

Abstract
A critical event in the apoptotic cascade is the proteolytic activation of procaspases to active caspases. The caspase auto-activating compound PAC-1 induces cancer cell apoptosis and exhibits antitumor activity in murine xenograft models when administered orally as a lipid-based formulation or implanted subcutaneously as a cholesterol pellet. However, high doses of PAC-1 were found to induce neurotoxicity, prompting us to design and assess a novel PAC-1 derivative called S-PAC-1. Similar to PAC-1, S-PAC-1 activated procaspase-3 and induced cancer cell apoptosis. However, S-PAC-1 did not induce neurotoxicity in mice or dogs. Continuous intravenous infusion of S-PAC-1 in dogs led to a steady state plasma concentration of ~10 µM for 24–72 hours. In a small efficacy trial of S-PAC-1, evaluation of six pet dogs with lymphoma revealed that S-PAC-1 was well-tolerated and that the treatments induced partial tumor regression or stable diseasein 4 / 6 subjects. Our results support this canine setting for further evaluation of small molecule procaspase-3 activators, including S-PAC-1, a compound that is an excellent candidate for further clinical evaluation as a novel cancer chemotherapeutic.
Introduction
Members of the caspase family of cysteine proteases are key players in both the initiation and execution of apoptosis. These enzymes exist in the cell as low activity zymogens (proenzymes) that are proteolytically activated to the mature, highly active enzyme. Most critical to apoptosis is the proteolytic conversion of procaspase-3 to caspase-3. As both the intrinsic and extrinsic apoptotic pathways converge to activate procaspase-3, and as caspase-3 has over 100 cellular substrates, the activation of procaspase-3 to caspase-3 is a pivotal and committed event in the apoptotic cascade. Interestingly, procaspase-3 is overexpressed in a variety of tumor histologies including breast cancer (1), colon cancer (2), lung cancer (3), lymphoma (4), neuroblastoma (5), melanoma (6) and liver cancer (7), suggesting that a small molecule that activates procaspase-3 could have selectivity for cancer cells versus normal cells.
In 2006, we reported the discovery of a small molecule, called PAC-1 (Figure 1A), which enhances procaspase-3 activity in vitro, induces death in cancer cells in culture, and has efficacy in multiple mouse xenograft models when administered orally as a lipid-based formulation or implanted subcutaneously as a cholesterol pellet (8). PAC-1 activates procaspase-3 in vitro through the chelation of inhibitory zinc ions (9), and derivative synthesis and evaluation reveal that the biological activity of PAC-1 is tied to having an intact ortho-hydroxy N-acyl hydrazone zinc-chelating motif (10). Evidence suggests that PAC-1 induces apoptotic death in cancer cells through the chelation of zinc from procaspase-3, most notably the colocalization of a fluorescent PAC-1 derivative with sites of cellular caspase-3 activity(10).
Figure 1.
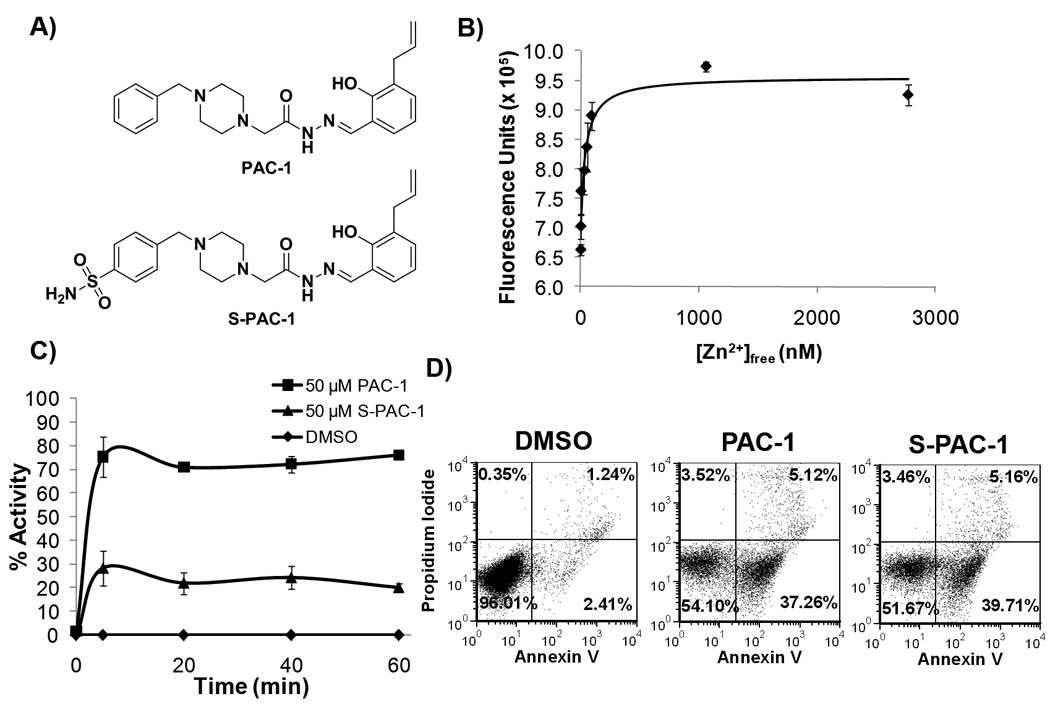
In vitro assessment of PAC-1 and S-PAC-1. A, Structures of PAC-1 and S-PAC-1. B, The formation curve of the Zn2+:S-PAC-1 complex as determined by EGTA titration. S-PAC-1 binds zinc with a Kd of 46±5 nM. Error bars represent SEM (n=3). C, PAC-1 and S-PAC-1 induce rapid relief of zinc-mediated inhibition of procaspase-3 (D3A). Using a chromogenic Ac-DEVD-pNA substrate, the activity of procaspase-3 (7.5 µM) was assessed in the presence of inhibitory zinc (10 µM) and vehicle, PAC-1 or S-PAC-1 (50 µM). Maximal activity is observed after a 5-minute incubation with compound. Error bars represent the SEM (n=3). D, PAC-1 and S-PAC-1 both induce apoptotic cell death in U-937 cells as demonstrated by a population of annexin V positive, PI negative cells. Data shown is representative of three separate experiments.
As the first procaspase-activating compound, experiments with PAC-1 can begin to define the potential of procaspase-3 activation as a viable anticancer strategy. To further develop PAC-1 as an experimental therapeutic for the treatment of cancer in humans, we sought to characterize the effect of this compound when administered intravenously and in more sophisticated in vivo tumor model systems, specifically, canines with spontaneous cancer. The evaluation of experimental therapeutics in pet dogs with cancer offers many advantages over murine xenograft models (11). Herein we report toxicity studies of intravenously administered PAC-1 in mice, and the discovery of a novel PAC-1 derivative (called S-PAC-1) that induces apoptosis in cancer cell lines in culture, is well-tolerated in mice and research dogs, and has moderate activity in a small trial of canine patients with spontaneous lymphoma. These results demonstrate the feasibility of S-PAC-1 administration to pet dogs with lymphoma as a means to evaluate the therapeutic potential of this class of compounds.
Materials and Methods
Cell Lines and Reagents
U-937, Jurkat, SK-MEL-5, Hela, MDA-MB-231 and EL4 cells were obtained from ATCC (authenticated by STR analysis) and maintained at low passage number. Two canine B-cell lymphoma lines (17–71 and GL-1) were provided by Dr. Steve Suter of NCSU. All cultures were maintained in RPMI-1640 media supplemented with 10% FBS and 1% Pen-Strep and grown at 37 °C and 5% CO2. PAC-1 was synthesized as previously described (8). S-PAC-1 was synthesized as described in the supplementary data. Ac-DEVD-pNA was synthesized as previously described (12). Chelex® treated Hepes-NaCl buffer is 50 mM Hepes, 300 mM NaCl and is treated with Chelex® resin for 1 hour prior to use.
EGTA Fluorescence Titration Assay
This assay was performed according to a developed protocol (13) exactly as previously reported (10).
Recombinant expression, purification, and evaluation of uncleavable procaspase-3 mutant (D3A)
Procaspase-3 D3A was expressed and purified exactly as previously reported (10).
Procaspase-3 Activation
Recombinantly expressed, zinc-free procaspase-3 D3A (7.5 µM) in Chelex® treated Hepes-NaCl was incubated in the presence of ZnSO4 (10 µM) and the basal activity was assessed by addition of Ac-DEVD-pNA substrate (200 µM) and monitored at 405 nm with a Spectramax plate reader (Molecular Devices, Sunny Vale, CA). After the basal activity was determined, DMSO, PAC-1 or S-PAC-1 was added to each sample to a final concentration of 50 µM. Activity of each stock was assessed as described above every 20 minutes. The slope of each data set was used to determine the activity of the protein. Protein activity was normalized to a percent activity at each time point using a zinc-free sample as 100% activity and the DMSO control as 0% activity controls.
Apoptosis and Cytotoxicity Assays
Induction of apoptosis was assessed exactly as previously reported(10). Cell death IC50 values were assessed by sulforhodamine B assay exactly as previously reported(10) or by MTS assay (Promega) according to the manufacturer’s suggested protocol.
Toxicity determinations of PAC-1 and S-PAC-1
PAC-1 and S-PAC-1 were formulated in 2-hydroxypropyl-β-cyclodextrin (HPβCD)(14) as described in the supplementary data. C57/BL6 mice were administered varying doses of either PAC-1 or S-PAC-1 via tail vein injection. Mice were monitored by observation for a period of 24 hours for signs of toxicity including sensitivity to touch, hypothermia, hunched posture, agitation, and rapid or depressed breathing.
Pharmacokinetics of S-PAC-1 in healthy research dogs and dogs with lymphoma
Four healthy male hound dogs weighing at least 30 kg were used for all preclinical pharmacokinetic studies. All dogs received a single dose of S-PAC-1 via IV injection. After a two week washout period three dogs received a dosing regimen predicted to achieve and maintain a steady state plasma concentration of 10 µM, that is, IV S-PAC-1 delivered as a constant rate infusion comprising a 10-minute loading dose, followed by a constant maintenance dose for 24 hours. Dogs with lymphoma were treated with IV S-PAC-1 in a constant rate infusion for either 24 hours or 72 hours. For all dogs and dosing strategies, blood was drawn from the femoral vein, centrifuged, and plasma stored at −80°C until analysis.
Toxicological Assessment of S-PAC-1 in healthy research dogs
Toxicity of S-PAC-1 administration was determined by serial weekly complete blood counts, serum biochemistry panels, and animal caregiver gastrointestinal toxicity observational scores adhering to the Veterinary co-operative oncology group common terminology criteria for adverse events (VCOG-CTCAE).
Antitumor assessment of S-PAC-1 in dogs with lymphoma
Caliper measurement was performed according to the RECIST method(15). Briefly, the longest linear length measurement was recorded for 4 pairs of peripheral lymph nodes (mandibular, prescapular, inguinal, and popliteal), with the summation of these values giving a RECIST score. In addition to caliper measurements for all four sets of peripheral lymph nodes, computed tomography (CT) scans were performed of the mandibular lymph nodes in every patient, allowing for accurate and objective measurement of maximal lymph node linear length.
Results
In vivo toxicity of PAC-1
In an effort to characterize the feasibility and tolerability of intravenously administered PAC-1, toxicity testing of PAC-1 (when administered in HPβCD via tail vein injection) was carried out with C57/BL6 mice and is summarized in Table S1. Mice that received PAC-1 exhibited transient neurotoxicity with onset occurring within 5 minutes of drug administration and resolution occurring within 2 hours. Mice that received only the HPβCD vehicle did not exhibit any clinically observable toxicity. The observed neurotoxicity of PAC-1 in mice did not appear to be species specific, as grand mal seizure activity in one healthy dog was elicited when administered 25 mg/kg of PAC-1 as a 5-minute intravenous infusion. Adverse side effects in this dog were transient and not life-limiting.
Given the known affinity of PAC-1 for zinc in vitro(9), and the data suggesting that PAC-1 binds cellular zinc (10), we hypothesized that the neurotoxicity observed in mice and dogs administered PAC-1 in HPβCD was caused by the chelation of intracellular zinc at NMDA receptors within the central nervous system (CNS). This hypothesis is consistent with data from in vivo studies of other zinc chelators that induce a neurological phenotype reminiscent of what we observed with PAC-1(16, 17). Indeed, in silico analysis of PAC-1 to predict the partitioning across the blood-brain barrier (BBB)(18) shows that PAC-1 has a calculated logBB of −0.07. This logBB would correlate to a partitioning ratio of 1.0:0.85 between the blood and the brain, suggesting that a significant amount of PAC-1 may be entering the CNS. Several studies have indicated that chelation of intracellular zinc stores relieves tonic suppression of NMDA receptors, resulting in neuronal hyperexcitation(17, 19).
Design of S-PAC-1
In an effort to overcome the undesirable side effect of neurotoxicity, we hypothesized that a derivative of PAC-1 that had a lower propensity to cross the BBB would exhibit decreased neurotoxicity and allow for dosing at higher concentrations. Based on the structure-activity relationship previously reported(10), it was predicted that a polar functional group installed on the benzyl ring of PAC-1 should decrease the predicted logBB while maintaining the activity of the parent compound. As such S-PAC-1 (Figure 1A), a sulfonamide derivative of PAC-1, was designed as a compound predicted to have a markedly decreased ability to cross the BBB (logBB of −1.26, providing a predicted blood:brain ratio of 1.0:0.055). See supplementary data for the synthetic route to S-PAC-1.
S-PAC-1 binds zinc in vitro
The activity of S-PAC-1 was characterized in several biochemical assays analogous to previously performed PAC-1 studies(9, 10). An EGTA competition titration experiment(13) was used to determine the binding constant for the S-PAC-1:Zn2+ complex. In the presence of EGTA, the changes in the fluorescence of the S-PAC-1:Zn2+ complex were used to plot a formation curve (Figure 1B). Using the known binding constant of EGTA, the free zinc concentration can be calculated and used to determine the binding constant of the S-PAC-1:Zn2+ complex. This complex has a Kd of 46 ± 5 nM compared to 52 ± 2 nM for PAC-1:Zn2+(10).
S-PAC-1 activates procaspase-3 in vitro
The ability of S-PAC-1 to activate recombinantly expressed procaspase-3 in the presence of exogenous zinc was assessed in vitro. To ensure that the enzymatic activity of the proenzyme was being monitored, a proteolytically uncleavable mutant of procaspase-3 was used in which the three aspartic acid cleavage site residues are mutated to alanine (D9A/D28A/D175A)(9, 20). This uncleavable form of procaspase-3 in not capable of processing to the mature active caspase-3, thus ensuring that any increase in activity observed is due to an increase in the activity of the proenzyme rather than autoproteolysis of the proenzyme. As shown in Figure 1C, S-PAC-1 rapidly (within 5 minutes) enhances the enzymatic activity of the proenzyme by relief of zinc-mediated inhibition, although to a lesser degree than PAC-1.
S-PAC-1 induces death in multiple cancer cell lines in culture
Having confirmed that S-PAC-1 chelates zinc and activates procaspase-3 in vitro, the antineoplastic activity of S-PAC-1 was assessed against a panel of human, canine, and murine cancer cell lines using the sulforhodamine B assay(21). The 72 hour IC50 values for PAC-1 and S-PAC-1 are reported in Table 1. Both PAC-1 and S-PAC-1 have micromolar cytotoxic IC50 values against all lymphoma cell lines tested regardless of the species of origin. U-937 cells treated with DMSO, 50 µM PAC-1 or 50 µM S-PAC-1 for 12 hours were assessed by annexin V/PI staining and analyzed by flow cytometry (Figure 1D). Both PAC-1 and S-PAC-1 treatment lead to a similar increase in the population of apoptotic cells (annexin V positive, PI negative).
Table 1.
Assessment of PAC-1 and S-PAC-1 cytotoxicity in cancer cell lines. Cells were exposed to compound for the time indicated, compound was washed out, and cell viability/biomass assessment was made after 72 hours.
| Cell line | Species | Origin | Exposure Time (h) |
S-PAC-1 72 hour IC50 (µM) |
PAC-1 72 hour IC50 (µM) |
|---|---|---|---|---|---|
| U-937 | Human | Lymphoma | 1 | >100 | -- |
| 3 | >100 | -- | |||
| 6 | >100 | -- | |||
| 9 | 20 ± 12 | -- | |||
| 12 | 9.7 ± 1.1 | -- | |||
| 24 | 5.9 ± 1.0 | -- | |||
| 48 | 5.6 ± 0.8 | -- | |||
| 72 | 6.4 ± 0.8 | 9.3 ± 0.5 | |||
| EL-4 | Mouse | Lymphoma | 72 | 7.1 ± 1.3 | 3.8 ± 0.9 |
| 17–71a | Dog | Lymphoma | 72 | 2.7 ± 0.8 | 2.5 ± 0.9 |
| GL-1 | Dog | Lymphoma | 72 | 7.1 ± 0.3 | 4.9 ± 0.3 |
| OSW | Dog | Lymphoma | 72 | 11.0 ± 0.9 | 8.6 ± 1.3 |
| Jurkat | Human | Leukemia | 72 | 4.5 ± 1.1 | 5.7 ± 2.8 |
| SK-Mel-5 | Human | Melanoma | 72 | 8.6 ± 1.3 | 11.5 ± 3.6 |
| HeLa | Human | Cervical | 72 | 28.4 ± 7.7 | 15.5 ± 3.8 |
| MDA-MB-231 | Human | Breast | 72 | 11.7 ± 5.3 | 9.9 ± 1.0 |
This cell line was assessed using the MTS cell viability assay. All other cell lines were assessed with the sulforhodamine B assay.
Error is SEM, n = 3
In an effort to determine an appropriate treatment strategy for the evaluation of S-PAC-1 in vivo, the time dependency of S-PAC-1 cytotoxicity was evaluated. U-937 cells were treated with S-PAC-1 for various lengths of time. After treatment with S-PAC-1, cells were washed to remove compound and were cultured in growth media without compound. Cell death was assessed at 72 hours for all treatment times. An IC50 value was determined for each exposure time and reported in Table 1. At times shorter than 6 hours, the IC50 value was greater than the highest concentration tested. Between 12 and 24 hours the IC50 value rapidly decreased to a minimum and showed little variation over the course of the subsequent 48 hours. These time dependency experiments suggest that S-PAC-1 will be most effective in vivo if cancer cells are exposed to the compound for at least 24 hours.
S-PAC-1 has no detectable neurotoxic effect in mice
Having confirmed the activity of S-PAC-1 in vitro and in cell culture, the toxicity of S-PAC-1 (when administered in HPβCD via tail vein injection) was assessed in C57/BL6 mice and is summarized in Table S1. S-PAC-1 exhibited no observable toxicity at any dose tested. Given the dramatically reduced neurotoxic effect of S-PAC-1, pharmacokinectic analysis was performed to compare the plasma concentrations achievable with PAC-1 and S-PAC-1. Mice were treated with 20 mg/kg PAC-1 (the dose at which significant neurological symptoms first appear), 50 mg/kg PAC-1 (the dose where acute neurological symptoms are present), and 350 mg/kg S-PAC-1 (the highest dose tested) via tail vein injection. Serum was analyzed by HPLC to provide the pharmacokinetic profiles for S-PAC-1 and PAC-1 (Figure 2A). At the PAC-1 dosage that induced mild neurotoxicity (20 mg/kg), peak plasma concentrations were ~50 µM. In contrast, a 350 mg/kg dose of S-PAC-1 provided peak plasma levels of ~3500 µM without any signs of neurotoxicity.
Figure 2.
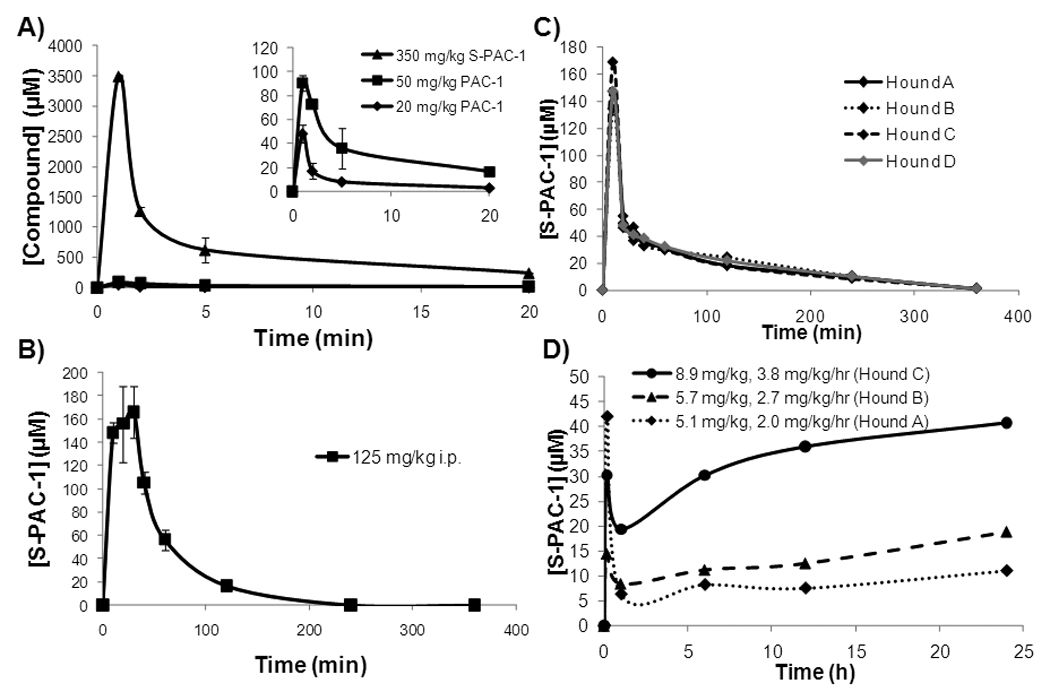
Serum concentrations of PAC-1 and S-PAC-1 in mice, and of S-PAC-1 in mice and dogs A, C57/BL6 mice were treated with 20 or 50 mg/kg PAC-1, or 350 mg/kg S-PAC-1 via tail vein injection. Mice were sacrificed at the times indicated, blood was drawn, and the concentration of drug was determined by HPLC. For PAC-1 error bars represent range (n=2), for S-PAC-1 error bars represent SEM (n=3). B, The serum concentration of S-PAC-1 after i.p. administration to mice. C57/BL6 mice were treated i.p. with 125 mg/kg S-PAC-1, sacrificed at the times indicated, blood was drawn, and S-PAC-1 serum concentrations determined by HPLC. Administration of 125 mg/kg S-PAC-1 provides a peak plasma concentration of ~170 µM and a half-life of ~1 hour. Error bars represent SEM (n=3). C, Four research hound dogs received a single intravenous dose of S-PAC-1 (25 mg/kg), blood was drawn at the time indicated, and S-PAC-1 serum concentration was determined by HPLC. D, Three healthy hound dogs were each administered a different dose of S-PAC-1 by continuous i.v. infusion. Dogs received an initial loading dose (mg/kg, indicated in inset) over the course of 10 minutes followed by a constant rate infusion (mg/kg/h) over the next 24 hours. Blood was drawn at the times indicated, and the S-PAC-1 serum concentration was determined by HPLC. Continuous infusion of S-PAC-1 was well-tolerated and successful in establishing a steady state serum concentration of S-PAC-1 in dogs.
S-PAC-1 has a short half-life in mice
S-PAC-1 (125 mg/kg) was administered to mice via i.p. injection and found to have a short half-life (~1 hour, Figure 2B); it was predicted that the mice would need to be treated with a 350 mg/kg dose of S-PAC-1 every two hours to achieve and maintain a minimum plasma concentration of 10 µM over a course of 24 hours. Although this frequent dosing regimen for S-PAC-1 (i.p. every 2 hours for 24 hours) was technically feasible, further evaluation of S-PAC-1 as a novel therapeutic agent using murine tumor models was not methodologically practical. As such we sought to further investigate S-PAC-1 in a larger mammalian experimental system, specifically healthy and spontaneous cancer-bearing dogs, which conferred greater practicality for the maintenance of S-PAC-1 steady state concentrations for prolonged periods of time.
Assessment of S-PAC-1 in research dogs
Healthy research hound dogs were utilized for pharmacokinetic and toxicity investigations of S-PAC-1. First, four research hound dogs were treated with 25 mg/kg S-PAC-1 (solublized in HPβCD) via IV injection over 10 minutes. In addition to pharmacokinetic analysis, the hematologic and non-hematologic tolerability of single-dose, intravenous S-PAC-1 administration was monitored in research dogs weekly for 4 consecutive weeks (Table S2A). As shown in Figure 2C, the peak plasma concentration resulting from this 25 mg/kg i.v. bolus dose was ~150 µM. From analysis of the pharmacokinetic profile, the half-life of S-PAC-1 in dogs was calculated to be 1.09±0.02 hours (Table 2). Additionally, single-dose, intravenous S-PAC-1 treatment was well-tolerated by all 4 research dogs and no short or long term adverse events were observed with these animals as a result of treatment.
Table 2.
Non-compartmental pharmacokinetic analysis of S-PAC-1 (25 mg/kg) in four healthy research dogs.
| Parameters | Hound A | Hound B | Hound C | Hound D |
|---|---|---|---|---|
| Body weight (kg) | 33.2 | 34.1 | 34.2 | 32.5 |
| Lambda_z half-life (h) | 1.05 | 1.11 | 1.11 | 1.09 |
| Tmax (h) | 0.17 | 0.17 | 0.17 | 0.17 |
| Cmax (ug/ml) | 69.86 | 63.71 | 80.07 | 69.83 |
| AUC0-inf h*ug/ml | 73.56 | 74.60 | 82.19 | 78.81 |
| AUMC0-inf h*h*ug/ml | 93.63 | 104.77 | 90.48 | 101.78 |
| Cls (ml/h/kg) | 340 | 335 | 304 | 317 |
| Vss (ml/kg) | 432.5 | 470.8 | 334.8 | 410 |
| MRT (h) | 1.27 | 1.40 | 1.10 | 1.29 |
Based on the half-life of 1.09±0.02 hours in dogs, continuous-rate infusion was explored in an attempt to maintain a steady-state serum concentration of S-PAC-1 during the course of the treatment. Three healthy research dogs were utilized to determine if S-PAC-1 could be safely administered via a continuous-rate infusion regimen, and to determine appropriate dosing levels to maintain plasma concentrations above ~10 µM. Each dog received a different dose of S-PAC-1 (Figure 2D) with an initial loading dose via IV infusion over the course of 10 minutes followed by a maintenance dose delivered by an infusion pump for an additional 24 hours. Each dog was observed throughout the course of their 24-hour infusions for adverse reactions and blood was drawn at intervals to assess the pharmacokinetic profile of S-PAC-1 treatment. In addition, following completion of S-PAC-1 infusion, research dogs were evaluated for hematologic and non-hematologic toxicity weekly for 4 consecutive weeks. During this period, no dogs exhibited hematological parameters outside of normal ranges (Table S2B) and no adverse events were reported by animal care staff.
S-PAC-1 administered as a 24-hour continuous-rate infusion can be safely given to research dogs and easily reaches micromolar steady-state plasma concentrations that correlate with dose escalation (Figure 2D). Based on these results it was predicted that a 7 mg/kg loading dose and 3 mg/kg/h constant rate infusion would be sufficient to achieve a steady state plasma concentration of ~10 µM.
Assessment of S-PAC-1 in dogs with lymphoma
Having confirmed the in vitro activity of S-PAC-1, shown that the compound can be safely administered via continuous-rate infusion, and that steady state plasma concentrations of S-PAC-1 of >10 µM may be achieved for a 24 hour duration, a small (n=6) clinical trial of client-owned pet dogs with spontaneous lymphoma was conducted. The aim of this trial was to demonstrate the feasibility of dosing S-PAC-1 in dogs with lymphoma, and to determine if therapeutic serum levels of compound could be achieved via this dosing regimen. Pet dogs presented or referred to the Small Animal Clinic at the UIUC College of Veterinary Medicine were considered for enrollment in the clinical trial (see supplementary data for inclusion criteria). Dogs that entered the study received treatment with S-PAC-1 as a single entity drug for 4 weeks and were followed for an additional 2 weeks after drug withdrawal.
Toxicity and pharmacokinetics of S-PAC-1 in canine lymphoma patients
Six patient dogs received treatment with S-PAC-1 via one of two treatment regimens (summarized in Table 3). Patients enrolled in the first treatment regimen (n=3) received a once-a-week 24 hour continuous infusion of S-PAC-1 for 4 weekly cycles. Patients enrolled in the second treatment regimen (n=3) received a 72 hour continuous infusion of S-PAC-1 every other week for 2 treatment cycles. Blood was collected during each S-PAC-1 treatment cycle for pharmacokinetic analysis. In addition, blood was collected from all pet dogs at each scheduled follow up visit to characterize hematologic and non-hematologic toxicity. Between administrations, patients were monitored by pet owners for gastrointestinal toxicity observational scores adhering the Veterinary Co-operative Oncology Group Common Terminology and Criteria for Adverse Events (VCOG_CTCAE)(22). All patients enrolled in either treatment regimen did not demonstrate any clinically significant hematologic or non-hematologic toxicity (Table S2C and D). Only minor adverse events were reported by pet owners such as self-limiting and localized irritation at the infusion site (n=4), transient loss of appetite (n=1), and mild diarrhea (n=3). All adverse reactions subsided within 48 hours of the end of each treatment cycle. Serum analysis indicated that all patients had measurable serum concentrations of S-PAC-1, as shown in Figure 3. In accordance with our prediction from the healthy research dogs, an infusion with a loading dose of 7 mg/kg and a constant rate infusion of 3 mg/kg/h was sufficient to achieve steady-state plasma concentrations of >10 µM in the majority of treatments.
Table 3.
Characteristics of the six pet dogs with lymphoma treated with S-PAC-1.
| Patient | Breed | Male/Female | Age | Weight (lb) |
Immunophenotype | Prior Therapy |
Treatment | Outcome |
|---|---|---|---|---|---|---|---|---|
| 1 | Mixed Breed | Castrated Male | 7 | 79 | B-Cell Lymphoma | CHOP | 24 h | PR |
| 2 | Labrador Retriever | Spayed Female | 7 | 89 | T-Cell Lymphoma | Naïve | 24 h | SD |
| 3 | Newfoundland | Intact Male | 4 | 160 | T-Cell Lymphoma | Naïve | 24 h | SD |
| 4 | Welch Corgi | Spayed Female | 8 | 30 | B-Cell Lymphoma | Naïve | 72 h | SD |
| 5 | Golden Retriever | Spayed Female | 7 | 80 | B-Cell Lymphoma | Naïve | 72 h | PD |
| 6 | MC Boxer | Castrated Male | 6 | 59 | T-cell Lymphoma | Naïve | 72 h | PD |
PD, Progressive Disease; PR, Partial Response; SD, Stable Disease
Figure 3.
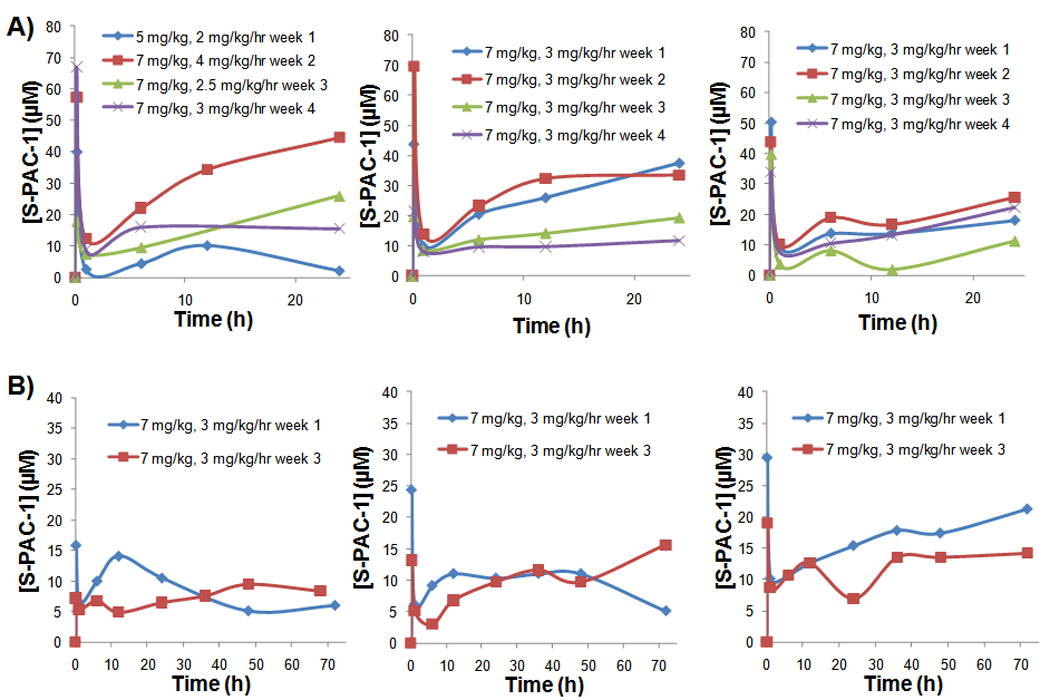
Serum concentrations of S-PAC-1 in six dogs with lymphoma enrolled in the clinical trial. Administration of S-PAC-1 as a constant rate infusion was generally successful in achieving the target plasma concentration (10 µM) for either 24 hours (A, dogs 1–3) or 72 hours (B, dogs 4–6). Data labels indicate doses (loading dose, mg/kg; continuous infusion dose, mg/kg/h) administered during the clinical trial. Blood was drawn at various times throughout drug administration and serum concentration was determined by HPLC.
Antitumor activity of S-PAC-1
Given the small number of patients included in this clinical study, antitumor activity of S-PAC-1 cannot be conclusively determined. However, of the 6 patients treated, one patient (patient 1) showed a partial response with a ~30% reduction in both caliper RECIST score and the mandibular lymph node measurements by CT scan over the course of the 4-week treatment (Figures 4A and B). During treatment, this dog received a 24-hour continuous intravenous infusion of S-PAC-1 once-a-week (total of 4 treatments). Following the four treatment cycles, drug administration was withdrawn and the dog was monitored by RECIST for 2 subsequent weeks. After discontinuation of S-PAC-1 treatment, the RECIST scores for this dog increased dramatically (day 36 and 42). In addition to this partial response, three patients showed stable disease (patients 2–4) while two patients showed disease progression (patients 5 and 6), as indicated in Table 3. The rapid increase in RECIST scores after drug withdrawal shown in patient 1 is representative of the effect of drug withdrawal in all patients.
Figure 4.
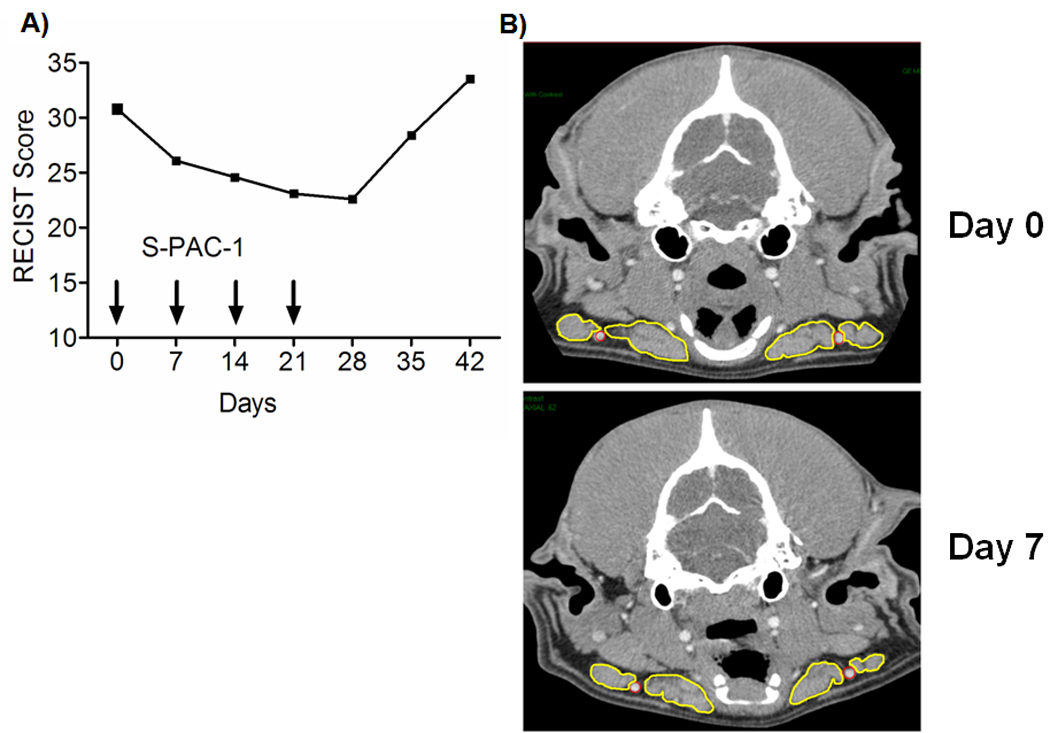
Activity of S-PAC-1 in patient 1. A, RECIST scores for patient 1 over the course of 7 weeks. Arrows indicate days during which patient received S-PAC-1 as a 24 hour continuous IV infusion (as detailed in text and Figure 3A). After drug withdrawal, tumor size increased rapidly (day 28 to 42). B, CT scans of the mandibular lymph nodes of patient 1 (outlined in yellow) demonstrate a clinically measurable decrease in tumor size one week after S-PAC-1 administration.
Analysis of lymph node aspirates from two dogs (patients 1 and 2) demonstrates the presence of the procaspase-3 drug target prior to treatment as well as an increase in cleaved caspase-3 after treatment with S-PAC-1 (Figure S1). Lymph node aspirates collected from these dogs with lymphoma consisted of greater than 90% malignant lymphocytes based upon cytologic evaluation. Based upon the predominance of malignant lymphocytes, the observed shift in cleaved caspase-3 after S-PAC-1 treatment (Figure S1) most likely represents the induction of apoptosis in malignant lymphocytes; however, a small percentage of this shift may be due to non-malignant lymphocytes and stromal cells.
Discussion
S-PAC-1 is the first compound in the PAC-1 class (and the first small molecule activator of procaspase-3) to be evaluated in a clinical trial of cancer patients. As such, the evaluation of S-PAC-1 in a clinical setting is proof-of-concept for the strategy of direct procaspase-3 activation as an anticancer therapy. Compounds in the PAC-1 class activate procaspase-3 via chelation of inhibitory zinc ions (10). Intracellular zinc is found principally in tightly-bound complexes in metalloproteases, zinc-finger domains, and other metal binding proteins; however, approximately 10% of cellular zinc is believed to exist in a loosely-bound, labile pool(23). A number of studies implicate labile zinc as an endogenous antiapoptotic regulator(24–26), and chelation of the loosely-bound pool of zinc is an emerging anticancer strategy(9, 10, 27). Thus, the evaluation of S-PAC-1 is also proof-of-concept for the chelation of the labile zinc pool as an anticancer therapy.
It is perhaps not surprising that high doses of PAC-1 induce neurotoxicity, given its zinc chelation properties and predicted BBB permeability. Zinc homeostasis is important in the CNS (28), and NMDA receptors require bound zinc to provide a tonic inhibition(17). NMDA receptors bind to zinc with a low affinity (Kd=5.5 µM)(29), which suggests that a zinc chelator with a higher affinity for zinc such as PAC-1 (Kd=52 nM) would be able to successfully sequester zinc from these receptors. Several studies indicate that intracellular zinc chelation results in hyperexcitation of these receptors(16), resulting in symptoms such as uncontrolled muscle movement and seizure(17, 30). One strategy for reducing the permeability of a compound through the BBB is to increase the polarity of the molecule(18). Addition of the sulfonamide functional group is a good candidate for such polarity increase as the aryl sulfonamide motif is common in a number of small molecule therapeutics. The dramatically different neurotoxic effects of S-PAC-1 and PAC-1 suggest that S-PAC-1 is considerably less BBB permeable, and may be the more attractive drug candidate.
Spontaneously arising cancers in pet dogs share many similarities with human cancers including histologic appearance, tumor genetics, molecular targets, biologic and clinical behavior, and response to therapy(31). Of these spontaneous canine cancers, multicentric lymphoma is the most common, occurring in 13 to 24 of every 100,000 dogs(32). The clinical progression and treatment of multicentric B-cell or T-cell canine lymphoma has many of the same characteristics of non-Hodgkin’s lymphoma in humans. Canine lymphoma and human non-Hodgkin’s lymphoma both respond clinically to the same cytotoxic drugs such as doxorubicin, vincristine and cyclophosphamide. These drugs are components of the CHOP treatment protocol, first line therapy for diffuse large B-cell lymphoma in humans(33). When administered to dogs, CHOP will induce complete clinical remission in approximately 90% of dogs diagnosed with lymphoma(34, 35). Similar to the human response, the majority of dogs who achieve remission with CHOP therapy will experience disease relapse(36). Given the commonalities between human and canine lymphoma, evaluation of S-PAC-1 in a canine lymphoma clinical trial may provide important translational information for the development of S-PAC-1 as a novel human therapeutic.
Although inconclusive in this study design, it is highly encouraging that four out of six patients achieved partial response or stable disease for 4 weeks in duration, as canine lymphoma is generally a rapidly progressive malignancy with dramatic enlargement of peripheral lymph nodes within weeks of disease diagnosis. As shown in Table 3, one dog enrolled in the current study (patient 1) was in remission for 5 months after CHOP therapy, and was recently diagnosed with recurrent lymphoma. Upon enrollment in the study, this dog showed a ~30% reduction in tumor size in response to S-PAC-1 treatment. This partial response is significant given the comparatively short treatment duration (4 weeks for S-PAC-1 vs. 19 weeks for CHOP), and that the dose of S-PAC-1 administered does not appear to be near the maximum tolerated dose.
In conclusion, we have discovered S-PAC-1 as a procaspase-activating compound that can be safely administered in vivo, and have identified pet dogs with lymphoma as a tractable model for the assessment of small molecule procaspase-3 activation as an anticancer strategy. Use of this large animal model allows drug administration via continuous rate IV infusion, something necessary with S-PAC-1 (due to its short half-life in vivo) and not practical in murine tumor models. S-PAC-1 induces apoptotic death in cultured cancer cells, with similar activity to the parent compound PAC-1. While high doses of PAC-1 (when administered in HPβCD) induce neurotoxicity in vivo, S-PAC-1 does not cause this effect at serum concentration ~70-fold higher. S-PAC-1 can be safely administered to mice, research dogs, and dogs with lymphoma, and shows encouraging clinical effect in this preliminary evaluation. Given the absence of neurotoxicity with S-PAC-1 and the large safety window observed in mice, it is anticipated that S-PAC-1 will prove to be safe at escalated doses, and thus its further evaluation in cancer patients is warranted.
Supplementary Material
Acknowledgement
We thank BioLine Rx for initial assistance with PAC-1 formulations. This research was supported by the National Institutes of Health (R01-CA120439) and UIUC. QPP was supported by a NIH Chemistry-Biology Interface Training Grant (NRSA 1-T32-GM070421) and by a predoctoral fellowship from the Medicinal Chemistry Division of the American Chemical Society. DCW was partially supported by NRSA F31-CA130138-01S1.
References
- 1.O'Donovan N, Crown J, Stunell H, et al. Caspase 3 in breast cancer. Clin Cancer Res. 2003;9:738–742. [PubMed] [Google Scholar]
- 2.Roy S, Bayly CI, Gareau Y, et al. Maintenance of caspase-3 proenzyme dormancy by an intrinsic "safety catch" regulatory tripeptide. Proc Natl Acad Sci. 2001;98:6132–6137. doi: 10.1073/pnas.111085198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krepela E, Prochazka J, Liul X, Fiala P, Kinkor Z. Increased expression of Apaf-1 and procaspase-3 and the functionality of intrinsic apoptosis apparatus in non-small cell lung carcinoma. Biol Chem. 2004;385:153–168. doi: 10.1515/BC.2004.034. [DOI] [PubMed] [Google Scholar]
- 4.Izban KF, Wrone-Smith T, Hsi ED, Schnitzer B, Quevedo ME, Alkan S. Characterization of the interleukin-1b-converting enzyme/Ced-3-family protease, caspase-3/CPP32, in Hodgkin's disease. Am J Pathol. 1999;154:1439–1447. doi: 10.1016/s0002-9440(10)65398-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakagawara A, Nakamura Y, Ikeda H, et al. High levels of expression and nuclear localization of interleukin-1 b converting enzyme (ICE) and CPP32 in favorable human neuroblastomas. Cancer Res. 1997;57:4578–4584. [PubMed] [Google Scholar]
- 6.Fink D, Schlagbauer-Wadl H, Selzer E, et al. Elevated procaspase levels in human melanoma. Melanoma Res. 2001;11:385–393. doi: 10.1097/00008390-200108000-00009. [DOI] [PubMed] [Google Scholar]
- 7.Persad R, Liu C, Wu T-T, et al. Overexpression of caspase-3 in hepatocellular carcinomas. Mod Pathol. 2004;17:861–867. doi: 10.1038/modpathol.3800146. [DOI] [PubMed] [Google Scholar]
- 8.Putt KS, Chen GW, Pearson JM, et al. Small-molecule activation of procaspase-3 to caspase-3 as a personalized anticancer strategy. Nat Chem Biol. 2006;2:543–550. doi: 10.1038/nchembio814. [DOI] [PubMed] [Google Scholar]
- 9.Peterson QP, Goode DR, West DC, Ramsey KN, Lee J, Hergenrother PJ. PAC-1 Activates Procaspase-3 in Vitro through Relief of Zinc-Mediated Inhibition. J Mol Biol. 2009;388:144–158. doi: 10.1016/j.jmb.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peterson QP, Hsu DC, Goode DR, Novotny CJ, Totten RK, Hergenrother PJ. Procaspase-3 Activation as an Anti-Cancer Strategy: Structure-Activity Relationship of PAC-1, and its Cellular Co-Localization with Procaspase-3. J Med Chem. 2009;52:5721–5731. doi: 10.1021/jm900722z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paoloni M, Khanna C. Translation of new cancer treatments from pet dogs to humans. Nat Rev Cancer. 2008;8:147–156. doi: 10.1038/nrc2273. [DOI] [PubMed] [Google Scholar]
- 12.Peterson QP, Goode DR, West DC, Botham RC, Hergenrother PJ. Preparation of the caspase-3/7 substrate Ac-DEVD-pNA by solution-phase peptide synthesis. Nat Protoc. 2010;5:294–302. doi: 10.1038/nprot.2009.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang S, Clark RJ, Zhu L. Highly sensitive fluorescent probes for zinc ion based on triazolyl-containing tetradentate coordination motifs. Org Lett. 2007;9:4999–5002. doi: 10.1021/ol702208y. [DOI] [PubMed] [Google Scholar]
- 14.Brewster ME, Simpkins JW, Hora MS, Stern WC, Bodor N. The potential use of cyclodextrins in parenteral formulations. J Parenter Sci Technol. 1989;43:231–240. [PubMed] [Google Scholar]
- 15.Padhani AR, Ollivier L. The RECIST (Response Evaluation Criteria in Solid Tumors) criteria: implications for diagnostic radiologists. Br J Radiol. 2001;74:983–986. doi: 10.1259/bjr.74.887.740983. [DOI] [PubMed] [Google Scholar]
- 16.María-Isabel D, José-Miguel B-I, Carlos C, Juan N, Ana-Isabel M-M, Francisco-José M-G. Neural Overexcitation and Implication of NMDA and AMPA Receptors in a Mouse Model of Temporal Lobe Epilepsy Implying Zinc Chelation. Epilepsia. 2006;47:887–899. doi: 10.1111/j.1528-1167.2006.00501.x. [DOI] [PubMed] [Google Scholar]
- 17.Domnguez MI, Blasco-Ibez JM, Crespo C, Marqus-Mar AI, Martnez-Guijarro FJ. Zinc chelation during non-lesioning overexcitation results in neuronal death in the mouse hippocampus. Neuroscience. 2003;116:791–806. doi: 10.1016/s0306-4522(02)00731-5. [DOI] [PubMed] [Google Scholar]
- 18.Clark DE. In silico prediction of blood-brain barrier permeation. Drug Discov Today. 2003;8:927–933. doi: 10.1016/s1359-6446(03)02827-7. [DOI] [PubMed] [Google Scholar]
- 19.Lavoie N, Peralta MR, 3rd, Chiasson M, et al. Extracellular chelation of zinc does not affect hippocampal excitability and seizure-induced cell death in rats. J Physiol. 2007;578:275–289. doi: 10.1113/jphysiol.2006.121848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bose K, Pop C, Feeney B, Clark AC. An uncleavable procaspase-3 mutant has a lower catalytic efficiency but an active site similar to that of mature caspase-3. Biochemistry. 2003;42:12298–12310. doi: 10.1021/bi034998x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protocols. 2006;1:1112–1116. doi: 10.1038/nprot.2006.179. [DOI] [PubMed] [Google Scholar]
- 22.Veterinary Co-operative Oncology Group. Common Terminology Criteria for Adverse Events (VCOG-CTCAE) following chemotherapy or biological antineoplastic therapy in dogs and cats v1.0. Vet Comp Oncol. 2004;2:195–213. doi: 10.1111/j.1476-5810.2004.0053b.x. [DOI] [PubMed] [Google Scholar]
- 23.Franklin RB, Milon B, Feng P, Costello LC. Zinc and zinc transporters in normal prostate and the pathogenesis of prostate cancer. Front Biosci. 2005;10:2230–2239. doi: 10.2741/1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aiuchi T, Mihara S, Nakaya M, Masuda Y, Nakajo S, Nakaya K. Zinc ions prevent processing of caspase-3 during apoptosis induced by geranylgeraniol in HL-60 cells. J Biochem (Tokyo) 1998;124:300–303. doi: 10.1093/oxfordjournals.jbchem.a022111. [DOI] [PubMed] [Google Scholar]
- 25.Chai F, Truong-Tran AQ, Ho LH, Zalewski PD. Regulation of caspase activation and apoptosis by cellular zinc fluxes and zinc deprivation: A review. Immunol Cell Biol. 1999;77:272–278. doi: 10.1046/j.1440-1711.1999.00825.x. [DOI] [PubMed] [Google Scholar]
- 26.Chimienti F, Seve M, Richard S, Mathieu J, Favier A. Role of cellular zinc in programmed cell death: temporal relationship between zinc depletion, activation of caspases, and cleavage of Sp family transcription factors. Biochem Pharmacol. 2001;62:51–62. doi: 10.1016/s0006-2952(01)00624-4. [DOI] [PubMed] [Google Scholar]
- 27.Huesca M, Lock LS, Khine AA, et al. A novel small molecule with potent anticancer activity inhibits cell growth by modulating intracellular labile zinc homeostasis. Mol Cancer Ther. 2009;8:2586–2596. doi: 10.1158/1535-7163.MCT-08-1104. [DOI] [PubMed] [Google Scholar]
- 28.Frederickson CJ, Koh J-Y, Bush AI. The neurobiology of zinc in health and disease. Nat Rev Neurosci. 2005;6:49–62. doi: 10.1038/nrn1671. [DOI] [PubMed] [Google Scholar]
- 29.Karakas E, Simorowski N, Furukawa H. Structure of the zinc-bound amino-terminal domain of the NMDA receptor NR2B subunit. EMBO J. 2009;28:3910–3920. doi: 10.1038/emboj.2009.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adler M, Dinterman RE, Wannemacher RW. Protection by the heavy metal chelator N,N,N',N'-tetrakis (2-pyridylmethyl)ethylenediamine (TPEN) against the lethal action of botulinum neurotoxin A and B. Toxicon. 1997;35:1089–1100. doi: 10.1016/s0041-0101(96)00215-2. [DOI] [PubMed] [Google Scholar]
- 31.Breen M, Modiano JF. Evolutionarily conserved cytogenetic changes in hematological malignancies of dogs and humans--man and his best friend share more than companionship. Chromosome Res. 2008;16:145–154. doi: 10.1007/s10577-007-1212-4. [DOI] [PubMed] [Google Scholar]
- 32.Dorn CR, Taylor DO, Schneider R. The epidemiology of canine leukemia and lymphoma. Bibl Haematol. 1970:403–415. doi: 10.1159/000391733. [DOI] [PubMed] [Google Scholar]
- 33.Kahl B. Chemotherapy combinations with monoclonal antibodies in non-Hodgkin's lymphoma. Semin Hematol. 2008;45:90–94. doi: 10.1053/j.seminhematol.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garrett LD, Thamm DH, Chun R, Dudley R, Vail DM. Evaluation of a 6-month chemotherapy protocol with no maintenance therapy for dogs with lymphoma. J Vet Intern Med. 2002;16:704–709. doi: 10.1892/0891-6640(2002)016<0704:eoacpw>2.3.co;2. [DOI] [PubMed] [Google Scholar]
- 35.Chun R, Garrett LD, Vail DM. Evaluation of a high-dose chemotherapy protocol with no maintenance therapy for dogs with lymphoma. J Vet Intern Med. 2000;14:120–124. doi: 10.1892/0891-6640(2000)014<0120:eoahcp>2.3.co;2. [DOI] [PubMed] [Google Scholar]
- 36.Rassnick KM, Mauldin GE, Al-Sarraf R, Mauldin GN, Moore AS, Mooney SC. MOPP chemotherapy for treatment of resistant lymphoma in dogs: a retrospective study of 117 cases (1989–2000) J Vet Intern Med. 2002;16:576–580. doi: 10.1892/0891-6640(2002)016<0576:mcftor>2.3.co;2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.