

Abstract
Moyamoya is a cerebrovascular angiopathy characterized by a progressive stenosis of the terminal part of the intracranial carotid arteries and the compensatory development of abnormal and fragile collateral vessels, also called moyamoya vessels, leading to ischemic and hemorrhagic stroke. Moyamoya angiopathy can either be the sole manifestation of the disease (moyamoya disease) or be associated with various conditions, including neurofibromatosis, Down syndrome, TAAD (autosomal-dominant thoracic aortic aneurysm), and radiotherapy of head tumors (moyamoya syndromes). Its prevalence is ten times higher in Japan than in Europe, and an estimated 6%–12% of moyamoya disease is familial in Japan. The pathophysiological mechanisms of this condition remain obscure. Here, we report on three unrelated families affected with an X-linked moyamoya syndrome characterized by the association of a moyamoya angiopathy, short stature, and a stereotyped facial dysmorphism. Other symptoms include an hypergonadotropic hypogonadism, hypertension, dilated cardiomyopathy, premature coronary heart disease, premature hair graying, and early bilateral acquired cataract. We show that this syndromic moyamoya is caused by Xq28 deletions removing MTCP1/MTCP1NB and BRCC3. We also show that brcc3 morphant zebrafish display angiogenesis defects that are rescued by endothelium-specific expression of brcc3. Altogether, these data strongly suggest that BRCC3, a deubiquitinating enzyme that is part of the cellular BRCA1 and BRISC complexes, is an important player in angiogenesis and that BRCC3 loss-of-function mutations are associated with moyamoya angiopathy.
Introduction
Moyamoya (MIM 252350, MIM 607151, MIM 608796) is a cerebrovascular angiopathy characterized by a progressive stenosis of the terminal part of the intracranial internal carotid arteries (ICA) and their proximal branches within the circle of Willis.1,2 This disease is associated with the compensatory development of abnormal, thin, and fragile collateral vessels (“moyamoya” vessels) at the base of the brain. Reduced cerebral blood flow and rupture of the fragile collateral vessels cause ischemic and hemorrhagic stroke in children and adults with moyamoya. The prevalence of this disorder has been estimated to be close to 3/100 000 in Japan and is ten times less prevalent in Europe.1,2
Histopathological analysis of affected carotids shows a marked decrease in the outer diameters of the carotid terminations, a fibrocellular thickening of the intima containing proliferating smooth-muscle actin-positive cells, luminal thrombosis, and thinning of the media vascular layer. Collateral moyamoya vessels display fragmented elastic lamina, thinned media in the vessel wall, and microaneurysms.3 Despite a great deal of investigation, the molecular etiology and pathogenesis of moyamoya angiopathy remains unclear.4
Moyamoya angiopathy can be associated with different conditions, including tuberculous meningitis, atherosclerosis, radiotherapy of head tumors, and various diseases of known genetic etiology, such as Down syndrome (MIM 190685), neurofibromatosis type 1 (MIM 162200), sickle cell disease (MIM 603903), and autosomal-dominant thoracic aortic aneurysm disease, also called TAAD (MIM 102620).1,5–7 These conditions are known as moyamoya syndromes. They are distinct from moyamoya disease (MMD), which occurs as an idiopathic disorder in which the sole manifestation of the disease is the moyamoya angiopathy.
MMD occurs predominantly in patients of Asian origin, and the highest prevalence of the disease is found in Japan, especially among pediatric patients.2 However, it can occur in people of all ethnicities and in all age groups. Although most cases of MMD appear to be sporadic, an estimated 6% −12% of all reported cases in Japan are familial. The inheritance has been suggested to follow an autosomal-dominant, autosomal-recessive, X-linked-recessive, or multifactorial inheritance pattern.8 Several whole-genome linkage studies have been aimed at identification of MMD genetic loci. Linkage analyses conducted in Japanese families suggested that MMD candidate loci might be located at 3p24.2-p26, 6q25, 8q23, 12p12, and 17q25.9–12 No mutated gene has yet been identified within those linked regions. Interestingly, a recent genome-wide association study suggested that RNF213 (MIM 613768), a gene that is located at 17q25 and encodes a ring finger protein, is an MMD susceptibility gene.13 Additionally, mutations in ACTA2 (MIM 102620), which encodes the vascular smooth-muscle-cell-specific isoform of α-actin, have been reported in rare MMD patients.14
Herein, we report on three unrelated families affected by an X-linked moyamoya syndrome in which an overlapping deletion at Xq28 removes MTCP1/MTCP1NB (MIM 300116) and BRCC3 (MIM 300617) and cosegregates with the affected phenotype. Morpholino-mediated knockdown of the BRCC3 ortholog in zebrafish led to defective angiogenesis that was rescued by endothelial-specific expression of brcc3, strongly suggesting that this deubiquitinating enzyme is a novel important player in angiogenesis.
Material and Methods
Patients and Families
Three families (F1–F3) including a total of ten males affected by a syndromic moyamoya condition were included in this study. All patients and family members provided written informed consent. This study was conducted in accordance with ethical recommendations in France for genetic disorders. The geneological trees are shown in Figure 1. Some of the clinical and magnetic resonance imaging (MRI) features of family F1 have previously been reported.6 Additional information on family F1 as well as clinical, biological, and MRI features of F2 family members and of two of the affected F3 family members is provided in the results section. Three additional unrelated families including two offspring showing pure, nonsyndromic MMD were also included in this study (F4: two brothers. F5: brother and sister. F6: two sisters).
Figure 1.
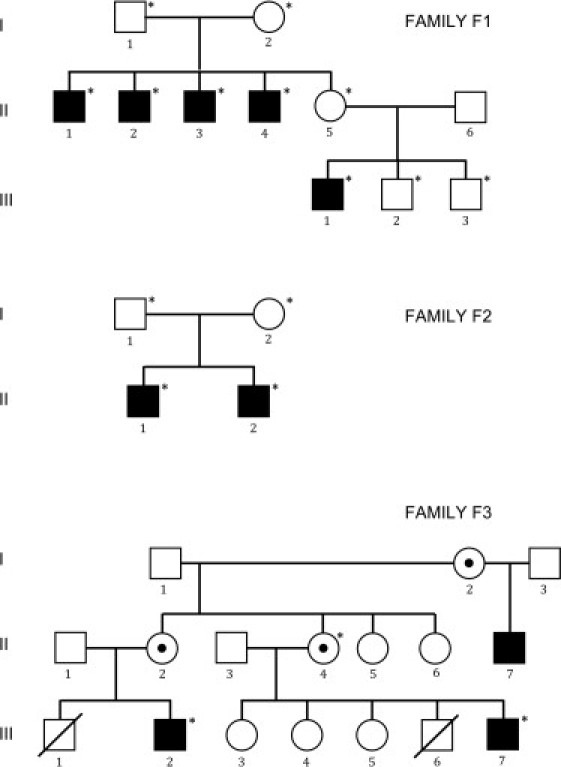
Genealogical Trees of Families F1, F2, and F3
Circles: females. Squares: males. Black-filled symbols: affected individuals. Empty symbols: healthy individuals. Members whose blood has been sampled are indicated by an asterisk.
DNA and RNA Extraction
Genomic DNA from probands and consenting relatives was extracted from peripheral blood leukocytes by standard procedures. F1–F3 family members whose blood was sampled are indicated in Figure 1. Total RNA was isolated from lymphoblastoid Epstein-Barr virus (EBV) cell lines via the Trizol method and transcribed into cDNA via dT primers. RNA was available for probands of families F1, F2, F4, and F5.
Microsatellite Markers and Chromosome X Haplotyping
Family F1 was genotyped with a panel of 19 microsatellite markers spanning the X chromosome (average genetic distance = 20 cM). Additional microsatellite markers, DXS984, DXS8106, DXS8045, DXS1193, and DXS8061, were used for Xq27-q28 haplotyping in family F1. Parametric multipoint and two-point linkage analyses were carried out with the MINX program of MERLIN v1.1.2 package; a recessive X-linked model with complete penetrance, disease allele frequency of 0.0001, and a phenocopy frequency of 0.0001 were assumed.
Copy-Number Analysis with Affymetrix SNP 6.0 Arrays
On the basis of the hypothesis that a deletion might be involved in the disease mechanisms, we used high-density Affymetrix SNP 6.0 arrays (Affymetrix Santa Clara, CA) to search for putative deletions located within the linked region on the X chromosome in family F1. Genomic DNA from the mother (F1 I-2) and two of her affected sons (F1 II-3 and F1 II-4) was used for genotyping and copy-number analysis. Sample processing and labeling were performed according to the manufacturer's instructions (Affymetrix Cytogenetics Copy Number Assay User Guide, P/N 702607 Rev. 2). Hybridization was performed with a GeneChip Hybridization oven 640 at 50°C and 60 rpm for 16 hr, washed with a GeneChip Fluidics Station 450, and scanned with a GeneChip Scanner 3000-7G. The image data were processed with the Affymetrix Genotyping Console (GTC v4.0) for determination of SNP call and copy-number variation.
UCSC databases, the HapMap project, and the Database of Genomic Variants were used for filtering copy-number polymorphisms within the region of interest.
STS Markers and Oligonucleotides Used for Walking PCR
To confirm the putative deletion in family F1 and refine the critical interval, we used a panel of 12 STS markers selected from NCBI build 36.3 or designed with Primer 3 software (Table S1, available online). Two of these STS markers, ECD13623 and ECD22603, closely flanked the putative deletion. Three of these STS markers (ECD07678, ECD07752, and ECD15347) map within FUNDC2, MTCP1/MTCP1NB, and BRCC3. Standard PCR was carried out with DNA from all affected males, two obligatory female carriers (F1 I-2 and F1 II-5), and healthy males (F1 III-2 and F1 III-3). Additional oligonucleotides were used for walking PCR in family F2 (Table S1).
Long-Range PCR and Sequencing
In order to identify the deletion breakpoints, we performed long-range PCR with primers ECD21451 (forward) and ECD02005R (reverse) in family F1 and STSR7 (forward) and BRCC3R4 (reverse) in families F2 and F3 (Table S1 and S2). Long-range PCR was carried out on DNA from all affected individuals and each obligatory carrier female. Unaffected spouses were included as negative controls. The reactions were carried out with the Expand 20 kbPLUS PCR system (Roche) according to the manufacturer's instructions. Purified PCR products were sequenced with BigDye Terminator 3.1 (Applied Biosystems, Foster City, CA) chemistry on an ABI 3130 DNA sequencing apparatus according to the manufacturer's instructions.
Screening for BRCC3 and MTCP1/MTCP1NB in Familial MMD Cases
All coding exons and flanking splice sites of BRCC3, MTCP1, and MTCP1NB were amplified and sequenced with the genomic DNA of probands from families F4–F6 (RefSeq accession numbers: BRCC3, NM_001018055.1; MTCP1, NM_001018025.3; and MTCP1NB, NM_001018024.2). Sequencing primers are shown in Table S2.
Immunoblot Analyses
A polyclonal antibody specific for human BRCC3 was purchased from ProSci (reference number 4331F). A polyclonal antibody specific for human p8MTCP1NB was kindly provided by M.H. Stern (Institut Curie, France).
Immunoblot analysis of p8MTCP1NB and BRCC3 expression in EBV-transformed lymphoblastoid cell lines from patients F1 II-2 and F2 II-2 and two healthy controls was conducted with 10 μg of total protein lysates.
In addition, 10 μg of protein lysates from HUVEC endothelial cells, 40 μg of total protein from human adult arterial tissue (Gentaur Molecular Products P1234013), and total protein from human adult vein tissue (Gentaur Molecular Products P1234020) were used in screens for p8MTCP1NB and BRCC3 protein levels.
Zebrafish Lines and Husbandry
EK strain wild-type fish were used for in situ hybridization, and both Tg(fli1-eGFP)y1 and Tg(flk1a-eGFP) were used in morpholino knockdown experiments.15 All lines were maintained according to established convention.16
brcc3 and mtcp1nb cDNA Cloning and In Situ Hybridization
Total RNA was isolated from 24 hpf wild-type zebrafish embryos with TRIzol (Invitrogen, Carlsbad, CA) and reversed transcribed with the SuperScript III First Strand Synthesis System (Invitrogen). brcc3 and mtcp1nb cDNAs were amplified from cDNA by PCR with the following primers:
brcc3-F: 5′-TTCGCGATGGCTGTCAACGCGGTGCATTT-3′
brcc3-R: 5′-TTACAATGCCGCCAGCTCTTGCGTCAGC-3′
mtcp1nb-F: 5′-AGCTCGAAGTCGGAGTTCATTTC-3′
mtcp1nb-R: 5′-GACAGTGTTTAATGATGTGTAGCAGCA-3′
The resulting PCR products were cloned into pCR®II-TOPO® (Invitrogen) according to manufacturer's protocol. Riboprobes were generated using the mMESSAGE mMACHINE® T7 Kit (Life Technologies Corp., Carlsbad, CA). Riboprobes were generated with the Roche DIG RNA Labeling Kit (Indianapolis, IN). Whole-mount in situ hybridization was performed as previously described.17
Morpholino, mRNA, and DNA Injections
Morpholinos were designed to disrupt splicing of brcc3 or mtcp1nb (Gene Tools, LLC. Philomath, OR). The brcc3 morpholino (5′-GTGATGCAGGAATAAAGCACATTCA-3′) or mtcp1nb morpholino (5′-AGCTGATAAACACAAAAGTCACACA-3′) was injected into one-cell-stage Tg(fli1-eGFP)y1 or Tg(flk1a-eGFP) embryos. The effectiveness of the brcc3 morpholino was verified by reverse transcriptase PCR (RT-PCR) aimed at detecting loss of the appropriately spliced gene product (data not shown). The brcc3 and mtcp1nb morpholinos were injected at doses of 9.5 pg and 4.0 pg, respectively. For mRNA rescue experiments, wild-type brcc3 was cloned into pCS-DEST and transcribed with the Ambion mMESSAGE mMACHINE SP6 Kit. brcc3 was injected (375 pg) into control or brcc3 morpholino-injected embryos.18 Injection of brcc3 mRNA into control animals had no effect on vessel growth (data not shown). Endothelium-specific rescue of brcc3 morphants was performed by mosaic expression of brcc3 via a 6.4 kb flk1 promoter.19 Detailed methods for construction of flk1-mCherry-2A-brcc3 and flk1-mCherry transgenes are provided in Figure S5. SceI homing endonuclease was coinjected with the mcherry-2a-brcc3 transgene (30 pg) so that mosaicism would be increased.20
Microscopy and Imaging
The vascular endothelium of injected Tg(fli1-eGFP)y1 and Tg(flk-eGFP) was illuminated with a 488 nm argon laser and visualized with an Olympus BX61WI confocal microscope. Embryos were mounted laterally in Nusieve GTG agarose (0.5% in Embryo water), and optical sections were collected through the trunk at 1.5 μm intervals. Confocal Z series were rendered into three-dimensional representations with Olympus Fluoview software. For quantitation of phenotypes, ten ISVs from each of 12 different injected embryos were scored for each control and experimental condition. Statistical significance was determined by a two-sided student's t test calculation at a 99.9% confidence interval.
Results
Clinical Features of Families F1–F3
Within families F1 and F2, a total of seven males showed a strikingly similar phenotype characterized by the association of a moyamoya angiopathy, a hypergonadotropic hypogonadism, and several multisystemic manifestations (Figure 1 and Figure 2). The main clinical, biological, and morphological features of this condition are summarized in Table 1.
Figure 2.
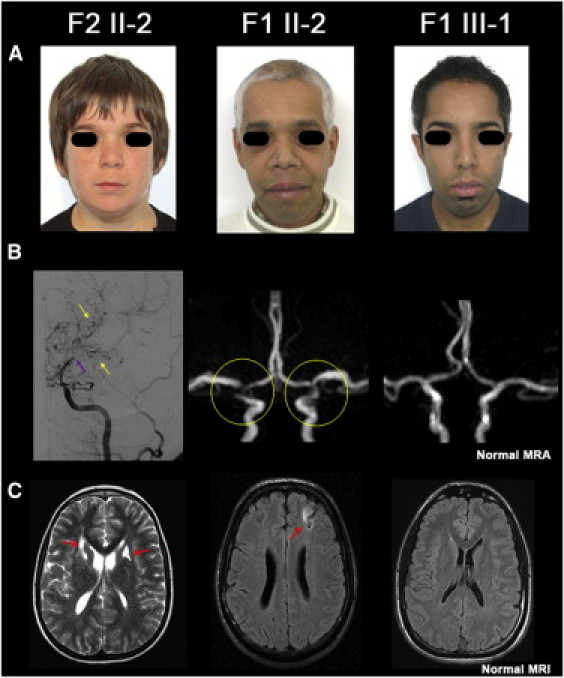
Facial Dysmorphy and Moyamoya Angiopathy of Cases F1 II-2, F1 III-1, and F2 II-2
(A) facial dysmorphy observed in three affected members from families F1 and F2. Hypertelorism is obvious in F1 III-1 and F2 II-2, and a long philtrum is observed in all three patients. Premature white hair locks are present in F1 II-2 and F1 III-1 (note that F1 III-1 dyed his hair).
(B and C) Moyamoya angiopathy. In case F2 II-2, conventional angiography showed an occlusion of a distal internal carotid artery (purple arrow), and MRI revealed neovascularization (yellow arrows) associated with bilateral small deep infarcts (red arrows). Magnetic resonance angiography (MRA) of case F1 II-2 showed a stenosis of the terminal portion of the internal carotid arteries and of the origin of anterior and middle cerebral arteries associated with a left cortical infarct on FLAIR images (red arrow). Cerebral MRA and MRI did not detect any moyamoya angiopathy nor parenchymal lesion in F1 III-1, the youngest patient of family F1.
Table 1.
Main Characteristics of Symptomatic Members of Families F1 and F2
|
Family 1 |
Family 2 |
||||||
|---|---|---|---|---|---|---|---|
| III-1 | II-1 | II-2 | II-3 | II-4 | II-2 | II-1 | |
| Gender | M | M | M | M | M | M | M |
| Current age (age at death), yr | 28 | (34) | 41 | 45 | 48 | 14 | 17 |
| Short stature of postnatal onset | yes | yes | yes | yes | yes | yes | yes |
| Facial dysmorphism | yes | yes | yes | yes | yes | yes | yes |
| Premature canities | yes | yes | yes | yes | yes | no | no |
| Azoospermia | yes | yes | yes | n.a. | n.a. | n.a. | n.a. |
| Hormonal testing | |||||||
| Age at testing, yr | 17 | 29 | 30 | 44 | 46 | 14 | 12.5 |
| Testis volume—Tanner Stage | n.a. | T1 | T2 | n.a. | n.a. | T1 | T1 |
| Testosterone, nmol/litera | 3.47 (↓) | 23.6 | 19.8 | 5.7 | 6.6 | 0.29 | 0.11 (↓) |
| LH plasma levels, IU/literb | 9 (↑) | 14 (↑) | 10 (↑) | 11 (↑) | 20.3 (↑) | 0.3 (↑) | 0.25 (↑) |
| FSH plasma levels, IU/literc | 71 (↑) | 37 (↑) | 36 (↑) | 38 (↑) | 48.6 (↑) | 2.4 | 4.1 (↑) |
| AMH, pmol/literd | n.a. | n.a. | n.a. | <0.35 | <0.35 | n.a. | 358 |
| Inhibin B, pg/mle | n.a. | <15 (↓) | <15 (↓) | <15 (↓) | <15 (↓) | n.a. | n.a. |
| IGF-1, mcg/literf | n.a. | n.a. | n.a. | n.a. | n.a. | 116(↓) | n.a. |
| Partial GH insufficiencyg | yes | yes | no | n.a. | n.a. | yes | yes |
| Stroke | no | yes | yes | yes | yes | yes | no |
| Age at neurological onset, yr | - | 22 | 35 | 34 | 4 | 10 | - |
| Moya-Moya angiopathyh | no | yes | yes | yes | yes | yes | yes |
| History of hypertension | no | no | no | no | no | yesi | no |
| Dilated Cardiomyopathyj | yes | yes | no | yes | no | no | no |
| Right aortic arch | n.a. | no | n.a. | yes | n.a. | n.a. | no |
| Early onset cataract | yes | no | yes | yes | yes | no | no |
Normal values: 5.4–30.4 for tests performed in family 1, 0.28 ± 0.01 for tests performed in family 2.
Normal values: 1–7 for family 1, 0.17–0.19 for family 2.
Normal values: 1.5–14 for family 1, 0.44–3.1 for family 2.
Normal values: 15–90 for family 1, 250–1500 for family 2.
Normal values: 80–270.
Normal values: 230–876.
The definition of complete and partial GH deficiency was a GH peak response of less than 3 and 10 ng/ml, respectively, after a stimulation test.
Moyamoya angiopathy on MR Angiography or Conventional Angiography.
No renal artery stenosis was detected on ultrasound examination.
The definition of dilated cardiomyopathy was an association of left ventricular enlargement with reduction of left ventricular fraction shortening. A down arrow indicates a decrease, and an up arrow indicates an increase.
In family 1, five male members related through a maternal lineage over two generations were clinically symptomatic, strongly suggesting an X-linked pattern of inheritance. Moyamoya angiopathy was responsible for cerebral infarcts or hemorrhages in four of these affected individuals, and the age of individuals at onset of acute neurological symptoms ranged from 4 to 32 years. A progressive worsening of arterial narrowing associated with additional strokes was observed in two affected members, F1 II-1 and F1 II-2, (Figure 3). One member (F1 III-1) had no intracranial artery abnormality, but he was only 27 years at the time of the last examination. Short stature of postnatal onset was constant and associated with azoospermia at adulthood. All patients had increased levels of gonadotropic hormones as a result of a primary gonadal failure. GH deficiency was partial when observed and was never associated with any pituitary gland abnormality detectable by MRI. Another hallmark observed in all affected members was a stereotyped facial dysmorphism with hypertelorism, long philtrum, mild ptosis, and premature white hair locks. The hands were small and had short and broad fingers. Involvement of the heart varied from isolated and asymptomatic left ventricular enlargement to severe nonspecific dilated cardiomyopathy responsible for heart failure and recurrent neurologic deficits due to low cardiac output. Other manifestations of the disease in this family included a right aortic arch in one affected individual and a nonspecific bilateral early-onset cataract in four affected individuals. High-resolution karyotyping performed for four of the patients was normal.
Figure 3.
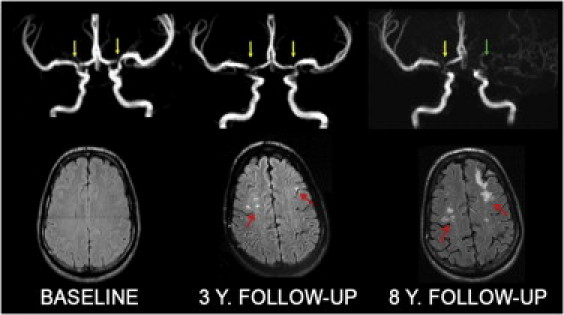
Progression of the Moyamoya Angiopathy in Case F1 II-2 over 8 Years
At baseline, MRA shows a bilateral stenosis of the terminal portion of the ICAs and the origin of ACAs and MCAs (yellow arrows). Three years later, a progression of the arterial stenoses is visible on MRA and punctuate ischemic lesions are detected on MRI (red arrows). A 8 years follow-up examination reveals a progression of the angiopathy with left ICA occlusion (green arrow) and larger ischemic lesions.
In family 2, two male siblings born from nonconsanguineous healthy parents with unremarkable family, prenatal, and perinatal histories were affected (Figure 1, Table 1). At the age of 12 years, they were both diagnosed with short stature and delayed puberty. Biologically, the results suggested a partial deficiency of growth hormone and a hypergonadotropic hypogonadism with preferential dysfunction of Sertoli cells (Table 1). Neither pituitary gland nor brain abnormalities were observed on a cerebral MRI scan. Case F2 II-1 received a substitutive therapy by rhGH from age 13. The growth rate remained low despite a high dose of rhGH (55 μg/kg/day). At age 17, the delayed puberty resolved progressively during treatment. He never had neurological symptoms, but cerebral MRI detected a moyamoya angiopathy with severe stenosis of the terminal portion of ICAs. Case F2 II-2 suffered from dyspraxia during childhood. At age 14, he had a diagnostic work-up prior to rhGH treatment. Imaging of the brain revealed a moyamoya angiopathy with occlusion of both distal ICA and an extensive neovascularization in the territory of lenticulostriate arteries. Arterial hypertension was suspected and confirmed by blood-pressure monitoring. Ultrasound examination excluded stenosis of the renal arteries. In both siblings, hypertelorism, mild bilateral ptosis, long philtrum, and flared nares were observed. Neither cataract nor dilated cardiomyopathy were present at the last examination (when the siblings were 17 and 14 years old).
In family 3, there were three affected males, including two maternal cousins for whom clinical information was available for the study (Figure 1). At the ages of 18 and 21, both cousins were diagnosed with short stature (160.0 cm). Neither received hormone therapy. A maternal family history of dwarfism was present. Both mothers measured 147.3 cm. Symptoms of delayed puberty or hypergonadotropic hypoganadism were not reported. Case F3 III-7 reported speech and behavioral developmental delays of 15–16 months. He was diagnosed with infantile febrile seizures at the age of 1 and epilepsy at the age of 10. He is hypertensive and has hypercholesterolemia, which is present among his immediate family members. He presented with right-hemisphere transient ischaemic attacks (TIAs), and an MR head scan demonstrated bilateral frontal watershed infarcts. Digital subtraction angiography showed bilateral supraclinoid internal carotid artery (ICA) occlusion and extensive moyamoya collateral vessels bilaterally (Figure S1). Case F3 III-2 had developmental delays in walking, speaking, and social behavior at an early age, was diagnosed with an enlarged heart at the age of 8, and suffered a myocardial infarction at the age of 21 as measured by elevated troponin levels. Subsequent cardiac catheterization showed significant coronary artery disease. He was also diagnosed with hypertension. He suffered left-hemisphere TIAs, and an MR head scan showed bilateral watershed infarcts. Digital subtraction angiography showed a right cervical ICA occlusion; reconstitution of the cavernous ICA from multiple collateral vessels, including moyamoya vessels; and in the left cervical ICA, multiple areas of irregularity and narrowing. These findings suggested prior occlusion with partial recanalization and occlusion of both vertebral artery origins with reconstitution from collateral vessels (Figure S2). He also had dysmorphic facial features, including a narrow cranial vault, thick eyebrows, wide nose, deeply set eyes, and low-set ears, as well as significant graying (over 75%) of the hair at the age of 26.
Cosegregation of an Xq28 Deletion in Affected Members of Family F1
The segregation of the disease in family F1 strongly suggested an X-linked recessive pattern of inheritance. To confirm this hypothesis, we conducted a genetic-linkage analysis in this family by using a panel of microsatellite markers spanning the X chromosome. A maximum multipoint point LOD score of 2.1 (maximum theoretical LOD score in family F1) was obtained at four closely linked markers, DXS8106, DXS8045, DXS1193, and DXS8061, mapping on Xq27–q28. Haplotype analysis mapped the gene as being distal to DXS984 (centromeric boundary) as the result of a recombination event in the affected individual III-1 (Figure S3). None of the telomeric markers, down to the telomere, detected any recombinant event. The physical size of the linked region has been estimated to be close to 15.5 Mb.
Because the pleiotropism of the clinical manifestations observed in family F1 was possibly consistent with a contiguous gene-deletion syndrome, we used an Affymetrix SNP 6.0 array to screen for putative loss of genetic material within the linked region on chromosome X. We analyzed three members, including the mother (F1 I-2) and two of her affected sons (F1 II-3 and F1 II-4).
Ten consecutive copy-number probes detected putative null alleles in the two affected sons, and hemizygosity for these markers was observed in the mother (Table S3). These consecutive probes spanned a 46.3 Mb interval bracketed by CN_923665 and CN_923676 copy-number probes (Table S3 and Figure 4C). Amplification of four STS markers, including ECD13623, ECD07678, ECD07752, and ECD15347, within this interval failed in all affected males of family F1, confirming the existence of a deletion within the linked interval. Flanking STS markers ECD21451 (cen) and ECD20005 (tel) were successfully amplified. We then used walking PCR with additional STS markers in combination with long-range PCR and sequencing to identify the centromeric and telomeric breakpoints. In brief, for all affected individuals we amplified a 3416 bp fragment that was absent in control individuals and present in the heterozygous state in the obligate female carriers. The sequencing of this PCR product revealed that the deletion size was 41007 bp long. Interestingly, a 32 bp insertion was detected at the breakpoint junction. This insert corresponded to a small part of the first intron of MTCP1/MTCP1NB.
Figure 4.
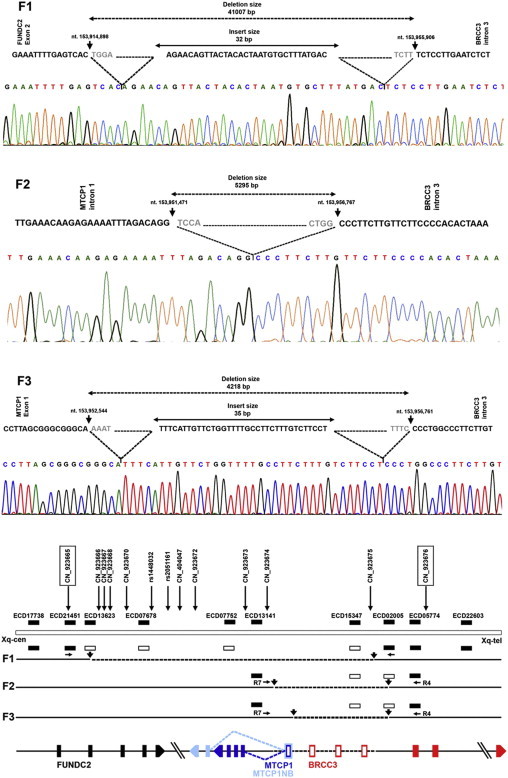
Xq28 Deletions in Families F1–F3
(F1) The deletion in family F1 begins after the 153,914,898th nucleotide of chromosome X (build 36.3) within the second exon of FUNDC2. It removes the remaining part of FUNDC2, MTCP1/MTCP1NB, and three exons of BRCC3. A fragment of 32 bp is inserted at the breakpoint junction.
(F2) The deletion identified in family F2 begins after the 153,951,471th nucleotide within intron 1 of MTCP1/MTCP1NB. It removes exon 1 of MTCP1 and three exons and part of intron 3 of BRCC3.
(F2) The deletion identified in family F3 begins after the 153,952,544th nucleotide, within exon 1 of MTCP1/MTCP1NB. It removes part of exon 1 of MTCP1 and three exons and part of intron 3 of BRCC3.
(Lower panel) Schematic view of the region involved in the deletion. Approximate positions of Affymetrix SNP 6.0 array probes and STS markers used for deletion mapping are shown. The copy-number probes flanking the deletion in family F1 are boxed. The STS markers showing the null alleles are represented as empty bars; the filled bars indicate that the sequence is not deleted. The dashed black lines show the deleted region in families F1–F3. Black arrows indicate the breakpoints. The genes interrupted by the deletion are represented as vertical, differently colored bars, and arrows indicate their transcriptional direction. The 3362 bp critical interval is represended by a dashed line, and the exons lying within this region are denoted by empty bars.
This deletion interrupted two genes, FUNDC2 and BRCC3, and deleted entirely MTCP1/MTCP1NB. The breakpoint within FUNDC2 occurred within exon 2, and the breakpoint in BRCC3 occurred within intron 3.
Altogether, these data established the existence of a40 kb Xq28 deletion that cosegegated with the affected phenotype in family F1.
Detection of Additional Xq28 Deletions in Families F2 and F3
Family F2 includes two affected brothers (F2 II-1 and F2 II-2) whose phenotypes were strikingly similar to the one observed in family F1. The blood of both brothers and their two parents was sampled. We failed to amplify the first exon of MTCP1/MTCP1NB or exons 1–3 of BRCC3 in proband F2 II-2 and his brother F2 II-1, strongly suggesting the existence of a genomic deletion. Walking PCR, long-range PCR, and sequencing identified a 5295 bp deletion in these two affected brothers. Their mother was heterozygous for this deletion. The deleted fragment removed 691 bp of intron 1 and exon 1 of MTCP1/MTCP1NB, exons 1–3 of BRCC3, and 1866 bp of intron 3 of BRCC3.
Family F3 includes three affected males. The blood of two of them (F3 III-2, F3 III-7) as well as that of the mother of F3 III-7 (F3 II-4) has been sampled. A 4218 bp deletion removing 197 bp of exon 1 of MTCP1/MTCP1NB, exons 1–3 of BRCC3, and 1860 bp of intron 3 of BRCC3 has been identified in both cousins and the carrier mother F3 II-4.
The Critical Region of Overlap between F1, F2, and F3 Deletions Includes MTCP1/MTCP1NB Exon 1 and the First Three Exons of BRCC3
The three deletions observed in families F1, F2, and F3 overlapped over an interval of 3362 bp, which is thus defined as a new critical interval that includes MTCP1/MTCP1NB exon 1 and the first three exons of BRCC3. FUNDC2 is located outside of this region, strongly suggesting that it is not involved in the disease.
MTCP1 was until recently annotated as a single locus expressing alternatively spliced transcripts; it is now annotated as two genes, MTCP1 and MTCP1NB, sharing exon 1 and encoding two distinct proteins, p13MTCP1 and p8MTCP1NB. The protein p13MTCP1 is not detected in normal tissues but is restricted to a rare form of mature-T-cell-proliferation leukemia. P8MTCP1NB encodes a mitochondrial 8 kDa protein of unknown function. BRCC3 encodes a 36 kDa ubiquitous deubiquitinating enzyme.
MTCP1 and BRCC3 Screening in Patients with Nonsyndromic Familial Moyamoya Disease
The screening of the coding regions and intron-exon boundaries of BRCC3, MTCP1, and MTCP1NB in the probands of the three pure MMD-unrelated F4–F6 families did not detect any deleterious mutation.
p8MTCP1NB and BRCC3 Analysis in Patient Lymphoblastoid Cell Lines and in Human Vascular Tissues
Immunoblot analysis of EBV lymphoblastoid cell lines from affected individuals F1 II-2 and F2 II-2 did not detect BRCC3 or p8MTCP1NB, whereas these proteins were readily detected in healthy control EBV cell lysates (Figure S5).
p8MTCP1NB and BRCC3 were also readily detectable in HUVEC endothelial cells as well as in arterial and venous lysates from normal human individuals (Figure S4).
Morpholino-Mediated Knockdown of brcc3 Leads to Defective Angiogenesis in Zebrafish
We decided to further examine the function of the genes inactivated by the deletions in families F1–F3 by using the zebrafish. The region syntenic to the portion of the genome removed by the overlapping deletion of these families is found on zebrafish chromosome 21. The human and zebrafish syntenic regions share similar organization; there is a small intergenic GC island located between the mtcp1nb and brcc3 loci, which are transcribed in opposite directions (Figure 5A). A mtcp1 homolog is not found in zebrafish. Mtcp1 homologs are present in both mammals and sauropsids but are not detected in amphibians, suggesting that Mtcp1 arose in the last common tetrapod ancestor shared by mammals and sauropsids (Figure S6). In situ hybridization of 24 hr postfertilization (hpf) zebrafish embryos reveals low-level ubiquitous expression of brcc3 ortholog with somewhat higher levels of expression in the head and in the ventral trunk adjacent to developing vessels (Figure 5B). Expression of mtcp1nb is not detectable by in situ hybridization in 24 hpf embryos (data not shown).
Figure 5.
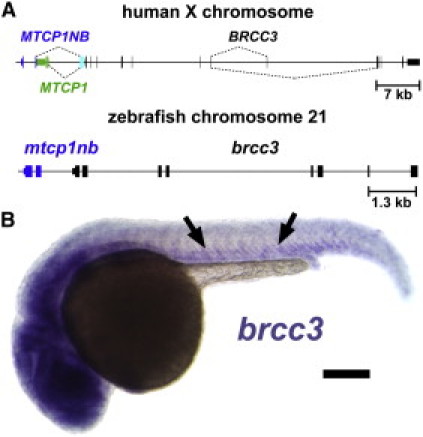
Zebrafish Orthologs of Moyamoya Candidate Genes
(A) Schematic drawing depicting the moyamoya deletion critical region in the human genome and comparing it to the syntenic region of the zebrafish genome. An untranslated exon (cyan) is alternatively spliced (dashed lines) into two transcripts encoding the unrelated MTCP1 (green) and MTCP1NB (blue) proteins. The mtcp1nb (blue) and brcc3 (black) loci are conserved in zebrafish, but mtcp1 is absent.
(B)The whole-mount in situ hybridization of a 24 hpf zebrafish embryo probed for brcc3, showing lower level ubiquitous expression with somewhat higher expression in the head and around the trunk axial vessels (arrows). The scale bar represents 200 μm.
The role of brcc3 and mtcp1nb in vascular development was analyzed by morpholino-mediated knockdown in Tg(fli1-eGFP)y1 transgenic zebrafish embryos, which permit easy visualization of blood vessels by fluorescence microscopy because of eGFP expression within endothelial cells of the transparent animal15 (Figure 6). Injection of morpholinos targeting brcc3 inhibits angiogenesis of trunk intersegmental vessels (Figure 6C). This defect is ameliorated by coinjection of wild-type brcc3 mRNA (Figures 6D and 6E), suggesting that defective angiogenesis in brcc3 morpholino-injected animals is due to a specific functional requirement for this gene. As noted above, brcc3 shows a low level of ubiquitous expression, but increased expression is noted in the vicinity of the developing axial blood vessels from which the intersegmental vessels sprout and grow. To determine whether the defects in brcc3 morpholino-injected animals reflect a cell-autonomous requirement for brcc3 function within endothelial cells, we generated flk1-mCherry-2A-brcc3 and flk1-mCherry constructs for endothelium-specific expression, driven by the flk1 promoter, of either brcc3 plus mCherry or mCherry alone. Injection of these constructs into zebrafish embryos results in mosaic endothelial expression of their respective transgenes (Figures 6F–6I). Expression of brcc3 in ISVs of injected Tg(fli1-eGFP)y1 animals is sufficient to rescue angiogenic sprouting and growth (arrows); in comparison, in transgenic flk1-mCherry-expressing endothelial cells (arrowheads), no rescue was observed (Figures 6F–6J). Unlike brcc3 morpholino-injected animals, mtcp1nb morpholino-injected animals showed no vascular phenotype at either 24 hpf or 48 hpf (data not shown). These results indicate that brcc3 ortholog function is required cell-autonomously within the endothelium for angiogenesis.
Figure 6.
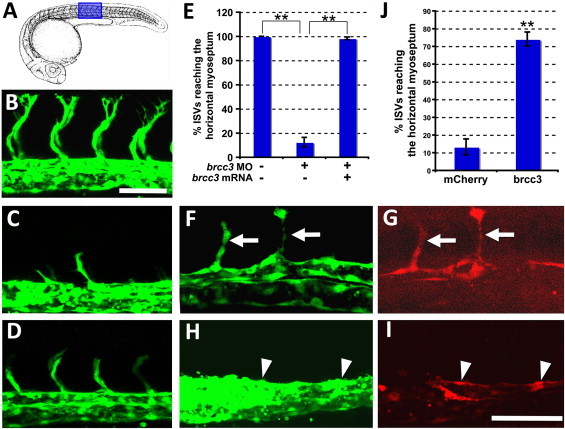
Loss of brcc3 Function in the Endothelium Results in Defective Angiogenesis
(A) Drawing of 24 hpf zebrafish embryo (adapted from Kimmel et al.37). The region shown in subsequent panels is highlighted in blue.
(B–D) Confocal imaging of intersegmental vessels (ISVs) in 24 hpf Tg(flk1a-eGFP) transgenic zebrafish embryos injected with either control morpholino (B), brcc3 morpholino (C), or brcc3 morpholino plus brcc3 mRNA (D).
(E) Quantitation of the percentage of ISVs that sprout and grow at least half-way up the trunk (to the horizontal myoseptum) in morpholino- and mRNA-injected animals.
(F–I) Confocal imaging of ISVs in 24 hpf Tg(flk1a-eGFP) transgenic zebrafish embryos injected with either flk1-mCherry-2A-brcc3 (F and G) or flk1a-mCherry (H and I) transgene DNA, showing EGFP-positive vessels (F and H) and mCherry-positive transgene-expressing cells (G and I).
(J) Quantitation of the percentage of ISVs that sprout and grow at least half-way up the trunk (to the horizontal myoseptum) in flk1a-mCherry-2A-brcc3 or flk1a-mCherry transgene-injected animals. For the quantitative data in panels (E) and (J), ten ISVs from each of 12 different injected embryos were scored for each control and experimental condition. Error bars show the standard error of the mean for these data. Scale bars represent 25 μm (B–D) and 25 μm (F–I).
Discussion
We report herein on three unrelated families affected by a disorder characterized by the association of a moyamoya angiopathy, short stature, and a stereotyped facial dysmorphism. In addition, other symptoms included a hypergonadotropic hypogonadism (7/9 patients), hypertension (3/9 patients), a dilated cardiomyopathy (3/9 patients), premature coronary heart disease (1/9 patients), premature hair graying (6/9 patients), and early bilateral acquired cataract (4/9 patients). In these three families, we identified three overlapping Xq28 deletions cosegregating with the affected phenotype. The critical interval includes exon 1 of MTCP1/MTCP1NB and exons 1–3 of BRCC3. This deletion leads to a complete loss of BRCC3 and MTCP1NB (the sole MTCP protein expressed in normal tissues) in affected patients. In order to investigate the in vivo consequences of the loss of each of these two proteins, we conducted morpholino knockdown experiments for both genes in zebrafish. Knockdown of mtcp1nb did not lead to any vascular phenotype at either 24 hpf or 48 hpf, whereas injection of morpholinos targeting brcc3 lead to defective angiogenesis, which was rescued by endothelial expression of brcc3.
The defective angiogenesis observed in brcc3 morphant zebrafish and the endothelial-specific brcc3 rescue of these defects strongly suggest that this protein plays an important role in vascular physiology and may therefore be involved in the moyamoya angiopathy of these patients. BRCC3 is a ubiquitously expressed K63-specific deubiquinating (DUB) enzyme containing a JAMM (JAB1/MPN/Mov34 metalloenzyme) domain.21–24 It is a member of two protein complexes, the nuclear BRCA1/Rap80/Abraxas/Merit40/BRCC45 DNA repair complex and the cytoplasmic BRISC complex.25 After cell irradiation, BRCC3 is readily detected at DNA double-strand breaks, where it interacts with abraxas and is thought to be a positive regulator of BRCA1 E3 ligase activity. It is implicated in G2 checkpoint and viability response through its DUB activity. How an alteration of DNA repair could possibly lead to moyamoya angiopathy would be entirely speculative at this point. However, several lines of evidence suggest a possible link between genomic maintenance and moyamoya. Irradiation of the neck is one of the most frequent environmental causes of moyamoya, and the clinical, radiological, and histological features of irradiation-induced cerebral vasculopathy and those of MMD are strikingly similar.5 In addition, moyamoya angiopathy is a frequent complication of MOPDII (MIM 210720, Majewski Osteodysplastic Primordial dwarfism) and Seckel syndrome (MIM 210600, 606744, 613676, 608664) which are recessive disorders caused by mutations of pericentrin, a protein involved in DNA-damage signaling.26–28 Other clinical features, such as early acquired cataract and premature hair graying, encountered in these patients are also present in several disorders caused by mutations in DNA repair genes.
BRCC3 is also a member of the BRISC complex (BRCC36 Isopeptidase Complex), a cytoplasmic complex containing BRCC3, BRCC45, MERIT40, and ABRO1, a complex whose function was until recently totally unknown.25,29,30 Recent data suggest that ABRO1 and the BRISC complex might have a cardioprotective function.31 Indeed, ABRO1 is strongly expressed in cardiomyocytes and is upregulated in myocardial infarction. Its induction is associated with a K63-linked deubiquitination of cardiomyocyte proteins associated with a cardio-protective effect. An exciting hypothesis is that loss of the integrity of the BRISC complex could lead to the cardiomyopathy and premature coronary heart disease detected in several of our patients.
In addition to moyamoya angiopathy, all patients have a short stature (and most have partial GH deficiency). Seven out of nine patients showed a hypergonadotropic hypogonadism, suggesting a preferential dysfunction of Sertoli cells. BRCC45 (also called BRE) is mostly expressed in the testis; there is a strong mRNA signal in germ cells and Sertoli cells.32 Because BRCC45 is a member of both BRCC3 complexes, either of these two complexes could also be involved in this endocrine phenotype. Interestingly, hypergonadotropic hypogonadism and infertility are important clinical features encountered in several chromosome-breakage and -instability syndromes, including Fanconi anemia.33
Because both MTCP1/MTCP1NB and BRCC3 are interrupted in all three families, a role for MTCP1/MTCP1NB loss in the disease phenotype cannot be ruled out at this point. MTCP1, also called c6.1B, was initially identified because of its involvement in the t(X;14)(q28;q11) translocation, which is associated with a rare subset of T cell prolymphocytic leukemia, involving proliferation of mature T cells.34–36 It leads to two types of transcripts, MTCP1 and MTCP1NB, with two distinct open reading frames. The longest transcript encodes p13MTCP1, a protein that is not found in any normal tissue but only in T cell prolymphocytic leukemic cells, in which the t(X;14) translocation event activates a cryptic promoter sequence within MTCP1 intron 1. p8MTCP1NB encodes a mitochondrial 8 kDa protein whose function is unknown and which is detected in various normal tissues, including arteries and veins, and is overexpressed in mature T cell leukemia cells. This unusually small 68 amino acid protein is expressed in the mitochondria. However, at present we have no idea as to the putative pathophysiological links between its loss and the phenotype, or part of the phenotype, reported herein.
In summary, we report herein on a novel X-linked syndromic moyamoya disorder whose investigation leads to the identification of a deletion removing two genes with a previously unsuspected role in vessel physiology. Several lines of evidence strongly suggest that one of these two genes, BRCC3, might play an important role in angiogenesis and vessel maintenance.
Acknowledgments
We thank affected individuals and their families for their participation in this reasearch program. We also thank the French Tanguy moyamoya association of patients. We are also indebted to N. Allili, M.G. Bousser, S. Chabrier, A. Gaudric, E. Houdart, V. Krivosic, D. Logeart, J.P. Metzger, P. Touraine, and A. Verloes for excellent clinical evaluation of some of the patients, to M.H. Stern for helpful discussions and the MTCP1NB polyclonal antibody, to Marc Nomaksteinsky for microsatellite analysis, and to Audrey Delaforge for excellent technical help in preparing the samples.
Research was supported by the INSERM and the Leducq Foundation (grant to E.T.L. and B.M.W.), Foundation Leducq Transatlantic Network of Excellence (07 CVD 02 Hemorrhagic Stroke), the intramural program of the Eunice Kenndy Shriver National Institute of Child Health and Human Development (NICHD; to B.M.W.), and the Huber Family Moyamoya Fund (G.K.S.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Database of Genomic Variants, http://project.tcag.ca/variation/
International HapMap Project, http://hapmap.ncbi.nlm.nih.gov/
NCBI BLAST, http://blast.ncbi.nlm.nih.gov/Blast.cgi/
NCBI Entrez Gene, http://www.ncbi.nlm.nih.gov/gene/ http://www.ncbi.nlm.nih.gov/gene
NCBI Single Nucleotide Polymorphism Database, http://www.ncbi.nlm.nih.gov/snp/
NCBI UniSTS, http://www.ncbi.nlm.nih.gov/unists/
Online Mendelian Inheritance in Man, http://www.omim.org/
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Scott R.M., Smith E.R. Moyamoya disease and moyamoya syndrome. N. Engl. J. Med. 2009;360:1226–1237. doi: 10.1056/NEJMra0804622. [DOI] [PubMed] [Google Scholar]
- 2.Kuroda S., Houkin K. Moyamoyadisease: Current concepts and future prospectives. Lancet Neurol. 2008;7:1056–1066. doi: 10.1016/S1474-4422(08)70240-0. [DOI] [PubMed] [Google Scholar]
- 3.Yamashita M., Oka K., Tanaka K. Histopathology of the brain vascular network in moyamoya disease. Stroke. 1983;14:50–58. doi: 10.1161/01.str.14.1.50. [DOI] [PubMed] [Google Scholar]
- 4.Achrol A.S., Guzman R., Lee M., Steinberg G.K. Pathophysiology and genetic factors in moyamoya disease. Neurosurg. Focus. 2009;26:E4. doi: 10.3171/2009.1.FOCUS08302. [DOI] [PubMed] [Google Scholar]
- 5.Desai S.S., Paulino A.C., Mai W.Y., Teh B.S. Radiation-induced moyamoya syndrome. Int. J. Radiat. Oncol. Biol. Phys. 2006;65:1222–1227. doi: 10.1016/j.ijrobp.2006.01.038. [DOI] [PubMed] [Google Scholar]
- 6.Hervé D., Touraine P., Verloes A., Miskinyte S., Krivosic V., Logeart D., Alili N., Laredo J.D., Gaudric A., Houdart E. A hereditary moyamoya syndrome with multisystemic manifestations. Neurology. 2010;75:259–264. doi: 10.1212/WNL.0b013e3181e8ee3f. [DOI] [PubMed] [Google Scholar]
- 7.Guo D.C., Papke C.L., Tran-Fadulu V., Regalado E.S., Avidan N., Johnson R.J., Kim D.H., Pannu H., Willing M.C., Sparks E. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am. J. Hum. Genet. 2009;84:617–627. doi: 10.1016/j.ajhg.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roder C., Nayak N.R., Khan N., Tatagiba M., Inoue I., Krischek B. Genetics of Moyamoya disease. J. Hum. Genet. 2010;55:711–716. doi: 10.1038/jhg.2010.103. [DOI] [PubMed] [Google Scholar]
- 9.Ikeda H., Sasaki T., Yoshimoto T., Fukui M., Arinami T. Mapping of a familial moyamoya disease gene to chromosome 3p24.2-p26. Am. J. Hum. Genet. 1999;64:533–537. doi: 10.1086/302243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Inoue T.K., Ikezaki K., Sasazuki T., Matsushima T., Fukui M. Linkage analysis of moyamoya disease on chromosome 6. J. Child Neurol. 2000;15:179–182. doi: 10.1177/088307380001500307. [DOI] [PubMed] [Google Scholar]
- 11.Sakurai K., Horiuchi Y., Ikeda H., Ikezaki K., Yoshimoto T., Fukui M., Arinami T. A novel susceptibility locus for moyamoya disease on chromosome 8q23. J. Hum. Genet. 2004;49:278–281. doi: 10.1007/s10038-004-0143-6. [DOI] [PubMed] [Google Scholar]
- 12.Mineharu Y., Liu W., Inoue K., Matsuura N., Inoue S., Takenaka K., Ikeda H., Houkin K., Takagi Y., Kikuta K. Autosomal dominant moyamoya disease maps to chromosome 17q25.3. Neurology. 2008;70:2357–2363. doi: 10.1212/01.wnl.0000291012.49986.f9. [DOI] [PubMed] [Google Scholar]
- 13.Kamada F., Aoki Y., Narisawa A., Abe Y., Komatsuzaki S., Kikuchi A., Kanno J., Niihori T., Ono M., Ishii N. A genome-wide association study identifies RNF213 as the first Moyamoya disease gene. J. Hum. Genet. 2011;56:34–40. doi: 10.1038/jhg.2010.132. [DOI] [PubMed] [Google Scholar]
- 14.Roder C., Peters V., Kasuya H., Nishizawa T., Wakita S., Berg D., Schulte C., Khan N., Tatagiba M., Krischek B. Analysis of ACTA2 in European moyamoya disease patients. Eur. J. Paediatr. Neurol. 2011;15:117–122. doi: 10.1016/j.ejpn.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 15.Lawson N.D., Weinstein B.M. In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev. Biol. 2002;248:307–318. doi: 10.1006/dbio.2002.0711. [DOI] [PubMed] [Google Scholar]
- 16.Westerfield M. University of Oregon Press; Eugene, OR: 2000. The zebrafish book. A guide for the laboratory use of zebrafish (Danio rerio) [Google Scholar]
- 17.Hauptmann G., Gerster T. Two-color whole-mount in situ hybridization to vertebrate and Drosophila embryos. Trends Genet. 1994;10:266. doi: 10.1016/0168-9525(90)90008-t. [DOI] [PubMed] [Google Scholar]
- 18.Villefranc J.A., Amigo J., Lawson N.D. Gateway compatible vectors for analysis of gene function in the zebrafish. Dev. Dyn. 2007;236:3077–3087. doi: 10.1002/dvdy.21354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi J., Dong L., Ahn J., Dao D., Hammerschmidt M., Chen J.N. FoxH1 negatively modulates flk1 gene expression and vascular formation in zebrafish. Dev. Biol. 2007;304:735–744. doi: 10.1016/j.ydbio.2007.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grabher C., Joly J.S., Wittbrodt J. Highly efficient zebrafish transgenesis mediated by the meganuclease I-SceI. Methods Cell Biol. 2004;77:381–401. doi: 10.1016/s0091-679x(04)77021-1. [DOI] [PubMed] [Google Scholar]
- 21.Dong Y., Hakimi M.A., Chen X., Kumaraswamy E., Cooch N.S., Godwin A.K., Shiekhattar R. Regulation of BRCC, a holoenzyme complex containing BRCA1 and BRCA2, by a signalosome-like subunit and its role in DNA repair. Mol. Cell. 2003;12:1087–1099. doi: 10.1016/s1097-2765(03)00424-6. [DOI] [PubMed] [Google Scholar]
- 22.Cooper E.M., Cutcliffe C., Kristiansen T.Z., Pandey A., Pickart C.M., Cohen R.E. K63-specific deubiquitination by two JAMM/MPN+ complexes: BRISC-associated Brcc36 and proteasomal Poh1. EMBO J. 2009;28:621–631. doi: 10.1038/emboj.2009.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang B., Elledge S.J. Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc. Natl. Acad. Sci. USA. 2007;104:20759–20763. doi: 10.1073/pnas.0710061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shao G., Patterson-Fortin J., Messick T.E., Feng D., Shanbhag N., Wang Y., Greenberg R.A. MERIT40 controls BRCA1-Rap80 complex integrity and recruitment to DNA double-strand breaks. Genes Dev. 2009;23:740–754. doi: 10.1101/gad.1739609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng L., Wang J., Chen J. The Lys63-specific deubiquitinating enzyme BRCC36 is regulated by two scaffold proteins localizing in different subcellular compartments. J. Biol. Chem. 2010;285:30982–30988. doi: 10.1074/jbc.M110.135392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Griffith E., Walker S., Martin C.A., Vagnarelli P., Stiff T., Vernay B., Al Sanna N., Saggar A., Hamel B., Earnshaw W.C. Mutations in pericentrin cause Seckel syndrome with defective ATR-dependent DNA damage signaling. Nat. Genet. 2008;40:232–236. doi: 10.1038/ng.2007.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brancati F., Castori M., Mingarelli R., Dallapiccola B. Majewski osteodysplastic primordial dwarfism type II (MOPD II) complicated by stroke: Clinical report and review of cerebral vascular anomalies. Am. J. Med. Genet. A. 2005;139:212–215. doi: 10.1002/ajmg.a.31009. [DOI] [PubMed] [Google Scholar]
- 28.Bober M.B., Khan N., Kaplan J., Lewis K., Feinstein J.A., Scott C.I., Jr., Steinberg G.K. Majewski osteodysplastic primordial dwarfism type II (MOPD II): expanding the vascular phenotype. Am. J. Med. Genet. A. 2010;152A:960–965. doi: 10.1002/ajmg.a.33252. [DOI] [PubMed] [Google Scholar]
- 29.Sowa M.E., Bennett E.J., Gygi S.P., Harper J.W. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu X., Kim J.A., Castillo A., Huang M., Liu J., Wang B. NBA1/MERIT40 and BRE interaction is required for the integrity of two distinct deubiquitinating enzyme BRCC36 containing complexes. J. Biol. Chem. 2011;286:11734–11745. doi: 10.1074/jbc.M110.200857. Published online January 31, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cilenti L., Balakrishnan M.P., Wang X.L., Ambivero C., Sterlicchi M., del Monte F., Ma X.L., Zervos A.S. Regulation of Abro1/KIAA0157 during myocardial infarction and cell death reveals a novel cardioprotective mechanism for Lys63-specific deubiquitination. J. Mol. Cell. Cardiol. 2011;50:652–661. doi: 10.1016/j.yjmcc.2010.12.015. Published online December 30, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Poon H.K., Chan J.Y.H., Lee K.H., Chow P.H. Tissue specific expression and sequence analysis of a stress responsive gene Bre in adult golden hamster (Mesocricetus auratus) Cell Tissue Res. 2004;316:305–313. doi: 10.1007/s00441-004-0871-0. [DOI] [PubMed] [Google Scholar]
- 33.Giri N., Batista D.L., Alter B.P., Stratakis C.A. Endocrine abnormalities in patients with Fanconi anemia. J. Clin. Endocrinol. Metab. 2007;92:2624–2631. doi: 10.1210/jc.2007-0135. [DOI] [PubMed] [Google Scholar]
- 34.Stern M.H., Soulier J., Rosenzwajg M., Nakahara K., Canki-Klain N., Aurias A., Sigaux F., Kirsch I.R. MTCP-1: A novel gene on the human chromosome Xq28 translocated to the T cell receptor alpha/delta locus in mature T cell proliferations. Oncogene. 1993;8:2475–2483. [PubMed] [Google Scholar]
- 35.Madani A., Choukroun V., Soulier J., Cacheux V., Claisse J.F., Valensi F., Daliphard S., Cazin B., Levy V., Leblond V. Expression of p13MTCP1 is restricted to mature T-cell proliferations with t(X;14) translocations. Blood. 1996;87:1923–1927. [PubMed] [Google Scholar]
- 36.Soulier J., Madani A., Cacheux V., Rosenzwajg M., Sigaux F., Stern M.H. The MTCP-1/c6.1B gene encodes for a cytoplasmic 8 kD protein overexpressed in T cell leukemia bearing a t(X;14) translocation. Oncogene. 1994;9:3565–3570. [PubMed] [Google Scholar]
- 37.Kimmel C.B., Ballard W.W., Kimmel S.R., Ullmann B., Schilling T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.