

Abstract
Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominant disorder characterized by endocrine tumors of parathyroids, pancreatic islets, and anterior pituitary. The MEN1 gene encodes a nuclear protein called menin. In MEN1 carriers inactivating mutations give rise to a truncated product consistent with menin acting as a tumor suppressor gene. However, the role of menin in tumorigenesis and its physiological functions are not known. Here, we show that menin inactivation by antisense RNA antagonizes transforming growth factor type β-mediated cell growth inhibition. Menin interacts with Smad3, and antisense menin suppresses transforming growth factor type β-induced and Smad3-induced transcriptional activity by inhibiting Smad3/4-DNA binding at specific transcriptional regulatory sites. These results implicate a mechanism of tumorigenesis by menin inactivation.
The multiple endocrine neoplasia type 1 (MEN1) gene, on chromosome 11q13 (1), and cDNA have been cloned (2, 3) and more than 300 independent germ-line and somatic mutations scattered throughout the protein coding region have been identified (4, 5). Somatic mutations have been found to a variable extent in sporadic endocrine tumors (see refs. 4 and 5 and references therein). Many of the mutations are clearly inactivating, giving rise to a truncated product. Consequently lack of menin caused by loss of both alleles leads to tumor development consistent with menin acting as a tumor suppressor gene. Furthermore, stable overexpression of menin in ras-transformed NIH 3T3 cells inhibits cell growth and tumor formation in nude mice (6). Although there is some evidence that menin function may be related to transcriptional regulation (7) or cell cycle control (8), the mechanism of action of menin in tumorigenesis and its physiological functions are not known.
The data of the present report provide evidence for a mechanism of tumorigenesis by menin inactivation. We demonstrate that menin inactivation by antisense RNA antagonizes transforming growth factor type β (TGF-β)-mediated cell growth inhibition. Furthermore, we show that menin interacts with the receptor-regulated Smad, Smad3, and antisense menin suppresses TGF-β-induced and Smad3-induced transcriptional activity by disrupting Smad3/4-DNA binding at specific transcriptional regulatory sites.
Materials and Methods
TGF-β, Antibodies, and cDNAs.
TGF-β1 was from Oncogene Science. The menin rabbit polyclonal antibody was raised against a decapentapeptide (synthesized by solid-phase chemistry at the Peptide Synthesis Facility of the Sheldon Biotechnology Centre of McGill University) corresponding to amino acids 476–489 of menin (a sequence that is completely conserved between human and rat) with an additional cysteine residue at the COOH terminus to conjugate it to keyhole limpet hemocyanin. The antiserum was immunoaffinity-purified before use, and by Western blot of a variety of cell lines of different species recognized human and rodent menin with similar affinity (8). The human menin cDNAs (untagged and flag-tagged) were constructed as described (8). For the antisense menin construct, human menin cDNA was cloned in an antisense orientation into the EcoRI site of pcDNA3.1(+). The menin NH2-terminal goat polyclonal antibody was raised against a decanonapeptide corresponding to amino acids 1–19 of menin, and the COOH-terminal goat polyclonal antibody was raised against a decanonapeptide corresponding to amino acids 591–610 of menin. These sequences are identical in human and rodent menin. These and the myc, Smad2/3, and Stat3 antibodies were from Santa Cruz Biotechnology. The flag antibodies were from Sigma. The myc-tagged Smad2 and Smad3, and flag-tagged Smad4 cDNA constructs were prepared as described (9).
Stable Transfections.
GH4C1 and GH3 cells were stably transfected with Lipofectamine (GIBCO). After 48 h, cells were passaged, and clones were selected in DMEM supplemented with G418 (0.6 mg/ml) (GIBCO) and 10% FBS.
Transient Transfections.
GH4C1, HepG2, Chinese hamster ovary, and COS-7 cells were seeded at 2.5 × 106 cells/100-mm tissue culture dish and incubated for 12–24 h. DNA was transfected by SuperFect (Qiagen, Mississauga, ON, Canada), and cells were harvested 48 h later.
Reverse Transcription–PCR.
Five-microgram RNA samples were reverse-transcribed and subjected to standard PCR procedures. Primers used were as follows: human menin, sense, 5′-GGAAGACGACGAGGAGATCTACA-3′; antisense, 5′-CAGTAGTTCAGAGGCCTTTGCGCT-3′; neomycin-resistance gene, sense, 5′-CAACCGCAATGTGCGTGAAG-3′, antisense, 5′-TGGCCACCAGTAGCTCCTTC-3′; glyceraldehyde-3-phosphate dehydrogenase, sense, 5′-CCCTTCATTGACCTCAACTACATGGT-3′, antisense, 5′-GAGGGGCCATCCACAGTCTTCTG-3′.
Cell Proliferation Assays.
Cell number estimations, [3H]thymidine incorporation (TdR), and the colorimetic tetrazolium salt MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] assays were done according to standard procedures.
3TPLux Transcription Assay.
HepG2 and GH4C1 cells were seeded at a density of 2 × 105 per 6-well plate. Cells were transfected 24 h after with 5 μg of the reporter plasmid (p3TP-Lux) together with the effector plasmids (3 μg) and the pCH110 plasmid expressing β-galactosidase (1 μg), using Lipofectamine. Fifteen hours later, the medium was changed to DMEM containing 4% FCS, and the cells were incubated for an additional 9 h. Thereafter, cells were cultured for 24 h in the absence or presence of TGF-β in DMEM containing 0.2% FCS. Cells were lysed, and the luciferase activity was measured and normalized to the relative β-galactosidase activity as described (9).
Immunoprecipitations.
Cell lysates were made and immunoprecipitated overnight at 4°C with anti-c-myc (9E10) or anti-flag antibody (M2) and 20 μl of proteinA-G agarose as described (9).
Western Blot Analysis.
Cell lysates were made, electrophoresed on SDS gels, and immunoblotted as described (8). Within any one experiment, identical amounts of protein (as assessed by Bradford assay) were loaded in each lane.
Subcellular Fractionation.
Trypsinized and washed Chinese hamster ovary or GH4C1 cells were collected by centrifugation. Subcellular fractionation and collection of nuclear pellets were as described (8).
Electrophoretic Mobility-Shift Assay (EMSA).
EMSAs were performed as described with probe A (10).
Statistical Analysis.
Results are expressed as mean ± SEM. Differences were assessed by one-way ANOVA or the unpaired t test, where appropriate. A P value less than 0.05 was considered significant.
Results and Discussion
Anterior Pituitary Cell Lines Stably Expressing Antisense Menin cDNA.
To explore the function of menin, GH4C1 cells, a rat anterior pituitary cell line, were stably transfected with an antisense menin cDNA (Fig. 1a). To detect the expression of menin, we used a specific polyclonal antibody that recognizes menin as a 69-kDa species (8). As shown in Fig. 1b, the expression of rat (endogenous) menin protein in antisense-menin-transfected GH4C1 cells is much less than in empty vector-transfected cells. By reverse transcription–PCR, the neomycin-resistance gene transcript is detected only in empty vector- or antisense-transfected cells, but not wild-type cells (data not shown), and human menin transcripts (derived from the antisense cDNA) are detected only in antisense menin-transfected cells (Fig. 1c). These findings indicate that the antisense menin mRNA is appropriately expressed and effectively reduces endogenous menin expression. Similar data were obtained in another GH4Cl cell clone as well as a clone of GH3 cells, another rat anterior pituitary cell line (data not shown).
Figure 1.

Endogenous menin expression is suppressed in GH4C1 cells stably transfected with antisense menin cDNA. (a) Scheme for construction of the antisense menin vector. MCS, multiple cloning site; Neomycin, neomycin resistance gene; PCMV, human cytomegalovirus immediate-early promoter/enhancer; E, EcoRI; S, SacI. The asterisk indicates the stop codon at position 611 in the menin cDNA. (b) Menin expression in empty vector-transfected (V)- and antisense menin (AS)-transfected GH4C1 cells was assessed by immunoblotting. Equivalent amounts of total protein were loaded in each lane. (c) Human menin antisense transcripts were assessed by reverse transcription–PCR. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (d and e) TGF-β stimulates menin expression in GH4C1 cells. Serum-starved cells were cultured (d) in the presence of various concentrations of TGF-β for 1 h, (e) in the presence of 100 pM TGF-β for the indicated times, or (f) in the presence of 100 pM TGF-β for 1 h. Total cell lysates (30 μg) were separated by SDS/PAGE, immunoblotted, and revealed with anti-menin antibody. As a control for protein loading, membranes were probed with an anti-Stat3 antibody.
Reduced Menin Expression Antagonizes TGF-β-Mediated Inhibition of Cell Proliferation.
TGF-β causes growth inhibition of most epithelial, endothelial, fibroblast, neuronal, lymphoid, and hematopoietic cells (11). TGF-β is considered to regulate the proliferation of pituitary cells by inhibiting their growth in an autocrine or paracrine manner (12). There is evidence that oncoproteins abrogate normal cellular growth control by blocking the TGF-β signaling system (13, 14). As menin is a putative tumor suppressor gene we hypothesized that menin also may be involved in the growth inhibitory actions of TGF-β in pituitary cells. We first examined whether TGF-β would affect the expression of menin in GH4C1 cells. As shown in Fig. 1d, TGF-β increases the expression of menin in a dose-dependent fashion. The stimulation of menin expression is a very rapid event, occurring within 30 min of TGF-β stimulation and reaching its maximum at 1–3 h (Fig. 1e). In serum-freed GH4C1 cells stably transfected with antisense menin TGF-β's ability to stimulate menin accumulation was blocked (Fig. 1f). These data raised the possibility that menin inactivation might interfere with TGF-β signaling pathways. We, therefore, examined the effects of menin inactivation on TGF-β-mediated cell growth inhibition by using the antisense RNA strategy. There is no difference in cell proliferation between empty vector-transfected and antisense menin-transfected GH4C1 cells (Fig. 2a). TGF-β significantly inhibits cell growth, as measured by cell number (Fig. 2b), TdR (Fig. 2c), and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide assay (Fig. 2d) in empty vector-transfected cells. However, the presence of the antisense menin clearly antagonizes these inhibitory effects of TGF-β on cell proliferation (Fig. 2 b–d). Forskolin, which activates the protein kinase A signaling pathway, inhibits cell growth in both empty vector-transfected and antisense menin-transfected cells, indicating that the protein kinase A signaling pathway is not disrupted in the menin-deficient cells. Similar results were obtained in two other independent clones of antisense menin-transfected GH4C1 and GH3 cells (data not shown). This finding demonstrates that reduced menin expression specifically antagonizes TGF-β-mediated inhibition of cell proliferation in endocrine cells.
Figure 2.

Antisense menin blocks TGF-β-induced inhibition of pituitary cell proliferation. (a) Serum-starved GH4C1 cells [empty vector (V)- or antisense menin (AS)-transfected] were cultured with 5% FBS for the indicated times. Then, TdR was measured 4 h after addition of 2.5 μCi/well of [3H]thymidine. (b) Serum-starved GH4C1 cells were cultured with or without 100 pM TGF-β or 10 μM forskolin for 72 h. Then, cell numbers were counted. (c) TdR was measured. (d) Colorimetric tetrazolium salt MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] assay was performed. Each value represents mean ± standard error. n = 4; *, P < 0.01, TGF-β- or forskolin-treated compared with control, and, shown as a bar, P < 0.05, TGF-β-treated (AS) versus V.
Menin Inactivation Inhibits TGF-β-Induced-Transcriptional Activity.
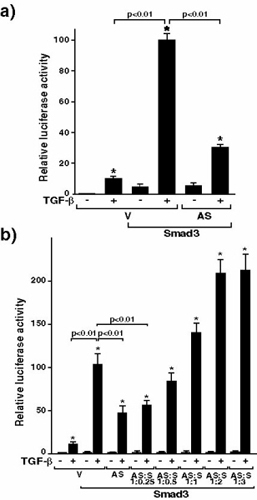
Menin is located primarily in the nucleus (7, 8) and is known to repress JunD-mediated transcriptional activity (15). It therefore seemed likely that menin would be a transcriptional regulator of other genes, and menin inactivation also might affect TGF-β-mediated transcriptional activity. To address this issue, we examined the effects of menin antisense on TGF-β-mediated transcriptional responses. HepG2 cells, a human hepatoma cell line in which TGF-β effects have been well described (16), were transfected with the TGF-β-responsive, plasminogen activator inhibitor 1 (PAI-1) promoter containing 3TP-Lux construct (17, 18). As shown in Fig. 3a, 200 pM TGF-β greatly increases luciferase activity in the absence of antisense menin cotransfection in HepG2 cells. However, antisense menin cotransfection, which reduces menin expression as assessed by immunoblotting (Fig. 3b), significantly blocks TGF-β-induced transcriptional activity. There is little effect of antisense menin on the basal promoter activity of 3TP-Lux. The specificity of the antisense menin was demonstrated by the restoration of transcriptional activity with cotransfection of increasing amounts of the sense menin construct (Fig. 3a). This demonstrates that menin inactivation specifically inhibits TGF-β-induced transcriptional activity.
Figure 3.

Antisense menin inhibits TGF-β-mediated transcriptional responses and menin specifically binds Smad3. (a) 3TP-Lux was transfected into HepG2 cells together with empty vector (V) or antisense menin (AS) either alone or with sense menin (S) and the cells were stimulated (+) or not (−) with 200 pM TGF-β. Relative luciferase activity was measured. Values of relative luciferase activity represent the mean ± standard error of three separate experiments. *, P < 0.01, TGF-β + versus − and, shown as a bar, P < 0.01, AS versus V. (b) Menin expression in empty vector (V)- and antisense menin (AS)-transfected HepG2 cells assessed by immunoblotting. (c–e) Association of menin and Smad proteins. W, Western blot; IP, immunoprecipitation. Menin was transfected into COS7 cells with the indicated myc-tagged Smad constructs (c and e) or flag-tagged Smad4 (d and e). Cell extracts were immunoprecipitated with anti-myc (c), anti-flag (d), or anti-Smad2/3 antibodies (e) followed by immunoblotting with menin (c–e) or anti-flag (d) antibodies. Expression of myc, flag, or menin was monitored.
Menin Interacts with Smad3.
The Smad family proteins are critical components of the TGF-β signaling pathways (19). TGF-β exerts growth inhibitory and transcriptional responses through the two receptor-regulated Smads, Smad2 and Smad3 (20). Receptor-mediated phosphorylation of Smad2 or Smad3 induces their association with the common partner Smad4 followed by translocation into the nucleus where these complexes activate transcription of specific genes (19). To identify the target through which antisense menin blocks the TGF-β signal pathway we examined whether menin would interact with the Smad proteins. COS cells were transiently transfected with the DNA coding for myc-tagged Smad2 or Smad3, or flag-tagged Smad4 (9, 21) in the presence or the absence of the menin cDNA. As shown in Fig. 3 c and d, menin specifically coimmunoprecipitates with Smad3 but not with Smad2 or Smad4, despite equal levels of expression of Smad proteins (Fig. 3 c and d Bottom). Smad2 and Smad4 or Smad3 and Smad4 were transfected into COS cells, and the cells stimulated with TGF-β. As shown in Fig. 3e, both Smad2 and Smad3 can coimmunoprecipate with Smad4 in response to TGF-β, confirming the functional ability of the Smad constructs in our cell system. Flag-tagged menin also was coimmunoprecipitated with Smad3 (data not shown). These findings indicate that menin interacts physically with Smad3.
To locate the Smad3 binding domain of menin, we examined the abilities of specific deletion mutants of menin to bind Smad3. The structures of these deletion constructs are shown in Fig. 4a. Menin Δ(1–40) and Menin Δ(278–477), which demonstrate nuclear localization, but not JunD binding (7, 15), retained the ability to bind Smad3 (Fig. 4b). This finding indicates that nuclear localization of menin, but not JunD binding with menin, is important for the interaction of menin and Smad3. All Smad proteins share structural conservation of amino-terminal and carboxyl-terminal domains, designated MH1 and MH2, respectively (19). Although the MH1 domain of Smad3 is important for DNA interaction, the MH2 domain is involved in interaction with other transcriptional regulators as well as in transcriptional activity of Smad3 (19). A mutant form of Smad3, in which the MH2 domain was removed (Smad3ΔC) (Fig. 4a), is unable to interact with menin (Fig. 4c), suggesting that the MH2 region is essential for the interaction of menin and Smad3.
Figure 4.

MH2 domain of Smad3 is required for menin interaction. (a) Schema of full-length menin and its deletion mutants, and Smad3 and its deletion mutant. (b) Structural requirement of menin for the interaction with Smad3. Myc-Smad3 was transfected into COS7 cells with the indicated menin deletion mutants. Total cell extracts were immunoprecipitated with anti-myc antibody followed by SDS/PAGE, immunoblotting, and staining with antibodies against amino- or carboxyl-terminal menin. Expression of myc or menin was monitored. (c) Structural requirement of Smad3 for the interaction with menin. Menin was transfected into COS7 cells with myc-Smad3 or myc-Smad3ΔC mutant. Cell extracts were immunoprecipitated with anti- myc antibodies followed by SDS/PAGE and immunoblotting with anti- menin antibody. Expression of myc or menin was monitored. W, Western blot; IP, immunoprecipitation.
Antisense Menin Inhibits Smad3-Mediated Transcriptional Activity.
It is known that Smad3 can activate the PAI-1 promoter (in 3TP-Lux) by itself (22). To explore the functional relationship of Smad3 and menin we examined whether antisense menin would affect Smad3-mediated transcriptional activity. As shown in Fig. 6a, which is published as supplemental material on the PNAS web site, www.pnas.org, Smad3 overexpression increased transcriptional activity itself and augmented TGF-β-induced transcriptional activity in HepG2 cells, and antisense menin significantly inhibited the Smad3-mediated transcriptional activity. In GH4C1 cells, antisense menin likewise inhibited the normal TGF-β-stimulated transcriptional activity (Fig. 6b). The specificity of the antisense menin effect was demonstrated by the rescue of the transcriptional activity with cotransfection of the sense menin construct in HepG2 cells (data not shown) or GH4C1 cells (Fig. 6b). These data suggest that menin functionally interacts with Smad3 and menin inactivation blocks Smad3-mediated transcriptional effects in the TGF-β pathway. The demonstration of menin-Smad3 complex formation indicates that menin plays an important role in supporting TGF-β and Smad3 transcriptional control of cell growth, and that menin inactivation disrupts TGF-β-mediated transcription and growth inhibition. Recent reports show that the oncoproteins, Ski1 and SnoN, inhibit the TGF-β signaling pathway through a Smad transcriptional corepressor (23, 24). It is conceivable therefore that menin also may block the effect of corepressors, allowing TGF-β signaling.
Antisense Menin Does Not Affect TGF-β-Induced Smad3/4 Oligomerization and Nuclear Translocation.
We next examined whether antisense menin would affect TGF-β-mediated Smad3 and Smad4 heterodimerization by coimmunoprecipitation studies. As shown in Fig. 5a, the TGF-β-induced heterodimeric complex formation between Smad2/4 or Smad3/4 is not affected by the presence of antisense menin, suggesting that the inhibitory effect of the antisense takes place downstream of this locus. Also, note the effectiveness of antisense menin in completely blocking the normal TGF-β stimulation of menin (Fig. 5a, W:α-menin). We then examined whether antisense menin would affect the translocation of the Smad3/4 complex into the nucleus. As shown in Fig. 5b, both Smad2 and Smad3 are transported into the nucleus from the cytoplasm with TGF-β stimulation in the absence of antisense menin. However, the presence of antisense menin does not affect TGF-β-mediated nuclear accumulation of Smad2 and Smad3. Together these data indicate that antisense menin does not affect TGF-β-induced Smad3/Smad4 oligomerization and nuclear translocation.
Figure 5.

Effect of antisense menin on Smad heterodimerization, Smad nuclear accumulation, and formation of Smad/DNA complexes. (a) Effect of antisense menin (AS) on Smad2/3 and Smad4 heterodimerization. Myc-tagged Smad2 or Smad3 and flag-tagged Smad4 constructs were transfected (as indicated) into COS7 cells with or without antisense menin. Transfected cells were treated (+) or not (−) with 200 pM TGF-β for 1 h after overnight serum starvation. Cell extracts were immunoprecipitated with anti-Smad2/3 antibody followed by immunoblotting with anti-flag antibody. Expression of myc, flag, or menin was monitored. W, Western blot; IP, immunoprecipitation. (b) Effect of antisense menin (AS) on Smad2 and Smad3 nuclear accumulation. Nuclear extracts of Chinese hamster ovary cells transfected or not with the antisense menin were cultured in the absence or presence of 200 pM TGF-β and immunoblotted with Smad2/3, Stat3, and menin antibodies. V, empty vector. (c) Effect of antisense menin on the Smad3/4-DNA complex. GH4C1 cells were transfected with myc-Smad3 and flag-Smad4 in the presence or absence of antisense menin. Nuclear extracts were subjected to EMSA; a 29-bp double-stranded oligonucleotide derived from the PAI-1 promoter sequence of 3TP-Lux was used as probe. The shifted band is indicated by the arrow; it was decreased in intensity by antisense menin and by anti-myc antibodies (*). Menin and Smad3/4 in the nuclear extracts were monitored.
Menin Inactivation Disrupts Smad3 Binding to DNA.
Smad3 and Smad4 are TGF-β-inducible DNA binding proteins (25, 26) and the Smad3/4 complex recognizes a specific binding site within the PAI-1 promoter in 3TP-Lux (10). Therefore, we examined whether antisense menin would affect the DNA binding ability of the Smad3/4 complex by using an EMSA with a probe derived from the PAI-1 promoter (10). As shown in Fig. 5c, levels of the Smad3/4-DNA complex are significantly decreased when antisense menin is expressed with Smad3 and Smad4. Furthermore, addition of myc antibody that binds the NH2 terminus of the epitope-tagged Smad3 close to the MH1 DNA binding domain blocked the formation of the Smad/DNA complex. Coexpression of sense menin restored the EMSA band (data not shown), and when untagged Smads were expressed the anti-myc antibody had no effect (data not shown). In addition, the gel shift responses were insensitive to cycloheximide (data not shown), indicating that new transcription/protein synthesis was not needed for the observed effects. These data indicate that antisense menin abrogates the interaction of the Smad3 complex with its binding sequence in DNA. Thus, menin seems to interact with the TGF-β pathway in the nucleus through Smad3 and inactivation of menin interrupts Smad3 binding to DNA, thereby blocking TGF-β signaling.
Implications of Menin/Smad Interactions.
Menin is a putative tumor suppressor gene and its inactivation specifically promotes endocrine tumor formation, including pituitary adenomas. Tumor suppressors often function as checkpoints, ensuring that the cell cycle is arrested when the cell is exposed to inhibitory growth factors such as TGF-β (27). Blockage of TGF-β signaling may disrupt the delicately balanced cellular steady state, pushing the cell toward inappropriate growth that ultimately results in tumor formation. Indeed, several reports have described inactivating mutations in genes encoding proteins known to be essential for the TGF-β signaling pathways, including Smad2 and Smad4 genes, in cancers of pancreas, biliary tract, colon, lung, head and neck, and hepatocellular carcinoma (see ref. 28 and the references therein). Thus, TGF-β may play a role in maintaining certain cells, including those of the pituitary, in the differentiated state and loss of this control in one fashion or another may contribute to dysregulated pituitary cell growth. Further study will be required to clarify why inactivation of the menin gene has a tumorigenic effect only in the limited subset of cell types affected in MEN1.
Menin binds the AP-1 transcription factor JunD and represses JunD-mediated transcriptional activity (15). As JunD can be considered to be antimitogenic and menin is a putative tumor suppressor, the significance of this observation is unclear at present. It was recently demonstrated (29) that Smad3 and Smad4 bind directly to Jun family members including JunD, and previous studies (30, 31) suggested that the Fra-2-JunD AP-1 complex and JunD homodimers regulate the activity of the murine laminin-α3A promoter and IL-6 expression, respectively, by TGF-β. Therefore, JunD by binding menin may play a role in TGF-β and Smad3 signaling pathways and thereby exert its cell growth control.
The present study suggests a mechanism by which menin functions as a tumor suppressor and documents that inactivation of a tumor suppressor causes blockade of the TGF-β signaling pathway.
Supplementary Material
Acknowledgments
We thank J. Massagué for 3TP-Lux and Y. Chen for Smad3ΔC cDNA. This work was supported in part by grants from the Canadian Institutes of Health Research (MT-14658 to J.-J.L., MT-5775 to D.G., and MT-9315 to G.N.H.). H.K. is the recipient of a fellowship, J.-J.L. a scholarship, and L.C. a doctoral fellowship from the Canadian Institutes of Health Research.
Abbreviations
- TGF-β
transforming growth factor type β
- MEN1
multiple endocrine neoplasia type 1
- PAI-1
plasminogen activator inhibitor type 1
- TdR
[3H]thymidine incorporation
- EMSA
electrophoretic mobility-shift assay
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Larsson C, Skogseid B, Oberg K, Nakamura Y, Nordenskjold M. Nature (London) 1988;332:85–87. doi: 10.1038/332085a0. [DOI] [PubMed] [Google Scholar]
- 2.Chandrasekharappa S C, Guru S C, Manickam P, Olufemi S E, Collins F S, Emmert-Buck M R, Debelenko L V, Zhuang Z, Lubenski I A, Liotta L A, et al. Science. 1997;276:404–407. doi: 10.1126/science.276.5311.404. [DOI] [PubMed] [Google Scholar]
- 3.The European Consortium on MEN1. Hum Mol Genet. 1997;6:1177–1183. doi: 10.1093/hmg/6.7.1177. [DOI] [PubMed] [Google Scholar]
- 4.Marx S J, Agarwal S K, Kester M B, Heppner C, Kim Y S, Skarulis M C, James L A, Goldsmith P K, Saggar S K, Park S Y, et al. Recent Prog Horm Res. 1999;54:397–439. [PubMed] [Google Scholar]
- 5.Pannett A A J, Thakker R V. Endocr Cancer. 1999;6:449–473. doi: 10.1677/erc.0.0060449. [DOI] [PubMed] [Google Scholar]
- 6.Kim Y S, Burns A L, Goldsmith P K, Heppner C, Park S Y, Chandrasekharappa S C, Collins F S, Spiegel A M, Marx S J. Oncogene. 1999;18:5936–5942. doi: 10.1038/sj.onc.1203005. [DOI] [PubMed] [Google Scholar]
- 7.Guru SC, Goldsmith P K, Burns A L, Marx S J, Spiegel A M, Collins F S, Chandrasekharappa S C. Proc Natl Acad Sci USA. 1998;95:1630–1634. doi: 10.1073/pnas.95.4.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaji H, Canaff L, Goltzman D, Hendy G N. Cancer Res. 1999;59:5097–5101. [PubMed] [Google Scholar]
- 9.Lebrun J J, Takabe K, Chen Y, Vale W. Mol Endocrinol. 1999;13:15–23. doi: 10.1210/mend.13.1.0218. [DOI] [PubMed] [Google Scholar]
- 10.Stroschein S L, Wang W, Luo K. J Biol Chem. 1999;274:9431–9441. doi: 10.1074/jbc.274.14.9431. [DOI] [PubMed] [Google Scholar]
- 11.Roberts A M, Sporn M B. In: Peptide Growth Factors and Their Receptors. Sporn M B, Roberts A B, editors. Berlin: Springer; 1990. pp. 421–427. [Google Scholar]
- 12.Asa S L, Ezzat S. Endocr Rev. 1998;19:798–827. doi: 10.1210/edrv.19.6.0350. [DOI] [PubMed] [Google Scholar]
- 13.Kurokawa M, Mitani K, Irie K, Matsuyama T, Takahashi T, Chiba S, Yazaki Y, Matsumoto K, Hirai H. Nature (London) 1998;394:92–96. doi: 10.1038/27945. [DOI] [PubMed] [Google Scholar]
- 14.Kretzschmar M, Doody J, Timokhina I, Massague J. Genes Dev. 1999;13:804–816. doi: 10.1101/gad.13.7.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Agarwal S K, Guru S C, Heppner C, Erdos M R, Collins R M, Park S Y, Saggar S, Chandrasekharappa S C, Collins F S, Spiegel A M, et al. Cell. 1999;96:143–152. doi: 10.1016/s0092-8674(00)80967-8. [DOI] [PubMed] [Google Scholar]
- 16.Labbe E, Silvestri C, Hoodless P A. Mol Cell. 1999;2:109–120. doi: 10.1016/s1097-2765(00)80119-7. [DOI] [PubMed] [Google Scholar]
- 17.Carcamo J, Weis F M, Ventura F, Wieser R, Wrana L, Attisano L, Massague J. Mol Cell Biol. 1994;14:3810–3821. doi: 10.1128/mcb.14.6.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wrana J L, Attisano L, Wieser R, Ventura F, Massague J. Nature (London) 1994;370:341–347. doi: 10.1038/370341a0. [DOI] [PubMed] [Google Scholar]
- 19.Massagué J. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 20.ten Dijke P, Miyazono K, Heldin C-H. Trends Biochem Sci. 2000;25:64–70. doi: 10.1016/s0968-0004(99)01519-4. [DOI] [PubMed] [Google Scholar]
- 21.Chen Y, Lebrun J J, Vale W. Proc Natl Acad Sci USA. 1996;93:12992–12997. doi: 10.1073/pnas.93.23.12992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y, Feng X, We R, Derynck R. Nature (London) 1996;383:168–172. doi: 10.1038/383168a0. [DOI] [PubMed] [Google Scholar]
- 23.Stroschein S L, Wang W, Zhou S, Zhou Q, Luo K. Science. 1999;286:771–774. doi: 10.1126/science.286.5440.771. [DOI] [PubMed] [Google Scholar]
- 24.Luo K, Stroschein S L, Wang W, Chen D, Martens E, Zhou S, Zhou Q. Genes Dev. 1999;13:2196–2206. doi: 10.1101/gad.13.17.2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yingling J M, Datto M B, Wong C, Frederick J P, Liberati N T, Wang X F. Mol Cell Biol. 1997;17:7019–7028. doi: 10.1128/mcb.17.12.7019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauther J M. EMBO J. 1998;17:3091–3100. doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu P P, Datto M B, Wang X F. Endocr Rev. 1998;19:349–363. doi: 10.1210/edrv.19.3.0333. [DOI] [PubMed] [Google Scholar]
- 28.Yakicier M C, Irmak M B, Romano A, Kew M, Ozturk M. Oncogene. 1999;18:4879–4883. doi: 10.1038/sj.onc.1202866. [DOI] [PubMed] [Google Scholar]
- 29.Liberati N T, Datto M B, Frederick J P, Shen X, Wong C, Rougier-Chapman E M, Wang X F. Proc Natl Acad Sci USA. 1999;96:4844–4849. doi: 10.1073/pnas.96.9.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Virolle T, Monthouel M N, Djabari Z, Ortonne J P, Meneguzzi G, Aberdam D. J Biol Chem. 1998;28:17318–17325. doi: 10.1074/jbc.273.28.17318. [DOI] [PubMed] [Google Scholar]
- 31.Eickelberg O, Pansky A, Mussmann R, Bihl M, Tamm M, Hildebrand P, Perruchoud A P, Roth M. J Biol Chem. 1999;274:12933–12938. doi: 10.1074/jbc.274.18.12933. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}