

Abstract
Vomeronasal sensitivity is important for detecting intraspecific pheromonal cues as well as environmental odorants and is involved in mating, social interaction, and other daily activities of many vertebrates. Two large families of seven-transmembrane G-protein–coupled receptors, V1rs and V2rs, bind to various ligands to initiate vomeronasal signal transduction. Although the macroevolution of V1r and V2r genes has been well characterized throughout vertebrates, especially mammals, little is known about their microevolutionary patterns, which hampers a clear understanding of the evolutionary forces behind the rapid evolutionary turnover of V1r and V2r genes and the great diversity in receptor repertoire across species. Furthermore, the role of divergent vomeronasal perception in enhancing premating isolation and maintaining species identity has not been evaluated. Here we sequenced 44 V1r genes and 25 presumably neutral noncoding regions in 14 wild-caught mice belonging to Mus musculus and M. domesticus, two closely related species with strong yet incomplete reproductive isolation. We found that nucleotide changes in V1rs are generally under weak purifying selection and that only ∼5% of V1rs may have been subject to positive selection that promotes nonsynonymous substitutions. Consistent with the low functional constraints on V1rs, 18 of the 44 V1rs have null alleles segregating in one or both species. Together, our results demonstrate that, despite occasional actions of positive selection, the evolution of V1rs is in a large part shaped by purifying selection and random drift. These findings have broad implications for understanding the driving forces of rapid gene turnovers that are often observed in the evolution of large gene families.
Keywords: Mus musculus, Mus domesticus, vomeronasal receptor, pheromone detection, V1r, evolution
Introduction
Olfaction plays a critical role in the daily life of vertebrates, such as prey detection, predator avoidance, mating, and territoriality (Mombaerts 1999). Two distinct nasal olfactory systems exist in most terrestrial vertebrates: the main olfactory system (MOS) and the vomeronasal system (VNS) (Dulac and Torello 2003; Grus and Zhang 2006). Although partially overlapping in function, the MOS appears to be mainly responsible for recognizing environmental odorants, whereas the VNS primarily detects pheromones, which constitute a poorly defined class of chemicals that are emitted and sensed by individuals of the same species to elicit sexual/social behaviors and physiological changes (Restrepo et al. 2004; Spehr et al. 2006). The MOS and the VNS are anatomically and neurologically separated; they use different receptors and have distinct signal transduction pathways (Dulac and Torello 2003; Grus and Zhang 2006). The VNS is of particular interest to evolutionists because of its high diversity in complexity among species (Grus et al. 2005; Young et al. 2005, 2010; Shi and Zhang 2007; Grus and Zhang 2008). Although the morphological components of the VNS are believed to first emerge in the common ancestor of tetrapods, its genetic components have been inferred to exist in the common ancestor of all vertebrates (Grus and Zhang 2006, 2009). Among tetrapods, the VNS varies from completely absent in birds, catarrhine primates (humans, apes, and Old World monkeys), most bats, and many cetaceans to rudimentary in amphibians to highly complex in murids, opossums, and the platypus (Zhang and Webb 2003; Grus et al. 2005, 2007; Grus and Zhang 2006; Shi and Zhang 2007, 2009; Zhao et al. 2011).
Vomeronasal sensitivity is mediated by two families of G-protein–coupled receptors known as V1rs and V2rs (Mombaerts 2004). In the genome of the laboratory mouse, there are about 190 putatively functional V1r and 70 V2r genes (Shi et al. 2005; Yang et al. 2005). Note that despite the availability of genome sequences, the gene numbers are only approximate due to among-strain variations and/or incomplete genomic sequencing (Zhang et al. 2004). The V1r and V2r gene repertoires, especially the former, have been examined in many mammalian genomes (Rodriguez and Mombaerts 2002; Rodriguez et al. 2002; Grus and Zhang 2004, 2008; Grus et al. 2005, 2007; Young et al. 2005, 2010; Shi and Zhang 2007; Young and Trask 2007). It was reported that the among-species size variation in V1r and V2r gene repertoires is among the highest of all mammalian gene families (Grus et al. 2005, 2007; Yang et al. 2005). This variation is not random, at least in the case of V1rs, because a clear positive correlation exists between the morphological complexity of the VNS and the number of putatively functional V1r genes (Grus et al. 2005, 2007). An evolutionary hallmark of V1rs and V2rs is the exceptionally rapid gene turnover that results in lineage-specific receptors. For example, between 187 mouse and 106 rat V1rs examined, only 18 are one-to-one orthologous (Grus and Zhang 2004, 2008), in sharp contrast to the genome-wide estimate that 86–94% of rat genes have one-to-one mouse orthologs (Gibbs et al. 2004). Despite the striking macroevolutionary diversity of V1rs and V2rs, the evolutionary forces acting on these genes are unclear due to the lack of knowledge about the population genetic dynamics of V1r and V2r genes. Specifically, it would be interesting to test whether V1rs and V2rs evolve by divergent selective pressures in sibling species because pheromones are by definition species specific (Brennan and Keverne 2004).
In this work, we study the microevolution of vomeronasal receptor genes in two closely related mouse species, Mus musculus (abbreviated as Mm) and Mus domesticus (Md). Mm is distributed from Eastern Europe to Japan, across Russia and northern China, whereas Md is common in Western Europe, Africa, and the near-East and was transported by humans to the Americas and Australia (Guenet and Bonhomme 2003). Mm and Md diverged within the last 500 thousand years (Salcedo et al. 2007). The two species form a narrow zone of hybridization through Central Europe that extends from the Jutland Peninsula to the Bulgarian coast of the Black Sea (Sage et al. 1993; Tucker 2007). Mice from the center of the hybrid zone have higher parasite loads than those from the edges of the hybrid zone (Sage et al. 1986; Moulia et al. 1991, 1993), indicative of reduced fitness due to hybrid inviability. Additional laboratory studies have documented reproductive incompatibility between Mm and Md. There is clear evidence of hybrid male sterility between the two species (Forejt and Ivanyi 1974; Forejt 1996; Alibert et al. 1997; Storchova et al. 2004; Britton-Davidian et al. 2005; Trachtulec et al. 2005; Vyskocilova et al. 2005). There is also evidence for limited female sterility in some crosses but not others (Forejt and Ivanyi 1974; Britton-Davidian et al. 2005). Evidence for partial premating isolation is also ample (Laukaitis et al. 1997; Smadja and Ganem 2002; Smadja et al. 2004). Although some authors regard Mm and Md as two subspecies of the species M. musculus (Tucker 2007), for simplicity, we treat them as two species that are in an early stage of divergence with a low degree of gene flow.
There are several reasons why we chose to study Mm and Md. First, the laboratory mouse, a mosaic of Mm, Md, and Mus castaneus (Frazer et al. 2007; Yang et al. 2007), is a model organism for studying vomeronasal sensitivity. A substantial amount of genetic, neurological, and behavioral data related to vomeronasal sensitivity is available for the laboratory mouse, allowing a more accurate interpretation of the evolutionary and population genetic data that we collect. Second, the genome sequence of the laboratory mouse is known, making the experimental design much easier. Third, mice represent those vertebrates with a relatively high level of vomeronasal sensitivity (Takami 2002; Grus et al. 2005). Thus, their vomeronasal sensitivities may be more important in determining organismal fitness and under stronger natural selection.
Avoiding the hybrid zone, we trapped seven wild Mm and seven wild Md in Czech Republic and France, respectively. Our present study focuses on V1rs because they have only one coding exon, making DNA amplification and sequencing much easier. Here, we report the microevolution of 44 V1rs and 25 presumably neutral noncoding regions in these 14 mice.
Materials and Methods
Seven M. musculus and seven M. domesticus individuals were collected from Czech Republic and France, respectively. Although the mice from each species were sampled from restricted geographic areas (supplementary table 1, Supplementary Material online), it should not affect our results because mice have little geographic differentiation (Salcedo et al. 2007). The identity of the mice was confirmed by sequencing a 683-nt segment of the mitochondrial cytochrome c oxidase subunit I (COXI) gene that is commonly used as a barcode for identifying animal species.
The liver genomic DNAs of the mice were extracted using the PUREGENE genomic DNA purification kit (Gentra Systems, Minneapolis, MN), following the manufacturer's instruction. Gene-specific primers for amplifying 44 V1r genes were designed according to the Mus musculus reference sequence from GenBank (supplementary table 2, Supplementary Material online). The protein-coding region of each V1r gene studied has 870–1,104 nt, which were completely amplified in our experiments. Polymerase chain reactions (PCRs) were performed with GoTaq DNA Polymerase (Promega Corp, Madison, WI) under conditions recommended by the manufacturer. PCR products were examined on 1.5% agarose gel. Samples showing duplicated electropherograms due to insertions/deletions were cloned with TOPO PCR cloning kit (Invitrogen, Carlsbad, CA) and sequenced with universal T7 and M13 primers using the Sanger method on an automatic DNA sequencer. Otherwise, the PCR products were enzymatically processed using calf intestinal phosphatase and exonuclease I (Exo I) (New England Biolabs, Ipswich, MA) before being sequenced bidirectionally with the gene-specific primers. Sequencher (GeneCodes, Ann Arbor, MI) and MEGA4 (Tamura et al. 2007) were used to edit and align the sequences. Twenty-five presumably neutral noncoding regions (supplementary table 3, Supplementary Material online), most with ∼1,000 nt, were also amplified and directly sequenced in the same 14 mice. Watterson's θ, nucleotide diversity π, Tajima's D, and Fu and Li's D* were computed using DnaSP (Librado and Rozas 2009). Tajima's test (Tajima 1989) and Fu and Li's test (Fu and Li 1993) were conducted by 10,000 coalescent simulations in DnaSP. Hudson–Kreitman–Aguade (HKA) test (Hudson et al. 1987) was conducted using a program written by J. Hey (http://lifesci.rutgers.edu/∼heylab/). The sequences reported in this article have been submitted to GenBank (accession numbers JF782602–JF783819, JF782044–JF782601, and JF783820–JF783959).
Results
Intraspecific Variations of V1rs and Noncoding Regions
Based on previous studies (Grus and Zhang 2004; Shi et al. 2005), there are at least 188 putatively functional V1r genes in the mouse genome (fig. 1). We carefully selected 44 of them for an in-depth study in 14 mice. These 44 genes were chosen to represent major lineages of mouse V1rs, to include genes with (14) and without (30) rat one-to-one orthologs, and to allow gene-specific amplification and sequencing (fig. 1). For comparison, we also sequenced 25 presumably neutral noncoding regions in these 14 mice. Five of these 25 noncoding regions were from a previous study (Baines and Harr 2007), and the sequenced segments are either in introns or in intergenic regions that are >5 kb upstream of coding regions. The remaining 20 noncoding regions were randomly picked from the genome, with the criteria that the regions are at least 200 kb away from any known gene. All sequenced noncoding regions are on autosomes, so are the sequenced V1r genes. The average length of the noncoding regions sequenced (936 nt) is similar to the average length of the V1rs sequenced (934 nt).
FIG. 1.—

An unrooted tree of 188 putatively functional mouse V1rs (Shi et al. 2005). Red branches denote the 44 V1rs surveyed in this study. Branches denoted with * have putatively functional rat V1r orthologs (Grus and Zhang 2008). Gene family names are from Rodriguez et al. (2002). The tree was reconstructed using the Neighbor-Joining method (Saitou and Nei 1987) with Poisson-corrected protein distances. The scale bar shows 0.1 amino acid substitution per site.
The basic population genetic parameters of individual V1rs and noncoding regions are presented in table 1 and table 2, respectively. Consistent with other population genetic studies (Salcedo et al. 2007), we found nucleotide diversity per site (π) at the 25 noncoding regions to be higher in Md (0.0021) than in Mm (0.0013), although the difference is not statistically significant (P = 0.15, two-tailed paired t-test; table 2). However, the opposite is found for V1rs, although the difference is again not significant (P = 0.09, two-tailed paired t-test; table 1). Compared with the noncoding regions, V1rs show an overall higher π in Mm (P = 0.24, two-tailed Mann–Whitney test) but a lower π in Md (P = 0.014, two-tailed Mann–Whitney test) (tables 1 and 2). In neither species is there a significant difference in π between V1rs with one-to-one rat orthologs and those without such orthologs (P > 0.2, two-tailed Mann–Whitney test; table 1).
Table 1.
Intra- and Interspecific Sequence Variations of 44 Mouse V1r Genes
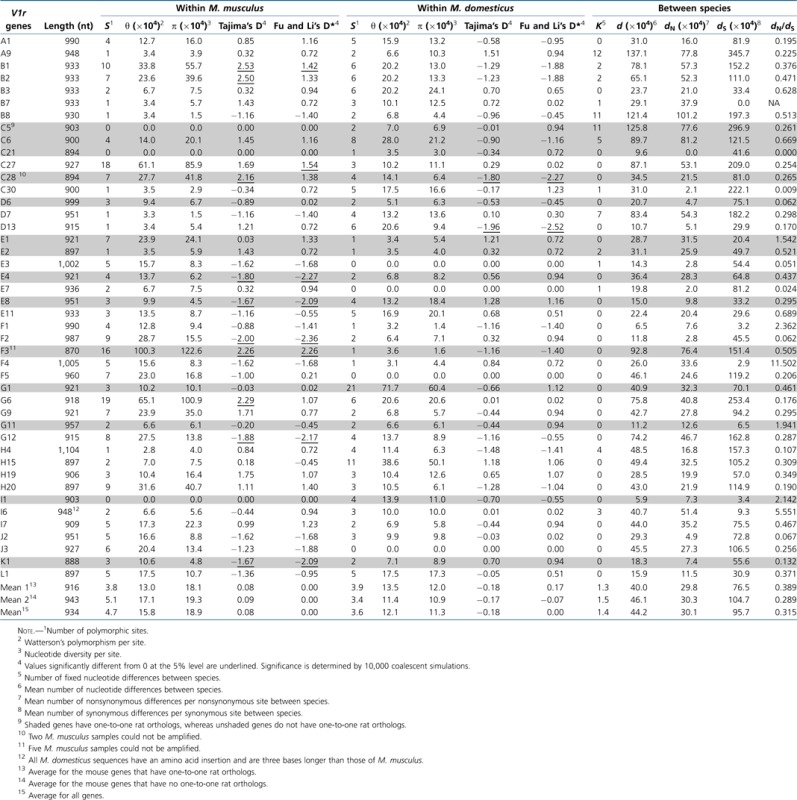 |
Table 2.
Intra- and Interspecific Sequence Variations of 25 Noncoding Regions in Mice
| Region name1 | Chromos. no. | Nucleotide position2 | length (nt)3 | Within M. musculus |
Within M. domesticus |
Between species |
|||||||||
| S4 | θ (×104)5 | π (×104)6 | Tajima's D7 | Fu and Li's D*7 | S4 | θ (×104)5 | π (×104)6 | Tajima's D7 | Fu and Li's D*7 | K8 | d (×104)9 | ||||
| 032 | 3 | 61421835–61422742 | 908 | 1 | 3.5 | 1.6 | −1.16 | −1.40 | 10 | 34.6 | 26.4 | −0.93 | 0.58 | 2 | 85.7 |
| con1 | 5 | 4750295–4751185 | 885 | 4 | 14.2 | 22.4 | 1.89 | 1.16 | 5 | 17.8 | 23.6 | 1.14 | 1.23 | 0 | 44.7 |
| con2 | 5 | 4177067–4178013 | 948 | 6 | 19.9 | 26.9 | 1.26 | 1.29 | 4 | 13.3 | 11.1 | −0.53 | 1.16 | 0 | 40.0 |
| 062 | 6 | 13370671–13371601 | 930 | 1 | 3.6 | 5.7 | 1.38 | 0.75 | 5 | 16.9 | 17.7 | 0.17 | 0.51 | 9 | 122.8 |
| 063 | 6 | 13335122–13336060 | 940 | 0 | 0.0 | 0.0 | 0.00 | 0.00 | 7 | 23.5 | 32.5 | 1.41 | 0.77 | 7 | 93.5 |
| 071 | 7 | 34198101–34199051 | 951 | 6 | 19.8 | 14.9 | −0.89 | −0.61 | 8 | 26.5 | 29.5 | 0.43 | 1.37 | 9 | 150.0 |
| 072 | 7 | 1485242–1486253 | 1,012 | 2 | 6.2 | 5.2 | −0.44 | 0.94 | 1 | 3.1 | 4.9 | 1.21 | 0.72 | 11 | 117.9 |
| 073 | 7 | 14736902–14737811 | 902 | 15 | 55.1 | 90.7 | 2.78 | 1.51 | 7 | 24.2 | 30.1 | 0.90 | 0.21 | 4 | 148.9 |
| 074 | 7 | 13161241–13162059 | 818 | 1 | 3.8 | 1.7 | −1.16 | −1.40 | 10 | 38.4 | 65.4 | 2.73 | 1.42 | 1 | 71.6 |
| con3 | 7 | 12877447–12878480 | 1,034 | 3 | 9.1 | 12.4 | 0.07 | 0.30 | 4 | 12.2 | 10.5 | −0.44 | −0.55 | 1 | 26.2 |
| 102 | 10 | 54958829–54959659 | 812 | 0 | 0.0 | 0.0 | 0.00 | 0.00 | 1 | 3.8 | 1.7 | −1.16 | −1.40 | 9 | 111.7 |
| 131 | 13 | 4938296–4939215 | 920 | 2 | 6.8 | 10.5 | 1.46 | 0.94 | 3 | 10.3 | 13.3 | 0.90 | 0.02 | 9 | 117.2 |
| con4 | 13 | 5177266–5178204 | 939 | 3 | 10.0 | 14.4 | 1.32 | 1.07 | 6 | 20.1 | 28.8 | 1.55 | 1.29 | 4 | 81.4 |
| 133 | 13 | 14942772–14943791 | 1,021 | 1 | 3.1 | 5.3 | 1.51 | 0.72 | 7 | 21.6 | 18.1 | −0.60 | 1.33 | 4 | 60.9 |
| con5 | 13 | 10340883–10341849 | 984 | 3 | 9.6 | 8.3 | −0.42 | −1.04 | 8 | 25.5 | 21.9 | −0.53 | 0.86 | 6 | 86.4 |
| con6 | 14 | 12999764–13000706 | 948 | 5 | 16.6 | 15.1 | −0.32 | 0.51 | 2 | 6.6 | 4.3 | −0.96 | −0.45 | 1 | 39.9 |
| con7 | 14 | 55603471–55604414 | 944 | 3 | 10.5 | 5.3 | −1.63 | −1.95 | 17 | 56.6 | 29.3 | −1.99 | −2.45 | 1 | 45.9 |
| con8 | 17 | 1418876–1419796 | 921 | 5 | 17.1 | 13.2 | −0.78 | −0.95 | 5 | 17.1 | 13.6 | −0.70 | −0.22 | 2 | 42.7 |
| 173 | 17 | 27883797–27884681 | 885 | 2 | 7.1 | 6.6 | −0.20 | −0.45 | 1 | 3.6 | 5.1 | 0.84 | 0.72 | 15 | 181.6 |
| con9 | 17 | 64305167–64306026 | 860 | 10 | 36.6 | 30.2 | −0.68 | −1.52 | 3 | 11.0 | 6.4 | −1.28 | −1.04 | 0 | 64.8 |
| GGH | 4 | 20166795–201667294 | 499 | 1 | 5.0 | 3.5 | −0.34 | 0.72 | 4 | 18.3 | 19.3 | 0.00 | 0.00 | 0 | 2.9 |
| MELK | 4 | 2162811–2163729 | 918 | 0 | 0.0 | 0.0 | 0.00 | 0.00 | 14 | 50.6 | 75.0 | 2.06 | 1.20 | 5 | 116.2 |
| NKD1 | 8 | 91403263–91404440 | 1,178 | 8 | 16.9 | 14.9 | −0.87 | −0.22 | 11 | 21.1 | 12.8 | 0.83 | 0.02 | 0 | 26.7 |
| PUM1 | 4 | 130022004–130023187 | 1,183 | 4 | 8.8 | 2.2 | 0.00 | 0.00 | 5 | 10.0 | 3.9 | −1.16 | −1.40 | 0 | 7.9 |
| SFRP1 | 8 | 2480594–2481312 | 1,071 | 1 | 3.0 | 5.0 | 1.43 | 0.72 | 12 | 30.1 | 24.3 | −0.75 | −0.68 | 10 | 122.0 |
| Mean | 936.4 | 3.5 | 11.7 | 12.6 | 0.17 | 0.04 | 160 | 20.7 | 21.2 | 0.13 | 0.21 | 4 | 80.4 | ||
NOTE.—1Most noncoding regions do not have standard names, and the names listed are the idendification numbers used in our laboratory.
Nucleotide positions in the mouse genome sequence of National Center for Biotechnology Information Build 37.
Length in M. musculus. Length in M. domesticus may be slightly different due to indels.
Number of polymorphic sites.
Watterson's polymorphism per site.
Nucleotide diversity per site.
Values significantly different from 0 at the 5% level are underlined. Significance is determined by 10,000 coalescent simulations.
Number of fixed nucleotide differences between species.
Mean number of nucleotide differences between species.
We applied Tajima's test of neutrality (Tajima 1989) to each of the V1rs (table 1) and noncoding regions (table 2). Note that the null hypothesis in Tajima's test is the Wright–Fisher model of strict neutrality. Thus, rejection of the null hypothesis may indicate one or more of the following: purifying selection, positive selection, and demographic changes. For both V1rs and noncoding regions, several loci show significantly negative or positive Tajima's D. For example, in Mm, five V1rs and two noncoding regions show significantly positive D (nominal P < 0.05), whereas five V1rs and one noncoding region show significantly negative D. In Md, zero V1r and two noncoding regions show significantly positive D, whereas two V1rs and one noncoding region show significantly negative D. In neither species is there a significant difference between V1rs and noncoding regions in the fraction of loci with significantly positive or negative D (P > 0.1 in all cases, χ2 test). We also compared the frequency distribution of Tajima's D between V1rs and noncoding regions (fig. 2a and 2b) but found no significant differences (P = 0.44 for Mm and 0.12 for Md, Kolmogorov–Smirnov test). We found similar results from comparing the distribution of Fu and Li's D* between V1rs and noncoding regions (P = 0.74 for Mm and 0.30 for Md, Kolmogorov–Smirnov test, fig. 2c and 2d).
FIG. 2.—

Frequency distributions of Tajima's D and Fu and Li's D* among 44 V1rs and 25 noncoding regions in mouse. (A) Tajima's D in M. musculus; (B) Tajima's D in M. domesticus; (C) Fu and Li's D* in M. musculus; and (D) Fu and Li's D* in M. domesticus.
Interspecific Divergences of V1rs and Noncoding Regions
The mean number of nucleotide difference per site between Mm and Md is 0.00442 for the V1rs (table 1) and 0.00804 for the noncoding regions (table 2). The mean nonsynonymous nucleotide difference per nonsynonymous site (dN) of the 44 V1rs divided by the mean synonymous difference per synonymous site (dS) of the same set of genes is 0.315. Seven V1rs have dN/dS >1, but none of them significantly exceeds 1 by Fisher's exact test (Zhang et al. 1997). The fraction of V1rs with a dN/dS ratio below 1 is significantly greater than 50% (P = 3 × 10−6, binomial test). These results indicate that the evolutionary divergence of V1rs is overall governed by purifying selection.
Combining the polymorphism and divergence data, we conducted several McDonald–Kreitman tests (McDonald and Kreitman 1991) by varying the consideration of the polymorphic data from one or both species (table 3). In all cases, nonsynonymous/synonymous ratio is lower for divergence than for polymorphism, although the differences are not statistically significant (table 3). These findings suggest that the evolution of mouse V1rs is largely neutral, with the presence of only weak purifying selection that hampers the fixation of some nonsynonymous changes. For instance, the nonsynonymous/synonymous ratio is 102/56 = 1.82 for intraspecific polymorphisms in Md but 32/31 = 1.03 for interspecific divergences (P = 0.068, Fisher's exact test). Consistent with the above interpretation, we found the nonsynonymous/synonymous ratios for polymorphisms and divergences to be more similar to each other when only derived alleles with frequencies equal to or greater than 2/14 are considered for polymorphisms. For instance, the nonsynonymous/synonymous ratio now becomes 62/44 = 1.41 for polymorphisms in Md, closer to the ratio of 1.03 for interspecific divergences (P = 0.76, Fisher's exact test).
Table 3.
Numbers of Synonymous and Nonsynonymous Sequence Variations in V1rs
| Nonsynonymous/synonymous |
Nonsynonymous/(noncoding + synonymous) |
||||||
| Nonsynonymous | Synonymous | Noncoding1 | Ratio | P value2 | Ratio | P value2 | |
| Polymorphisms in M. musculus | 111 | 97 | 80 | 1.144 | 0.774 | 0.627 | <0.001 |
| Polymorphisms in M. domesticus | 102 | 56 | 144 | 1.821 | 0.068 | 0.510 | <0.001 |
| High-frequency3 polymorphisms in M. musculus | 78 | 77 | 65 | 1.013 | 1.000 | 0.549 | <0.001 |
| High-frequency3 polymorphisms in M. domesticus | 62 | 44 | 104 | 1.409 | 0.330 | 0.419 | 0.01 |
| Fixed differences between the two species | 32 | 31 | 110 | 1.032 | 0.227 | ||
NOTE.—1Variations in the 25 noncoding regions.
P value is from Fisher's exact test in comparison with fixed differences.
Frequency of the derived allele is equal to or greater than 2 of 14.
Because of the relatively small numbers of synonymous polymorphisms and substitutions in our V1r data, we augmented this dataset with the 25 noncoding regions to enhance the statistical power of the McDonald–Kreitman test. That is, we lumped synonymous and noncoding changes and compared them with nonsynonymous changes (table 3). The results are consistent with those from the comparison between synonymous and nonsynonymous changes, but the difference between polymorphism and divergence becomes statistically more significant (table 3). For example, the nonsynonymous/(noncoding+synonymous) ratio is 0.510 for intraspecific polymorphisms in Md, significantly higher than that (0.227) for interspecific divergences (P < 0.001, Fisher's exact test). Together, the various McDonald–Kreitman tests demonstrate the overall action of purifying selection hampering the spread and fixation of nonsynonymous changes in V1rs. We did not perform McDonald–Kreitman tests for individual V1rs because of the low numbers of synonymous and nonsynonymous changes in each V1r and the consequent low statistical power.
We examined d/θ for each V1r in each species, where d is the average nucleotide difference per site between an Mm allele and an Md allele and θ is Watterson's estimate of polymorphism per site in a species (table 4). Because some sequences have no polymorphic sites, we used the actual number of polymorphic site plus 1 in calculating θ for each V1r gene or noncoding region. In Mm, the mean d divided by mean θ is 2.59 for V1rs, whereas the corresponding ratio is 7.30 for the noncoding regions. In Md, the ratios are 3.63 and 4.20 for V1rs and noncoding regions, respectively. Thus, overall, V1rs have lower divergence-to-polymorphism ratios than noncoding regions, indicative of purifying selection on V1rs. When each gene is examined separately by the HKA test (Hudson et al. 1987), however, three genes (A9, B8, and C5) show significantly greater d/θ than the 25 noncoding regions in both Mm and Md and three additional genes (F3, F5, and J3) show significantly greater d/θ than noncoding regions only in Md (table 4). Because 44 tests were conducted in each species, some of the significant cases (on average 2.2 cases per species) may be artifacts of multiple testing. After examining the fixed differences between Mm and Md, we believe that F3, F5, and J3 are probably false positives because they lack any fixed differences, whereas A9, B8, and C5 are likely to be true positives because they each contain at least five fixed nonsynonymous differences between the two species, and the statistical significance of the HKA test is high (P < 0.0025 for each gene in each species). Even after the conservative Bonferroni correction, A9 remains significant in both species and C5 remains significant in Mm. Thus, it is likely that a small fraction of V1rs has been subject to positive selection in the divergence of Mm and Md.
Table 4.
Comparison Between V1rs and the 25 Noncoding Regions by the HKA Test
|
M. musculus |
M. domesticus |
|||
| V1r genes | d/θ1 | P value2 | d/θ1 | P value2 |
| A1 | 1.95 | 2.6E-03 | 1.63 | 3.5E-01 |
| A9 | 20.67 | 1.5E-04 | 13.78 | 1.4E-04 |
| B1 | 2.11 | 5.0E-04 | 3.31 | 9.1E-01 |
| B2 | 2.41 | 4.4E-03 | 2.76 | 8.5E-01 |
| B3 | 2.35 | 1.0E-01 | 1.01 | 1.5E-02 |
| B7 | 4.32 | 8.5E-01 | 2.16 | 8.7E-01 |
| B8 | 17.95 | 1.6E-03 | 11.96 | 2.4E-03 |
| C5 | 36.12 | 3.2E-10 | 12.04 | 2.1E-03 |
| C6 | 5.13 | 3.3E-01 | 2.85 | 8.3E-01 |
| C21 | 2.73 | 9.5E-01 | 1.36 | 9.9E-01 |
| C27 | 1.35 | 2.9E-07 | 6.42 | 5.9E-01 |
| C28 | 1.09 | 1.2E-07 | 1.96 | 6.9E-01 |
| C30 | 4.44 | 7.7E-01 | 1.48 | 2.4E-01 |
| D6 | 1.65 | 1.7E-03 | 2.20 | 9.6E-01 |
| D7 | 12.61 | 7.0E-02 | 5.04 | 8.6E-01 |
| D13 | 1.56 | 1.8E-01 | 0.45 | 1.7E-05 |
| E1 | 1.05 | 5.9E-08 | 4.20 | 9.7E-01 |
| E2 | 4.43 | 7.7E-01 | 4.43 | 9.5E-01 |
| E3 | 0.76 | 4.5E-09 | 4.54 | 4.0E-01 |
| E4 | 2.14 | 4.5E-03 | 3.56 | 9.8E-01 |
| E7 | 1.97 | 4.2E-02 | 5.91 | 1.6E-01 |
| E8 | 1.14 | 3.4E-05 | 0.91 | 2.8E-02 |
| E11 | 1.66 | 1.9E-03 | 1.11 | 4.9E-02 |
| F1 | 0.41 | 2.2E-11 | 1.02 | 9.5E-01 |
| F2 | 0.37 | 1.7E-14 | 1.24 | 6.4E-01 |
| F3 | 0.87 | 7.2E-10 | 12.83 | 1.5E-03 |
| F4 | 1.38 | 1.8E-05 | 4.15 | 9.7E-01 |
| F5 | 1.76 | 1.4E-03 | 14.08 | 4.2E-06 |
| G1 | 2.99 | 1.2E-01 | 0.54 | 9.3E-06 |
| G6 | 1.11 | 1.1E-08 | 3.16 | 9.0E-01 |
| G9 | 1.56 | 2.5E-05 | 4.16 | 9.6E-01 |
| G11 | 1.14 | 6.4E-04 | 1.14 | 5.0E-01 |
| G12 | 2.40 | 3.2E-03 | 4.32 | 9.2E-01 |
| H4 | 8.52 | 4.2E-01 | 3.41 | 9.4E-01 |
| H15 | 4.69 | 5.2E-01 | 1.17 | 1.6E-02 |
| H19 | 2.05 | 1.2E-02 | 2.05 | 8.4E-01 |
| H20 | 1.23 | 3.2E-07 | 3.07 | 9.6E-01 |
| I1 | 1.70 | 1.0E+00 | 0.34 | 3.6E-05 |
| I6 | 4.09 | 4.7E-01 | 3.07 | 9.6E-01 |
| I7 | 2.12 | 2.8E-03 | 4.24 | 9.6E-01 |
| J2 | 1.48 | 2.2E-05 | 2.21 | 8.8E-01 |
| J3 | 1.91 | 5.4E-04 | 13.40 | 1.8E-04 |
| K1 | 1.29 | 1.4E-04 | 1.72 | 8.9E-01 |
| L1 | 0.76 | 4.5E-09 | 0.76 | 3.0E-03 |
| All V1r genes | 2.59 | 1.5E-96 | 3.63 | 6.9E-05 |
| 25 noncoding regions | 7.30 | 4.20 | ||
NOTE.—1Interspecific divergence per site divided by Watterson's polymorphism per site. The actual number of polymorphc site plus 1 was used in calculating Watterson's polymorphism per site. Values of d/θ that are significantly greater than expected from the 25 noncoding regions are underlined.
P values are from chi-squares tests.
Abundant Segregating Null Alleles of V1rs
We observed a large number of V1r genes that have segregating null alleles in either one or both mouse species based on the occurrences of single nucleotide polymorphisms and/or insertions/deletions (indels) that introduce premature stop codons. For two V1r genes (C28 and F3), amplification was unsuccessful in some but not all mouse individuals even after extensive experimentation with multiple primer sets including those within coding regions, suggesting that the two genes may have been partially or entirely deleted in these individuals. We thus regard these cases as null alleles as well. In total, 14 V1rs harbor null alleles in Mm and 7 in Md (table 5). Given these numbers, we should expect 14 × 7/44 = 2.23 V1rs to have segregating null alleles in both species if V1r pseudogenization in the two species is independent. Consistent with this expectation, two V1rs harbor segregating null alleles in both species, and the pseudogenization events were independent because the null alleles in the two species were generated by different open reading frame (ORF)–disrupting mutations (table 5). In addition to the prevalence of pseudogenized V1rs, the frequencies of the null alleles are not particularly low (table 5), especially in Mm, suggesting the lack of strong selection preventing the null alleles from spreading through the populations. This finding is consistent with an overall low purifying selection acting on V1rs and provides a microevolutionary explanation for the rapid gene turnover observed at the macroevolutionary time scale.
Table 5.
Mouse V1rs With Segregating Null Alleles1
| V1rs | M. musculus | M. domesticus |
| A9 | 3 (deletion) | |
| B1 | 8 (deletion) | |
| B2 | 1 (deletion) | |
| C6 | 4 (SNP) | |
| C27 | 5 (SNP) + 1 (deletion) | |
| C28 | 4 (no amplification) | 1 (SNP) |
| E1 | 2 (deletion) | |
| F1 | 1 (deletion) + 2 (deletion) | |
| F3 | 10 (no amplification) + 2 (SNP) | |
| F5 | 1 (deletion) | |
| H4 | 1 (insertion) | |
| H15 | 6 (SNP) | |
| I6 | 2 (insertion) | 1 (insertion) + 1 (SNP) |
| I7 | 2 (deletion) | 1 (insertion) |
| J2 | 2 (SNP) | |
| J3 | 1 (SNP) | |
| K1 | 1 (SNP) + 2 (deletion) | |
| L1 | 1 (SNP) |
NOTE.—Numbers in the table are the numbers of null alleles (of 14 per species) for V1rs that harbor null alleles in the species. The type of null mutations is also indicated.
Discussion
In this study, we characterized the intra- and interspecific sequence variations of 44 V1r genes and 25 noncoding regions in two closely related Mus species. Both intraspecific polymorphisms and interspecific divergences are generally reduced in V1rs compared with the noncoding regions, suggesting that the overall force in V1r evolution is purifying selection. The strength of the purifying selection, however, is relatively weak. This is reflected by a ratio of approximately 0.32 between the mean nonsynonymous substitution rate and mean synonymous substitution rate of the 44 V1rs. A similar ratio (0.34) is obtained when only fixed differences between Mm and Md are considered. In comparison, this ratio is on average ∼0.11 when all 11,503 mouse–rat orthologous genes are compared. The Rat Genome Sequencing Project Consortium reported that 15% of mouse–rat orthologous genes have a dN/dS ratio greater than 0.28, but we found that 57% of V1rs belong to this category, the difference being highly significant (P < 10−6, binomial test). The overall weak purifying selection is also reflected by the high fraction of V1r loci that have segregating null alleles in either one or both Mus species examined. It is likely that a sizable proportion of V1r genes are not functionally constrained in each mouse species, consistent with the previous observation of virtually neutral variations of V1r gene copy number within and between species (Nozawa et al. 2007; Zhang 2007). In other words, the seemingly rapid V1r gene turnover is at least in part caused by neutral genomic drift (Nozawa et al. 2007). We did not find any V1r that has been duplicated in one of the two Mus species since their separation, but this is attributable to our intentional avoidance of studying V1rs with closely related paralogs to ease gene-specific amplification. In fact, in our preliminary study, one gene (E13) appeared to be duplicated in some individuals, but it was removed from the subsequent study due to the difficulty in designing gene-specific primers. Our findings of weak purifying selection in the microevolution of V1rs is generally consistent with a recent interspecific comparison of a subset of V1rs between the laboratory mouse and M. spretus (Kurzweil et al. 2009), which diverged from each other much earlier than the separation between Mm and Md. Kurzweil et al. sequenced a genomic segment of M. spretus that harbors two subfamilies of V1rs, including 18 genes. They observed that 1 of 11 genes in subfamily a and 2 of 7 genes in subfamily b have been lost in M. spretus, whereas 2 genes in subfamily a are becoming pseudogenes in M. musculus. They also identified two orthologous pairs that have been subject to positive selection. However, they did not analyze population genetic dynamics of V1rs because no intraspecific polymorphism data were collected. Our data thus complement theirs in providing necessary information for inferring the microevolutionary forces acting on V1rs.
It should be noted that, across mammals, there is a strong positive correlation between the morphological (and presumably physiological) complexity of the VNS in a species and the number of intact V1r genes the species has (Grus et al. 2005, 2007). Furthermore, during the evolutionary transition of vertebrates from water to land, there was a ∼50-fold increase in the ratio of the number of V1rs, which likely bind to airborne ligands, to that of V2rs, which likely bind to water-soluble ligands (Shi and Zhang 2007). Thus, it is likely that the evolution of the V1r repertoire is also subject to positive selection. Indeed, using the HKA test, we detected positive selection for nucleotide substitutions in ∼5% of the mouse V1rs surveyed after controlling for multiple testing. Extrapolating this result to all V1rs suggests that there are ∼10 V1rs that have adaptive differences between the two Mus species compared. Although we have no knowledge about the number of pheromonal differences between the two species, it is not unlikely that they differ by no more than a dozen pheromones. Recently, Karn et al. suggested that a particular V1r gene, Vmn1r67 (or E10 in our nomenclature), experienced adaptive divergences among Mus species and that it might be responsible for detecting the androgen-binding protein, a species-specific cue for species recognition (Karn et al. 2010). Although this gene is not included in our 44 V1rs, positive selection on this gene would not be inconsistent with our estimate of ∼5% of positively selected V1rs in the divergence between Mm and Md. In the future, it will be interesting to confirm their finding in the wild mice used here. An earlier paper compared two assemblies of mouse genome sequences, which were acquired from different mouse inbred lines, and reported an overall dN/dS ratio between the two assemblies to be 1.13 for V1rs (Zhang et al. 2004). The authors interpreted this finding as evidence for positive selection acting on most V1rs without actually testing whether the dN/dS ratio is significantly greater than 1, which is required for establishing positive selection. In our study, we found the mean dN/dS ratio for polymorphisms to be ∼0.5 when all V1r polymorphisms in the 14 mice sequenced here are considered. It remains to be seen whether this disparity is due to the difference between the genes we sampled and the rest of V1rs, sequencing errors in draft genome sequences, or the differences between the wild mice and inbred laboratory mice. Furthermore, it will be important to reexamine the polymorphism and divergence of mouse V1rs at sites important for binding to ligands when such sites are identified.
It is important to note that the sample size (seven mice per species) is relatively small in our study, making population genetic tests of positive selection less powerful. Nonetheless, multiple observations from our data are consistent with one another in supporting the conclusion that weak purifying selection is the predominant force in mouse V1r evolution. Furthermore, the McDonald–Kreitman test also strongly rejects the strict neutrality in support of purifying selection rather than positive selection. Thus, it is unlikely that the observed paucity of positive selection in V1rs is an artifact of our small sample. We focused our analysis exclusively on coding regions rather than on regulatory regions because all V1rs are expressed in the VNS and are not expected to have important evolutionary changes in gene regulation. A previous analysis of the promoter regions of V1rs supports this view (Stewart and Lane 2007). One limitation of our study is that we focused exclusively on point mutations and small indels, whereasthe macroevolution of V1rs is also known to be characterized by gene duplication, deletion, and possibly gene conversion. In the future, it would be especially interesting to examine the dynamics of copy number variations for V1rs as has been analyzed for odorant receptor genes (Nozawa et al. 2007; Zhang 2007; Hasin et al. 2008; Young et al. 2008).
Evolutionary changes of large gene families appear to contribute disproportionately to genomic evolution because several of the largest gene families in eukaryotic genomes evolve rapidly (Shiu et al. 2004; Nei and Rooney 2005; Niimura 2009; Shi and Zhang 2009). It will be interesting to study whether our findings on the microevolution of V1rs extend to other large gene families.
Supplementary Material
Supplementary tables S1–S3 are available at Genome Biology and Evolution online (http://www.oxfordjournals.org/our_journals/gbe/).
Acknowledgments
We thank John Baines and Bettina Harr at University of Cologne for the primer sequences used for amplifying some noncoding regions. We thank Soochin Cho, Nathan Pearson, and two anonymous reviewers for valuable comments. This work was supported by a research grant from the U.S. National Institutes of Health. S.H.P. was in part supported by a visiting fellowship from Korea University.
References
- Alibert P, FelClair F, Manolakou K, BrittonDavidian J, Auffray JC. Developmental stability, fitness, and trait size in laboratory hybrids between European subspecies of the house mouse. Evolution. 1997;51:1284–1295. doi: 10.1111/j.1558-5646.1997.tb03975.x. [DOI] [PubMed] [Google Scholar]
- Baines JF, Harr B. Reduced X-linked diversity in derived populations of house mice. Genetics. 2007;175:1911–1921. doi: 10.1534/genetics.106.069419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan PA, Keverne EB. Something in the air? New insights into mammalian pheromones. Curr Biol. 2004;14:R81–R89. doi: 10.1016/j.cub.2003.12.052. [DOI] [PubMed] [Google Scholar]
- Britton-Davidian J, Fel-Clair F, Lopez J, Alibert P, Boursot P. Postzygotic isolation between the two European subspecies of the house mouse: estimates from fertility patterns in wild and laboratory-bred hybrids. Biol J Linn Soc Lond. 2005;84:379–393. [Google Scholar]
- Dulac C, Torello AT. Molecular detection of pheromone signals in mammals: from genes to behaviour. Nat Rev Neurosci. 2003;4:551–562. doi: 10.1038/nrn1140. [DOI] [PubMed] [Google Scholar]
- Forejt J. Hybrid sterility in the mouse. Trends Genet. 1996;12:412–417. doi: 10.1016/0168-9525(96)10040-8. [DOI] [PubMed] [Google Scholar]
- Forejt J, Ivanyi P. Genetic studies on male sterility of hybrids between laboratory and wild mice (Mus musculus L.) Genet Res. 1974;24:189–206. doi: 10.1017/s0016672300015214. [DOI] [PubMed] [Google Scholar]
- Frazer KA, et al. A sequence-based variation map of 8.27 million SNPs in inbred mouse strains. Nature. 2007;448:1050–1053. doi: 10.1038/nature06067. [DOI] [PubMed] [Google Scholar]
- Fu YX, Li WH. Statistical tests of neutrality of mutations. Genetics. 1993;133:693–709. doi: 10.1093/genetics/133.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs RA, et al. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature. 2004;428:493–521. doi: 10.1038/nature02426. [DOI] [PubMed] [Google Scholar]
- Grus WE, Shi P, Zhang J. Largest vertebrate vomeronasal type 1 receptor gene repertoire in the semiaquatic platypus. Mol Biol Evol. 2007;24:2153–2157. doi: 10.1093/molbev/msm157. [DOI] [PubMed] [Google Scholar]
- Grus WE, Shi P, Zhang YP, Zhang J. Dramatic variation of the vomeronasal pheromone receptor gene repertoire among five orders of placental and marsupial mammals. Proc Natl Acad Sci U S A. 2005;102:5767–5772. doi: 10.1073/pnas.0501589102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grus WE, Zhang J. Rapid turnover and species-specificity of vomeronasal pheromone receptor genes in mice and rats. Gene. 2004;340:303–312. doi: 10.1016/j.gene.2004.07.037. [DOI] [PubMed] [Google Scholar]
- Grus WE, Zhang J. Origin and evolution of the vertebrate vomeronasal system viewed through system-specific genes. BioEssays. 2006;28:709–718. doi: 10.1002/bies.20432. [DOI] [PubMed] [Google Scholar]
- Grus WE, Zhang J. Distinct evolutionary patterns between chemoreceptors of 2 vertebrate olfactory systems and the differential tuning hypothesis. Mol Biol Evol. 2008;25:1593–1601. doi: 10.1093/molbev/msn107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grus WE, Zhang J. Origin of the genetic components of the vomeronasal system in the common ancestor of all extant vertebrates. Mol Biol Evol. 2009;26:407–419. doi: 10.1093/molbev/msn262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenet JL, Bonhomme F. Wild mice: an ever-increasing contribution to a popular mammalian model. Trends Genet. 2003;19:24–31. doi: 10.1016/s0168-9525(02)00007-0. [DOI] [PubMed] [Google Scholar]
- Hasin Y, et al. High-resolution copy-number variation map reflects human olfactory receptor diversity and evolution. PLoS Genet. 2008;4:e1000249. doi: 10.1371/journal.pgen.1000249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR, Kreitman M, Aguade M. A test of neutral molecular evolution based on nucleotide data. Genetics. 1987;116:153–159. doi: 10.1093/genetics/116.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karn RC, Young JM, Laukaitis CM. A candidate subspecies discrimination system involving a vomeronasal receptor gene with different alleles fixed in M. m. domesticus and M. m. musculus. PLoS One. 2010;5:e12638. doi: 10.1371/journal.pone.0012638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurzweil VC, Getman M, Green ED, Lane RP. Dynamic evolution of V1R putative pheromone receptors between Mus musculus and Mus spretus. BMC Genomics. 2009;10:74. doi: 10.1186/1471-2164-10-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laukaitis CM, Critser ES, Karn RC. Salivary androgen-binding protein (ABP) mediates sexual isolation in Mus musculus. Evolution. 1997;51:2000–2005. doi: 10.1111/j.1558-5646.1997.tb05121.x. [DOI] [PubMed] [Google Scholar]
- Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- Mombaerts P. Molecular biology of odorant receptors in vertebrates. Annu Rev Neurosci. 1999;22:487–509. doi: 10.1146/annurev.neuro.22.1.487. [DOI] [PubMed] [Google Scholar]
- Mombaerts P. Genes and ligands for odorant, vomeronasal and taste receptors. Nat Rev Neurosci. 2004;5:263–278. doi: 10.1038/nrn1365. [DOI] [PubMed] [Google Scholar]
- Moulia C, et al. Wormy mice in a hybrid zone—a genetic-control of susceptibility to parasite infection. J Evol Biol. 1991;4:679–687. [Google Scholar]
- Moulia C, Le Brun N, Dallas J, Orth A, Renaud F. Experimental evidence of genetic determinism in high susceptibility to intestinal pinworm infection in mice: a hybrid zone model. Parasitology. 1993;106(Pt 4):387–393. doi: 10.1017/s0031182000067135. [DOI] [PubMed] [Google Scholar]
- Nei M, Rooney AP. Concerted and birth-and-death evolution of multigene families. Annu Rev Genet. 2005;39:121–152. doi: 10.1146/annurev.genet.39.073003.112240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niimura Y. On the origin and evolution of vertebrate olfactory receptor genes: comparative genome analysis among 23 chordate species. Genome Biol Evol. 2009;1:34–44. doi: 10.1093/gbe/evp003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozawa M, Kawahara Y, Nei M. Genomic drift and copy number variation of sensory receptor genes in humans. Proc Natl Acad Sci U S A. 2007;104:20421–20426. doi: 10.1073/pnas.0709956104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restrepo D, Arellano J, Oliva AM, Schaefer ML, Lin W. Emerging views on the distinct but related roles of the main and accessory olfactory systems in responsiveness to chemosensory signals in mice. Horm Behav. 2004;46:247–256. doi: 10.1016/j.yhbeh.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Rodriguez I, Del Punta K, Rothman A, Ishii T, Mombaerts P. Multiple new and isolated families within the mouse superfamily of V1r vomeronasal receptors. Nat Neurosci. 2002;5:134–140. doi: 10.1038/nn795. [DOI] [PubMed] [Google Scholar]
- Rodriguez I, Mombaerts P. Novel human vomeronasal receptor-like genes reveal species-specific families. Curr Biol. 2002;12:R409–R411. doi: 10.1016/s0960-9822(02)00909-0. [DOI] [PubMed] [Google Scholar]
- Sage R, Atchley W, Capanna E. House mice as models in systematic biology. Syst Biol. 1993;42:523–561. [Google Scholar]
- Sage RD, Heyneman D, Lim KC, Wilson AC. Wormy mice in a hybrid zone. Nature. 1986;324:60–63. doi: 10.1038/324060a0. [DOI] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Salcedo T, Geraldes A, Nachman MW. Nucleotide variation in wild and inbred mice. Genetics. 2007;177:2277–2291. doi: 10.1534/genetics.107.079988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi P, Bielawski JP, Yang H, Zhang YP. Adaptive diversification of vomeronasal receptor 1 genes in rodents. J Mol Evol. 2005;60:566–576. doi: 10.1007/s00239-004-0172-y. [DOI] [PubMed] [Google Scholar]
- Shi P, Zhang J. Comparative genomic analysis identifies an evolutionary shift of vomeronasal receptor gene repertoires in the vertebrate transition from water to land. Genome Res. 2007;17:166–174. doi: 10.1101/gr.6040007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi P, Zhang J. Extraordinary diversity of chemosensory receptor gene repertoires among vertebrates. In: Meyerhof W, Korsching S, editors. Chemosensory systems in mammals, fishes, and insects. Berlin (Germany): Springer; 2009. pp. 1–23. [Google Scholar]
- Shiu SH, Karlowski WM, Pan R, Tzeng YH, Mayer KF, Li WH. Comparative analysis of the receptor-like kinase family in Arabidopsis and rice. Plant Cell. 2004;16:1220–1234. doi: 10.1105/tpc.020834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smadja C, Catalan J, Ganem G. Strong premating divergence in a unimodal hybrid zone between two subspecies of the house mouse. J Evol Biol. 2004;17:165–176. doi: 10.1046/j.1420-9101.2003.00647.x. [DOI] [PubMed] [Google Scholar]
- Smadja C, Ganem G. Subspecies recognition in the house mouse: a study of two populations from the border of a hybrid zone. Behav Ecol. 2002;13:312–320. [Google Scholar]
- Spehr M, Spehr J, Ukhanov K, Kelliher KR, Leinders-Zufall T, Zufall F. Parallel processing of social signals by the mammalian main and accessory olfactory systems. Cell Mol Life Sci. 2006;63:1476–1484. doi: 10.1007/s00018-006-6109-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart R, Lane RP. V1R promoters are well conserved and exhibit common putative regulatory motifs. BMC Genomics. 2007;8:253. doi: 10.1186/1471-2164-8-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storchova R, Gregorova S, Buckiova D, Kyselova V, Divina P, Forejt J. Genetic analysis of X-linked hybrid sterility in the house mouse. Mamm Genome. 2004;15:515–524. doi: 10.1007/s00335-004-2386-0. [DOI] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takami S. Recent progress in the neurobiology of the vomeronasal organ. Microsc Res Tech. 2002;58:228–250. doi: 10.1002/jemt.10094. [DOI] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Trachtulec Z, Mihola O, Vlcek C, Himmelbauer H, Paces V, Forejt J. Positional cloning of the Hybrid sterility 1 gene: fine genetic mapping and evaluation of two candidate genes. Biol J Linn Soc. 2005;84:637–641. [Google Scholar]
- Tucker PK. Systematics of the genus Mus. In: Fox J, et al., editors. Mouse in Biomedical Research, 2nd ed. Boston: Elsevier Press. p. 13--23. In: Fox J, et al., editors. 2007. [Google Scholar]
- Vyskocilova M, Trachtulec Z, Forejtv J, Pialek J. Does geography matter in hybrid sterility in house mice? Biol J Linn Soc Lond. 2005;84:663–674. [Google Scholar]
- Yang H, Bell TA, Churchill GA, Pardo-Manuel de Villena F. On the subspecific origin of the laboratory mouse. Nat Genet. 2007;39:1100–1107. doi: 10.1038/ng2087. [DOI] [PubMed] [Google Scholar]
- Yang H, Shi P, Zhang YP, Zhang J. Composition and evolution of the V2r vomeronasal receptor gene repertoire in mice and rats. Genomics. 2005;86:306–315. doi: 10.1016/j.ygeno.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Young JM, Endicott RM, Parghi SS, Walker M, Kidd JM, Trask BJ. Extensive copy-number variation of the human olfactory receptor gene family. Am J Hum Genet. 2008;83:228–242. doi: 10.1016/j.ajhg.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JM, Kambere M, Trask BJ, Lane RP. Divergent V1R repertoires in five species: amplification in rodents, decimation in primates, and a surprisingly small repertoire in dogs. Genome Res. 2005;15:231–240. doi: 10.1101/gr.3339905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JM, Massa HF, Hsu L, Trask BJ. Extreme variability among mammalian V1R gene families. Genome Res. 2010;20:10–18. doi: 10.1101/gr.098913.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JM, Trask BJ. V2R gene families degenerated in primates, dog and cow, but expanded in opossum. Trends Genet. 2007;23:212–215. doi: 10.1016/j.tig.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Zhang J. The drifting human genome. Proc Natl Acad Sci U S A. 2007;104:20147–20148. doi: 10.1073/pnas.0710524105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Kumar S, Nei M. Small-sample tests of episodic adaptive evolution: a case study of primate lysozymes. Mol Biol Evol. 1997;14:1335–1338. doi: 10.1093/oxfordjournals.molbev.a025743. [DOI] [PubMed] [Google Scholar]
- Zhang J, Webb DM. Evolutionary deterioration of the vomeronasal pheromone transduction pathway in catarrhine primates. Proc Natl Acad Sci U S A. 2003;100:8337–8341. doi: 10.1073/pnas.1331721100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Rodriguez I, Mombaerts P, Firestein S. Odorant and vomeronasal receptor genes in two mouse genome assemblies. Genomics. 2004;83:802–811. doi: 10.1016/j.ygeno.2003.10.009. [DOI] [PubMed] [Google Scholar]
- Zhao H, Xu D, Zhang S, Zhang J. Widespread losses of vomeronasal signal transduction in bats. Mol Biol Evol. 2011;28:7–12. doi: 10.1093/molbev/msq207. [DOI] [PMC free article] [PubMed] [Google Scholar]