

Abstract
Background
Xylose isomerase (XI) catalyses the isomerisation of xylose to xylulose in bacteria and some fungi. Currently, only a limited number of XI genes have been functionally expressed in Saccharomyces cerevisiae, the microorganism of choice for lignocellulosic ethanol production. The objective of the present study was to search for novel XI genes in the vastly diverse microbial habitat present in soil. As the exploitation of microbial diversity is impaired by the ability to cultivate soil microorganisms under standard laboratory conditions, a metagenomic approach, consisting of total DNA extraction from a given environment followed by cloning of DNA into suitable vectors, was undertaken.
Results
A soil metagenomic library was constructed and two screening methods based on protein sequence similarity and enzyme activity were investigated to isolate novel XI encoding genes. These two screening approaches identified the xym1 and xym2 genes, respectively. Sequence and phylogenetic analyses revealed that the genes shared 67% similarity and belonged to different bacterial groups. When xym1 and xym2 were overexpressed in a xylA-deficient Escherichia coli strain, similar growth rates to those in which the Piromyces XI gene was expressed were obtained. However, expression in S. cerevisiae resulted in only one-fourth the growth rate of that obtained for the strain expressing the Piromyces XI gene.
Conclusions
For the first time, the screening of a soil metagenomic library in E. coli resulted in the successful isolation of two active XIs. However, the discrepancy between XI enzyme performance in E. coli and S. cerevisiae suggests that future screening for XI activity from soil should be pursued directly using yeast as a host.
Background
The soil habitat is an immensely diverse environment. One gram of soil may harbour up to ten billion bacteria belonging to more than 4,000 to 7,000 different species [1], although this value varies according to the soil type [2]. However, only 1% of the soil bacterial flora can be cultivated under standard laboratory conditions [3]. Metagenomics, which consists of the extraction, cloning and analysis of the entire genetic material in a given habitat [4,5], has emerged as a tool for assessing the genetic information from uncultivable microorganisms. Metagenomic libraries are a powerful source for discovering new biological activities and have already been successfully used to isolate novel hydrolytic enzymes such as amylases, cellulases and xylanases [6,7].
The screening of metagenomic libraries, which is a critical step for the successful isolation of new and improved biological activities, can be performed by sequence homology or activity-based assays [8]. In sequence-based screening, the isolation of novel proteins is based on homology searches. Although it impedes the discovery of entirely new gene sequences, it is an efficient method of isolating enzymes with different levels of similarity from previously identified genes. For instance, sequence-based screening from the metagenome of a hindgut microbiota of a wood-feeding higher termite identified 700 domains with homology to the glycoside-hydrolase catalytic site [9]. In activity-based assays, metagenomic libraries are constructed in expression vectors to allow for the isolation of biological activities from entirely new gene sequences that are also successfully expressed in the chosen host.
Activity-based screening methods have been categorized into chromogenic, product detection and growth test assays [10]. Chromogenic assays are commonly used for the detection of hydrolases from metagenomic libraries [6,7], whereas a product detection method has been utilized for the identification of enzymes from a soil metagenome producing porphyrin intermediates [11]. Although it is a high-throughput method, growth-based screening has not been frequently described, as it may be difficult to develop and implement a selection protocol that clearly discriminates growth from nongrowth.
Metagenomic libraries have not yet been utilized for the isolation of novel genes involved in xylose catabolism. Xylose is the second most abundant sugar in nature, and the production of cost-efficient lignocellulosic bioethanol requires its complete fermentation [12]. One xylose catabolic pathway is found in naturally xylose-utilizing bacteria [13] and some fungi [14,15] and includes the isomerisation of xylose to xylulose via xylose isomerase (XI). High ethanol yields from xylose have been reported for Saccharomyces cerevisiae strains harbouring actively expressed XI genes [15-18].
In this study, the possibility of isolating xylose catabolic enzymes from a soil metagenomic library was evaluated. Both sequence-based and growth-based screening were utilized. Each method resulted in the isolation of a novel gene sequence coding for a XI. Restoration of growth on xylose was verified in both Escherichia coli and S. cerevisiae strains by overexpression of the isolated genes in strains lacking the initial xylose catabolic pathway. The advantages and disadvantages of the two screening methods are discussed.
Results
Isolation of XI genes from soil metagenome
A soil metagenomic library with an estimated size of 1.26 × 105 clones was constructed in plasmid pRSETB (Table 1) using DNA extracted from garden compost and screened for XI activity using either sequence analysis or growth assays.
Table 1.
Plasmids and strains used in this study
| Plasmids/strains | Relevant characteristics | Source |
|---|---|---|
| Plasmids | ||
| pGEM-T Easy | PCR product cloning, ampR | Promega (Madison, WI, USA) |
| pRSETB | ampR, T7p-T7t | Invitrogen (Carlsbad, CA, USA) |
| p426TEF | URA3 TEFp-CYC1t | [43] |
| pRESTB-XIPiromyces | AmpR, T7p-xiPiromyces-T7t | This study |
| pRSETB-Xym1 | AmpR, T7p-xym1-T7t | This study |
| pRSETB-Xym2 | AmpR, T7p-xym2-T7t | This study |
| p426TEF-XiPiromyces | URA3, p426TEFp-XiPiromyces-CYC1t | This study |
| p426TEF-Xym1 | URA3, p426TEFp-xym1-CYC1t | This study |
| p426TEF-Xym2 | URA3, p426TEFp-xym2-CYC1t | This study |
| Escherichia coli strains | ||
| ElectroTen-Blue | KanR, TetR | Stratagene (La Jolla, California, USA) |
| HB101 | ara-14, proA2, xyl-5, leuB6, thi-1 | Takara Bio (Otsu, Shiga, Japan) |
| DH5α | SmR | Life Technologies (Rockville, MD, USA) |
| TMB2010 | HB101 + pRSETB | This study |
| TMB2011 | HB101 + pRSETB-xiPiromyces | This study |
| TMB2012 | HB101 + pRSETB-Xym1 | This study |
| TMB2013 | HB101 + pRSETB-Xym2 | This study |
| Saccharomyces cerevisiae strains | ||
| TMB3044 | CEN.PK 2-1C Δgre3, his3::PGK1p-XKS1-PGK1t,TAL1::PGK1p-TAL1-PGK1t,TKL1::PGK1p-TKL1-PGK1t, RKI1::PGK1p-RKI1-PGK1t, RPE1::PGK1p-RPE1-PGK1t,ura3 | [26] |
| TMB3363 | TMB3044, p426TEF | This study |
| TMB3359 | TMB3044, p426TEF-XiPiromyces | This study |
| TMB3364 | TMB3044, p426TEF-Xym1 | This study |
| TMB3365 | TMB3044, p426TEF-Xym2 | This study |
For the sequence-based screening, degenerate primers were designed based on the alignment of amino acid sequences of known XIs from soil microorganisms such as Streptomyces sp. and Rhizobium sp. Amino acid sequences of XI genes whose expression has been attempted in S. cerevisiae, such as the ones from Piromyces sp. [14] and E. coli [19], were also included (Figure 1). The first polymerase chain reaction (PCR) was performed with primers DF1 and DR2 (Table 2) and with the soil metagenomic library used as a template. A fragment of approximately 750 bp was amplified. The purified fragment was then utilized as the template for a nested PCR with internal primers DF2 and DR1. A single fragment of about 350 bp was obtained, purified from an agarose gel and cloned into the pGEM-T Easy Vector System (Promega, Madison, WI, USA) (Table 1). Sequence analysis revealed that this fragment belonged to a gene coding for a XI. The complete gene sequence was then obtained by PCR utilizing the metagenomic library as the template and primers designed to the sequence of the known 350-bp fragment and in plasmid pRSETB. The gene sequence encoded a 443-amino acid protein with 83% identity with Sorangium cellulosum XI and was designated xym1.
Figure 1.

Conserved regions utilized for the construction of the degenerate primers. The choice of regions was based on multiple alignments of XI amino acid sequences. The microorganisms chosen, in presented order, are Piromyces sp., Streptomyces diastaticus, Escherichia coli, Pseudomonas sp., Rhizobium sp., Streptomyces coelicolor, Saccharophagus sp., Enterobacter sp., Mesorhizobium loti and Clostridium sp.
Table 2.
Primers useda
| Primer | Sequence |
|---|---|
| DF1 | 5' AAGCT(A/C/G/T)GG(A/C/G/T)G(C/T)(A/C/G/T)CCT T(C/T)(C/T)TATTGTTT(C/T)CACGAC 3' |
| DR2 | 5' TCCTCC(A/C/G/T)TGCTG(C/T)(T/A)(T/A)GCCGCC(A/C/G/T)CC(A/C/G/T)CG(A/C/G/T)AG(A/G)AT 3' |
| DF2 | 5' GGT(A/G)T(A/C/G/T)AA(A/G)CT(A/C/G/T)CTCTGGGG(A/C/G/T)CAGGCCAA(C/T)CT(A/C/G/T)TT(C/T) 3' |
| DR1 | 5' (A/G)TG(C/T)TTCTGGGG(C/T)TC(C/T)TGCGGCTTGGGCTC (A/C/G/T)A(T/G) 3' |
| pRSETBF | 5' GGTGGACAGCAAATGGGTCGG 3' |
| pRSETBR | 5' GGGCTTTGTTAGCAGCCGGATC 3' |
| XYM1F | 5' CCTGGATCCATGAGCGTTGTTCTTGGCGACAAAG 3' |
| XYM1R | 5' TGGGTCGACTTACCGGATCCACCGGTTCATAATG 3' |
| XYM2F | 5'CAGGAATTCATGAAACTTACCGTAGGAGACAAGG 3' |
| XYM2R | 5' TGGGTCGACTTAAATAAACCTGCTAATCAAATTCTCAATATAC 3' |
aRestriction sites, when present, are underlined.
For the activity screening, the metagenomic library was transferred to the host strain HB101 (Table 1) that has a nonfunctional xylA, which allows selection for growth on xylose. After bacterial transformation, cells were plated in defined media supplemented with xylose and incubated at 37°C for four to ten days. Positive (pRSETB-Xi Piromyces) and negative (pRSETB) controls were also transformed into the screening strain and plated in selective media to validate the screening method. Plasmids were extracted and sequenced from colonies that grew on the selective media. All extracted plasmids contained identical inserted fragments with an insert size of approximately 2.5 kb. Sequencing analysis revealed a complete open reading frame that corresponded to a XI gene named xym2. xym2 encoded a protein of 442 amino acids with 73% similarity to the XI from Robiginitalea biformata and 67% identity with xym1-encoded XI. A phylogenetic tree constructed using MEGA4 software [20] and 22 different XI amino acid sequences revealed that xym1-encoded XI was most similar to XIs from the Proteobacteria, while xym2-encoded XI was more similar to the XIs from members of the Bacterioides phylum (Figure 2).
Figure 2.

Neighbour-joining tree showing the phylogenetic positions of xym1- and xym2-encoded XIs based on conserved amino acids of different XIs. Microorganism sources of XIs were chosen based on sequence similarity and/or active expression in Saccharomyces cerevisiae. The phylogenetic tree was constructed with MEGA software using an unweighted group method with arithmetic mean (UPGMA) bootstrapped with 6,000 interactions.
Growth evaluation in E. coli
The xym1 and xym2 genes and the positive control xylA gene from Piromyces [14] were each cloned into the multicopy vector pRSETB, generating plasmids pRSETB-Xym1, pRSETB-Xym2 and pRSETB-XiPiromyces (Table 1). Strain HB101 was transformed with each of the three constructs, as well as the negative control pRSETB, to generate the four strains: TMB2010 (negative control), TMB2011 (Piromyces xylA), TMB2012 (xym1) and TMB2013 (xym2).
Growth was evaluated for the four strains in SM3 liquid media with xylose. Growth with xylose as the sole carbon source was reestablished for all strains overexpressing any of the XI-encoding genes (Figure 3). No significant differences in growth rates and final Optical density (OD) were observed between the positive control strain, TMB2011, and the strains overexpressing xym1 and xym2 that were isolated from the metagenomic library (Figure 3).
Figure 3.

Aerobic growth of E. coli recombinant strains carrying pRSETB-Xym1 (filled triangle), pRSETB-Xym2 (filled circle), pRSETB-XIPiromyces (filled square), or the empty plasmid pRSETB (filled diamond). All strains were cultivated at 37°C in SM3 media supplemented with 30 g/L xylose.
Growth evaluation in S. cerevisiae
XI is a key enzyme for the construction of recombinant S. cerevisiae strains for xylose conversion to ethanol. However, despite many efforts [19,21-24], only a few XI genes have been successfully expressed in S. cerevisiae at sufficient levels to allow growth on xylose [15,16,25]. Therefore, xym1 and xym2 were overexpressed in S. cerevisiae to assess growth in a recombinant S. cerevisiae strain lacking the initial xylose catabolic pathway. xym1, xym2 and the xylA gene from Piromyces sp. (positive control) were cloned in the multicopy vector p426TEF to generate plasmids p246TEFXiPiromyces, p426TEF-Xym1 and p426TEF-Xym2 (Table 1). The S. cerevisiae screening strain TMB3044 that has been optimised for efficient pentose utilization, but which lacks the initial conversion step from xylose to xylulose [26], was transformed with the three constructed plasmids in addition to the empty plasmid to generate strains TMB3363(empty plasmid), TMB3359(XiPiromyces), TMB3364 (xym1) and TMB3365 (xym2). No growth was observed for the negative control. In contrast, overexpression of both xym1 and xym2 enabled growth on xylose in S. cerevisiae (Figure 4). However, the strains carrying either xym1or xym2 grew at a lower growth rate (0.021 hour-1 for both strains) than the positive control harbouring Piromyces sp XI (0.069 hour-1 ± 0.006) (Figure 4).
Figure 4.

Aerobic growth of S. cerevisiae recombinant strains carrying p426TEF-Xym1 (filled triangle), p426TEF-Xym2 (filled circle), p426TEF-XiPiromyces (filled square) or the empty plasmid p426TEF (filled diamond). All strains were cultivated at 30°C in mineral media supplemented with 50 g/L xylose.
Evaluation of XI-specific activities in both hosts
XI-specific activities were compared in cell extracts of E. coli and S. cerevisiae cells grown in defined medium with glucose. For each microorganism, positive (Piromyces sp. XI) and negative (empty vector) control strains were included for comparison with the strains overexpressing either xym1 or xym2.
In S. cerevisiae, the background activity measured in the negative control (0.195 U/mg protein-1) was twice as high as that measured in E. coli (0.059 U/mg protein-1) (Figure 5). Strains overexpressing the positive control or xym1 had about the same specific XI activity in both E. coli and S. cerevisiae. For the xym2-overexpressing strain, the specific activity (0.420 U/mg protein-1) was in the same range as the ranges for the xym1-overexpressing strain and the positive control in E. coli. In contrast, the specific activity in the xym2-overexpressing strain was at the level of the background activity in S. cerevisiae (0.195 U/mg protein-1) (Figure 5).
Figure 5.
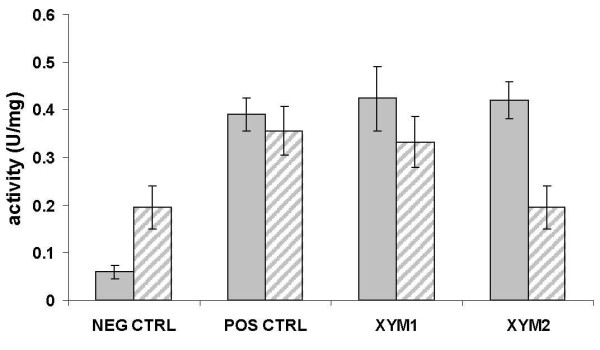
XI specific activity (U/mg protein-1) in E. coli (gray) and S. cerevisiae (stripped) strains overexpressing xym1, xym2 and Piromyces sp. XI gene. The background activity was obtained from the same strains carrying the respective empty vectors.
Discussion
In this study, for the first time, two screening methods were established in parallel for the isolation of novel XI-encoding genes from a soil metagenomic library. The sequence-based method using degenerate primers deduced from the alignment of amino acid sequences of several XIs from soil microorganisms rapidly resulted in the isolation of the first XI-encoding gene xym1 from a metagenomic library. Growth-based screening was also used for the first time to isolate the XI-encoding gene xym2 from soil metagenomic library using an E. coli strain lacking XI activity. The two different methods resulted in the isolation of two XI-encoding genes that shared 67% identity at the protein level and could both complement E. coli and S. cerevisiae strains lacking a xylose catabolic enzyme.
Some examples where the sequence-based screening method has already been used include (1) the identification of genes responsible for the hydrolytic dehalogenation of 4-chlorobenxoate from a metagenomic library constructed from a denitrifying, degrading consortium [27] and (2) the isolation of a polyketide synthase gene from a metagenome constructed from marine sediment in the East China Sea [28]. It is a high-throughput screening method, but one disadvantage is that it selects for gene sequences with close homology to those utilized for the design of the degenerate primers. This was notably exemplified by the isolation of closely related chitinase genes from cultured and uncultured marine bacteria [29].
In contrast, activity-based screening permits the identification of previously unknown gene sequences, as the screening is based on the gene product only. One disadvantage, however, is that activity-based screening that relies on chromogenic or fluorogenic assays often require high-throughput equipment, such as colony pickers, that transfer colonies from plates to the assay system. When possible, it is therefore advisable to develop a growth-based screening method because the method is simple (each library can be plated on a limited number of plates, as very few clones will grow) and enables high-throughput (only the clones displaying the desired activity are able to grow). This method is limited in that a host strain or an experimental setup must be designed that does not enable the host to grow unless the desired activity is present.
Screening by growth has previously been used to isolate genes from a soil metagenomic library conferring Na+(Li+)/H+ antiporter activity to an antiporter-deficient E. coli host strain [30]. Novel DNA polymerase activities have also been isolated from glacial ice metagenomic libraries using a cold-sensitive E. coli strain that harboured a mutation in the DNA polymerase domain generating lethality at temperatures below 20°C [31]. Finally, a novel D-3-hydroxybutyrate short-chain dehydrogenase with less than 35% similarity to known proteins with similar activity has been isolated from soil metagenomic libraries using a mutant of Sinorhizobium meliloti that was unable to synthesize D-3-hydroxybutyrate [32]. In the present study, xym1- and xym2-encoded XIs displayed strong similarity in amino acid sequence, although they were isolated by using different methodologies. This may be explained by the fact that most of the XI sequences that were used to identify homologous regions for the sequence-based screening originated from organisms that were know to be present in soil.
Screening for XI-encoding genes by growth versus nongrowth was successfully implemented in E. coli, since the expression of xym1 and xym2 genes could restore growth in xylose minimal media with the same growth rate observed for the positive control overexpressing the Piromyces XI gene. However, growth was restored when the same genes were transferred to the heterologous host S. cerevisiae, where the activity is required for xylose fermentation to ethanol, the growth rate was four times lower than that observed for Piromyces XI. Expression of XI-encoding genes in S. cerevisiae is a complex issue [33]. The first one to be expressed at high levels in S. cerevisiae was Piromyces XI [18], and it occurred after many unsuccessful attempts and limited success with several other bacterial and plant XIs [19,21-24]. Successful expression of XI in S. cerevisiae and its growth rate on xylose are not always correlated to sequence similarity or enzymatic activity (Table 3). In our case, xym1-encoded XI had the same XI activity as Piromyces sp XI, but the growth rate was fourfold lower. In contrast, the specific activity of the xym2-encoded XI was in the range of the background activity, but the growth rate was identical to the strain expressing xym1. The growth rate was also in the same range when the Xanthomonas campestris XI gene was expressed, although the reported activity range was about 20 times lower than that for the xym1-overexpressing strain (Table 3). Finally, a high activity level and growth rate were achieved when overexpressing a codon-optimised XI gene from Clostridium phytofermentans that shared only 54% similarity with Piromyces sp. at the protein level (Table 3) [16]. Differences in kinetic properties may also explain the discrepancies among all these results. For instance, the affinity for xylose may be much higher in xym2- than in xym1-encoded XI, so that the lower rate of conversion would be compensated for by a higher affinity for the substrate. Alternatively, the novel XI may be more inhibited by xylitol than the one from Piromyces sp. or C. phytofermentans. We were not able to measure Km or Ki for the new XIs, because the activities were below the detection limit. Transport may also explain differences in behaviour between S. cerevisiae and E. coli, since active xylose transporters are present in E. coli [34,35] but missing in S. cerevisiae.
Table 3.
Reported specific activity and maximum specific growth rate for XI genes cloned in Saccharomyces cerevisiaea
| Source of XI | Activity (U/mg protein) | Aerobic growth rate on xylose (hour-1) | Source | Identity with xym1 XI | Identity with xym2 XI | Identity with Piromyces XI |
|---|---|---|---|---|---|---|
| Metagenome xym1b | 0.33 ± 0.05 | 0.02 | This study | 100% | 67% | 61% |
| Metagenome xym2b | 0.20 ± 0.04 | 0.02 | This study | 67% | 100% | 63% |
| Piromyces sp.b,c | 0.35 | 0.07 | This study | 61% | 63% | 100% |
| Piromyces sp. | 0.3 to 1.1 | 0.22 | [18] | 61% | 63% | 100% |
| Piromyces sp.c | 0.5 to 0.8 | 0.02 | [17] | 61% | 63% | 100% |
| Piromyces sp.c | 0.0538 | 0.056 | [16] | 61% | 63% | 100% |
| Orpinomyces sp. | 1.6 | 0.01 | [15] | 61% | 61% | 94% |
| Xanthomonas campestris | 0.016 | 0.02 | [44] | 64% | 63% | 61% |
| Bacteroides thetaiotaomicron | 0.14 | n.m. | [45] | 61% | 64% | 83% |
| Clostridium phytofermentansc | 0.0344 | 0.057 | [16] | 54% | 52% | 54% |
| Clostridium thermosulfurogenes | n.m. | n.d. | [46] | 51% | 49% | 49% |
| Escherichia coli | n.m. | n.d. | [19] | 50% | 52% | 48% |
| Bacillus subtilis | n.m. | n.m. | [47] | 48% | 48% | 47% |
| Lactobacillus pentosus | n.d. | n.d. | [47] | 48% | 47% | 46% |
| Thermus thermophilus | 0.0007 to 0.012 | n.m. | [48] | 31% | 30% | 29% |
an.m., not measured; n.d., not detected. Sequence identity with xym1, xym2 and Piromyces XI gene is also given at the protein level. bGlucose-grown cells. cCodon-optimised gene.
E. coli has been the preferred choice as the host for screening metagenomic libraries [6] because numerous molecular tools are available and E. coli genetics are well elucidated. In our case, E. coli had the additional advantage that it is naturally able to utilize xylose, allowing the isolation of the desired activity without any other limitation up- or downstream of XI. The use of only one host, however, may limit the exploitation of microbial diversity for a given habitat due to, for instance, differences in codon utilization and/or mRNA and protein stability among different hosts. E. coli was, for instance, estimated to express only 40% of genes from diverse microbial origin [36]. In a previous study where growth-based screening was utilized for the isolation of novel D-3-hydroxybutyrate short-chain dehydrogenases from the soil metagenomic library, 25 positive clones were initially isolated when the library was screened in Sinorhizobium melioti; however, only one clone complemented the same phenotype in E. coli [32]. Screening of a soil metagenomic library performed in six different bacterial hosts identified only one case where the molecule-producing clone could be isolated using two different hosts [37].
Further screening of the soil metagenomic library described in this study should be performed in S. cerevisiae to investigate more suitable XIs for ethanol production from xylose. This is complicated by the fact that, whereas in bacterial metagenomic libraries large fragments are cloned and theoretically all genes within the fragment are transcribed and translated, only the closest gene to the methylated cap on the 5' terminus of the mRNA is translated in yeast [38]. Construction of future libraries for use in yeast will have to combine the cloning of small fragments corresponding to single gene size with an efficient transformation protocol to accommodate the larger colony numbers required to represent the same soil population. Alternatively, construction and screening of metagenomic cDNA libraries may be attempted; however, neither of these strategies has yet been reported.
Conclusions
In the present study, a soil metagenomic library was successfully constructed and screened for the first time by using sequence- and growth-based methods to isolate genes conferring xylose isomerase activity. It enabled the isolation of xym1 and xym2 genes, respectively, that shared 67% identity at the protein level. Both genes could restore growth in E. coli and S. cerevisiae strains lacking the initial xylose catabolic pathway. However, the limited growth rates in S. cerevisiae highlight the importance of screening for XI activity in this host instead.
Methods
Plasmids and strains
Plasmids and strains used in this study are listed in Table 1. Strains were stored as recommended by the manufacturer or as 20% glycerol stocks in liquid media at -80°C.
Cultivation conditions
E. coli strains were plated in Luria broth (LB) media plates (10 g/L Tryptone, 5 g/L yeast extract and 10 g/L NaCl, agar 15 g/L) and grown at 37°C overnight. Antibiotics were added when necessary to a final concentration of 30 μg/mL kanamycin and 50 μg/mL ampicillin and streptomycin. Single E. coli colonies were inoculated in LB media (10 g/L Bacto Tryptone, 5 g/L yeast extract and 10 g/L NaCl) for approximately 12 hours at 37°C in an incubator with constant agitation at 180 rpm (Boule Medical AB, Stockholm, Sweden).
For the metagenomic library screening, cells were plated in defined SM3 media [39] supplemented with 11 g/L xylose, 15 g/L agar, 1 mM isopropyl-β-D-thiogalactoside (IPTG) and 50 μg/ml ampicillin.
S. cerevisiae strains were grown on yeast nitrogen base (YNB) media plates (6.7 g/L Difco YNB without amino acids; Becton Dickinson, Sparks, MD, USA) supplemented with 20 g/L glucose and 20 g/L agar.
For growth kinetics on xylose, E. coli recombinant strains were pregrown in 20 mL of LB media at 180 rpm and 30°C until the end of the exponential phase. Cells were washed in distilled water and transferred to 500-mL Erlenmeyer flasks containing 50 mL of SM3 media supplemented with 30 g/L xylose and 1 mM IPTG at a starting OD620nm = 0.1.
S. cerevisiae strains were pregrown in 500-mL Erlenmeyer flasks containing 50 mL of YNB medium supplemented with 20 g/L glucose at 180 rpm and 30°C for approximately 12 hours. Cells were washed in distilled water and inoculated at OD620nm = 0.2 in 100 mL of YNB medium supplemented with 50 g/L xylose in 1-L Erlenmeyer flasks and grown at 180 rpm and 30°C.
Standard molecular procedures
Standard DNA manipulation techniques were used [40]. Restriction and ligation enzymes were purchased from Fermentas (St. Leon-Rot, Germany). Primers were purchased from Eurofins MWG Operon (Ebersberg, Germany). DNA extraction from agarose gels and purification of PCR products were performed using the QIAquick extraction kit (Qiagen, Hilder, Germany). Plasmid DNA was prepared using the Bio-Rad Miniprep Kit (Bio-Rad, Hercules, CA, USA). Sequencing was performed at Eurofins MWG Operon. If not otherwise stated, bacterial transformation was performed by heat shock with competent cells prepared by using the Inoue Method for Preparation and Transformation of Competent E. coli as previously described [41]. Yeast transformation was performed as previously described [42].
Construction of the soil metagenomic library
Soil DNA was extracted from the upper layer of a garden in Höör, southern Sweden, during Spring 2008. DNA extraction was performed using the PowerSoil DNA Isolation Kit (MoBio, Carlsbad, CA, USA). Isolated DNA was digested with BamHI and MboI. Agarose gel electrophoresis was performed for purification of DNA fragments ranging between 2.0 and 6.0 kb to generate a small insert library with an average size of 4.0 kb. Vector pRSETB was digested with BamHI and treated with alkaline phosphatase to prevent plasmid self-ligation. DNA fragments were ligated into pRSETB. Bacterial transformation was performed with ElectroTen-Blue competent cells (Stratagene, La Jolla, CA, USA) according to the manufacturer's instructions. The library size was estimated on the basis of the total number of E. coli clones minus the ones with empty plasmid.
Cloning and expression of xym1, xym2 and XI from Piromyces sp
The isolated isomerase genes were cloned both into E. coli and S. cerevisiae expression vectors. The xym1 gene was amplified with primers XYM1F and XYM1R, which contain restriction sites for BamHI and SalI, respectively (Table 2). After PCR, both the amplified gene and plasmids pRSETB and p426TEF were digested. Plasmids with inserts obtained after ligation and bacterial transformation were sequenced and named pRSETB-Xym1 and p426TEF-Xym1, respectively (Table 1). Cloning of the xym2 gene followed the same procedure as for xym1, with the exception that EcoRI and SalI restriction sites were used. xym1 and xym2 sequences will be publicly available when the corresponding patent application is published.
A codon-optimised version of the XI-encoding gene from Piromyces sp. was amplified by PCR utilizing plasmid YeplacHXT-XI as template [26]: forward primer 5' GGAGGATCCATGGCTAAGGAATATTTTCCACAAATTC 3' and reverse primer 5' TTGGAATTCTTACTGATACATTGCAACAATAGCTTCG 3'. Restriction sites for BamHI and EcoRI are underlined in the forward and reverse primers, respectively. Plasmid p426TEF [43], pRSETB (Invitrogen, Carlsbad, CA, USA) and the PCR product were digested with BamHI and EcoRI and then ligated. Clones with inserts were sequenced prior to yeast transformation.
Enzyme activities
S. cerevisiae strains were grown in YNB medium supplemented with 20 g/L glucose at 30°C to the early stationary phase. E. coli strains were pregrown in LB media supplemented with 100 μg/ml ampicillin for about 5 hours. After that, cells were washed with water and inoculated for an initial OD620 nm = 0.2 in SM3 medium supplemented with 10 g/L glucose and 1 mM IPTG at 37°C overnight.
Yeast and bacteria strains were harvested by centrifugation at 5,000 × g for 5 minutes and washed once with distilled water. Wet cells were suspended (0.5 mg/mL) in Y-per yeast or B-per bacteria extraction protein reagent (Pierce Biotechnology, Rockford, IL, USA) in a 2-mL Eppendorf tube and incubated on a turning table at room temperature for 50 minutes. Cell debris was spun down for 5 minutes at 16,100 × g (Z160M table centrifuge; HERMLE Labortechnik, Wehingen, Germany). Protein concentration was determined using Coomassie Protein Assay Reagent (Pierce Biotechnology). Bovine serum albumin was used to determine the standard curve.
XI activity was determined at 30°C using sorbitol dehydrogenase as previously described [18]. Assays were performed in a U-2000 spectrophotometer (Hitachi, Tokyo, Japan). Enzyme assays were performed in three biological replicates.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
NSP planned and carried out the experimental work and wrote the manuscript. MFGG participated in the study design and its coordination and critically read and commented on the manuscript. Both authors reviewed and approved the final version of the manuscript.
Contributor Information
Nádia Skorupa Parachin, Email: Nadia.Skorupa_Parachin@tmb.lth.se.
Marie F Gorwa-Grauslund, Email: Marie-Francoise.Gorwa@tmb.lth.se.
Acknowledgements
We thank Prof. Bärbel Hahn-Hägerdal for her valuable comments, Catherine Paul for critically reading the manuscript and Birgit Johansson for providing fresh soil for DNA extraction. This work was financed by the Swedish Research Council (VR).
References
- Rosselló-Mora R, Amann R. The species concept for prokaryotes. FEMS Microbiol Rev. 2001;25:39–67. doi: 10.1111/j.1574-6976.2001.tb00571.x. [DOI] [PubMed] [Google Scholar]
- Torsvik V, Øvreås L, Thingstad TF. Prokaryotic diversity: magnitude, dynamics, and controlling factors. Science. 2002;296:1064–1066. doi: 10.1126/science.1071698. [DOI] [PubMed] [Google Scholar]
- Handelsman J, Rondon MR, Brady SF, Clardy J, Goodman RM. Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chem Biol. 1998;5:R245–R249. doi: 10.1016/S1074-5521(98)90108-9. [DOI] [PubMed] [Google Scholar]
- Torsvik V, Øvreås L. Microbial diversity and function in soil: from genes to ecosystems. Curr Opin Microbiol. 2002;5:240–245. doi: 10.1016/S1369-5274(02)00324-7. [DOI] [PubMed] [Google Scholar]
- Schmeisser C, Steele H, Streit WR. Metagenomics, biotechnology with non-culturable microbes. Appl Microbiol Biotechnol. 2007;75:955–962. doi: 10.1007/s00253-007-0945-5. [DOI] [PubMed] [Google Scholar]
- Steele HL, Jaeger KE, Daniel R, Streit WR. Advances in recovery of novel biocatalysts from metagenomes. J Mol Microbiol Biotechnol. 2009;16:25–37. doi: 10.1159/000142892. [DOI] [PubMed] [Google Scholar]
- Ferrer M, Beloqui A, Timmis KN, Golyshin PN. Metagenomics for mining new genetic resources of microbial communities. J Mol Microbiol Biotechnol. 2009;16:109–123. doi: 10.1159/000142898. [DOI] [PubMed] [Google Scholar]
- Lorenz P, Eck J. Metagenomics and industrial applications. Nat Rev Microbiol. 2005;3:510–516. doi: 10.1038/nrmicro1161. [DOI] [PubMed] [Google Scholar]
- Warnecke F, Luginbühl P, Ivanova N, Ghassemian M, Richardson TH, Stege JT, Cayouette M, McHardy AC, Djordjevic G, Aboushadi N, Sorek R, Tringe SG, Podar M, Martin HG, Kunin V, Dalevi D, Madejska J, Kirton E, Platt D, Szeto E, Salamov A, Barry K, Mikhailova N, Kyrpides NC, Matson EG, Ottesen EA, Zhang X, Hernández M, Murillo C, Acosta LG. et al. Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature. 2007;450:560–565. doi: 10.1038/nature06269. [DOI] [PubMed] [Google Scholar]
- Reymond JL, Babiak P. Screening systems. Adv Biochem Eng Biotechnol. 2007;105:31–58. doi: 10.1007/10_2006_032. [DOI] [PubMed] [Google Scholar]
- Kim JS, Lim HK, Lee MH, Park JH, Hwang EC, Moon BJ, Lee SW. Production of porphyrin intermediates in Escherichia coli carrying soil metagenomic genes. FEMS Microbiol Lett. 2009;295:42–49. doi: 10.1111/j.1574-6968.2009.01577.x. [DOI] [PubMed] [Google Scholar]
- Sassner P, Galbe M, Zacchi G. Techno-economic evaluation of bioethanol production from three different lignocellulosic materials. Biomass Bioenergy. 2008;32:422–430. doi: 10.1016/j.biombioe.2007.10.014. [DOI] [Google Scholar]
- Chen WP. Glucose isomerase (a review) Process Biochem. 1980;15:30–35. [Google Scholar]
- Harhangi HR, Akhmanova AS, Emmens R, van der Drift C, de Laat WT, van Dijken JP, Jetten MS, Pronk JT, Op den Camp HJ. Xylose metabolism in the anaerobic fungus Piromyces sp. strain E2 follows the bacterial pathway. Arch Microbiol. 2003;180:134–141. doi: 10.1007/s00203-003-0565-0. [DOI] [PubMed] [Google Scholar]
- Madhavan A, Tamalampudi S, Ushida K, Kanai D, Katahira S, Srivastava A, Fukuda H, Bisaria VS, Kondo A. Xylose isomerase from polycentric fungus Orpinomyces: gene sequencing, cloning, and expression in Saccharomyces cerevisiae for bioconversion of xylose to ethanol. Appl Microbiol Biotechnol. 2009;82:1067–1078. doi: 10.1007/s00253-008-1794-6. [DOI] [PubMed] [Google Scholar]
- Brat D, Boles E, Wiedemann B. Functional expression of a bacterial xylose isomerase in Saccharomyces cerevisiae. Appl Environ Microbiol. 2009;75:2304–2311. doi: 10.1128/AEM.02522-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karhumaa K, Garcia-Sanchez R, Hahn-Hägerdal B, Gorwa-Grauslund MF. Comparison of the xylose reductase-xylitol dehydrogenase and the xylose isomerase pathways for xylose fermentation by recombinant Saccharomyces cerevisiae. Microb Cell Fact. 2007;6:5. doi: 10.1186/1475-2859-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuyper M, Harhangi HR, Stave AK, Winkler AA, Jetten MS, de Laat WT, den Ridder JJ, Op den Camp HJ, van Dijken JP, Pronk JT. High-level functional expression of a fungal xylose isomerase: the key to efficient ethanolic fermentation of xylose by Saccharomyces cerevisiae? FEMS Yeast Res. 2003;4:69–78. doi: 10.1016/S1567-1356(03)00141-7. [DOI] [PubMed] [Google Scholar]
- Sarthy AV, McConaughy BL, Lobo Z, Sundstrom JA, Furlong CE, Hall BD. Expression of the Escherichia coli xylose isomerase gene in Saccharomyces cerevisiae. Appl Environ Microbiol. 1987;53:1996–2000. doi: 10.1128/aem.53.9.1996-2000.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Amore R, Hollenberg CP. Xylose isomerase from Actinoplanes missouriensis: primary structure of the gene and the protein. Nucleic Acids Res. 1989;17:7515. doi: 10.1093/nar/17.18.7515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moes CJ, Pretorius IS, Zyl WH. Cloning and expression of the Clostridium thermosulfurogenes D-xylose isomerase gene (xyLA) in Saccharomyces cerevisiae. Biotechnol Lett. 1996;18:269–274. [Google Scholar]
- Walfridsson M, Bao X, Anderlund M, Lilius G, Bülow L, Hahn-Hägerdal B. Ethanolic fermentation of xylose with Saccharomyces cerevisiae harboring the Thermus thermophilus xylA gene, which expresses an active xylose (glucose) isomerase. Appl Environ Microbiol. 1996;62:4648–4651. doi: 10.1128/aem.62.12.4648-4651.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gárdonyi M, Hahn-Hägerdal B. The Streptomyces rubiginosus xylose isomerase is misfolded when expressed in Saccharomyces cerevisiae. Enzyme Microb Technol. 2003;32:252–259. doi: 10.1016/S0141-0229(02)00285-5. [DOI] [Google Scholar]
- Kuyper M, Winkler AA, van Dijken JP, Pronk JT. Minimal metabolic engineering of Saccharomyces cerevisiae for efficient anaerobic xylose fermentation: a proof of principle. FEMS Yeast Res. 2004;4:655–664. doi: 10.1016/j.femsyr.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Karhumaa K, Hahn-Hägerdal B, Gorwa-Grauslund MF. Investigation of limiting metabolic steps in the utilization of xylose by recombinant Saccharomyces cerevisiae using metabolic engineering. Yeast. 2005;22:359–368. doi: 10.1002/yea.1216. [DOI] [PubMed] [Google Scholar]
- Chae JC, Song B, Zylstra GJ. Identification of genes coding for hydrolytic dehalogenation in the metagenome derived from a denitrifying 4-chlorobenzoate degrading consortium. FEMS Microbiol Lett. 2008;281:203–209. doi: 10.1111/j.1574-6968.2008.01106.x. [DOI] [PubMed] [Google Scholar]
- Jiao YL, Wang LH, Dong XY, Chen YF, Zong Y, Gao Y, Ren N, Guo AY, Zhang XQ, Jiao BH. Isolation of new polyketide synthase gene fragments and a partial gene cluster from East China Sea and function analysis of a new acyltransferase. Appl Biochem Biotechnol. 2008;149:67–78. doi: 10.1007/s12010-007-8053-7. [DOI] [PubMed] [Google Scholar]
- Cottrell MT, Wood DN, Yu L, Kirchman DL. Selected chitinase genes in cultured and uncultured marine bacteria in the α- and γ-subclasses of the proteobacteria. Appl Environ Microbiol. 2000;66:1195–1201. doi: 10.1128/AEM.66.3.1195-1201.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majerník A, Gottschalk G, Daniel R. Screening of environmental DNA libraries for the presence of genes conferring Na+(Li+)/H+ antiporter activity on Escherichia coli: characterization of the recovered genes and the corresponding gene products. J Bacteriol. 2001;183:6645–6653. doi: 10.1128/JB.183.22.6645-6653.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon C, Herath J, Rockstroh S, Daniel R. Rapid identification of genes encoding DNA polymerases by function-based screening of metagenomic libraries derived from glacial ice. Appl Environ Microbiol. 2009;75:2964–2968. doi: 10.1128/AEM.02644-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Meek DJ, Panchal P, Boruvka N, Archibald FS, Driscoll BT, Charles TC. Isolation of poly-3-hydroxybutyrate metabolism genes from complex microbial communities by phenotypic complementation of bacterial mutants. Appl Environ Microbiol. 2006;72:384–391. doi: 10.1128/AEM.72.1.384-391.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn-Hägerdal B, Karhumaa K, Jeppsson M, Gorwa-Grauslund MF. Metabolic engineering for pentose utilization in Saccharomyces cerevisiae. Adv Biochem Eng Biotechnol. 2007;108:147–177. doi: 10.1007/10_2007_062. [DOI] [PubMed] [Google Scholar]
- Davis EO, Henderson PJ. The cloning and DNA sequence of the gene xylE for xylose-proton symport in Escherichia coli K12. J Biol Chem. 1987;262:13928–13932. [PubMed] [Google Scholar]
- Kurose N, Watanabe K, Kimura A. Nucleotide sequence of the gene responsible for D-xylose uptake in Escherichia coli. Nucleic Acids Res. 1986;14:7115–7123. doi: 10.1093/nar/14.17.7115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabor EM, Alkema WB, Janssen DB. Quantifying the accessibility of the metagenome by random expression cloning techniques. Environ Microbiol. 2004;6:879–886. doi: 10.1111/j.1462-2920.2004.00640.x. [DOI] [PubMed] [Google Scholar]
- Craig JW, Chang FY, Kim JH, Obiajulu SC, Brady SF. Expanding small-molecule functional metagenomics through parallel screening of broad-host-range cosmid environmental DNA libraries in diverse proteobacteria. Appl Environ Microbiol. 2010;76:1633–1641. doi: 10.1128/AEM.02169-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin B. Genes VII. New York: Oxford University Press; 2000. [Google Scholar]
- Rothen SA, Sauer M, Sonnleitner B, Witholt B. Growth characteristics of Escherichia coli HB101[pGEc47] on defined medium. Biotechnol Bioeng. 1998;58:92–100. doi: 10.1002/(SICI)1097-0290(19980405)58:1<92::AID-BIT10>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritch E, Maniatis T. Molecular Cloning: A Laboratory Manual. 2. Cold Spring Harbor: Cold Spring harbor Laboratory Press; 1989. [Google Scholar]
- Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. 3. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- Gietz RD, Schiestl RH, Willems AR, Woods RA. Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast. 1995;11:355–360. doi: 10.1002/yea.320110408. [DOI] [PubMed] [Google Scholar]
- Mumberg D, Müller R, Funk M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene. 1995;156:119–122. doi: 10.1016/0378-1119(95)00037-7. [DOI] [PubMed] [Google Scholar]
- Ejiofor CG. MSc thesis. Lund University, Department of Applied Microbiology; 2004. Molecular tools for improving xylose fermentation in xylose isomerase expressing yeasts. [Google Scholar]
- Winkler AA, Kuyper SM, De Laat WT, Van Dijken JP, Pronk JT. Metabolic Engineering of Xylose Fermenting Eukaryotic Cells. Geneva: World Intellectual Property Organization; 2006. http://www.wipo.int/pctdb/en/wo.jsp?WO=2006009434&IA=NL2005000516&DISPLAY=DESC International Patent Application WO/2006/009434. Published 26 January 2006. [Google Scholar]
- Hallborn J. PhD thesis. Lund University, Department of Applied Microbiology; 1995. Metabolic engineering of Saccharomyces cerevisiae: expression of genes involved in pentose metabolism. [Google Scholar]
- Amore R, Wilhelm M, Hollenberg CP. The fermentation of xylose: an analysis of the expression of Bacillus and Actinoplanes xylose isomerase genes in yeast. Appl Microbiol Biotechnol. 1989;30:351–357. doi: 10.1007/BF00296623. [DOI] [Google Scholar]
- Lönn A, Träff-Bjerre KL, Cordero Otero RR, van Zyl WH, Hahn-Hägerdal B. Xylose isomerase activity influences xylose fermentation with recombinant Saccharomyces cerevisiae strains expressing mutated xylA from Thermus thermophilus. Enzyme Microb Technol. 2003;32:567–573. doi: 10.1016/S0141-0229(03)00024-3. [DOI] [Google Scholar]