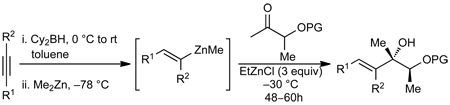
(E)-Di- and (E)-Trisubstituted Allylic Alcohol Procedure. General Procedure C
A dry 10 mL Schlenk flask, which was evacuated under vacuum and backfilled with N2 (g) three times, was charged with dicyclohexylborane (Cy2BH) (125 mg, 0.7 mmol) and toluene (1.5 mL). The solution was cooled to 0 °C followed by slow addition of alkyne (0.7 mmol). After 5 min the reaction was warmed to room temperature and stirred for an additional 15 min. The solution was cooled to −78 °C and dimethylzinc (Me2Zn) (0.4 mL, 2M in toluene) was added. After stirring at −78 °C for 30 min, the reaction flask was warmed to −30 °C and EtZnCl (1.05 mmol) was added under a steady flow of N2 (g). Immediately thereafter the ketone (0.35 mmol, in 0.2 mL toluene) was added. The reaction mixture stirred at −30 °C and was monitored by TLC until completion (usually 36–48 h). The reaction mixture was quenched with saturated aq. NH4Cl (2 mL) and 2N HCl (1 mL) followed by addition of 5 mL Et2O. The organic layer was separated and the aqueous layer was extracted successively with Et2O (2 × 5 mL). The combined organic layers were successively washed with aq. NaHCO3, brine, dried over MgSO4, and filtered. The filtrate was concentrated in vacuo and purified by column chromatography on silica gel.
(Z)-Disubstituted Allylic Alcohol Procedure. General Procedure D
A dry 10 mL Schlenk flask, which was evacuated under vacuum and backfilled with N2 (g) three times, was charged with diethylborane (0.88 mL, 1M toluene) and 1 mL dry THF. After cooling the flask to 0 °C, the bromoalkyne (0.88 mmol) was added dropwise and the flask was stirred at room temperature for 20 min. t–BuLi (0.51 mL, 1.7 M in pentane) was added to the flask at −78 °C and stirred for 35 minutes, warmed to room temperature and stirred for an additional 40 min. The volatiles were then removed under vacuum over 30 minutes and the residue redissolved in toluene (2 mL). Dimethylzinc (0.44 mL, 2M in toluene) was slowly added to the reaction mixture at −78 °C and stirred for 30 min. The EtZnCl (136 mg, 1.05 mmol) was then added followed by the ketone solution (0.35 mmol, in 0.2 mL toluene). The reaction mixture was stirred at −30 °C and monitored by TLC until completion (usually 36–48 h). The reaction mixture was quenched with saturated aq. NH4Cl (2 mL) and 2N HCl (1 mL) followed by addition of 5 mL Et2O. The organic layer was separated and the aqueous layer was extracted successively with Et2O (2×5 mL). The combined organic layers were successively washed with aq. NaHCO3 and brine, dried over MgSO4, and filtered. The filtrate was concentrated in vacuo and purified column chromatography on silica gel.
3. Results and Discussion
To develop a synthetically useful method for the chelation-controlled addition of organozinc reagents to α-silyloxy ketones, we envisioned employing a variety of alkyl and vinyl zinc nucleophiles. We were most concerned about additions with dialkylzinc reagents, because they are considerably less reactive than mixed alkyl vinylzinc and divinylzinc reagents. Nonetheless, several dialkylzinc reagents are commercially available, providing a good starting point and facilitating the reaction optimization process.
3.1. Optimization of Diethylzinc Addition to α-Silyloxy Ketone 1a
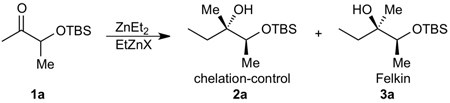
Initially, we examined the reaction of diethylzinc with racemic TBS-protected 3-hydroxybutanone (1a). As shown in Table 1, in the absence of EtZnX Lewis acids the reaction of diethylzinc (1.5 equiv) with TBS-protected 1a provided < 5% of the addition product after several days at 0 °C. Furthermore, the product consisted of a 1:1 mixture of diastereomers, as determined by GC analysis (entry 1). We therefore added 2.0 equiv. EtZnCl and conducted the reaction in toluene for 24 h at rt (entry 2). Under these conditions good conversion to the expected addition product was observed with >20:1 diastereoselectivity. Unfortunately, 18% of the starting ketone was converted to aldol product (9% yield, entry 2). In an attempt to minimize the aldol side product, the reaction temperature was decreased to 0 °C and the reaction conducted in both toluene and methylene chloride (entries 3 and 4). Although both reactions exhibited good conversion and excellent diastereoselectivity (>20:1), in the reaction in toluene 16% of the starting ketone was converted to aldol product compared to 30% in dichloromethane. It is noteworthy that solvents containing oxygen (THF, Et2O, and even t-BuOMe) are not compatible with our chelation-controlled additions, because they effectively bind to the Lewis acidic RZnX and cause complete loss of diastereoselectivity. Lowering the reaction temperature to −15 °C resulted in a decrease in the amount of ketone converted to aldol side product (from 16% in entry 4 to 4% in entry 5). Use of EtZnBr under the same conditions resulted in high diastereoselectivity (>20:1), but lower conversion and increased aldol production (entry 6). Employing EtZnOTf gave low conversion and significantly reduced diastereoselectivity (entry 7). Raising the amount of EtZnCl to 3.0 and 3.5 equiv. resulted in up to 97% conversion after 48 h and gave the chelation-controlled addition product with >20:1 dr (entries 8–9).
Table 1.
Optimization of Diethylzinc Addition to TBS-protected 2-hydroxybutanone
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| entry | X | 1:EtZnX: Et2Zn | solvent | temp (°C) | time (h) | dr(2:3) | 1a | aldolb | 2 + 3b |
| 1 | – | 1.0:0: 1.5 | toluene | −15 | 72 | 1:1 | >95 | – | <5 |
| 2 | CI | 1.0:2.0: 1.5 | toluene | rt | 24 | >20:1 | 5 | 18 | 77 |
| 3 | CI | 1.0:2.0: 1.5 | CH2Cl2 | 0 | 24 | >20:1 | 3 | 30 | 67 |
| 4 | CI | 1.0:2.0: 1.5 | toluene | 0 | 24 | >20:1 | 3 | 16 | 81 |
| 5 | CI | 1.0:2.0: 1.5 | toluene | −15 | 24 | >20:1 | 13 | 4 | 83 |
| 6 | Br | 1.0:2.0: 1.5 | toluene | −15 | 24 | >20:1 | 24 | 10 | 65 |
| 7 | OTf | 1.0:2.0: 1.5 | toluene | −15 | 24 | 6:1 | 93 | - | 7 |
| 8 | CI | 1.0:3.0: 1.5 | toluene | −15 | 48 | >20:1 | 5 | 2 | 93 |
| 9 | CI | 1.0:3.5: 1.5 | toluene | −15 | 48 | >20:1 | 1 | 2 | 97 |
Remaining starting material, conversion, and dr (Cram-chelation:Felkin addition) based on GC analysis.
Conversion of 2 mol of starting ketone to 1 mol of aldol side product, as by GC analysis.
The relative configuration of the products in Table 1 were assigned by 1) synthesis of both diastereomeric products through addition of EtMgBr to the ketone 1a and 2) by comparison of the addition products derived from EtMgBr and EtZnCl/Et2Zn to the 1H NMR spectra of these diastereomers reported in the literature.59 The major product formed in the addition of EtMgBr to ketone 1a in Et2O was the Felkin addition product 3a, which corresponded to the minor diastereomer formed in our organozinc additions in Table 1.
3.2. Addition of Dialkylzinc Reagents to α-Silyloxy Ketones
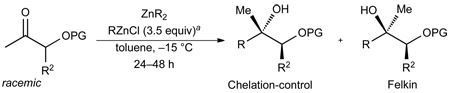
The optimized conditions in Table 1, entry 9, were applied to the addition of diethyl- and dibutylzinc to TBS and TES-protected α-silyloxy ketones. Entry 1 contains the optimized ethyl addition from Table 1, which has been included for the purpose of comparison. With the same ketone substrate 3.5 equiv of dibutylzinc were required for consumption of the ketone in 48 h. We speculate that more dibutylzinc was needed to promote addition due to a more facile formation of aldol side products. Nonetheless, the product formed with >20:1 diastereoselectivity and was isolated in 86% yield after chromatographic purification (entry 2). The TES protected analogue underwent addition with both diethyl- and dibutylzinc to afford the product with >20:1 dr in 96 and 78% yield (entries 3 and 4).
To probe the impact of the ketone structure, we varied the substituents on the 3-position of the substrates. The α-methyl substituent of the TBS or TES–protected α–hydroxy ketones were replaced with Ph and Bn groups (Table 2, entries 6–10). Increasing the size of the substituent from methyl to phenyl and benzyl should make chelation less favorable. If chelation-control remains the dominant pathway, however, the diastereoselectivity is expected to be higher due to the larger size of phenyl and benzyl over methyl (Figure 1). The reactions with TBS- and TES-protected phenyl substituted ketones proceeded with excellent diastereoselectivity (>20:1) and products were isolated in 68–85% yield. Additionally, there was no erosion of ee when enantiopure ketones were employed (entry 5, see Supporting Information for details). Addition of diethylzinc to the Bn substituted ketone occurred with >20:1 diastereoselectivity, albeit in slightly diminished yield due to a more sluggish reaction.
Table 2.
Diastereoselective Alkyl Additions to Silyl-Protected α–Silyloxy Ketones
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| entry | ketone | ZnR2b | yield (%) | drc | |||||
| 1 | 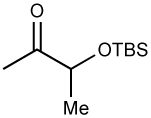 |
ZnEt2 | 91 | >20:1 | |||||
| 2 | ZnBu2 | 86 | >20:1 | ||||||
| 3 | 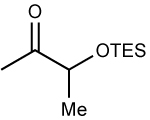 |
ZnEt2 | 96 | >20:1 | |||||
| 4 | ZnBu2 | 78 | >20:1 | ||||||
| 5 | 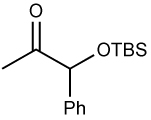 |
ZnEt2 | 88 | >20:1 | |||||
| 6 | ZnBu2 | 81 | >20:1 | ||||||
| 7 | 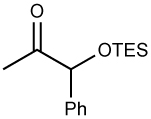 |
ZnEt2 | 85 | >20:1 | |||||
| 8 | ZnBu2 | 78 | >20:1 | ||||||
| 9 | 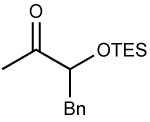 |
ZnEt2 | 68 | >20:1 | |||||
Equiv RZnX relative to ketone.
1.5 equiv ZnEt2 or 3.5 equiv Zn(n-Bu)2 were used.
dr (Cram-chelation:Felkin) determined by 1H NMR or GC analysis of crude products. Relative stereochemistry was determined by comparison to corresponding Grignard or organolithium addition.
For the substrates in Table 2, the additions also were preformed with the corresponding Grignard or organolithium reagents to prepare the diastereomeric mixture enriched in the Felkin addition product (see Supporting Information for details). In each case, the major product formed in the Grignard and organolithium additions had a diastereomeric relationship with those generated in Table 2 using organozinc reagents.
To further explore the reactivity and substrate scope with respect to the ketone electrophile, we replaced the α’-methyl substituent with n-butyl. The addition of diethylzinc to the corresponding n-butyl ketone proved to be significantly slower, giving less than 50% conversion even at rt. This considerable difference in reactivity can be attributed to the lower electrophilicity of the n-butyl ketones and the greater steric hindrance. After examining several reaction temperatures, we found that the reaction of diethylzinc with TBS-protected 2-hydroxyheptan-3-one at 50 °C resulted in full consumption of the starting ketone (Table 3, entry 1). The chelation-controlled product was obtained with excellent diastereoselectivity, albeit in moderate yield (54%) due to the formation of aldol side products. Dimethylzinc is known to react much slower than diethylzinc with aldehydes and ketones.60 However, the reaction of dimethylzinc with TBS-protected 2-hydroxyheptan-3-one under our optimized conditions gave rise to the expected addition product in 81% yield and >20:1 dr. Moreover, there was no erosion of ee when enantiopure ketones were employed (see Supporting Information for details). The corresponding TES-protected ketone underwent addition with diethyl-and dimethylzinc to furnish the chelation-controlled addition products with >20:1 dr and 51–86% yield (entries 3 and 4). We have also prepared the α-phenyl derivatives and subjected them to the diastereoselective addition reactions. The yields and dr of the addition products (entries 5 and 6) mirrored those observed in entries 1–4.
Table 3.
Diastereoselective Alkyl Additions to Butyl-substituted Ketones
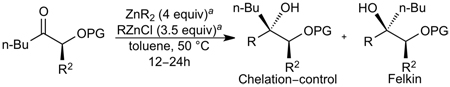 | ||||
|---|---|---|---|---|
| entry | chiral ketone | ZnR2 | yield (%) | drb |
| 1 | 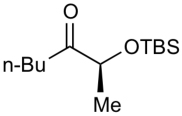 |
ZnEt2 | 54 | >20:1 |
| 2 | ZnMe2 | 81 | >20:1 | |
| 3 | 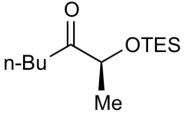 |
ZnEt2 | 51 | >20:1 |
| 4 | ZnMe2 | 86 | >20:1 | |
| 5 | 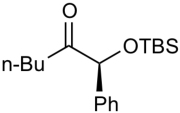 |
ZnEt2 | 56 | >20:1 |
| 6 | ZnMe2 | 77 | >20:1 | |
Equiv relative to ketone.
dr (Cram-chelation:Felkin) determined by 1H NMR or GC analysis of crude products. Relative stereochemistry was determined by comparison to corresponding Grignard or organolithium addition products.
The results in Tables 2 and 3 can be compared to prior reports by the Reetz and Eliel groups. In pioneering work Reetz28 demonstrated that MeTiCl3 underwent addition to silyl protected α-hydroxy ketones with dr as high as 85:15 favoring the chelation-controlled addition product (Scheme 2). The generality of the method has not been expanded beyond addition of methyl groups.
Scheme 2.

Chelation-Controlled Methyl Addition to α–Silyloxy Ketones by Reetz and Co-workers.
The dramatic increase in the rate of dialkylzinc additions to ketones (Tables 1 and 2) in the presence of EtZnX can be compared the seminal investigations by Eliel and Frye.22–24 These researchers studied the reaction of MgMe2 in THF with silyl-protected 3-hydroxy-2-butanone at −70 °C (Table 4). Smaller silyl groups, such as TMS, resulted in very high diastereoselectivities favoring the chelation-controlled product (entry 1, dr = 99:1). The diastereoselectivity steadily decreased with increasing size of the silyl group.23 Ultimately, with the TIPS protected ketone substrate, the Felkin addition product prevailed (entry 5, 58:42) leading to the conclusion that the dominant pathway with this substrate was the non-chelation-controlled addition. This conclusion was supported by a relative rate study in which a two hundred-fold rate acceleration with the TMS protected substrate over the TIPS derivative was observed. Despite the conceptual innovation of this study, it was not developed into a synthetically useful method and scope remains limited to MgMe2 and protected chiral α-hydroxy propiophenones.
Table 4.
Addition of Dimethylmagnesium to Silyloxy Ketones by Eliel, Frye and Coworkers23
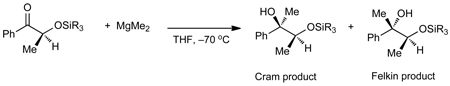 | |||
|---|---|---|---|
| Entry | SiR3 | Relative rate | dra |
| 1 | SiMe3 | ~100 | 99 : 1 |
| 2 | SiEt3 | 8 | 96 : 4 |
| 3 | Si(t-Bu)Me2 | 2.5 | 88 : 12 |
| 4 | Si(t-Bu)Ph2 | 0.8 | 63 : 37 |
| 5 | Si(i-Pr)3 | 0.5 | 42 : 58 |
Dr = Cram chelation:Felkin
3.3. Addition of Vinylzinc Reagents to α-Silyloxy Ketones
Tertiary allylic alcohols are key synthetic intermediates61,62 and are common structural motifs in natural products.47 To increase the utility of our method, we explored the diastereoselective addition of vinylzinc reagents to α–silyloxy ketones (Table 2). Utilizing Oppolzer’s procedure,63–66 (E)-vinylzinc reagents were generated in situ. Thus, hydroboration of alkynes with HBCy2 was followed by B to Zn transmetallation with Me2Zn to form the (E)–vinylzinc intermediates. In the presence of EtZnCl the vinylzinc reagents added to α–silyloxy ketones.
After screening various non-coordinating solvents and reaction temperatures in a fashion similar to that used in Table 1, we found that optimal diastereoselectivities and yields were obtained at −30 °C in toluene. As shown in Table 5, the use of terminal alkynes and 3 equiv EtZnCl with both TBS and TES-protected 3–hydroxybutanone led to the generation of tertiary (E)–disubstituted allylic alcohols with very high dr’s (≥18:1) favoring the chelation–controlled addition products (Table 5, entries 1–3 and 6–8). Equally noteworthy is the use of an enyne to produce the syn–dienyl alcohol in 83% yield with 18:1 dr (entry 3). Additionally, internal alkynes can be utilized in the reaction to afford (E)-trisubstituted allylic alcohols with >20:1 dr and 65–91% yield (entries 4, 5, 9, and 10).
Table 5.
Diastereoselective Generation of (E)-Di- and Trisubstituted Allylic Alcohols
 | |||||
|---|---|---|---|---|---|
| entry | PG | alkynea | yield (%) | drb | major product |
| 1 | TBS | 88 | >20:1 | 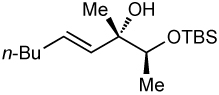 |
|
| 2c | 81 | >20:1 | 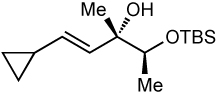 |
||
| 3 | 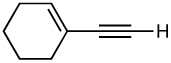 |
83 | 18:1 | 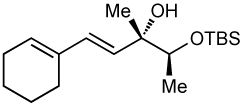 |
|
| 4d | 78 | >20:1 | 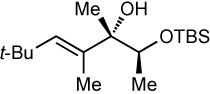 |
||
| 5d | 65 | >20:1 | 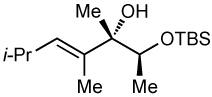 |
||
| 6 | TES | 91 | >20:1 | 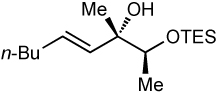 |
|
| 7 | 68 | >20:1 | 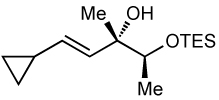 |
||
| 8 | 64 | >20:1 | 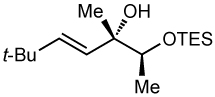 |
||
| 9d | 81 | >20:1 | 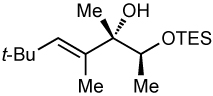 |
||
| 10d | 80 | >20:1 | 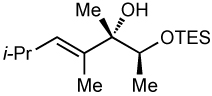 |
||
2 Equiv vinyzinc reagent relative to ketone was employed.
dr determined by 1H NMR of the crude product and refers to the ratio of Cram-chelation:Felkin addition products.
Reaction on 5 mmol scale.
Relative stereochemistry was determined by comparison to corresponding vinyllithium addition.
2.5 equiv vinylzinc reagent was employed relative to the ketone.
A prerequisite to the development of practical and useful methods is the demonstration of scalability. When the reaction in entry 2 was conducted using cyclopropylacetylene on a 5 mmol scale, the chelation-controlled allylic alcohol product was obtained in 81% yield with >20:1 dr.
We next applied our method to the addition of vinylzinc reagents to α–silyloxy ketones bearing a phenyl on the carbinol. As illustrated in Table 6, the reactions proceed with excellent diastereoselectivities, favoring the chelation–controlled products. It is noteworthy that despite the increased acidity of the benzylic C–H’s, the major product is derived from addition rather than deprotonation. Furthermore, there was no erosion of ee when enantiopure ketones were employed (entry 4, see Supporting Information for details).
Table 6.
Additions to α-Phenyl Silyloxy Ketones.
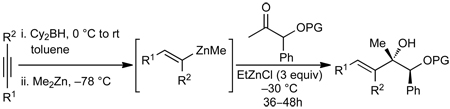 | |||||
|---|---|---|---|---|---|
| entry | PG | alkynea | yield (%) | drb | major product |
| 1 | TES | 71 | >20:1 | 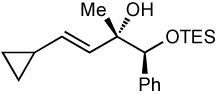 |
|
| 2 | 83 | >20:1 | 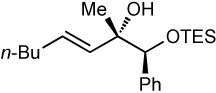 |
||
| 3c | 63 | >20:1 | 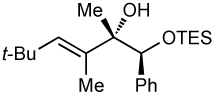 |
||
| 4d | TBS | 74 | >20:1 | 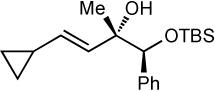 |
|
| 5 | 76 | >20:1 | 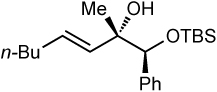 |
||
| 6c | 58 | >20:1 | 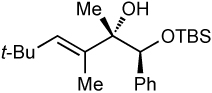 |
||
2 equiv vinyzinc reagent generated relative to ketone.
dr determined by 1H NMR of the crude product and refers to the ratio of Cram-chelation:Felkin addition products.
3 equiv vinylzinc reagent generated relative to ketone.
Relative stereochemistry determined by comparison to corresponding vinyllithium addition (see Supporting Information).
Our method of accessing 1,2-syn allylic diols containing (E)-di- and trisubstituted double bonds is complementary to elegant studies by Roush and coworkers that allow access to the unsubstituted analogues.67 As illustrated in Scheme 3, these authors recently reported the hydroboration of an allylboronate with diisopinocamphenylborane followed by subsequent aldehyde allylation and oxidative workup. The 1,2-syn diol products were isolated with excellent dr (>20:1) and ee (80–92%).
Scheme 3.

Synthesis of 1,2-syn Diols via Allene Hydroboration/Allylation by Roush and coworkers67
Given that (E)-vinylzinc reagents underwent highly diastereoselective additions to α-silyloxy ketones, we turned our attention to addition of the more sterically demanding (Z)-vinylzinc reagents. We recently developed methods for the (Z)-vinylation of aldehydes.43,68 As shown in Scheme 4, the (Z)-vinylzinc reagent is generated from the 1-bromo-1-alkyne by initial hydroboration with diethylborane in THF.43 To promote the formation of the (Z)-vinylborane, t-BuLi69 was added as a hydride source to initiate the 1,1-metallate rearrangement and generate the (Z)-vinylborane intermediate.
Scheme 4.

Proposed Mechanism for the Generation of (Z)-disubstituted Vinylzinc Reagents.
We next applied our (Z)-vinylation to the addition to α-silyloxy ketones (Scheme 5). Given the ability of ethereal solvents to bind tightly to RZnX, a solvent switch was necessary after the formation of the (Z)-vinylborane reagent. Thus, the THF was removed under vacuum and fresh toluene was added to the flask. The (Z)-vinylborane was then treated with Me2Zn, EtZnCl, and the α–silyloxy ketone (Scheme 5). The (Z)-allylic alcohol was obtained with >20:1 dr as a single double bond isomer.
Scheme 5.

Diastereoselective (Z)-Vinylation of an α-Silyloxy Ketone
3.4. Proposed Transition State
It is important to note that the diastereoselectivities for the chelation-controlled addition reactions in Tables 2, 3, 5, and 6 are exceptionally high, rivaling those observed with benzyl and alkyl protected α-hydroxy ketones.70–73 To rationalize the excellent levels of diastereoselectivity observed with the variety of organozinc nucleophiles and ketone substrates employed in this study, we propose a transition state structure that involves coordination of the chloride of the RZnCl to the dialkyl- or vinylzinc reagent (Figure 2). Coordination of RZnCl by ZnR2 is anticipated to increase the Lewis acidity of ClZnR and favor chelation of the substrate. Coordination of the chloride to the dialkylzinc reagent also increases the donor ability of the dialkylzinc, allowing a directed addition to the carbonyl carbon of the ketone. Further studies are necessary to determine the validity of this mechanistic proposal.
Figure 2.

Proposed transition state to rationalize the extremely high diastereoselectivities observed in this study.
4. Conclusions
In summary, we have developed the first general, highly diastereoselective method for the chelation-controlled addition of organozinc reagents to silyl-protected α-hydroxy ketones, solving a long-standing deficiency in organic synthesis. It is important to note that the exceptional diastereoselectivities outlined herein with silyl-protected α-hydroxy ketones are competitive with results reported with benzyl-protected α-hydroxy ketones. Previous methods for addition of organometallic reagents to silyl-protected α-hydroxy ketones generated Felkin addition products in the vast majority of cases, as outlined in most graduate level organic and physical organic chemistry textbooks.74–77 The results outlined herein, in combination with our chelation-controlled diastereoselective additions to silyl-protected α-hydroxy aldehydes,43,44,46 are expected to result in a paradigm shift in the addition of organometallic reagents to silyl-protected α-hydroxy carbonyl compounds.
Our method provides a new synthetic tool to generate 1,2–syn diols and (E)–di-, (E)–tri-, and (Z)-disubstituted tertiary allylic alcohols with excellent dr’s. These results, combined with the compatibility of organozinc reagents with a wide array of functionality, make this method useful for the synthesis of complex molecules and natural products.
Supplementary Material
Acknowledgment
This paper is dedicated to Professor Eusebio Juaristi on the occasion of his 60th birthday for his leadership and outstanding contributions to chemistry in Mexico. We thank the NSF (CHE-0848467) and NIH (National Institute of General Medical Sciences GM58101) for partial support of this work. We are also grateful to Professor Marisa Kozlowski (University of Pennsylvania) for helpful discussions. G.R.S thanks UNCF-Merck and NOBCChE/GlaxoSmithKline programs, ACS Organic Division, and Amgen for graduate scholarships. G.D.K. thanks TUBITAK for graduate scholarships. We would also like to thank the NIH for the purchase of a Waters LCTOF-Xe Premier ESI mass spectrometer (Grant No. 1S10RR23444-1).
Footnotes
Supporting Information Available: Procedures and full characterization of new compounds are available free of charge via the Internet at http://pubs.acs.org
References Cited
- 1.Corey EJ, Cheng X-M. The Logic of Chemical Synthesis. New York: Wiley; 1989. [Google Scholar]
- 2.Nicolaou KC, Snyder SA. Classics in Total Synthesis II : More Targets, Strategies, Methods. Weinheim, Germany: Wiley-VCH; 2003. [Google Scholar]
- 3.Nicolaou KC, Sorensen EJ. Classics in Total Synthesis. Weinheim, Germany: VCH; 1996. [Google Scholar]
- 4.Koskinen A. Asymmetric Synthesis of Natural Products. Chichester, NY: Wiley; 1993. [Google Scholar]
- 5.Guillarme S, Ple K, Banchet A, Liard A, Haudrechy A. Chem. Rev. 2006;106:2355. doi: 10.1021/cr0509915. [DOI] [PubMed] [Google Scholar]
- 6.Reetz MT. Angew. Chem., Int. Ed. 1984;23:556. [Google Scholar]
- 7.Mengel A, Reiser O. Chem. Rev. 1999;99:1191. doi: 10.1021/cr980379w. [DOI] [PubMed] [Google Scholar]
- 8.Keck GE, Abbott DE, Wiley MR. Tetrahedron Lett. 1987;28:139. [Google Scholar]
- 9.Keck GE, Boden E. Tetrahedron Lett. 1984;25:265. [Google Scholar]
- 10.Lodge EP, Heathcock CH. J. Am. Chem. Soc. 1987;109:3353. [Google Scholar]
- 11.Lodge EP, Heathcock CH. J. Am. Chem. Soc. 1987;109:2819. [Google Scholar]
- 12.Smith RJ, Trzoss M, Bühl M, Bienz S. Eur. J. Org. Chem. 2002:2770. [Google Scholar]
- 13.Cee VJ, Cramer CJ, Evans DA. J. Am. Chem. Soc. 2006;128:2920. doi: 10.1021/ja0555670. [DOI] [PubMed] [Google Scholar]
- 14.Evans DA, Siska SJ, Cee VJ. Angew. Chem., Int. Ed. 2003;42:1761. doi: 10.1002/anie.200350979. [DOI] [PubMed] [Google Scholar]
- 15.Cornforth JW, Cornforth RH, Mathew KK. J. Chem. Soc. 1959:112. [Google Scholar]
- 16.Paddon-Row MN, Rondan NG, Houk KN. J. Am. Chem. Soc. 1982;104:7162. [Google Scholar]
- 17.Evans DA, Cee VJ, Siska SJ. J. Am. Chem. Soc. 2006;128:9433. doi: 10.1021/ja061010o. [DOI] [PubMed] [Google Scholar]
- 18.Gung BW, Xue X. Tetrahedron: Asymmetry. 2001;12:2955. [Google Scholar]
- 19.Wuts PGM, Greene TW. Greene's Protective Groups in Organic Synthesis. 4 ed. Hoboken, NJ: Wiley; 2007. [Google Scholar]
- 20.Cram DJ, Kopecky KR. J. Am. Chem. Soc. 1959;81:2748. [Google Scholar]
- 21.Anh N, Eisenstein O. Nouv. J. Chim. 1977;1:61. [Google Scholar]
- 22.Frye SV, Eliel EL, Cloux R. J. Am. Chem. Soc. 1987;109:1862. [Google Scholar]
- 23.Chen X, Hortelano ER, Eliel EL, Frye SV. J. Am. Chem. Soc. 1992;114:1778. [Google Scholar]
- 24.Chen X, Hortelano ER, Eliel EL, Frye SV. J. Am. Chem. Soc. 1990;112:6130. [Google Scholar]
- 25.Evans DA, Allison BD, Yang MG, Masse CE. J. Am. Chem. Soc. 2001;123:10840. doi: 10.1021/ja011337j. [DOI] [PubMed] [Google Scholar]
- 26.Evans DA, Allison BD, Yang MG. Tetrahedron Lett. 1999;40:4457. [Google Scholar]
- 27.Evans DA, Halstead DP, Allison BD. Tetrahedron Lett. 1999;40:4461. [Google Scholar]
- 28.Reetz MT, Huellmann M. J. Chem. Soc., Chem. Commun. 1986:1600. doi: 10.1021/ja00269a044. [DOI] [PubMed] [Google Scholar]
- 29.Marshall AJ, Eidam P. Org. Lett. 2004;6:445. doi: 10.1021/ol0363568. [DOI] [PubMed] [Google Scholar]
- 30.Marshall JA, Bourbeau MP. Org. Lett. 2003;5:3197. doi: 10.1021/ol034918h. [DOI] [PubMed] [Google Scholar]
- 31.Williams DR, Kiryanov AA, Emde U, Clark MP, Berliner MA, Reeves JT. Angew. Chem., Int. Ed. 2003;42:1258. doi: 10.1002/anie.200390322. [DOI] [PubMed] [Google Scholar]
- 32.El-Sayed E, Anand NK, Carreira EM. Org. Lett. 2001;3:3017. doi: 10.1021/ol016431j. [DOI] [PubMed] [Google Scholar]
- 33.Rodriguez-Escrich C, Olivella A, Urpi F, Vilarrasa J. Org. Lett. 2007;9:989. doi: 10.1021/ol063035y. [DOI] [PubMed] [Google Scholar]
- 34.Roush WR, Palkowitz AD, Ando K. J. Am. Chem. Soc. 1990;112:6348. [Google Scholar]
- 35.Kolakowski RV, Williams LJ. Tetrahedron Lett. 2007;48:4761. [Google Scholar]
- 36.Sharon O, Monti C, Gennari C. Tetrahedron. 2007;63:5873. [Google Scholar]
- 37.Georges Y, Ariza X, Garcia J. J. Org. Chem. 2009;74:2008. doi: 10.1021/jo8025753. [DOI] [PubMed] [Google Scholar]
- 38.Soai K, Shimada C, Takeuchi M, Itabashi M. J. Chem. Soc., Chem. Commun. 1994;1994:567. [Google Scholar]
- 39.Jeon S-J, Chen YK, Walsh PJ. Org. Lett. 2005;7:1729. doi: 10.1021/ol050255n. [DOI] [PubMed] [Google Scholar]
- 40.Walsh PJ, Kozlowski MC. Fundamentals of Asymmetric Catalysis. Sausalito: University Science Books; 2008. [Google Scholar]
- 41.Soai K, Hatanaka T, Yamashita T. J. Chem. Soc. Perkin. Trans. 1. 1992:927. [Google Scholar]
- 42.Watanabe M, Komota M, Nishimura M, Araki S, Butsugan YJ. Chem. Soc., Perkin Trans. 1. 1993:2193. [Google Scholar]
- 43.Jeon S-J, Fisher EL, Carroll PJ, Walsh PJ. J. Am. Chem. Soc. 2006;128:9618. doi: 10.1021/ja061973n. [DOI] [PubMed] [Google Scholar]
- 44.Li H, Carroll PJ, Walsh PJ. J. Am. Chem. Soc. 2008;130:3521. doi: 10.1021/ja077664u. [DOI] [PubMed] [Google Scholar]
- 45.Kerrigan MH, Jeon S-J, Chen Y, Salvi L, Carroll PJ, Walsh PJ. J. Am. Chem. Soc. 2009;131:8434. doi: 10.1021/ja809821x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stanton GR, Johnson CN, Walsh PJ. J. Am. Chem. Soc. 2010;132:4399. doi: 10.1021/ja910717p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gao D, O'Doherty GA. Org. Lett. 2010;12:4. doi: 10.1021/ol101340n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hayashi Y, Yamaguchhi H, Toyoshima M, Okado K, Toyo T, Shoji M. Org. Lett. 2008;10:4. doi: 10.1021/ol800195g. [DOI] [PubMed] [Google Scholar]
- 49.Shibahara S, Fujino M, Tashiro Y, Okamoto N, Esumi T, Takahashi K, Ishihara J, Hatekeyama S. Synthesis. 2009;17:19. [Google Scholar]
- 50.Trost BM, Frederisksen MU, Papillon JPN, Harrington PE, Shin S, Shireman BT. J. Am. Chem. Soc. 2005;127:3666–3667. doi: 10.1021/ja042435i. [DOI] [PubMed] [Google Scholar]
- 51.Wang Y-G, Kobayashi Y. Org. Lett. 2002;4:4615–4618. doi: 10.1021/ol020193q. [DOI] [PubMed] [Google Scholar]
- 52.Boger DL, Ichikawa S, Zhong W. J. Am. Chem. Soc. 2001;123:4161–4167. doi: 10.1021/ja010195q. [DOI] [PubMed] [Google Scholar]
- 53.Dorta E, Díaz-Marrero AR, Cueto M, D'Croz L, Maté JL, Darias J. Tetrahedron Lett. 2004;45:7065–7068. doi: 10.1021/ol049287l. [DOI] [PubMed] [Google Scholar]
- 54.Hatano MM, T, Ishihara K. Chem. Commun. 2010;46:3. doi: 10.1039/c0cc01301c. [DOI] [PubMed] [Google Scholar]
- 55.Chiu P, Metz P, et al. Adv. Synth. Catal. 2009;351:5. [Google Scholar]
- 56.Fabicon RM, Richey HG. Organometallics. 2001;20:4018. [Google Scholar]
- 57.Guerrero A, Hughes DL, Bochmann M. Organometallics. 2006;25:1525. [Google Scholar]
- 58.Gopalsamuthiram V, Predeus AV, Huang RH, Wulff WD. J. Am. Chem. Soc. 2009;131:2. doi: 10.1021/ja905990u. [DOI] [PubMed] [Google Scholar]
- 59.Molander GA, Estévez-Braun AM. B. Soc. Chim. Fr. 1997;134:8. [Google Scholar]
- 60.Kitamura M, Okada S, Suga S, Noyori R. J. Am. Chem. Soc. 1989;111:4028. [Google Scholar]
- 61.Katsuki T, Martin VS. Org. React. 1996:1. [Google Scholar]
- 62.Hoveyda AH, Evans DA, Fu GC. Chem. Rev. 1993;93:1307. [Google Scholar]
- 63.Srebnik M. Tetrahedron Lett. 1991;32:2449. [Google Scholar]
- 64.Oppolzer W, Radinov RN. Helv. Chim. Acta. 1992;75:170. [Google Scholar]
- 65.Oppolzer W, Radinov RN, El-Sayed E. J. Org. Chem. 2001;66:4766. doi: 10.1021/jo000463n. [DOI] [PubMed] [Google Scholar]
- 66.Oppolzer W, Radinov RN, Brabander JD. Tetrahedron Lett. 1995;36:2607. [Google Scholar]
- 67.Chen M, Handa M, Roush WR. J. Am. Chem. Soc. 2009;131:14602–14603. doi: 10.1021/ja904599h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Salvi L, Jeon S-J, Fisher EL, Carroll PJ, Walsh PJ. J. Am. Chem. Soc. 2007;129:16119. doi: 10.1021/ja0762285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Campbell JB, Jr, Molander GA. J. Organomet. Chem. 1978;156:71. [Google Scholar]
- 70.Overman LE, Lin N-H. J. Org. Chem. 1985;50:2. [Google Scholar]
- 71.Williams DR, White FH. J. Org. Chem. 1987;52:13. [Google Scholar]
- 72.Crimmins MT, Zuccarello JL, Cleary PA, Parrish JD. Org. Lett. 2006;8:4. doi: 10.1021/ol0526625. [DOI] [PubMed] [Google Scholar]
- 73.Crimmins MT, Long A. Org. Lett. 2005;7:4. doi: 10.1021/ol0515107. [DOI] [PubMed] [Google Scholar]
- 74.Clayden J, Greeves NIn. Organic Chemistry. New York: Oxford University Press; 2001. p. 889. [Google Scholar]
- 75.Bruckner R. Advanced Organic Chemistry: Reaction Mechanisms. San Diego: Academic Press; 2002. p. 315. [Google Scholar]
- 76.Carey FA, Sundberg RJ. Advanced Organic Chemistry: Part A: Structure and Mechanisms. 5 ed. New York: Springer; 2007. p. 179. [Google Scholar]
- 77.Anslyn EV, Dougherty DA. Modern Physical Organic Chemistry. Salslito: University Science Books; 2006. p. 564. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.