

Abstract
Toll-like receptors (TLR) -7 and -8 are thought to play an important role in immune activation processes underlying the pathophysiology of HIV and several clinically important autoimmune diseases. Based on our earlier findings of TLR7-antagonistic activity in a 3H imidazoquinoline, we sought to examine a pilot library of 3H imidazoquinolines for dual TLR7/8 antagonists, since they remain a poorly explored chemotype. Two-dimensional NOE experiments were employed to unequivocally characterize the compounds. A quinolinium compound 12, bearing p-methoxybenzyl substituents on N3 and N5 positions was identified as a lead. Compound 12 was found to inhibit both TLR7 and TLR8 at low micromolar concentrations. Our preliminary results suggest that alkylation with electron-rich substituents on the quinoline N5, or conversely, elimination of the fixed charge of the resultant quaternary amine on the quinolinium may yield more active compounds.
Keywords: Toll-like receptor, TLR7, TLR8, Imidazoquinoline, NOESY, HIV, Autoimmune diseases
Introduction
Chronic immune activation is a hallmark of several infectious and autoimmune diseases. Dysregulated cellular and humoral immune responses in progressive HIV infection,1;2 for instance, leads to accelerated turnover of CD4+ lymphocytes, thereby providing a milieu for HIV replication.3 The engagement of toll-like receptor-7 (TLR7) by single-stranded viral RNA (ssRNA) has recently been reported to play a central role in immune activation-driven HIV replication.4 Progressive CD4+ T lymphocyte depletion in non-human primate models is highly correlated with TLR7-mediated interferon-α (IFN-α) production by plasmacytoid dendritic cells, and antagonists of TLR7 have been shown to inhibit immune activation.4 In contrast to the predominantly CD4+ T lymphocyte-driven activation by TLR7- and -8 mediated recognition of HIV ssRNA, autoreactive B lymphocytes are thought to play an important role in the sustained generation of autoantibodies directed against both cytosolic and nuclear components contributing to the pathophysiology of disease states such as Systemic Lupus Erythematosis and Sjögren’s syndrome.5–10 TLR7 and TLR8 are thus logical targets for pharmacological intervention, and inhibitors for these endosomal receptors are being actively studied for potential use in the therapy of such autoimmune diseases.6;11;12
Whereas small molecule agonists of TLR7 are well known,13–15 the only known class of TLR7 antagonists, until recently, were single-stranded phosphorothioate oligonucleotides.16–19 En route to the synthesis of a TLR7-agonistic imidazoquinoline, gardiquimod, we synthesized its 3H regioisomer, which was found to be a weak TLR7 antagonist.20 A des-amino precursor of the 3H regioisomer (4a, Scheme 1) was more potent as a TLR7 antagonist, with an IC50 value of 7.5 μM;20 negligible TLR8-antagonstic activity, however, was observed with this compound. Given the potential value of a detailed examination of this poorly explored chemotype toward obtaining leads for novel, and more potent TLR7/8 dual antagonists, we undertook the syntheses and evaluation of a preliminary library of 3H imidazoquinolines with the aim of identifying potential chemotypes capable of inhibiting both TLR7 and TLR8.
Scheme 1.

Syntheses of derivatives of 4a and N3-substituted 3H imidazoquinolines.
Reagents: i. 2-(tert-Butoxycarbonyl(ethyl)amino)acetic acid, HATU, DMF (b) NaOH/H2O, EtOH; ii. DBU, 2,2-dimethyloxirane; iii. CF3COOH; iv. 3-Chloroperoxybenzoic acid, CH2Cl2, CHCl3, MeOH; v. (a) Benzoyl isocyanate, CH2Cl2 (b) CF3COOH. vi. 1-(Chloromethyl)-4-methoxybenzene, THF, 80 oC (b) CF3COOH; vii. 3-Bromo-1-propanol, DMF, 80 °C (b) CF3COOH; viii. Propargyl bromide, THF, 90 oC (b) CF3COOH; ix. Methyl iodide, DBU, THF (b) CF3COOH.
Results and Discussion
Our point of departure was 4a (Scheme 1), a 4-desamino, 3H imidazoquinoline with a 2-methyl-propan-2-ol substituent at N3, and a 2-(ethylamino)methylene substituent at C2.20 We first synthesized the quinoline N-oxide and C4-N-benzoyl derivatives (10, and 11, respectively; Scheme 1). Upon finding that neither compound showed any appreciable activity (Table 1), we elected to first examine a small subset of compounds with varying substituents at N3 (5a, 6a, 7a, 8a; Scheme 1). These compounds also were found to be either inactive (6a) or of low potency (5a, 7a, 8a; Table 1).
Table 1.
TLR7- and TLR8-antagonistic activities of the title compounds. ND denotes no significant activity detected at the highest concentration tested (250 μM). All samples were tested in duplicate, except for 7b, 7d, and 12 which were assayed in quadruplicate. SD values are given for these latter compounds.
| Structure | Compound Number | TLR7 Antagonistic Activity (μM) | TLR8 Antagonistic Activity (μM) |
|---|---|---|---|
|
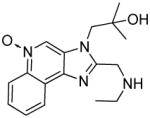
10 | ND | 46.5 |
|
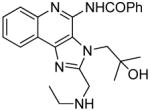
11 | ND | 21.17 |
|
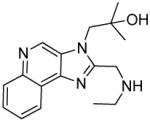
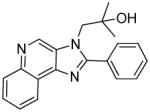
4a | 7.5 | 56.31 |
|
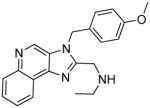
5a | 28.02 | 59.64 |
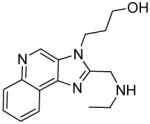
|
6a | ND | ND |
|
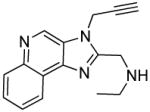
7a | 26.1 | 28.13 |
|
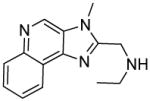
8a | 30.44 | 35.43 |
|
4b | ND | 38.42 |
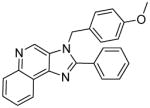
|
5b | ND | ND |
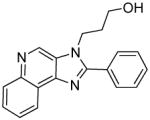
|
6b | ND | 37.24 |
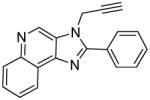
|
7b | 14.85 ± 0.74 | 13.71 ± 0.75 |
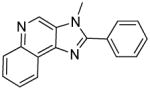
|
8b | 21.63 | 25.81 |
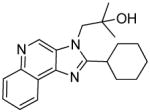
|
4c | ND | 38.73 |
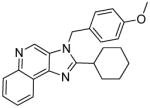
|
5c | 19.9 | 40.01 |
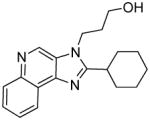
|
6c | ND | 20.32 |
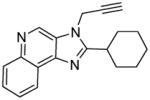
|
7c | 21.87 | 21.12 |
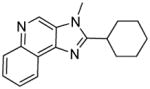
|
8c | 22.04 | 24.3 |
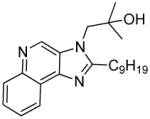
|
4d | 14.4 | 52.94 |
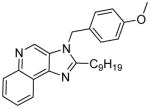
|
5d | 19.44 | 46.4 |
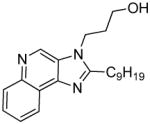
|
6d | ND | 4.81 |
|
7d | 8.58 ± 0.43 | 10.07 ± 0.50 |
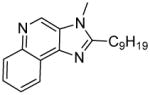
|
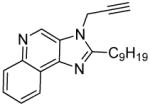
8d | 10.14 | 68.8 |
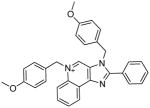
|
12 | 2.79 ± 0.12 | 4.55 ± 0.13 |
We next attempted generating a small subset (15 compounds) in which the substituents at N3 and C2 were combinatorially varied. Since this was an exploratory library, we chose the C2 substituents from among planar, aromatic (phenyl), a cycloaliphatic (cyclohexyl), and a long-chain aliphatic (nonyl) groups (Scheme 2), while preserving the N3 substituents that we had used in Scheme 1. Alkylation of the 3 series of compounds afforded, as expected, three sets of regioisomers. In order to unambiguously characterize the position of the alkyl groups in these isomers, we correlated the crystal structure of 4b with its 2D-NOESY spectrum (Fig. 1). The NOESY spectrum showed diagnostic NOEs between the methylene protons on N3 (atom 20) with the quinoline proton (atom 9), as well as the phenyl protons (atoms 15, 19). Also seen, as would be expected, are NOEs between the methylene protons on N3 (atom 20) with the terminal dimethyl protons on the N3 substituent (Fig. 1). NOESY spectra for compounds 6b, 7d, and 8c (Supporting Data) with differing C2 and N3 substituents are consistent with the regioselectivity observed with the alkylation reactions.
Scheme 2.

Syntheses of N3 and C2 modified 3H imidazoquinoline compounds.
Reagents: i. Polyphosphoric acid, R1-COOH, 180 oC; ii. DBU, 2,2-dimethyloxirane; iii. 1-(Chloromethyl)-4-methoxybenzene, DBU, THF, 80 °C; iv. 3-Bromo-1-propanol, DBU, DMF, 80 °C; v. Propargyl bromide, DBU, THF, 90 °C; vi. Methyl iodide, DBU, THF.
Fig. 1.

Crystal structure (top) and 2D-NOESY spectrum (bottom) of Compound 4b.
Most of these compounds displayed modest activity, with exceptions being the C2-nonyl-substituted 7d and 8d compounds exhibiting low micromolar TLR7-inhibitory activity. 7d was also found to be TLR8-antagonistic (IC50: 10 μM, Table 1).
During the synthesis of 5b, one of the side-products, 12, corresponded in mass- and NMR-spectral characteristics to a bis-alkylated compound. This was isolated and was found to be the best-in-class in both TLR7- and TLR8-antagonism assays, with IC50 values of 2.79 and 4.55 μM, respectively (Table 1). We verified that the inhibition was competitive by classic Schild analyses; the IC50 values for 12 were determined to be 2.79 μM and 0.82 μM, at agonist (gardiquimod) concentrations of 1 μg/mL and 250 ng/mL, respectively. In order to determine whether 12 was a quinolinium or imidazolinium species, we synthesized the mono-alkylated quinolinium 13 and the 1H regioisomer 17 (Scheme 3). The dialkyl species 12 was obtained using an excess of 1-(chloromethyl)-4-methoxybenzene and DBU as a base in THF at 150°C, whereas 13 was obtained in the absence of DBU and in DMF at 120°C. The 1H regioisomer 17 was synthesized by pre-installing the p-methoxybenzyl substituent on N1 (Scheme 3). We characterized the monoalkylated regioisomers 5b, 13, and 17 (Scheme 3) via 2D-NOESY experiments (Fig. 2).
Scheme 3.

Syntheses of analogues of 5b.
Reagents: i. 1-(Chloromethyl)-4-methoxybenzene, DBU, THF, 150 °C; ii. 1-(Chloromethyl)-4-methoxybenzene, DMF, 120 °C; iii. N,N-diethylpropan-2-amine, toluene:2-propanol (4:1), 70 °C; iv. (a) H2, Pd/C, 60 psi, MeOH; (b) Benzoic acid, HBTU, Et3N, DMAP, DMF.
Fig. 2.

2D-NOESY spectra of Compounds 5b, 13, 17 and 12.
Compounds 5b, 13, and 17 are all monoalkylated species, with the p-methoxybenzyl substituent at the N3 (imidazole), N5 (quinolinium), N1 (imidazole), respectively. Each of these compounds showed characteristic and diagnostic NOE crosspeaks. In 5b (as in 4b, discussed earlier), the NOESY spectrum showed NOEs between the methylene protons on N3 (atom 20) with the quinoline proton (atom 9), as well as the phenyl protons (atoms 15, 19); compound 13 showed NOEs between the methylene protons on N5 (atom 20) with the quinoline protons (atoms 9 and 6); compound 17 was distinguished by crosspeaks between phenyl protons (atoms 15, 19) and the quinoline proton (atom 3) (Fig. 2). The clear NOE patterns helped establish unequivocally that the additional p-methoxybezyl substituent in compound 12 was on the quinoline nitrogen (N5) because of NOEs similar to both 13 and 5b (Fig. 2).
Compound 12, to our considerable surprise, showed the highest potency in simultaneously inhibiting both TLR7 and TLR8 (Table 1, Fig. 3). This was unexpected since the quinolinium compound with its fixed charge is generally thought to be relatively membrane-impermeant and, as discussed earlier, both TLR7 and TLR8 are compartmentalized in the endosome.
Fig. 3.

2D-Scatter plot of TLR7 (abscissa) and TLR8 (ordinate) -antagonistic activities of the title compounds. Compounds that do not show significant inhibitory activity have been omitted.
In conclusion, a pilot library of 3H imidazoquinolines have been synthesized, characterized, and evaluated for biological activity. Although possessing modest activity, a dual TLR7/TLR8 antagonist, 12, has been identified with micromolar potencies. These preliminary results have been instructive in that they already point to strategies for improvement in potency. For instance, the monoalkylated compounds 7b and 7d, bearing propargyl groups on N3, ranked next in potency to 12, are attractive leads for additional alkylation with electron-rich substitutents on the quinoline N5. As mentioned earlier, the quaternary amine on the quinolinium of 12 may deter optimal trans-membrane transport and concentration in the endosomal compartment, and carbocyclic analogues may be evaluated to carefully examine the effect of the fixed charge.
Experimental Section
All of the solvents and reagents used were obtained commercially and used as such unless noted otherwise. Moisture- or air-sensitive reactions were conducted under nitrogen atmosphere in oven-dried (120 °C) glass apparatus. The solvents were removed under reduced pressure using standard rotary evaporators. Flash column chromatography was carried out using RediSep Rf ‘Gold’ high performance silica columns on CombiFlash Rf instrument unless otherwise mentioned, while thin-layer chromatography was carried out on silica gel CCM pre-coated aluminum sheets. Microwave reactions were done in Synthos 3000 instrument (Anton Paar). Purity for all final compounds was confirmed to be greater than 97% by LC-MS using a Zorbax Eclipse Plus 4.6 mm × 150 mm, 5 μm analytical reverse phase C18 column with H2O-isopropanol or H2O-CH3CN gradients and an Agilent ESI-TOF mass spectrometer (mass accuracy of 3 ppm) operating in the positive ion acquisition mode. Compounds 2, 3a, 4a and 14 were synthesized as published by us earlier.20 All reported yields are unoptimized.
Synthesis of Compound 10: 2-((ethylamino)methyl)-3-(2-hydroxy-2-methylpropyl)-3H-imidazo[4,5-c]quinoline 5-oxide
Compound 3a (145 mg, 0.36 mmol) was dissolved in 10 mL of anhydrous dichloromethane/chloroform (1:1) and 1 mL of anhydrous MeOH. 3-Chloro-peroxybenzoic acid (188 mg, 1.09 mmol) was added and the reaction mixture was refluxed for 30 minutes. The solvent was then removed under vacuum and the residue was purified using column chromatography (4% MeOH/dichloromethane) to obtain the compound 9 (100 mg, 66%). Compound 9 (28 mg, 0.07 mmol) was then dissolved in 5 mL of trifluoroacetic acid and stirred for 30 minutes. The solvent was then removed and the residue was washed with diethyl ether to obtain the compound 10 (40 mg, 95%). 1H NMR (400 MHz, MeOD) δ 9.46 (s, 1H), 8.77 – 8.67 (m, 1H), 8.66 – 8.55 (m, 1H), 8.00 – 7.86 (m, 2H), 4.81 (s, 2H), 4.41 (s, 2H), 3.39 (q, J = 7.3, 2H), 1.48 (t, J = 7.3, 3H), 1.31 (s, 6H). 13C NMR (101 MHz, MeOD) δ 151.64, 139.07, 137.10, 129.59, 129.53, 128.75, 127.64, 122.22, 122.06, 119.15, 70.58, 54.56, 43.12, 43.07, 26.06, 10.09. MS (ESI) calculated for C17H22N4O2, (M + H)+: 315.1816; observed: 315.1764.
Synthesis of Compound 11: N-(2-((ethylamino)methyl)-3-(2-hydroxy-2-methylpropyl)-3H-imidazo[4,5-c]quinolin-4-yl)benzamide
To a solution of 9 (68 mg, 0.16 mmol) in 5 mL of anhydrous dichloromethane, was added benzoylisocyanate (36 mg, 0.25 mmol) and the reaction mixture was refluxed for 30 minutes. The solvent was then removed under vacuum and the residue was purified using column chromatography (35 % EtOAc/dichloromethane) to obtain tert-butyl (4-benzamido-3-(2-hydroxy-2-methylpropyl)-3H-imidazo[4,5-c]quinolin-2-yl)methyl(ethyl)carbamate (30 mg, 35%), which was N-Boc deprotected by stirring in 5 mL of trifluoroacetic acid for 30 minutes The solvent was then removed and the residue was washed with diethyl ether to obtain the compound 11 (35 mg, 95%). 1H NMR at 50°C (400 MHz, MeOD) δ 8.42 (d, J = 8.1 Hz, 1H), 8.25 (d, J = 7.3 Hz, 2H), 7.81 – 7.73 (m, 1H), 7.72 – 7.65 (m, 1H), 7.57 (t, J = 6.7 Hz, 2H), 7.50 (t, J = 7.4 Hz, 2H), 5.19 (s, 2H), 4.80 (s, 2H), 3.35 (q, J = 7.3 Hz, 2H), 1.45 (t, J = 7.3 Hz, 3H), 1.31 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 178.99, 155.88, 151.18, 145.60, 137.98, 133.28, 131.79, 129.13, 128.93, 128.15, 125.10, 122.17, 118.43, 117.99, 70.60, 56.37, 45.68, 44.04, 27.42, 14.81. MS (ESI) calculated for C24H27N5O2, (M + H)+: 418.2238; observed: 418.2137.
Synthesis of Compound 5a: N-((3-(4-methoxybenzyl)-3H-imidazo[4,5-c]quinolin-2-yl)methyl)ethanamine
To a solution of 2 (50 mg, 0.15 mmol) in 1 mL of anhydrous THF, were added DBU (47 mg, 0.31 mmol) and 1-(chloromethyl)-4-methoxybenzene (96 mg, 0.61 mmol). The solution was then heated in a sealed vessel at 80 °C for 1 hour. After cooling to room temperature, and removing solvent under vacuum, the residue was dissolved in EtOAc and washed with water, dried over sodium sulfate, concentrated and purified using column chromatography (1% MeOH/dichloromethane) to obtain the compound tert-butyl ethyl ((3-(4- methoxybenzyl)-3H-imidazo [4,5-c]quinolin-2-yl)methyl)carbamate, which was dissolved in 5 mL of trifluoroacetic acid and stirred for 30 minutes. The solvent was then removed and the residue was washed with diethyl ether to obtain the compound 5a (18 mg, 21%). 1H NMR (400 MHz, MeOD) δ 9.46 (s, 1H), 8.85 – 8.71 (m, 1H), 8.28 (d, J = 8.0, 1H), 8.07 – 7.93 (m, 2H), 7.31 (d, J = 8.8 Hz, 2H), 7.04 – 6.95 (m, 2H), 5.77 (s, 2H), 4.81 (s, 2H), 3.80 (s, 3H), 3.40 (q, J = 7.3 Hz, 2H), 1.48 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, MeOD) δ 160.28, 153.97, 147.68, 133.33, 130.79, 129.07, 128.77, 128.59, 125.90, 122.61, 122.29, 121.52, 114.47, 54.42, 47.95, 43.33, 42.91, 10.00. MS (ESI) calculated for C21H22N4O, (M + H)+: 347.1866; observed: 347.1890.
Synthesis of Compound 6a: 3-(2-((ethylamino)methyl)-3H-imidazo[4,5-c]quinolin-3-yl)propan-1-ol
To a solution of 2 (50 mg, 0.15 mmol) in 1 mL of anhydrous THF, were added DBU (47 mg, 0.31 mmol) and 3-bromopropan-1-ol (32 mg, 0.23 mmol). The solution was then heated in a sealed vessel at 80 °C for an hour. After cooling to room temperature, and removing solvent under vacuum, the residue was dissolved in EtOAc and washed with water, dried over sodium sulfate, concentrated and purified using column chromatography to obtain tert-butyl ethyl((3-(3-hydroxypropyl)-3H-imidazo[4,5-c]quinolin-2-yl)methyl)carbamate (9 mg, 15%), which was dissolved in 8 mL of trifluoroacetic acid and stirred for 30 minutes. The solvent was then removed and the residue was washed with diethyl ether to obtain compound 6a (11 mg, 95%). 1H NMR (400 MHz, MeOD) δ 9.24 (s, 1H), 8.60 – 8.52 (m, 1H), 8.21 – 8.09 (m, 1H), 7.81 – 7.67 (m, 2H), 4.68 (t, J = 6.8 Hz, 2H), 4.31 (s, 2H), 3.58 – 3.50 (m, 2H), 2.88 (q, J = 7.2 Hz, 2H), 2.28 – 2.14 (m, 2H), 1.25 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, MeOD) δ 154.25, 143.61, 136.25, 128.49, 128.17, 127.45, 126.88, 121.74, 121.20, 57.03, 44.36, 43.42, 40.91, 32.25, 13.04. MS (ESI) calculated for C16H20N4O, (M + H)+: 285.1710; observed: 285.1752.
Synthesis of Compound 7a: N-((3-(prop-2-ynyl)-3H-imidazo[4,5-c]quinolin-2-yl) methyl)ethanamine
To a solution 2 (65 mg, 0.2 mmol) in 1 mL of anhydrous THF, were added DBU (61 mg, 0.4 mmol) and 80% propargyl bromide in toluene (119 mg, 1.0 mmol). The solution was then heated in a sealed vessel at 90 °C for 30 minutes. After cooling to room temperature, and removing the solvent under vacuum, the residue was dissolved in EtOAc and washed with water, dried over sodium sulfate, concentrated and purified using column chromatography to obtain tert-butyl ethyl((3-(prop-2-ynyl)-3H-imidazo[4,5-c]quinolin-2-yl)methyl)carbamate, which was dissolved in 5 mL of trifluoroacetic acid and stirred for 30 minutes. The solvent was then removed and the residue was washed with diethyl ether to obtain the compound 7a (23 mg, 43%). 1H NMR (400 MHz, MeOD) δ 9.74 (s, 1H), 8.77 (dd, J = 8.1, 1.1 Hz, 1H), 8.30 (d, J = 8.1 Hz, 1H), 8.08 – 7.92 (m, 2H), 5.54 (d, J = 2.6 Hz, 2H), 4.95 (s, 2H), 3.45 (q, J = 7.2 Hz, 2H), 3.27 (t, J = 2.5 Hz, 1H), 1.52 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, MeOD) δ 153.17, 147.34, 137.95, 133.55, 130.69, 129.01, 128.14, 123.11, 122.21, 121.49, 76.40, 75.07, 43.38, 42.62, 34.26, 10.02. MS (ESI) calculated for C16H16N4, (M + H)+: 265.1448; observed: 265.1553.
Synthesis of Compound 8a: N-((3-methyl-3H-imidazo[4,5-c]quinolin-2-yl)methyl) ethanamine
To a solution of 2 (50 mg, 0.15 mmol) in 1 mL of anhydrous THF, were added DBU (47 mg, 0.31 mmol) and iodomethane (33 mg, 0.23 mmol). The solution was stirred for an hour and the solvent was then removed under vacuum. The residue was dissolved in EtOAc, washed with water, dried over sodium sulfate, concentrated and purified using column chromatography (30% EtOAc/dichloromethane) to obtain tert-butyl ethyl((3-methyl-3H- imidazo[4,5-c]quinolin-2-yl) methyl)carbamate (7 mg, 20%), which was dissolved in 5 mL of trifluoroacetic acid and stirred for 30 minutes. The solvent was then removed and the residue was washed with diethyl ether to obtain the compound 8a (13 mg, 95%). 1H NMR (400 MHz, MeOD) δ 9.72 (s, 1H), 8.79 (d, J = 7.9 Hz, 1H), 8.30 (d, J = 8.2 Hz, 1H), 8.14 – 7.91 (m, 2H), 4.91 (s, 2H), 4.18 (s, 3H), 3.45 (q, J = 7.3 Hz, 2H), 1.52 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, MeOD) δ 154.89, 147.95, 136.50, 132.91, 130.97, 129.23, 129.14, 122.38, 121.87, 121.36, 43.38, 42.48, 30.57, 10.01. MS (ESI) calculated for C14H16N4, (M + H)+: 241.1448; observed: 241.1476.
Synthesis of Compound 3b: 2-phenyl-3H-imidazo[4,5-c]quinoline
To a mixture of 1 (450 mg, 2.83 mmol) and benzoic acid (691 mg, 5.66 mmol), was added polyphosphoric acid (approx. 25 mL) and heated to 180 °C for one hour. The reaction mixture was then slowly cooled to room temperature and the polyphosphoric acid was slowly neutralized with ammonium hydroxide until the pH was around 8. The compound was then extracted using EtOAc and the EtOAc fraction was then washed with water, dried over sodium sulfate, concentrated and purified using column chromatography (4% MeOH/dichloromethane) to afford the compound 3b (315 mg, 45%). 1H NMR (400 MHz, MeOD) δ 9.07 (s, 1H), 8.44 (d, J = 6.9 Hz, 1H), 8.18 – 8.16 (m, 1H), 8.16 – 8.15 (m, 1H), 8.11 – 8.06 (m, 1H), 7.72 – 7.62 (m, 2H), 7.60 – 7.52 (m, 3H). 13C NMR (101 MHz, MeOD) δ 143.22, 130.45, 129.06, 128.84, 128.14, 127.48, 126.74, 126.63, 121.39. MS (ESI) calculated for C16H11N3, (M + H)+: 246.1026; observed: 246.1025.
Compounds 3c and 3d were synthesized similarly as described for compound 3b
3c (409 mg, 58%)
1H NMR (500 MHz, MeOD) δ 8.93 (s, 1H), 8.25 (s, 1H), 7.98 (t, J = 8.2 Hz, 1H), 7.63 – 7.52 (m, 2H), 3.02 – 2.87 (m, 1H), 2.09 – 2.01 (m, 2H), 1.83 (dd, J = 10.4, 7.3 Hz, 2H), 1.69 (dd, J = 14.8, 8.0 Hz, 1H), 1.67 – 1.59 (m, 2H), 1.46 – 1.35 (m, 2H), 1.34 – 1.22 (m, 1H). 13C NMR (126 MHz, MeOD) δ 144.54, 129.56, 128.68, 127.95, 122.57, 40.03, 32.89, 27.16, 26.91. MS (ESI) calculated for C16H17N3, (M + H)+: 252.1495; observed: 252.1565.
3d (355 mg, 38%)
1H NMR (500 MHz, MeOD) δ 8.93 (s, 1H), 8.22 (s, 1H), 8.00 (d, J = 8.5 Hz, 1H), 7.62 – 7.53 (m, 2H), 2.91 (t, J = 7.7 Hz, 2H), 1.83 – 1.75 (m, 2H), 1.36 – 1.22 (m, 4H), 1.14–1.17 (m, 8H), 0.75 (t, J = 6.9 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 158.61, 144.52, 129.56, 128.72, 128.00, 122.49, 33.02, 30.55, 30.41, 30.37, 30.32, 29.92, 29.41, 23.74, 14.45. MS (ESI) calculated for C19H25N3, (M + H)+: 296.2121; observed: 296.2178.
Synthesis of Compound 4b: 2-methyl-1-(2-phenyl-3H-imidazo[4,5-c]quinolin-3-yl)propan-2-ol
To a solution of 3b (50 mg, 0.2 mmol) in 1 mL of 2,2-dimethyloxirane, was added DBU (62 mg, 0.41 mmol) and the solution was heated under microwave conditions (600 W, 80 °C, 1h). After cooling to room temperature, and removing the solvent under vacuum, the residue was dissolved in EtOAc and washed with water, dried over sodium sulfate, concentrated and purified using column chromatography (4% MeOH/dichloromethane) to obtain the compound 4b (24 mg, 38%). 1H NMR (400 MHz, MeOD) δ 9.34 (s, 1H), 8.65 – 8.51 (m, 1H), 8.17 – 8.13 (m, 1H), 7.87 – 7.79 (m, 2H), 7.78 – 7.69 (m, 2H), 7.67 – 7.63 (m, 3H), 4.59 (s, 2H), 1.00 (s, 6H). 13C NMR (101 MHz, MeOD) δ 155.90, 143.40, 143.02, 139.02, 130.15, 129.95, 129.85, 129.76, 128.72, 128.04, 127.46, 126.80, 121.65, 121.37, 70.71, 55.09, 26.19. MS (ESI) calculated for C20H19N3O, (M + H)+: 318.1601; observed: 318.1631.
Compounds 4c and 4d were synthesized similarly as described for compound 4b
4c (42 mg, 65%)
1H NMR (400 MHz, MeOD) δ 9.19 (s, 1H), 8.70 – 8.56 (m, 1H), 8.14 – 8.05 (m, 1H), 7.74 – 7.61 (m, 2H), 4.43 (s, 2H), 3.33 (dt, J = 3.3, 1.6 Hz, 1H), 2.07 – 1.80 (m, 7H), 1.62 – 1.42 (m, 3H), 1.32 (s, 6H). 13C NMR (101 MHz, MeOD) δ 162.55, 143.37, 143.26, 137.65, 128.97, 127.93, 127.12, 126.50, 121.56, 121.47, 70.32, 53.88, 36.16, 31.55, 26.26, 25.84, 25.53. MS (ESI) calculated for C20H25N3O, (M + H)+: 324.2070; observed: 324.2084.
4d (11 mg, 18%)
1H NMR (400 MHz, MeOD) δ 9.17 (s, 1H), 8.58 (d, J = 6.4 Hz, 1H), 8.14 – 8.09 (m, 1H), 7.69 (dd, J = 6.8, 3.2 Hz, 2H), 4.40 (s, 2H), 3.13 (t, J = 6.4 Hz, 2H), 2.02 – 1.88 (m, 2H), 1.49 (d, J = 6.3 Hz, 2H), 1.39 (d, J = 15.1 Hz, 2H), 1.38 – 1.20 (m, 14H), 0.92 – 0.86 (m, 3H). 13C NMR (101 MHz, MeOD) δ 158.89, 143.39, 143.19, 137.44, 129.30, 127.99, 127.27, 126.65, 121.52, 121.46, 70.72, 54.40, 31.66, 29.27, 29.22, 29.15, 29.05, 27.97, 27.32, 26.51, 22.37, 13.24. MS (ESI) calculated for C23H33N3O, (M + H)+: 368.2696; observed: 368.2733.
Synthesis of Compound 5b: 3-(4-methoxybenzyl)-2-phenyl-3H-imidazo[4,5-c]quinoline
To a solution of 3b (50 mg, 0.2 mmol) in 1 mL of anhydrous THF, were added DBU (62 mg, 0.41 mmol) and 1-(chloromethyl)-4-methoxybenzene (128 mg, 0.82 mmol). The solution was then heated in a sealed vessel at 80 °C for an hour. After cooling to room temperature, and removing the solvent under vacuum, the residue was dissolved in EtOAc and washed with water, dried over sodium sulfate, concentrated and purified using column chromatography (8% EtOAc/dichloromethane) to obtain the compound 5b (36 mg, 33%). 1H NMR (400 MHz, CDCl3) δ 8.92 (s, 1H), 8.73 – 8.69 (m, 1H), 8.24 – 8.20 (m, 1H), 7.82 – 7.77 (m, 2H), 7.74 – 7.69 (m, 2H), 7.59 – 7.53 (m, 3H), 7.06 (d, J = 8.8 Hz, 2H), 6.90 – 6.84 (m, 2H), 5.60 (s, 2H), 3.80 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 159.52, 154.86, 144.69, 144.54, 136.63, 130.42, 129.67, 129.62, 129.55, 129.11, 129.01, 128.30, 127.66, 127.53, 127.45, 126.86, 122.42, 121.87, 114.64, 55.33, 48.60. MS (ESI) calculated for C24H19N3O, (M + H)+: 366.1601; observed: 366.1491.
Compounds 5c and 5d were synthesized similarly as described for compound 5b
5c (9 mg, 12%)
1H NMR (400 MHz, CDCl3) δ 8.88 (s, 1H), 8.70 – 8.60 (m, 1H), 8.18 (dd, J = 6.6, 2.9 Hz, 1H), 7.72 – 7.60 (m, 2H), 7.04 (d, J = 8.8 Hz, 2H), 6.92 – 6.80 (m, 2H), 5.50 (s, 2H), 3.79 (s, 3H), 2.96 (s, 1H), 2.00 – 1.67 (m, 7H), 1.41 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 159.52, 135.86, 129.47, 127.60, 127.07, 126.42, 122.31, 121.95, 114.57, 55.33, 47.06, 36.74, 31.84, 26.25, 25.61. MS (ESI) calculated for C24H25N3O, (M + H)+: 372.2070; observed: 372.1990.
5d (21 mg, 30%)
1H NMR (400 MHz, CDCl3) δ 8.89 (s, 1H), 8.66 – 8.58 (m, 1H), 8.22 – 8.15 (m, 1H), 7.73 – 7.62 (m, 2H), 7.05 (d, J = 8.7 Hz, 2H), 6.91 – 6.82 (m, 2H), 5.47 (s, 2H), 3.79 (s, 3H), 3.05 – 2.95 (m, 2H), 1.87 (dt, J = 15.6, 7.7 Hz, 2H), 1.49 – 1.38 (m, 2H), 1.33 – 1.26 (m, 10H), 0.89 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 159.57, 156.76, 144.46, 144.11, 135.74, 129.54, 128.59, 127.69, 127.33, 127.14, 126.59, 122.20, 121.75, 114.57, 55.32, 47.36, 31.85, 29.54, 29.42, 29.29, 29.25, 28.28, 27.86, 22.66, 14.11. MS (ESI) calculated for C27H33N3O, (M + H)+: 416.2696; observed: 416.2560.
Synthesis of Compound 6b: 3-(2-phenyl-3H-imidazo[4,5-c]quinolin-3-yl)propan-1-ol
To a solution of 3b (60 mg, 0.25 mmol) in 2 mL of anhydrous DMF, were added DBU (62 mg, 0.41 mmol) and 3-bromopropan-1-ol (139 mg, 1.0 mmol). The solution was then heated in a sealed vessel at 80 °C for 2 hours. After cooling to room temperature, and removing the solvent under vacuum, the residue was dissolved in EtOAc and washed with water, dried over sodium sulfate, concentrated and purified using column chromatography (20% acetone/dichloromethane) to obtain the compound 6b (13 mg, 18%). 1H NMR (400 MHz, MeOD) δ 9.30 (s, 1H), 8.60 (dt, J = 5.0, 2.2 Hz, 1H), 8.21 – 8.17 (m, 1H), 7.89 – 7.85 (m, 2H), 7.79 – 7.71 (m, 2H), 7.67 (dd, J = 4.2, 2.4 Hz, 3H), 4.74 – 4.60 (m, 2H), 3.55 (t, J = 4.6 Hz, 2H), 2.12 – 2.03 (m, 2H). 13C NMR (101 MHz, MeOD) δ 155.37, 143.79, 143.53, 136.59, 130.46, 129.65, 129.38, 129.15, 128.78, 128.22, 127.59, 126.93, 121.70, 121.40, 58.02, 42.26, 32.70. MS (ESI) calculated for C19H17N3O, (M + H)+: 304.1444; observed: 304.1440.
Compounds 6c and 6d were synthesized similarly as described for compound 6b
6c (12 mg, 16%)
1H NMR (400 MHz, MeOD) δ 9.18 (s, 1H), 8.66 – 8.59 (m, 1H), 8.12 (dt, J = 4.8, 2.8 Hz, 1H), 7.72 – 7.67 (m, 2H), 4.61 (t, J = 7.2 Hz, 2H), 3.64 (dd, J = 12.7, 7.0 Hz, 2H), 3.27 – 3.16 (m, 1H), 2.15 – 2.09 (m, 2H), 2.05 – 1.85 (m, 8H), 1.66 – 1.39 (m, 2H). 13C NMR (101 MHz, MeOD) δ 161.46, 143.61, 135.86, 128.04, 127.90, 127.22, 126.61, 121.63, 121.44, 57.72, 40.52, 35.95, 32.95, 31.62, 25.82, 25.49. MS (ESI) calculated for C19H23N3O, (M + H)+: 310.1914; observed: 310.1927.
6d (11 mg, 22%)
1H NMR (400 MHz, CDCl3) δ 9.12 (s, 1H), 8.58 (dt, J = 17.0, 8.1 Hz, 1H), 8.17 (dt, J = 18.1, 9.6 Hz, 1H), 7.78 – 7.55 (m, 2H), 4.53 (t, J = 6.8 Hz, 2H), 3.72 (t, J = 5.6 Hz, 2H), 3.11 – 2.96 (m, 2H), 2.22 – 2.03 (m, 2H), 1.94 (dt, J = 15.6, 7.7 Hz, 2H), 1.54 – 1.43 (m, 2H), 1.35 – 1.28 (m, 10H), 0.89 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 156.89, 144.17, 144.08, 135.51, 129.19, 128.53, 127.16, 126.59, 122.15, 121.79, 58.16, 40.70, 32.85, 31.86, 29.62, 29.46, 29.36, 29.26, 28.43, 27.50, 22.66, 14.10. MS (ESI) calculated for C22H31N3O, (M + H)+: 354.2540; observed: 354.2401.
Synthesis of Compound 7b: 2-phenyl-3-(prop-2-ynyl)-3H-imidazo[4,5-c]quinoline
To a solution of 3b (50 mg, 0.21 mmol) in 1 mL of anhydrous THF, were added DBU (62 mg, 0.41 mmol) and 80% propargyl bromide in toluene (98 mg, 0.82 mmol). The solution was then heated in a sealed vessel at 90 °C for an hour. After cooling to room temperature, and removing the solvent under vacuum, the residue was dissolved in EtOAc and washed with water, dried over sodium sulfate, concentrated and purified using column chromatography (35% EtOAc/dichloromethane) to obtain the compound 7b (6 mg, 10%). 1H NMR (400 MHz, CDCl3) δ 9.31 (s, 1H), 8.72 – 8.67 (m, 1H), 8.30 – 8.25 (m, 1H), 7.97 – 7.90 (m, 2H), 7.69 – 7.77 (m, 2H), 7.67 – 7.60 (m, 3H), 5.15 (d, J = 2.5, 2H), 2.59 (t, J = 2.5 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 144.90, 136.03, 130.66, 129.72, 129.69, 129.17, 128.63, 127.60, 126.95, 121.90, 75.11, 35.35. MS (ESI) calculated for C19H13N3, (M + H)+: 284.1182; observed: 284.1046.
Compounds 7c and 7d were synthesized similarly as described for compound 7b
7c (8 mg, 12%)
1H NMR (400 MHz, CDCl3) δ 9.16 (s, 1H), 8.65 – 8.56 (m, 1H), 8.28 – 8.11 (m, 1H), 7.75 – 7.60 (m, 2H), 5.09 (d, J = 2.5 Hz, 2H), 3.00 (m, 1H), 2.48 (t, J = 2.5 Hz, 1H), 2.11 (m, 2H), 2.04 – 1.89 (m, 6H), 1.47 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 159.48, 144.62, 135.35, 129.54, 127.76, 127.20, 126.51, 121.95, 76.47, 74.50, 36.58, 33.42, 31.62, 26.20, 25.65. MS (ESI) calculated for C19H19N3, (M + H)+: 290.1652; observed: 290.1740.
7d (10 mg, 15%)
1H NMR (400 MHz, CDCl3) δ 9.16 (s, 1H), 8.65 – 8.53 (m, 1H), 8.29 – 8.16 (m, 1H), 7.76 – 7.61 (m, 2H), 5.07 (d, J = 2.5 Hz, 2H), 3.13 – 2.97 (m, 2H), 2.48 (t, J = 2.5 Hz, 1H), 1.96 (dt, J = 15.5, 7.7 Hz, 3H), 1.57 – 1.46 (m, 2H), 1.46 – 1.38 (m, 2H), 1.30 (dd, J = 5.8, 4.2 Hz, 7H), 0.90 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 156.03, 144.63, 144.11, 135.27, 129.59, 127.89, 127.30, 126.70, 122.10, 121.76, 76.17, 74.63, 33.73, 31.86, 29.51, 29.44, 29.31, 29.25, 28.17, 27.67, 22.66, 14.11. MS (ESI) calculated for C22H27N3, (M + H)+: 334.2278; observed: 334.2291.
Synthesis of Compound 8b: 3-methyl-2-phenyl-3H-imidazo[4,5-c]quinoline
To a solution of 3b (40 mg, 0.16 mmol) in 1 mL of anhydrous THF, were added DBU (73 mg, 0.48 mmol) and iodomethane (114 mg, 0.8 mmol). The solution was stirred for 30 minutes and the solvent was then removed under vacuum to obtain the residue, which was dissolved in EtOAc, washed with water, dried over sodium sulfate, concentrated and purified using column chromatography (20% EtOAc/dichloromethane) to obtain the compound 8b (15 mg, 36%). 1H NMR (400 MHz, CDCl3) δ 8.92 – 8.85 (m, 1H), 8.75 (s, 1H), 8.53 (dd, J = 8.3, 1.4 Hz, 2H), 7.83 – 7.67 (m, 3H), 7.55 – 7.44 (m, 3H), 4.29 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 170.40, 154.97, 139.30, 135.84, 134.27, 134.02, 129.75, 129.35, 128.61, 128.17, 126.36, 124.91, 121.36, 116.61, 43.88. MS (ESI) calculated for C17H13N3, (M + H)+: 260.1182; observed: 260.1188.
Compounds 8c and 8d were synthesized similarly as described for compound 8b
8c (8 mg, 12%)
1H NMR (400 MHz, CDCl3) δ 9.03 (s, 1H), 8.70 – 8.57 (m, 1H), 8.25 – 8.13 (m, 1H), 7.74 – 7.57 (m, 2H), 3.97 (s, 3H), 3.00 – 2.94 (m, 1H), 2.17 – 1.87 (m, 7H), 1.57 – 1.37 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 160.11, 144.46, 143.99, 135.12, 129.50, 128.88, 126.96, 126.37, 122.25, 121.97, 36.55, 31.38, 30.24, 26.24, 25.68. MS (ESI) calculated for C17H19N3, (M + H)+: 266.1652; observed: 266.1666.
8d (21 mg, 40%)
1H NMR (500 MHz, CDCl3) δ 9.03 (s, 1H), 8.60 – 8.56 (m, 1H), 8.19 (dt, J = 8.0, 4.3, 1H), 7.73 – 7.61 (m, 2H), 3.96 (s, 3H), 3.08 – 2.93 (m, 2H), 1.90 (dt, J = 15.6, 7.8, 2H), 1.48 (dt, J = 15.0, 7.0, 2H), 1.44 – 1.35 (m, 2H), 1.35 – 1.20 (m, 8H), 0.88 (t, J = 7.0, 3H). 13C NMR (126 MHz, CDCl3) δ 156.38, 143.89, 143.52, 134.55, 128.98, 128.49, 126.67, 126.14, 121.63, 121.31, 31.39, 30.06, 29.08, 28.99, 28.87, 28.80, 27.74, 27.22, 22.20, 13.65. MS (ESI) calculated for C20H27N3, (M + H)+: 310.2278; observed: 310.2306.
Synthesis of Compound 12: 3,5-bis(4-methoxybenzyl)-2-phenyl-3H-imidazo[4,5-c]quinolin-5-ium
To a solution of 3b (50 mg, 0.2 mmol) in 1 mL of anhydrous THF, were added DBU (91 mg, 0.61 mmol) and 1-(chloromethyl)-4-methoxybenzene (159 mg, 1.02 mmol), and the solution was heated under microwave conditions (600 W, 150 °C, 0.5 h). After cooling to room temperature, and removing the solvent under vacuum, the residue was purified using column chromatography (10% MeOH/dichloromethane) to obtain the compound 5b (21 mg, 22%). 1H NMR (400 MHz, MeOD) δ 9.63 (s, 1H), 8.96 (d, J = 7.8 Hz, 1H), 8.55 (d, J = 9.1 Hz, 1H), 8.13 – 8.08 (m, 1H), 8.07 – 8.02 (m, 1H), 8.02 – 7.99 (m, 2H), 7.77 – 7.68 (m, 3H), 7.24 (d, J = 8.8 Hz, 2H), 7.10 (d, J = 8.7 Hz, 2H), 6.94 (d, J = 8.7 Hz, 2H), 6.89 (d, J = 8.7 Hz, 2H), 6.20 (s, 2H), 5.86 (s, 2H), 3.82 (s, 3H), 3.79 (s, 3H). 13C NMR (101 MHz, MeOD) δ 163.08, 160.44, 159.97, 150.30, 135.98, 135.52, 132.27, 131.96, 129.73, 129.40, 129.10, 129.03, 128.45, 127.98, 127.57, 126.49, 124.96, 123.80, 122.18, 119.33, 114.43, 114.39, 59.82, 54.46, 54.43, 49.46. MS (ESI) calculated for C32H28N3O2+, (M+): 486.2176; observed: 486.2140.
Synthesis of Compound 13: 5-(4-methoxybenzyl)-2-phenyl-3H-imidazo[4,5-c]quinolin-5-ium
To a solution of 3b (20 mg, 0.08 mmol) in 1 mL of anhydrous THF, was added 1-(chloromethyl)-4-methoxybenzene (19 mg, 0.12 mmol) and the solution was heated in a sealed vessel at 120 °C for an 12–14 hours. After cooling to room temperature and removing the solvent under vacuum, the residue was subjected to column chromatography (8% MeOH/dichloromethane) to obtain the compound 13 (12 mg, 40%). 1H NMR (400 MHz, MeOD) δ 9.28 (s, 1H), 8.92 – 8.84 (m, 1H), 8.41 (d, J = 7.0 Hz, 2H), 8.24 – 8.14 (m, 1H), 7.84 – 7.74 (m, 2H), 7.54 (dq, J = 14.2, 7.0 Hz, 3H), 7.23 (d, J = 8.6 Hz, 2H), 6.93 (d, J = 8.7 Hz, 2H), 6.05 (s, 2H), 3.76 (s, 3H). 13C NMR (101 MHz, MeOD) δ 159.99, 138.62, 137.14, 134.14, 133.69, 129.64, 129.32, 128.37, 128.00, 127.75, 126.66, 123.91, 121.38, 118.59, 114.25, 58.48, 54.35. MS (ESI) calculated for C24H20N3O+, (M+): 366.1601; observed: 366.1760.
Synthesis of Compound 16: N-(4-methoxybenzyl)-3-nitroquinolin-4-amine
To a solution of 14 (147 mg, 0.71 mmol) in 4:1 mixture of toluene and 2-propanol were added N,N-diethylpropan-2-amine (0.14 mL, 6 mmol) and 15 (121 mg, 0.88 mmol). The reaction mixture was heated to 70 °C for half an hour until a solid started to precipitate. The reaction mixture was then cooled, filtered and washed with toluene/2-propanol (7:3), ether and cold water. The residue was dried at 80 °C to obtain the compound 16 (200 mg, 91%). 1H NMR (400 MHz, CDCl3) δ 9.82 (s, 1H), 9.41 (s, 1H), 8.34 (d, J = 8.4, 1H), 8.03 (d, J = 8.3, 1H), 7.79 (dd, J = 11.2, 4.1 Hz, 1H), 7.48 (dd, J = 11.3 Hz, 4.2, 1H), 7.34 (d, J = 8.6 Hz, 2H), 6.97 (d, J = 8.6 Hz, 2H), 5.07 (d, J = 5.4 Hz, 2H), 3.85 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 159.76, 150.82, 150.64, 147.43, 132.72, 130.46, 128.83, 128.68, 126.99, 126.20, 125.52, 119.22, 114.72, 55.37, 52.74. MS (ESI) calculated for C17H15N3O3, (M + H)+: 310.1186; observed: 310.1268.
Synthesis of Compound 17: 1-(4-methoxybenzyl)-2-phenyl-1H-imidazo[4,5-c]quinoline
Compound 16 (120 mg, 0.39 mmol) was dissolved in MeOH and hydrogenated over Pd/C as a catalyst at 60 psi hydrogen pressure for 4 hours. The solution was then filtered using celite, followed by evaporation of the solvent under reduced pressure to afford N4-(4-methoxybenzyl) quinoline-3,4-diamine (85 mg, 94%). N4-(4-methoxybenzyl)quinoline-3,4-diamine (85 mg, 0.31 mmol), benzoic acid (41 mg, 0.34 mmol), HBTU (129 mg, 0.34 mmol), triethylamine (34 mg, 0.34 mmol) and a catalytic amount of DMAP were dissolved in 5 mL of DMF and stirred for 10–12 hours. The solvent was then removed under vacuum. The residue was dissolved in EtOAc and washed with water, dried over sodium sulfate and concentrated under reduced pressure to obtain the residue, which was dissolved in 10 mL of ethanol, and a solution of excess of sodium hydroxide in 1 mL of water was added. The reaction mixture was refluxed for 5–6 hours and then the solvent was removed to obtain the residue, which was purified using column chromatography (4% MeOH/dichloromethane) to obtain the compound 17 (82 mg, 80%). 1H NMR (400 MHz, CDCl3) δ 9.47 (s, 1H), 8.31 (d, J = 7.7 Hz, 1H), 7.94 (d, J = 7.7 Hz, 1H), 7.71 (dd, J = 8.1 Hz, 1.4, 2H), 7.68 – 7.62 (m, 1H), 7.58 – 7.42 (m, 4H), 7.10 (d, J = 8.7 Hz, 2H), 6.95 – 6.91 (m, 2H), 5.81 (s, 2H), 3.82 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 159.35, 154.84, 145.54, 137.06, 130.86, 130.33, 129.46, 128.91, 127.76, 127.07, 126.71, 126.52, 120.53, 117.76, 114.91, 55.30, 50.12. MS (ESI) calculated for C24H19N3O, (M + H)+: 366.1601; observed: 366.1785.
2D-NOESY experiments
The 2D-NOESY experiments were performed on the Bruker Avance 400 or Avance AV-III 500 NMR instruments. Compounds were dissolved in appropriate deuterated solvents and experiments were performed with mixing time (d8) of 0.5 sec (400 MHz) or 0.7 sec (500 MHz). The data generated was processed using MestReNova 6.2.1 (Mestrelab Research S.L.).
TLR-7/8 antagonism assay
A reporter gene assay using TLR715;20;21 (or TLR8)-dependent NF-κB induction was used. The inhibition of induction of NF-κB, a key transcriptional activator of the innate immune system, was quantified using human embryonic kidney 293 cells stably transfected with plasmids encoding TLR7 as well as an NF-κB reporter gene coupled to secreted alkaline phosphatase (sAP) (InvivoGen, San Diego, CA), and were maintained in HEK-Blue™ Selection medium containing zeocin and normocin. Stable expression of secreted alkaline phosphatase (sAP) under control of NF-κB/AP-1 promoters is inducible by the gardiquimod (TLR7 agonist) or CL075 (TLR8 agonist), and extracellular sAP in the supernatant is proportional to NF-κB induction. HEK-Blue-7 (or HEK-Blue-8) cells were incubated at a density of ~105 cells/mL in a volume of 80 μL/well, in 384-well, flat-bottomed, cell culture-treated microtiter plates until confluency was achieved, and subsequently stimulated with 1μg/mL of gardiquimod or CL075. Concurrent to stimulation, serially diluted concentrations of test compounds were added to the cell medium using a rapid-throughput, automated protocol employing a Bio-Tek P2000 liquid handler and left to incubate overnight. sAP was assayed spectrophotometrically using an alkaline phosphatase-specific chromogen (present in HEK-detection medium as supplied by the vendor) at 620 nm.
Supplementary Material
Abbreviations
- CD
cluster of differentiation
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- DMAP
4-dimethylaminopyridine
- DMF
N,N-dimethylformamide
- ESI-TOF
electrospray ionization-time of flight
- HATU
2-(1H-7-azabenzo-triazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate
- HEK
human embryonic kidney
- HIV
human immunodeficiency virus
- IC50
half-maximal inhibitory concentration
- IFN-α
interferon-α
- NF-κB
nuclear factor-kappa B
- NOE
nuclear overhauser effect
- 2D-NOESY
two-dimensional nuclear overhauser effect spectroscopy
- sAP
secreted alkaline phosphatase
- ssRNA
single stranded RNA
- THF
tetrahydrofuran
- TLR
toll-like receptor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Boasso A, Shearer GM. Clin Immunol. 2008;126:235–242. doi: 10.1016/j.clim.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Douek DC, Roederer M, Koup RA. Annu Rev Med. 2008;60:471–484. doi: 10.1146/annurev.med.60.041807.123549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pantaleo G, Graziosi C, Demarest JF, Butini L, Montroni M, Fox CH, Orenstein JM, Kotler DP, Fauci AS. Nature. 1993;362:355–358. doi: 10.1038/362355a0. [DOI] [PubMed] [Google Scholar]
- 4.Mandl JN, Barry AP, Vanderford TH, Kozyr N, Chavan R, Klucking S, Barrat FJ, Coffman RL, Staprans SI, Feinberg MB. Nat Med. 2008;14:1077–1087. doi: 10.1038/nm.1871. [DOI] [PubMed] [Google Scholar]
- 5.Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ, Christensen SR, Shlomchik MJ, Viglianti GA, Rifkin IR, Marshak-Rothstein A. J Exp Med. 2005;202:1171–1177. doi: 10.1084/jem.20050630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rifkin IR, Leadbetter EA, Busconi L, Viglianti G, Marshak-Rothstein A. Immunol Rev. 2005;204:27–42. doi: 10.1111/j.0105-2896.2005.00239.x. [DOI] [PubMed] [Google Scholar]
- 7.Alspaugh MA, Talal N, Tan EM. Arthritis Rheum. 1976;19:216–222. doi: 10.1002/art.1780190214. [DOI] [PubMed] [Google Scholar]
- 8.Richez C, Blanco P, Rifkin I, Moreau JF, Schaeverbeke T. Joint Bone Spine. 2010;78:124–130. doi: 10.1016/j.jbspin.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 9.Avalos AM, Busconi L, Marshak-Rothstein A. Autoimmunity. 2010;43:76–83. doi: 10.3109/08916930903374618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Santiago-Raber ML, Dunand-Sauthier I, Wu T, Li QZ, Uematsu S, Akira S, Reith W, Mohan C, Kotzin BL, Izui S. J Autoimmun. 2010;34:339–348. doi: 10.1016/j.jaut.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 11.O’Neill LA, Bryant CE, Doyle SL. Pharmacol Rev. 2009;61:177–197. doi: 10.1124/pr.109.001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hennessy EJ, Parker AE, O’Neill LA. Nat Rev Drug Discov. 2010;9:293–307. doi: 10.1038/nrd3203. [DOI] [PubMed] [Google Scholar]
- 13.Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K, Horiuchi T, Tomizawa H, Takeda K, Akira S. Nat Immunol. 2002;3:196–200. doi: 10.1038/ni758. [DOI] [PubMed] [Google Scholar]
- 14.Hood JD, Warshakoon HJ, Kimbrell MR, Shukla NM, Malladi S, Wang X, David SA. Hum Vaccin. 2010;6:1–14. doi: 10.4161/hv.6.4.10866. [DOI] [PubMed] [Google Scholar]
- 15.Shukla NM, Malladi SS, Mutz CA, Balakrishna R, David SA. J Med Chem. 2010;53:4450–4465. doi: 10.1021/jm100358c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robbins M, Judge A, Liang L, McClintock K, Yaworski E, MacLachlan I. Mol Ther. 2007;15:1663–1669. doi: 10.1038/sj.mt.6300240. [DOI] [PubMed] [Google Scholar]
- 17.Wang D, Bhagat L, Yu D, Zhu FG, Tang JX, Kandimalla ER, Agrawal S. J Med Chem. 2009;52:551–558. doi: 10.1021/jm8014316. [DOI] [PubMed] [Google Scholar]
- 18.Yu D, Wang D, Zhu FG, Bhagat L, Dai M, Kandimalla ER, Agrawal S. J Med Chem. 2009;52:5108–5114. doi: 10.1021/jm900730r. [DOI] [PubMed] [Google Scholar]
- 19.Hamm S, Latz E, Hangel D, Muller T, Yu P, Golenbock D, Sparwasser T, Wagner H, Bauer S. Immunobiology. 2010;215:559–569. doi: 10.1016/j.imbio.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 20.Shukla NM, Kimbrell MR, Malladi SS, David SA. Bioorg Med Chem Lett. 2009;19:2211–2214. doi: 10.1016/j.bmcl.2009.02.100. [DOI] [PubMed] [Google Scholar]
- 21.Shukla NM, Mutz CA, Ukani R, Warshakoon HJ, Moore DS, David SA. Bioorg Med Chem Lett. 2010;20:6384–6386. doi: 10.1016/j.bmcl.2010.09.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.