

Abstract
Epidemiological, animal, and cell studies have demonstrated that nickel compounds are human carcinogens. The mechanisms of their carcinogenic actions remain to be investigated. p63, a close homologue of the p53 tumor suppressor protein, has been linked to cell fate determination and/or maintenance of self renewing populations in several epithelial tissues, including skin, mammary gland, and prostate. ΔNp63, a dominant negative isoform of p63, is amplified in a variety of epithelial tumors including squamous cell carcinomas and carcinomas of the prostate and mammary gland. The present study shows that nickel suppressed ΔNp63 expression in a short-time treatment (up to 48 hours). Nickel treatment caused activation of NF-κB. Blockage of NF-κB partially reversed nickel-induced ΔNp63 suppression. Nickel decreased interferon regulatory factor (IRF) 3 and IRF7, IKKε, and Sp100. Over-expression of IRF3 increased ΔNp63 expression suppressed by nickel. Nickel was able to activate p21, and its activation was offset by over-expression of ΔNp63. In turn, elevated p63 expression counteracted the ability of nickel to restrict cell growth. The present study demonstrated that nickel decreased interferon regulatory proteins IRF3 and IRF7, and activated NF- κB, resulting in ΔNp63 suppression and then p21 up-regulation. ΔNp63 plays an important role in nickel induced cell proliferation.
Keywords: Nickel chloride, ΔNp63, NF-κB, Interferon regulatory factor (IRF), p21, Cell proliferation, Keratinocytes
Introduction
Human exposure to nickel occurs predominantly in mining, refining, alloy production, electroplating, and welding. Exposure of workers to nickel compounds can produce a variety of adverse effects on human health, such as nickel allergy in the form of contact dermatitis, lung fibrosis, cardiovascular and kidney diseases, and cancer of the respiratory tract (Uddin et al. 2007). Epidemiological studies demonstrate increased mortality from cancers of the lung and nasal cavities in nickel-containing dusts and fumes (Roberts et al. 1989). Nickel induced human carcinogenesis may be due to its multiple mechanisms, including both genetic and epigenetic routes (Salnikow and Costa 2000; Clevers 2004; Zhao et al. 2004).
For many years, p53 has been considered the prototypical tumor suppressor and remains the subject of intensive research. p53 responds to cellular stress such as DNA damage and hypoxia and plays important roles in regulating cell cycle, cell differentiation, genomic stability, and apoptosis (Levine 1997; Oren 1999). p63, a member of p53 family, shares extensive homology to p53 and produces multiple transcripts with varying functions (Osada et al. 1998; Schmale and Bamberger 1997; Trink et al. 1998; Yang et al. 1998). p63 family includes two major classes of proteins, TA63, which contains the N-terminal transactivation (TA) domain, and N-terminal truncated (ΔN) p63, which lacks the transactivation domain (Yang et al. 2002). Alternative splicing at the 3′ end of the transcripts generates three different C-termini: α, β and γ (Candi et al. 2007). p63 exhibits a rather tissue-specific distribution in that its expression is higher in the basal layer of stratified epithelia, including the epidermis (Yang et al. 1998). The most highly expressed p63 isoform in the epidermis is ΔNp63α (Yang et al. 1998). This isoform lacks 5′ region that exhibits extensive homology to the transactivation domain of p53. ΔNp63α inhibits p53 transcriptional activity. Deficiency of ΔNp63α resulted in severe limb, craniofacial, and epithelial defects, leading to death shortly after birth (Mills et al. 1999; Yang et al. 1999). Newborns lacking of functional p63 display abnormal epidermis and hair follicles with a thin single cell layer covering the body.
p63 is essential to normal epidermal development and is homologue of the p53 tumor suppressor. It has been linked to cell fate determination and/or maintenance of self-renewing populations in several epithelial tissues, including skin, mammary gland, and prostate (Yang et al. 2002). ΔNp63 is over-expressed in a variety of epithelial tumors including oral and skin squamous cell carcinomas (Westfall and Pietenpol 2004). While elevated p63 expression can promote cell proliferation (King et al. 2006; Koster et al. 2004), the underlying mechanisms is still unknown. In this study we investigated the role of p63 in nickel-reduced cell proliferation using epidermal cells.
Materials and methods
Cells and chemicals
Spontaneously immortalized human keratinocytes (HaCats) were cultured in monolayers at 37°C, 5% CO 2 using Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS), 2 mM L-glutamine, and 25 μg of gentamicin/mL. Human primary keratinocytes (HPKs) were purchased from Lonza (Walksville, MD). The cells were cultured in the growth medium containing BPE (Bovine Pitutary Extract), hEGF, Insulin, Hydrocortisone, and GA-1000 (Gentamicin, Amphotericin B) according to the manufacture’s manual. Nickel chloride (Ni2+) was purchased from Sigma (St. Louis, MO).
Viruses
Human cDNA for p63α was obtained by RT-PCR cloning in pCDNA3.1 (Invitrogen) according to the manufactory manual. Proper expression was confirmed by transient transfection in 293 cells and immunoblotting with p63 pan-antibodies (Santa Cruz). ΔNp63 cDNA insert was then sub-cloned into the BamHI site of the PINCO retroviral vector (Nocentini et al. 1997) and was used to make retrovirus in 293 GPG cells. Expression of the retrovirally-transduced p63 cDNA was verified by immunoblotting of infected human keratinocytes with anti-p63 antibodies.
ΔNp63 siRNA cloning
To knock down the specific genes of interest, artificial miRNA sequences were designed using BLOCK-iT RNAi Designer (Invitrogen). The oligonucleotides were synthesized, annealed, and inserted into the pcDNA6.2-GW/miR vector (BLOCK-iT Pol II miR RNAi Expression Vector, Invitrogen) according to the protocol provided by the manufacturer. The artificial miRNA sequences inserted into the BLOCK-iT Pol II miR RNAi Expression Vector are as follow: ΔNp63, 5′-TGCTGTTTGATGCGCTGTTGC TTATTGTTTTGGCCACTGACTGACAATAAGCAAGCGCATCAAA-3′. The expression of the vectors was confirmed by Western blot.
Plasmids
The human ΔNp63 promoter region (1.477 kb) was cloned by PCR using human genomic DNA as template. The following primers were used to amplify nucleotides -−477 to −4 from the initiation codon, plus an additional BglII or HindIII restriction site: BglII-5′-AGCCAATGACCACGTCATCC-3′, reverse HindIII 5′-AGCTGTAAGATTGATCAA TGC-3′. The PCR product was then cloned into the pGL3 basic vector (Promega) digested by BglII-HindIII. The construct was verified by sequencing. pCR-FLAG-IKKβ-KM (K44A) was from Dr. Hiroyasu Nakano (Juntendo University, Japan). NF-κB luciferase reporter was purchased from Clontech (Mountain View, CA). p21 luciferase reporter and pCMV4-IRF3 overexpression vector were from Addgene Inc (Cambridge, MA).
Promoter activity assays
Transient transfection promoter activity assays were performed as previously described (Rangarajan et al. 2001; Talora et al. 2002), using co-transfection with a TK-renilla reporter for internal normalization. Total quantities of plasmid DNA were kept constant by adding appropriate amounts of empty vectors without inserts. Transfected cells were harvested at 24 hrs after transfection and relative luciferase activities were normalized for Renilla luciferase activity. All conditions were tested in triplicate wells, and experiments were repeated a minimum of three times.
Analysis of gene expression
Total RNA preparations (1–2 μg) were used in a reverse transcriptase reaction with random primers, followed by real time PCR with gene-specific primers, using an Icycler IQ™ Real-Time detection System (Bio-Rad) according to the manufacturer’s recommendations, with SYBR Green (Bio-Rad) for detection. ΔNp63 primers, forward 5′-CGTGTCCTTCCAGCAGTC-3′, reverse 5′-GCAATTTGGCAGTAGAGTT-3′. IRF3 primers, forward 5′-GTGGCCTGGGTGAACAAGAG, reverse 5′-CTGGAAGATTCCG AAATCCTCC. IRF7 primers, forward 5′-GAGCCGTACCTGTCACCCT, reverse 5′-GGGCCGTATAGGAACGTGC. IKKε primers, forward 5′-ACAAGGCCCGCAACAAGAAA, reverse 5′-TTGACGATGTTCTGGTGGTTC. Sp100 primers, forward 5′-TGAGGTGTGCAA CAAATGGG-3′, reverse 5′-AAGATGCAACTCCACGGGTTC-3′. p21 primers, forward 5′-CCTGTCACTGTCTTGTACCCT, reverse 5′-GCGTTTGGAGTGGTAGAAATCT. GAPDH primers, forward 5′-ATCATCAGCAATGCCTCC, reverse 5′-AGTCCTTCCACGATACCAA. Each sample was tested in triplicate and results were normalized using amplification of the same cDNAs with human GAPDH primer.
Western blotting
After designed treatments, both HaCat and HKC cells were collected and washed with PBS twice. The cells were lysed in RIPA buffer (150 mM NaCl, 100 mM Tris at pH8.0, 1% Triton X-100, 1% deoxycholic acid, 0.1% SDS, 5 mM EDTA and 10 mM NaF, 2mM DTT, 0.5mM PMSF, 1 mM sodium vanadate, 1ug/ml leupeptin). 30 μg of protein was loaded into 4–12% NuPAGE Bis-Tris gel (Invitrogene). After blocking with 5% milk, the membrane was probed with various antibodies and developed with ECL (Pierce).
Cell growth analysis
HaCat cells were infected with recombinant retroviruses expressing either a full-length ΔNp63α cDNA together with GFP (Pinco-ΔNp63α) or GFP alone (Pinco). Two days after infection, GFP-positive cells were purified by cell sorting using flow cytometry and then replated to normal cell culture. After 3 days of further cultivation, cells were trypsinized, counted, and split into triplicate dishes. The cells were treated with 100 μM nickel or remained untreated as control. The fresh medium with 100 μM nickel was added every 3 days. After 3 weeks of further cultivation, clonogenic growth was evaluated by staining of dishes and counting of macroscopically visible colonies (containing >50 cells).
Statistical Analysis
Statistical analyses were performed using the one-way ANOVA. Results were expressed as average values ± SD. The difference was considered significant if p < 0.05.
Results
ΔNp63 expression is down-modulated by short term nickel treatment
The molecular mechanism involved in control of p63 expression in growing versus differentiating keratinocytes is not known. Real time PCR study showed that ΔNp63 mRNA expression was sharply down-modulated by treatment with nickel in both immortalized human keratinocytes and primary human keratinocytes (Figs 1A and 1B). Similar down-modulation was found by immunoblotting at the protein level (Fig. 1C). It displayed a dose- and time-dependant manner. Phosphorylations of both p53ser15 and p73 were activated after nickel treatment, while the total protein level of actin remained essentially the same (Fig.1D). The analysis of 10-kb nucleotide sequence of human ΔNp63 promoter found that ΔNp63 promoter contains multiple binding sites of NF-κB-binding and interferon-responsive factors (IRF) (Nguyen et al. 2006). In the present study, the 10-kb ΔNp63 promoter assay was conducted to measure transcriptional activity of ΔNp63. The results showed that nickel treatment caused a dose-dependant decrease in ΔNp63 activity (Fig.1E). While the major isoform expressed in keratinocytes after birth is ΔNp63α (Yang et al. 1998), the TAp63, another isoform of p63, was also expressed in these cells at very low levels that were not quantifiable by real time PCR (data not shown).
Fig. 1.

Negative control of p63 expression in keratinocytes by nickel treatment. (A) and (B), Down-modulation of p63 mRNA expression by nickel. Both HaCat and HPK cells were treated with different doses of nickel for 24 hr (A), and treated with 1.0 mM for different time (B). ΔNp63 mRNA levels were quantified by real time RT-PCR. Values are expressed as relative arbitrary units, after internal normalization for GAPDH mRNA expression. (C) Down-modulation of p63 protein expression by nickel. Both HaCat and HPK cells were treated with nickel for different times and doses. The cells then were analyzed for ΔNp63 protein level by immunoblotting with the corresponding antibodies. Immunoblotting for β-actin was used for equal loading control. (D) Up-regulation of both p53 and p73 by nickel treatment. HaCat cells were treated with nickel for 24 hr. The cells were harvested, p53ser15, phospho-p73 and β-actin expression were analyzed by immunoblotting. (E) Down-modulation of p63 activity in keratinocytes in response to nickel treatment. Both the HaCat and HPK cells were transfected with 0.5 μg of Np63 luciferase reporter DNA followed by nickel treatment. The cells were harvested for the measurement of luciferase activity after 24 hr. Values are expressed as relative units after internal normalization for protein concentration. *, p<0.05 compared to control without nickel treatment (one-way ANOVA test).
Nickel suppresses ΔNp63 expression through negative regulation of the interferon signaling pathway
To gain further insights into regulation of p63 expression, we analyzed a 10 kb nucleotide sequence of the human ΔNp63α promoters for common transcription factor-binding motifs. The presence of multiple NF-κB-binding sites, interferon-stimulated responsive elements (ISRE) (Levy et al. 1988) and binding sites for interferon-responsive factors (IRF) (Taniguchi et al. 2001) was found to be a characteristic of both promoters (Nguyen et al. 2006). Nickel exposure caused activation of NF-κB (Cruz et al. 2004). The activation of NF-κB by nickel resulted in significant modulation of cellular and tissue responses (Denkhaus and Salnikow 2002). Moreover, various nickel induced allergic effects and contact skin hypersensitivity in industrialized countries are able to be interpreted as the activation of NF-κB (Goebeler et al. 1995). However, its possible impact on the interferon signaling pathway in this cell type has not been reported. Among NF-κB suppressing genes in human cells were the ones for IRF7, a key regulator of the interferon-dependent transcription cascade with oncogenic potential (Zhang and Pagano 2002; Honda et al. 2005), and Sp100, an essential component of nuclear bodies (NBs) (Moller et al. 2003). IRF7 physically interacts and functionally overlaps with IRF3 (Servant et al. 2002). IKKε is a key kinase that positively regulates the interferon response (Fitzgerald et al. 2003). Fig. 2A showed that nickel treatment caused an increase of NF-κB activities. RT–PCR analysis showed that IRF3, IRF7, IKKε and Sp100 expression levels were significantly decreased in nickel treated HaCat cells (Figs. 2B–2E).
Fig. 2.


Role of NF-κB and interferon signaling pathways in nickel-induced down-regulation of ΔNp63 expression. (A) Induction of NF-κB transcriptional activity by nickel. HaCat cells were transfected with 0.5 μg of NF-κB-responsive reporter (pNF-κB-luc) with increasing amounts of nickel as indicated. Cells were collected after 24 hr treatment. The promoter activity was measured as described in Materials and Methods. (B), (C), (D), and (E) Down-modulation of endogenous interferon-responsive genes IRF3, IRF7, IKKε, and Sp100 by nickel treatment. HaCat cells were treated with different doses of nickel for 24 hr, followed by determination of mRNA expression levels of IRF3 (B), IRF7 (C), IKKε (D), and Sp100 (E) by real-time PCR analysis. (F) Induction of p63 expression by inhibition of NF-κB. HaCat cells were transfected with IKKβ-KM, either alone or in combination with 1 mM of nickel as indicated. ΔNp63 mRNA levels were determined by real-time PCR. (G) Partial restoration of IRF3 on suppression of p63 expression by nickel. HaCat cells were transfected with 20 μg of IRF3 expression vector or empty vector for 4 hr. Then 1 mM nickel was added as indicated. ΔNp63 mRNA levels were determined by real-time PCR. *, p<0.05 compared to control without nickel treatment (one-way ANOVA test). #, p<0.05 compared to nickel treatment (one-way ANOVA test).
To assess whether the observed changes in both NF-κB and interferon signaling pathways contribute to down-modulated p63 expression by nickel, HaCat cells were transfected with expressing vector IKKβ kinase mutant (IKKβ-KM), which functions as inhibitor of NF-κB, or the full-length IRF3 protein. IRF3 is a key downstream mediator of the interferon response (Honda and Taniguchi 2006; Zhang et al. 2004). Expression of IKKβ-KM resulted in a partial restoration of nickel-reduced p63 expression (Fig. 2F), indicating that the NF-κB pathway functions as a negative regulator of p63 in keratinocytes under nickel stimulation. Transfection with IRF3 caused a partial restoration of p63 expression reduced by nickel treatment (Fig. 2G).
Down-modulation of ΔNp63 expression by nickel promotes cell proliferation
It has been reported that down-modulation of ΔNp63 expression may be essential for its ability to decrease cell proliferation (Pellegrini et al. 2001). The clonogenic behavior of keratinocytes provides a widely used assay for their growth potential (Rochat et al. 1994). We examined whether nickel treatment was able to suppress colonogenicity and if it did, whether this suppression depended on p63 down-modulation. HaCat cells were infected with a recombinant retrovirus expressing the ΔNp63 gene together with GFP (Pinco-ΔNp63) or a retrovirus expressing GFP alone (Pinco, empty vector). In each case, GFP-positive keratinocytes were purified by sorting and subsequently by treatment with 100 μM nickel. HaCat cells infected with the p63 retrovirus formed essentially the same colonies as cells infected with the retrovirus control, indicating that p63 over-expression per se is not sufficient to increase the number of clonogenic keratinocytes (Fig. 3). However, while treatment with nickel caused a drastic drop in number of clonogenic cells, a much lesser reduction was observed with cells that had been previously transduced with the ΔNp63 retrovirus (Fig. 3), indicating the role of p63 in maintenance of the keratinocyte growth potential under nickel stimulation.
Fig. 3.
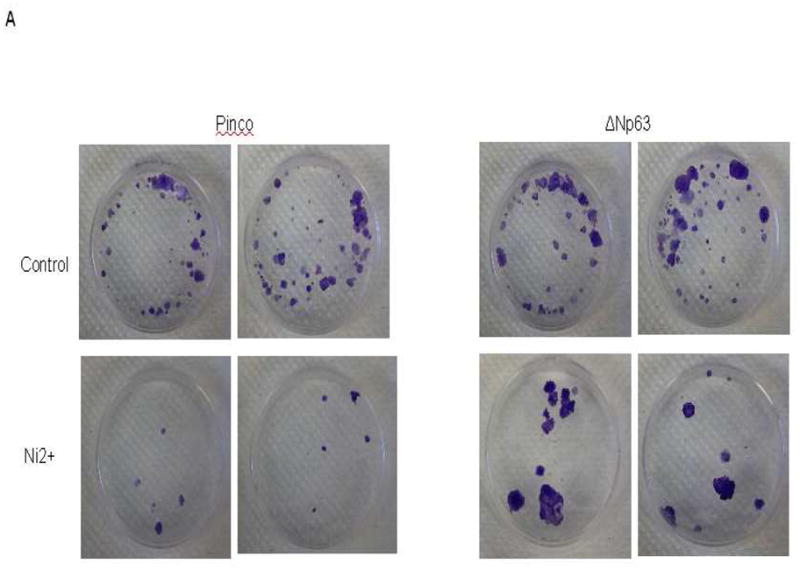

Over-expression of ΔNp63 expression restores nickel induced cell growth inhibition. The detailed procedure is described in the “Materials and Methods”. (A) The clonogenic growth was evaluated by staining of dishes. (B) Quantification of colony formation. *, p<0.05 compared to control without nickel treatment (one-way ANOVA test). #, p<0.05 compared to nickel treatment with Pinco vector (one-way ANOVA test).
Nickel treatment causes growth arrest through induction of AP-1 and NFAT by direct p38-dependent mechanisms (Ding et al. 2009; Huang et al. 2001). Figs. 4A and 4B showed that nickel treatment increased p21 expression at both mRNA and protein levels. Over-expression of ΔNp63 decreased p21 protein level in both untreated and nickel treated cells (Fig. 4B). Similarly, in transient transfection assays, the ability of nickel to induce the 2.4-kb promoter of the p21 gene was suppressed by over-expression of ΔNp63 in a dose-dependent fashion (Fig. 4C). This suppression may result from the demonstrated ability of ΔNp63α to bind directly to p53/p63-binding sites in the p21 promoter (Westfall et al. 2003), thereby interfering with nickel induced p21 up-regulation. However, induction of a minimal p21 promoter region not containing the binding site for p53/p63 was also suppressed by ΔNp63, to a similar extent as the full-length promoter (Fig. 4D). These results show that nickel induced up-regulation of p21, leading to decreased cell proliferation. ΔNp63 is a negative regulator of p21. The inhibitory effect of ΔNp63 on p21 promoter may be through either p53 binding site or other site beyond p53.
Fig. 4.

Counteracting effects of ΔNp63 on the nickel induced p21WAF1/Cip1 activation. (A) Induction of p21 by nickel. HaCat cells were treated with different doses of nickel for 24 hr. The cells were harvested for analysis of p21 mRNA level by real time PCR. (B–D) Countering effects of ΔNp63 on nickel induced p21activation. HaCat cells were transfected with ΔNp63 followed by nickel treatment. Immunobloting was used to measure p21 expression (B). HaCat cells were transiently transfected with reporter plasmids containing the 2.4-kb promoter region of the p21 gene (C), and a minimal region of the p21 promoter devoid of p53-binding sites but containing a fully conserved RBP-binding site (Rangarajan et al. 2001) (D). The cells were treated with 1 mM nickel, plus/minus an expression vector for ΔNp63 in increasing amounts as indicated. After 24 hr promoter activity were measured. *, p<0.05 compared to control (one-way ANOVA test). #, p<0.05 compared to nickel treatment (one-way ANOVA test).
Control p21 by endogenous ΔNp63
To assess whether p21 is under negative control of endogenous p63, HaCat cells were transfected with miRNA for the coding region of human p63 versus a scrambled miRNA control. This approach caused 80%–90% reduction in p63 mRNA and p63 protein levels 2–3 days after transfection. In cells treated with nickel, the p63 miRNAs caused an additional p63 reduction (Figs. 5A and 5B). p21 expression was up-regulated in cells with p63 knockdown (Fig. 5C). This knockdown resulted in a substantial increase in p21 expression under basal conditions, with a much higher induction in response to nickel treatment (Fig. 5C).
Fig. 5.

Control of p21 by endogenous ΔNp63. (A–B) Knockdown of endogenous ΔNp63 expression. HaCat cells were transfected with miRNA human ΔNp63 or miRNA control. Parallel cultures were treated with1 mM nickel for 24 hr after miRNA transfection. Cells were analyzed for measurement of p63 expression by real-time PCR (A) or immunoblotting with the corresponding antibodies (B). (C) Up-regulation of p21 expression as a consequence of ΔNp63 knockdown. The procedure is the same as (A). HaCat cells were harvested for analysis of p21 mRNA level by real-time PCR. *, p<0.05 compared to control (one-way ANOVA test). #, p<0.05 compared to nickel treatment (one-way ANOVA test).
Discussion
Although the molecular mechanisms by which nickel compounds cause cancer are still under investigation, recent studies show that the carcinogenic actions of nickel compound include oxidative stress, genomic DNA damage, epigenetic effects, and the regulation of gene expression by activation of certain transcription factors related to corresponding signal transduction pathways (Lu et al. 2005). It has been reported that nickel caused reactive oxygen species (ROS) generation, leading to cell transformation (Costa et al. 1994; Huang et al. 1994). Nickel was able to activate NF-κB (Goebeler et al. 1995) and NFAT (Huang et al. 2001), induce inflammation mediators such as COX-2 and TNF-α (Ding et al. 2009), and increase HIF-α, VEGF, and angiogenesis (Ouyang et al. 2009).
The present study investigated the role of p63 in the epidermal nickel response. First, we found that ΔNp63 isoform was predominantly expressed in the epidermis. ΔNp63 expression was dramatically reduced at both transcription and translation levels in response to nickel treatment. p53, a tumor suppress gene, was activated upon nickel treatment. Similarly, another member of p53 family, p73 was also up-regulated by nickel. Nickel was able to activate NF-κB. Inhibition of NF-κB partially restored ΔNp63 mRNA level reduced by nickel treatment. Furthermore, nickel treatments caused decreases in IRF3, IR7, IKKε, and Sp100 expressions in a dose-dependent manner. Over-expression of IRF3 reversed nickel induced suppression of ΔNp63 mRNA level. Cell proliferation assay showed that nickel treatment reduced the cell growth. Over-expression of ΔNp63 counteracted the effect of nickel, causing an increase of cell proliferation compared to nickel treatment only. The present study found that p21, a downstream regulatory protein of p53, played a role in ΔNp63 signaling. Nickel increased p21 activity, and ΔNp63 eliminated the p21 activity induced by nickel. Blockade of ΔNp63 dramatically increased p21 expression.
It has been shown that in p63 family, ΔNp63α is the most abundantly expressed isotype in tumor cells. This protein has been implicated in cell proliferation and oncogenic growth (Crook et al. 2000; Hibi et al. 2000; Park et al. 2000). The studies of p63 null mice have demonstrated that p63 plays a critical role in regulating cell proliferation and in mediating epidermal stem cell renewal during early development (Yang et al. 1999). These findings raise the intriguing possibility that p63 functions as a regulator of cell growth in normal tissues and in cancer (Patturajan et al. 2002). The results in the present study showed that short time (up to 48 hr) nickel treatment caused a decrease of ΔNp63 at both transcription and translation levels, and promoter activity. However, our preliminary results showed that 8 weeks and 16 weeks exposure to low dose nickel (100 μM) increased Np63 expression in HaCat cells (data not shown). Over-expression of ΔNp63 synergistically promoted nickel induced cell transformation and tumorigenesis. This is consistent with the study that low dose arsenic treatment in normal RWPE cells caused a decrease of ΔNp63 in early stage and dramatic increase of ΔNp63 in late stage due to the cells being transformed (Tokar et al. 2010). It has been observed that normal cell self-renewal was initially lost during early transformation, potentially due to aberrant differentiation, and then regained with malignant transformation, consistent with distorted self-renewal common to cancer cells (Pardal et al. 2003). Another study has also shown that ΔNp63α is reduced in response to UV-B treatment (Liefer et al. 2000). Nickel is able to cause DNA damage. When a cell incurs DNA damage, p63 is down-regulated, allowing p53 to step into action, protecting the cells from damaged DNA. Deregulated p63 expression would interfere with this protective role of p53, possibly by competing with p53 for binding to DNA targets and/or exclusion from the nucleus and degradation via the actions of newly synthesized p63.
ROS is one of the important determinants in the regulation of cell signaling pathways involved in proliferation, apoptosis, transformation, and senescence (Tokar et al. 2010; Westfall et al. 2005; Dalton et al. 1999). Many of these effects contribute to nickel induced carcinogenesis (Chen and Shi 2002). It was reported that p63−/− MEFs (mouse embryo fibroblasts) had significantly lower levels of ROS compared with cells from wild-type or heterozygous littermates (Ellisen et al. 2002). Over-expression of ΔNp63α in the p63−/− MEFs led to a marked enhancement of H2O2-induced ROS (Ellisen et al. 2002). 100 J/m2 of UV exposure to primary human keratinocytes decreased ΔNp63α expression level, concordant with increased ROS generation (Westfall et al. 2005). Our preliminary data showed that nickel treatment was able to cause ROS generation in HaCat cells (data not shown). Pretreatment with ROS scavengers, SOD and catalase increased ΔNp63 expression reduced by nickel. In addition, we also found that nickel treatment increased SOD2 expression. Inhibition of SOD2 expression partially restored ΔNp63 expression reduced by nickel. Those indicate that ΔNp63 may act as one of the target genes of ROS, promoting cell proliferation.
NF-κB is sequestered in the cytoplasm in an inactive form binding to the I-κBα inhibitor. NF-κB activation requires the I-κB kinase (IKK) to mediate I-κBα phosphorylation, an event leading to I-κBα degradation and consequently freeing NF-κB to translocate to the nucleus for regulating the transcription of its target genes (Hayden and Ghosh 2004; Karin and Greten 2005). The mechanism by which p63 initiates or directs the differentiation program and the key downstream targets involved are still poorly understood. p63 has been linked with many genes that have been shown to be important for promoting differentiation, a key protein being IKKα (Truong and Khavari 2007). IKKα was identified as a direct downstream target of p63 (Candi et al. 2006). IKKα is a component of the multi-unit IKK complex that phosphorylates the inhibitor molecule IκB, thereby targeting it for ubiquitination and degradation. This relieves the inhibitory unit of NF-κB, resulting in its activation (Zandi et al. 1997). There are several NF-κB binding sites in both mouse and human promoters of ΔNp63 (Nguyen et al. 2006). Since NF-κB is activated in keratinocytes differentiation and is involved in carcinogenesis, it is likely that NF-κB is involved in inhibition of ΔNp63 by nickel. Blockade of NF-κB by expressing of IKKβ kinase mutant reversed nickel induced suppression of ΔNp63, indicating that ΔNp63 is a downstream protein which is negatively regulated by NF-κB in response to nickel treatment.
In addition to NF-κB binding sides on ΔNp63 promoter, there are binding sites for interferon responsive elements, where a synergistic multi-protein complex is formed by NF-κB subunits and Interferon regulatory factor 3 and 7 (IRF3/IRF7), two key mediators of the interferon response (Nguyen et al. 2006). IRF3 and IRF7 participate in the formation of a large protein complex called an IFN-β enhancesome/DRAF1 that are also NF-κB, AP-1, and CREB binding protein (CBP)/p300 to activate transcription of IFN-β gene (Honda and Taniguchi 2006; Wathelet et al. 1998). IRF3 is believed to play a role in DNA damage-induced apoptosis as IRF3 protein is phosphorylated and translocates from the cytoplasm to the nucleus in response to DNA damage (Kim et al. 1999). In addition, IRF3 may function as a tumor suppressor gene (Savitsky et al.). An induction of IFN-γ was observed in nickel contacting patients (Bordignon et al. 2008). In vitro study also showed that treatment with nickel sulfate induced secretion of IFN-gamma by splenic natural killer cells (Kim et al. 2009). It has been reported that nickel compounds are able to up-regulate both TNFα and NF-κB (Huang et al. 1994). However, the regulation of interferon pathway remains unclear. It has been reported that up-regulation of IKKε depends on NF-κB activity (Hemmi et al. 2004; Kravchenko et al. 2003; Shimada et al. 1999). The mechanistic role of IKKε in NF-κB activation is still not fully understood. It is possible that IKKε-dependant activation of NF-κB targets genes in certain cells is mediated through IRFs. IRF3 is required for the activation of certain NF-κB target genes (Covert et al. 2005). Our data suggested that IKKε, IRF3 and IRF7 mRNA levels decreased after nickel treatment. Over-expression of IRF3 itself indeed reduced ΔNp63 mRNA level. Treatment of the cells by nickel together with IRF3 over-expression restored ΔNp63 expression reduced by nickel. These results demonstrate that IRF3 is involved in nickel-induced suppression of ΔNp63 expression.
Abundant genetic evidence indicates that one of p63 functions is to maintain regenerative capacity of base epithelia at several sites throughout the body. Down-modulation of ΔNp63 expression by nickel treatment may be important for the cell growth inhibition. The clonogenic assay provides a widely used method to determine the cell growth potential (Rochat et al. 1994). Using this method, we have shown that treatment with the low dose of 100 μM nickel chloride (NiCl2) for 2 weeks caused a dramatic growth inhibition compared to control without nickel treatment. Moreover, over-expression of ΔNp63 restored cell growth inhibited by nickel, indicating that ΔNp63 counteracts the ability of nickel to restrict cell growth.
Induction of p21 protein by nickel is responsible for the cell cycle arrest, whereas p63 may function as an inhibitor of p21 (Nguyen et al. 2006). The results from present study indicate that nickel treatment caused increase of p21 promoter activity. Nickel was still able to activate truncated p21 without p53 response elements. ΔNp63 counteracted the effect of nickel, causing the reduction of p21 activity. The reduction effect of ΔNp63 was p53-independent. It has been reported that ΔNp63 binds to the p21 other than p53 binding sites. Loss of ΔNp63α-dependent transcriptional repression of p21WAF1 correlates with ΔNp63α down-regulation during keratinocyte differentiation (Craig et al. 2010). Consistently, the present study shows that inhibition of ΔNp63 increased nickel-induced p21 expression. Inhibition of ΔNp63 alone caused a decrease of p21 mRNA expression. There are two modes of transcriptional suppression by ΔNp63. First, ΔNp63 can compete with p53 activity (or transactivating p63 isoforms) for DNA target sites (Yang et al. 1998; Davison et al. 1999). A second mode of inhibition might involve the formation of transactivation-incompetent heterocomplexes between DNA binding domains of p53 and the ΔNp63 (Ratovitski et al. 2001). Our results suggested that reduction of ΔNp63 by nickel may act on its downstream genes other than p53 binding sites.
In summary, our results showed that in keratinocytes nickel is able to decrease IRF3 and IRF7, and then activate NF-κB. The activation of NF-κB suppressed ΔNp63, resulting in increase of p21 expression and decrease in cell growth.
Acknowledgments
This research is supported by NIH grant R01ES015518.
Footnotes
Conflict of Interest
This manuscript will not be submitted to any other scientific journal prior to the decision by Toxicology and Applied Pharmacology. Each listed author on the manuscript is aware of and agrees to the contents of the manuscript, including the authorship. None of the listed authors has any financial or other interests that could be of conflict.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Uddin AN, Burns FJ, Rossman TG, Chen H, Kluz T, Costa M. Dietary chromium and nickel enhance UV-carcinogenesis in skin of hairless mice. Toxicol Appl Pharmacol. 2007;221(3):329–338. doi: 10.1016/j.taap.2007.03.030. [DOI] [PubMed] [Google Scholar]
- Roberts RS, Julian JA, Muir DC, Shannon HS. A study of mortality in workers engaged in the mining, smelting, and refining of nickel. II: Mortality from cancer of the respiratory tract and kidney. Toxicol Ind Health. 1989;5(6):975–993. doi: 10.1177/074823378900500606. [DOI] [PubMed] [Google Scholar]
- Salnikow K, Costa M. Epigenetic mechanisms of nickel carcinogenesis. J Environ Pathol Toxicol Oncol. 2000;19(3):307–318. [PubMed] [Google Scholar]
- Clevers H. At the crossroads of inflammation and cancer. Cell. 2004;118(6):671–674. doi: 10.1016/j.cell.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Zhao J, Yan Y, Salnikow K, Kluz T, Costa M. Nickel-induced down-regulation of serpin by hypoxic signaling. Toxicol Appl Pharmacol. 2004;194(1):60–68. doi: 10.1016/j.taap.2003.08.011. [DOI] [PubMed] [Google Scholar]
- Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88(3):323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- Oren M. Regulation of the p53 tumor suppressor protein. J Biol Chem. 1999;274(51):36031–36034. doi: 10.1074/jbc.274.51.36031. [DOI] [PubMed] [Google Scholar]
- Osada M, Ohba M, Kawahara C, Ishioka C, Kanamaru R, Katoh I, et al. Cloning and functional analysis of human p51, which structurally and functionally resembles p53. Nat Med. 1998;4(7):839–843. doi: 10.1038/nm0798-839. [DOI] [PubMed] [Google Scholar]
- Schmale H, Bamberger C. A novel protein with strong homology to the tumor suppressor p53. Oncogene. 1997;15(11):1363–1367. doi: 10.1038/sj.onc.1201500. [DOI] [PubMed] [Google Scholar]
- Trink B, Okami K, Wu L, Sriuranpong V, Jen J, Sidransky D. A new human p53 homologue. Nat Med. 1998;4(7):747–748. doi: 10.1038/nm0798-747. [DOI] [PubMed] [Google Scholar]
- Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dotsch V, et al. p63, a p53 homolog at 3q27–29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell. 1998;2(3):305–316. doi: 10.1016/s1097-2765(00)80275-0. [DOI] [PubMed] [Google Scholar]
- Yang A, Kaghad M, Caput D, McKeon F. On the shoulders of giants: p63, p73 and the rise of p53. Trends Genet. 2002;18(2):90–95. doi: 10.1016/s0168-9525(02)02595-7. [DOI] [PubMed] [Google Scholar]
- Candi E, Dinsdale D, Rufini A, Salomoni P, Knight RA, Mueller M, et al. TAp63 and DeltaNp63 in cancer and epidermal development. Cell Cycle. 2007;6(3):274–285. doi: 10.4161/cc.6.3.3797. [DOI] [PubMed] [Google Scholar]
- Mills AA, Zheng B, Wang XJ, Vogel H, Roop DR, Bradley A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature. 1999;398(6729):708–713. doi: 10.1038/19531. [DOI] [PubMed] [Google Scholar]
- Yang A, Schweitzer R, Sun D, Kaghad M, Walker N, Bronson RT, et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature. 1999;398(6729):714–718. doi: 10.1038/19539. [DOI] [PubMed] [Google Scholar]
- Westfall MD, Pietenpol JA. p63: Molecular complexity in development and cancer. Carcinogenesis. 2004;25(6):857–864. doi: 10.1093/carcin/bgh148. [DOI] [PubMed] [Google Scholar]
- King KE, Ponnamperuma RM, Gerdes MJ, Tokino T, Yamashita T, Baker CC, et al. Unique domain functions of p63 isotypes that differentially regulate distinct aspects of epidermal homeostasis. Carcinogenesis. 2006;27(1):53–63. doi: 10.1093/carcin/bgi200. [DOI] [PubMed] [Google Scholar]
- Koster MI, Kim S, Mills AA, DeMayo FJ, Roop DR. p63 is the molecular switch for initiation of an epithelial stratification program. Genes Dev. 2004;18(2):126–131. doi: 10.1101/gad.1165104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nocentini G, Giunchi L, Ronchetti S, Krausz LT, Bartoli A, Moraca R, et al. A new member of the tumor necrosis factor/nerve growth factor receptor family inhibits T cell receptor-induced apoptosis. Proc Natl Acad Sci U S A. 1997;94(12):6216–6221. doi: 10.1073/pnas.94.12.6216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangarajan A, Talora C, Okuyama R, Nicolas M, Mammucari C, Oh H, et al. Notch signaling is a direct determinant of keratinocyte growth arrest and entry into differentiation. EMBO J. 2001;20(13):3427–3436. doi: 10.1093/emboj/20.13.3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talora C, Sgroi DC, Crum CP, Dotto GP. Specific down-modulation of Notch1 signaling in cervical cancer cells is required for sustained HPV-E6/E7 expression and late steps of malignant transformation. Genes Dev. 2002;16(17):2252–2263. doi: 10.1101/gad.988902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen BC, Lefort K, Mandinova A, Antonini D, Devgan V, Della Gatta G, et al. Cross-regulation between Notch and p63 in keratinocyte commitment to differentiation. Genes Dev. 2006;20(8):1028–1042. doi: 10.1101/gad.1406006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DE, Kessler DS, Pine R, Reich N, Darnell JE., Jr Interferon-induced nuclear factors that bind a shared promoter element correlate with positive and negative transcriptional control. Genes Dev. 1988;2(4):383–393. doi: 10.1101/gad.2.4.383. [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Ogasawara K, Takaoka A, Tanaka N. IRF family of transcription factors as regulators of host defense. Annu Rev Immunol. 2001;19:623–655. doi: 10.1146/annurev.immunol.19.1.623. [DOI] [PubMed] [Google Scholar]
- Cruz MT, Goncalo M, Figueiredo A, Carvalho AP, Duarte CB, Lopes MC. Contact sensitizer nickel sulfate activates the transcription factors NF-kB and AP-1 and increases the expression of nitric oxide synthase in a skin dendritic cell line. Exp Dermatol. 2004;13(1):18–26. doi: 10.1111/j.0906-6705.2004.00105.x. [DOI] [PubMed] [Google Scholar]
- Denkhaus E, Salnikow K. Nickel essentiality, toxicity, and carcinogenicity. Crit Rev Oncol Hematol. 2002;42(1):35–56. doi: 10.1016/s1040-8428(01)00214-1. [DOI] [PubMed] [Google Scholar]
- Goebeler M, Roth J, Brocker EB, Sorg C, Schulze-Osthoff K. Activation of nuclear factor-kappa B and gene expression in human endothelial cells by the common haptens nickel and cobalt. J Immunol. 1995;155(5):2459–2467. [PubMed] [Google Scholar]
- Zhang L, Pagano JS. Structure and function of IRF-7. J Interferon Cytokine Res. 2002;22(1):95–101. doi: 10.1089/107999002753452700. [DOI] [PubMed] [Google Scholar]
- Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434(7034):772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- Moller A, Sirma H, Hofmann TG, Staege H, Gresko E, Ludi KS, et al. Sp100 is important for the stimulatory effect of homeodomain-interacting protein kinase-2 on p53-dependent gene expression. Oncogene. 2003;22(54):8731–8737. doi: 10.1038/sj.onc.1207079. [DOI] [PubMed] [Google Scholar]
- Servant MJ, Tenoever B, Lin R. Overlapping and distinct mechanisms regulating IRF-3 and IRF-7 function. J Interferon Cytokine Res. 2002;22(1):49–58. doi: 10.1089/107999002753452656. [DOI] [PubMed] [Google Scholar]
- Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4(5):491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- Honda K, Taniguchi T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol. 2006;6(9):644–658. doi: 10.1038/nri1900. [DOI] [PubMed] [Google Scholar]
- Zhang JY, Green CL, Tao S, Khavari PA. NF-kappaB RelA opposes epidermal proliferation driven by TNFR1 and JNK. Genes Dev. 2004;18(1):17–22. doi: 10.1101/gad.1160904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini G, Dellambra E, Golisano O, Martinelli E, Fantozzi I, Bondanza S, et al. p63 identifies keratinocyte stem cells. Proc Natl Acad Sci U S A. 2001;98(6):3156–3161. doi: 10.1073/pnas.061032098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochat A, Kobayashi K, Barrandon Y. Location of stem cells of human hair follicles by clonal analysis. Cell. 1994;76(6):1063–1073. doi: 10.1016/0092-8674(94)90383-2. [DOI] [PubMed] [Google Scholar]
- Ding J, Huang Y, Ning B, Gong W, Li J, Wang H, et al. TNF-alpha induction by nickel compounds is specific through ERKs/AP-1-dependent pathway in human bronchial epithelial cells. Curr Cancer Drug Targets. 2009;9(1):81–90. doi: 10.2174/156800909787313995. [DOI] [PubMed] [Google Scholar]
- Huang C, Li J, Costa M, Zhang Z, Leonard SS, Castranova V, et al. Hydrogen peroxide mediates activation of nuclear factor of activated T cells (NFAT) by nickel subsulfide. Cancer Res. 2001;61(22):8051–8057. [PubMed] [Google Scholar]
- Westfall MD, Mays DJ, Sniezek JC, Pietenpol JA. The Delta Np63 alpha phosphoprotein binds the p21 and 14-3-3 sigma promoters in vivo and has transcriptional repressor activity that is reduced by Hay-Wells syndrome-derived mutations. Mol Cell Biol. 2003;23(7):2264–2276. doi: 10.1128/MCB.23.7.2264-2276.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Shi X, Costa M, Huang C. Carcinogenic effect of nickel compounds. Mol Cell Biochem. 2005;279(1–2):45–67. doi: 10.1007/s11010-005-8215-2. [DOI] [PubMed] [Google Scholar]
- Costa M, Salnikow K, Cosentino S, Klein CB, Huang X, Zhuang Z. Molecular mechanisms of nickel carcinogenesis. Environ Health Perspect. 1994;102(Suppl 3):127–130. doi: 10.1289/ehp.94102s3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Zhuang Z, Frenkel K, Klein CB, Costa M. The role of nickel and nickel-mediated reactive oxygen species in the mechanism of nickel carcinogenesis. Environ Health Perspect. 1994;102(Suppl 3):281–284. doi: 10.1289/ehp.94102s3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang W, Zhang D, Li J, Verma UN, Costa M, Huang C. Soluble and insoluble nickel compounds exert a differential inhibitory effect on cell growth through IKKalpha-dependent cyclin D1 down-regulation. J Cell Physiol. 2009;218(1):205–214. doi: 10.1002/jcp.21590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crook T, Nicholls JM, Brooks L, O’Nions J, Allday MJ. High level expression of deltaN-p63: a mechanism for the inactivation of p53 in undifferentiated nasopharyngeal carcinoma (NPC)? Oncogene. 2000;19(30):3439–3444. doi: 10.1038/sj.onc.1203656. [DOI] [PubMed] [Google Scholar]
- Hibi K, Trink B, Patturajan M, Westra WH, Caballero OL, Hill DE, et al. AIS is an oncogene amplified in squamous cell carcinoma. Proc Natl Acad Sci U S A. 2000;97(10):5462–5467. doi: 10.1073/pnas.97.10.5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park BJ, Lee SJ, Kim JI, Lee CH, Chang SG, Park JH, et al. Frequent alteration of p63 expression in human primary bladder carcinomas. Cancer Res. 2000;60(13):3370–3374. [PubMed] [Google Scholar]
- Patturajan M, Nomoto S, Sommer M, Fomenkov A, Hibi K, Zangen R, et al. DeltaNp63 induces beta-catenin nuclear accumulation and signaling. Cancer Cell. 2002;1(4):369–379. doi: 10.1016/s1535-6108(02)00057-0. [DOI] [PubMed] [Google Scholar]
- Tokar EJ, Diwan BA, Waalkes MP. Arsenic exposure transforms human epithelial stem/progenitor cells into a cancer stem-like phenotype. Environ Health Perspect. 2010;118(1):108–115. doi: 10.1289/ehp.0901059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cell biology to cancer. Nat Rev Cancer. 2003;3(12):895–902. doi: 10.1038/nrc1232. [DOI] [PubMed] [Google Scholar]
- Liefer KM, Koster MI, Wang XJ, Yang A, McKeon F, Roop DR. Down-regulation of p63 is required for epidermal UV-B-induced apoptosis. Cancer Res. 2000;60(15):4016–4020. [PubMed] [Google Scholar]
- Westfall MD, Joyner AS, Barbieri CE, Livingstone M, Pietenpol JA. Ultraviolet radiation induces phosphorylation and ubiquitin-mediated degradation of DeltaNp63alpha. Cell Cycle. 2005;4(5):710–716. doi: 10.4161/cc.4.5.1685. [DOI] [PubMed] [Google Scholar]
- Dalton TP, Shertzer HG, Puga A. Regulation of gene expression by reactive oxygen. Annu Rev Pharmacol Toxicol. 1999;39:67–101. doi: 10.1146/annurev.pharmtox.39.1.67. [DOI] [PubMed] [Google Scholar]
- Chen F, Shi X. Signaling from toxic metals to NF-kappaB and beyond: not just a matter of reactive oxygen species. Environ Health Perspect. 2002;110(Suppl 5):807–811. doi: 10.1289/ehp.02110s5807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellisen LW, Ramsayer KD, Johannessen CM, Yang A, Beppu H, Minda K, et al. REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species. Mol Cell. 2002;10(5):995–1005. doi: 10.1016/s1097-2765(02)00706-2. [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18(18):2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5(10):749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- Truong AB, Khavari PA. Control of keratinocyte proliferation and differentiation by p63. Cell Cycle. 2007;6(3):295–299. doi: 10.4161/cc.6.3.3753. [DOI] [PubMed] [Google Scholar]
- Candi E, Terrinoni A, Rufini A, Chikh A, Lena AM, Suzuki Y, et al. p63 is upstream of IKK alpha in epidermal development. J Cell Sci. 2006;119(Pt 22):4617–4622. doi: 10.1242/jcs.03265. [DOI] [PubMed] [Google Scholar]
- Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell. 1997;91(2):243–252. doi: 10.1016/s0092-8674(00)80406-7. [DOI] [PubMed] [Google Scholar]
- Wathelet MG, Lin CH, Parekh BS, Ronco LV, Howley PM, Maniatis T. Virus infection induces the assembly of coordinately activated transcription factors on the IFN-beta enhancer in vivo. Mol Cell. 1998;1(4):507–518. doi: 10.1016/s1097-2765(00)80051-9. [DOI] [PubMed] [Google Scholar]
- Kim T, Kim TY, Song YH, Min IM, Yim J, Kim TK. Activation of interferon regulatory factor 3 in response to DNA-damaging agents. J Biol Chem. 1999;274(43):30686–30689. doi: 10.1074/jbc.274.43.30686. [DOI] [PubMed] [Google Scholar]
- Savitsky D, Tamura T, Yanai H, Taniguchi T. Regulation of immunity and oncogenesis by the IRF transcription factor family. Cancer Immunol Immunother. 59(4):489–510. doi: 10.1007/s00262-009-0804-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordignon V, Palamara F, Cordiali-Fei P, Vento A, Aiello A, Picardo M, et al. Nickel, palladium and rhodium induced IFN-gamma and IL-10 production as assessed by in vitro ELISpot-analysis in contact dermatitis patients. BMC Immunol. 2008;9:19. doi: 10.1186/1471-2172-9-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Huh K, Lee KY, Yang JM, Kim TJ. Nickel induces secretion of IFN-gamma by splenic natural killer cells. Exp Mol Med. 2009;41(4):288–295. doi: 10.3858/emm.2009.41.4.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmi H, Takeuchi O, Sato S, Yamamoto M, Kaisho T, Sanjo H, et al. The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J Exp Med. 2004;199(12):1641–1650. doi: 10.1084/jem.20040520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravchenko VV, Mathison JC, Schwamborn K, Mercurio F, Ulevitch RJ. IKKi/IKKepsilon plays a key role in integrating signals induced by pro-inflammatory stimuli. J Biol Chem. 2003;278(29):26612–26619. doi: 10.1074/jbc.M303001200. [DOI] [PubMed] [Google Scholar]
- Shimada T, Kawai T, Takeda K, Matsumoto M, Inoue J, Tatsumi Y, et al. IKK-i, a novel lipopolysaccharide-inducible kinase that is related to IkappaB kinases. Int Immunol. 1999;11(8):1357–1362. doi: 10.1093/intimm/11.8.1357. [DOI] [PubMed] [Google Scholar]
- Covert MW, Leung TH, Gaston JE, Baltimore D. Achieving stability of lipopolysaccharide-induced NF-kappaB activation. Science. 2005;309(5742):1854–1857. doi: 10.1126/science.1112304. [DOI] [PubMed] [Google Scholar]
- Craig AL, Holcakova J, Finlan LE, Nekulova M, Hrstka R, Gueven N, et al. DeltaNp63 transcriptionally regulates ATM to control p53 Serine-15 phosphorylation. Mol Cancer. 2010;9:195. doi: 10.1186/1476-4598-9-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davison TS, Vagner C, Kaghad M, Ayed A, Caput D, Arrowsmith CH. p73 and p63 are homotetramers capable of weak heterotypic interactions with each other but not with p53. J Biol Chem. 1999;274(26):18709–18714. doi: 10.1074/jbc.274.26.18709. [DOI] [PubMed] [Google Scholar]
- Ratovitski EA, Patturajan M, Hibi K, Trink B, Yamaguchi K, Sidransky D. p53 associates with and targets Delta Np63 into a protein degradation pathway. Proc Natl Acad Sci U S A. 2001;98(4):1817–1822. doi: 10.1073/pnas.98.4.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]