

Abstract
The allantoin degradation pathway in E. coli has long been thought to involve a putative novel oxamate transcarbamylase (OXTCase) that converts oxaluric acid to oxamate and carbamyl phosphate (CP), a substrate for carbamate kinase (CK). In the genome sequence of E. coli, the only gene that could encode a novel transcarbamylase is the ygeW gene. However, the recombinant protein has no transcarbamylase activity with oxamate, allantoin, or twenty five other related compounds as potential substrates. The crystal structures of this transcarbamylase with unknown function (UTCase) has been determined and refined at 2.0 Å resolution, providing structural insights into its possible function. Like N-acetyl-L-ornithine transcarbamylase and N-succinyl-L-transcarbamylase, UTCase has a deep 31 trefoil knot close to the active site, in contrast to aspartate transcarbamylase and ornithine transcarbamylase which do not have a knot. A Blast search of completed genomes indicates that 52 species including one non-bacterial species, Trichomonas vaginalis G3, have the ygeW gene. Gene context analysis and the structure of UTCase suggest that it is probably an ancestral catabolic transcarbamylase.
Keywords: ygeW gene, transcarbamylase, purine degradation pathway, knotted protein
INTRODUCTION
Purine degradation plays an essential role in nitrogen metabolism in most organisms. Uric acid is the final product of purine catabolism in humans, anthropoid apes, birds, uricotelic reptiles, and almost all insects1. Elevated levels of uric acid in blood (hyperuricemia) cause human diseases such as gout, kidney stones and renal failure2. Although no enzyme has been identified that further degrades uric acid in humans, it can be oxidized to produce allantoin by free-radical attack3. Indeed, elevated levels of allantoin are found in patients with rheumatoid arthritis, chronic lung disease, bacterial meningitis and non-insulin-dependent diabetes mellitus4. In other mammals, some insects and gastropods, uric acid is enzymatically degraded to the more soluble allantoin through the sequential action of three enzymes: urate oxidase, 5-hydroxyisourate (HIU) hydrolase and 2-oxo-4-hydroxy-4-carboxy-5-ureidoimidazoline (OHCU) decarboxylase (Supporting Information Fig. S1)5. Therefore, an elective treatment for acute hyperuricemia is the administration of urate oxidase6. Many organisms, including plants, some fungi and several bacteria, are able to catabolize allantoin to release nitrogen, carbon and energy4. In Arabidopsis thaliana and Eschrichia coli, S-allantoin has recently been shown to be degraded to glycolate and urea by four enzymes: allantoinase, allantoate amidohydrolase, ureidoglycine aminohydrolase and ureidoglycolate amidohydrolase7.
Approximately sixty years ago, oxamate was identified as one of the end products of allantoin degradation in Streptococcus allantoicus8 and further studies demonstrated that oxamate formation is catalyzed by an oxamate transcarbamylase (OXTCase)9–14. Interestingly, in contrast to all known transcarbamylases, OXTCase requires bivalent ions such as Mg2+ or Mn2+ for its activity 9,10,12,13. OXTCase has also been found in Proteus rettgeri, group D streptococci, and E. coli. Recent studies reexamining the ability of E. coli to utilize purine as a nitrogen source proposed a purine catabolic pathway in E. coli, with OXTCase encoded by the ygeW gene15. Although allantoin can be used by E. coli as the sole nitrogen source under anaerobic conditions16, purines are not sufficient for aerobic growth15. In these catabolic processes, the function of OXTCase is assumed to generate carbamyl phosphate (CP), which is used to produce ATP from oxalurate by carbamate kinase (CK) (Supporting Information Fig. S1)17,18. However, since no recombinant protein has been shown yet to have OXTCase activity, OXTCase is still classified as an orphan enzyme19.
To characterize the ygeW encoded protein, we cloned the gene from E. coli genomic DNA, purified the protein to homogeneity, determined its crystal structure to 2.0 Å resolution, and tested its activity with oxamate, allantoin, and various other potential substrates. The crystal structure is consistent with the encoded protein being a novel transcarbamylase, and analysis showed both similarities and differences compared to all other known transcarbamylases. An interesting feature of the structure is a 31 trefoil knot, similar to those found in N-succinyl-L-ornithine transcarbamylase (SOTCase)20,21 and N-acetyl-L-ornithine transcarbamylase (AOTCase)22. However, lack of activity with any of the potential substrates tested and the structure of the binding site suggests that the second substrate is larger than either oxalurate or allantoin, and remains to be identified. Thus the protein will be referred here forth to as UTCase (transcarbamylase with unknown function.)
MATERIALS AND METHODS
Cloning, expression, and purification
The ygeW gene was PCR amplified from E. coli genomic DNA (ATCC10798), using hot-started turbo pfu DNA polymerase and the primer 5’-GACATATGATGAAAACTGTTAATGAGC-3’ and 5’-GCGGATCCTTATTTCACGCGTTCTTGC-3’. The PCR products were cloned into a pET28a vectors (Novagen) and transformed into E. coli BL21(DE3) cells (Invitrogen) for overexpression. The protein was prepared using the Overnight Express Auto-induction system 1 (Novagen) according to the manufacturer’s protocol and purified by Histrap Ni-affinity column (GE Healthcare) and then by Hightrap DEAE column (GE Healthcare) using an AKTA FPLC system. The purified protein was stored in a buffer containing 50 mM NaCl, 20 mM Tris-HCl (pH 8.0), 1 mM EDTA and 5 mM β-mercaptoethanol. The His6 tag was removed by adding 50 units of thrombin to about 10 mg of protein that was incubated at 277 K overnight. Protein concentration was determined by the Bradford method using BioRad protein assay dye reagent with bovine serum albumin as a standard23. The protein was obtained in high yield and to >95% purity verified by SDS-PAGE.
The selenomethionine (Se-Met) substituted protein was prepared using Overnight Expression Auto-induction System 2 (Novagen) as described previously22,24 and purified in the same way as the wild-type protein. On average, more than 80% of methionines were substituted by Se-Met as determined by MALDI-TOF/TOF mass spectrometry as described previously22,24.
Activity assay
Carbamylation products can be detected conveniently using a colorimetric assay that detects the formation of a ureido group25, and this method has been successfully used to detect the carbamylaspartate for aspartate transcarbamylase (ATCase) activity25, citrulline for ornithine transcarbamylase (OTCase)26, N-acetylcitrulline for AOTCase24 and N-succinylcitrulline for SOTCase20. However, the 5-amino-S-hydantoin could not be tested as a substrate using colorimetric assay in the forward reaction (carbamylaton) and oxalurate could not be tested as a substrate in the reverse reaction because of their unavailability. Instead, we tested whether the enzyme can use allantoin, which is a stable intermediate or end product in many organisms in ureido degradation pathway, as the substrate in the reverse reaction, using arsenolysis as described previously27. The standard assay mixture (100 µl) contained 100 mM imidazole buffer, pH 6.8, 40 mM S-allantoin, 40 mM arsenate, 2 mM MgCl2 and 100 µg enzyme. The reaction was started by addition of allantoin, allowed to proceed for 2 h at 310 K, and terminated by addition of 100 µl of 30% trichloroacetic acid. The products were analyzed using an Agilent LC-MS 1100 in the selected-ion-monitoring mode with positive polarity. The mass ions of the protonated compounds of interest were monitored using m/z = 159 for allantoin, and m/z = 116 for 5-amino-allantoin. As a negative control, parallel reactions were performed in the same manner but without arsenate or enzyme. As a positive control, the assay was carried on in the same manner, but with N-acetylcitrulline as substrate and AOTCase from X. campestris as enzyme24.
Cross-link experiment and analytical gel chromatography
Protein cross-linking was carried out with 16 µg bis(sulfosuccinimidyl) suberate (BS3; Pierce) and 2.8 µg of protein in 25 µl of buffer containing 20 mM phosphate, 300 mM NaCl, 10% glycerol, 10 mM β-mercaptoethanol pH 7.4. The mixture was incubated on ice for 15 min and the reaction was stopped by adding 5 µl 1M Tris-HCl, pH 7.4. The resulting covalent adducts were analyzed by 4–12% SDS-PAGE. E. coli OTCase was used as a control. The molecular weight of UTCase in solution was determined with a Superdex 200 HR 10/30 column (Amersham Pharmacia Biotech) as previously described28.
Crystallization and Data Collection
The purified protein was concentrated to ~25 mg/ml with an Amicon-Y30 membrane concentrator (Millipore). The index screening kit was used for crystallization tests (Hampton Research) at 291 K. After optimization, hexagonal prism crystals grew to 0.2 mm within a week in a buffer containing 0.2 M MgCl2 0.1 M HEPES pH 7.5, 20% PEG 3350 (w/v). Se-Met derivative crystals were grown under conditions similar to the wild-type crystals, but with 30 mM S-allantoin added. While most wild-type crystals could only diffracted to about 3.0 Å, the Se-Met substituted crystals diffracted beyond 2.0 Å resolution.
Before data collection, a crystal was dipped quickly into a well solution supplemented by 25% (v/v) ethylene glycerol. The cryoprotected crystal was frozen by plunging it directly into liquid nitrogen. The SAD dataset at wavelength 0.97933 Å was collected from a single crystal at 100 K on beamline Southeast Regional Collaborative Access Team 22-ID equipped with MAR300 CCD at the Advanced Photon Source (APS) Argonne National Laboratory. The data were indexed, integrated and scaled with the XDS package29. The data collection statistics were listed in Table 1.
Table 1.
Data Collection and Refinement Statistics
| Data collection | |
|---|---|
| Space group | P63 |
| Resolution range (Å) | 30-2.0 (2.05-2.0) |
| Unit-cell parameters (Å) | a = b = 78.25 |
| c =100.10 | |
| Measurements | 216,335 |
| Unique reflections | 23,411 (1,686) |
| Redundancy | 9.2 (8.3) |
| Completeness (%) | 99.5 (96.1) |
| <I/σ(I)> | 15.5 (5.8) |
| Rmerg(%)b | 9.1 (40.8) |
| Refinement | |
| No. of protein atoms | 2,993 |
| No. of water atoms | 148 |
| RMSD of bond lengths (Å) | 0.007 |
| RMSD of bond angle (°) | 1.4 |
| Rwork (%)c | 17.5 (20.2) |
| Rfree (%)d | 22.6 (23.9) |
Figures in brackets apply to the highest-resolution shell.
Rmerg = ΣhΣi|I(h,i)-<I(h)>|/ΣhΣiI(h,i), where I(h,i) is the intensity of the ith observation of reflection h, and <I(h)> is the average intensity of redundant measurements of reflection h.
Rwork= Σh|Fobs−Fcalc|/Σh Fobs.
Rfree = Σh|Fobs – Fcalc|/Σh Fobs for 10% of the reserved reflections.
Structure Determination and Refinement
Although the molecular replacement solution could be found for data sets from the wild-type protein using either OTCase (PDB code 1DUV) or AOTCase (PDB code 3KZK) as a searching model and program Phaser30,31, the electron density was poor and difficult to interpret, because of the limited resolution and absence of non-crystallographic symmetry. To avoid model bias and time-consuming model building based on a poor density map, the experimental phasing method was used to determine structure. The Se-Met substituted protein structure was solved using the SAD protocol of Auto-Rickshaw: the EMBL-Hamburg automated crystal structure determination platform32. The input diffraction data were prepared and converted for use in Auto-Rickshaw using programs in the CCP4 suite33. The structure-factor amplitudes of anomalous scatterers, the so-called FA values, were calculated using the program SHELXC34. Based on an initial analysis of the data, the maximum resolution for substructure determination and initial phase calculation was set to 2.0 A. The 11 heavy atoms were found using the program SHELXD34. The correct hand for the substructure was determined using the programs ABS35 and SHELXE34. Initial phases were calculated after density modification using the program SHELXE34. 81.73% of the model was built using the program ARP/wARP36,37 and the initial model with the electron density map was viewed and adjusted using the program COOT38. The model was subjected to several cycles of refinement using program PHENIX39 and adjustment using the program COOT38. The refinement statistics for the final refined model are given in Table 1. The final model consisted of 381 amino acids and 148 water molecules. Most residues were well defined in the electron density map except residues 90–94, 114–120 and 149–151, which were not included in the final model. All the main-chain, side-chain and planar group parameters assessed with PROCHECK40 were comparable to or better than those in structures at similar resolution in the PDB. The atomic coordinates and structure factors have been deposited in the Protein Data Bank (accession number 3Q98).
RESULTS AND DISCUSSION
The ygeW gene encodes a knotted ancestral transcarbamylase
As in all other known transcarbamylases, the monomer of UTCase consists of two domains: the CP domain (CPD) and the second substrate domain (SSD), which are linked by two helices (H6 and H14) (Fig. 1A). Both the CPD and SSD consist of a parallel β-strand core flanked by α-helices on both sides. However, UTCase has four more helices than any other known transcarbamylase. The extra helices at both the N-terminal (H1, residues 2–14) and C-terminal (H15, residues 380–390) ends of the peptide chain form a triple-helix bundle with the second N-terminal helix (H2, residues 32–51), present in all known transcarbamylases. The other two extra helices (H10 and H11, residues 275–286 and 290–306, respectively) form an extended 240’s loop positioned so as to be able to interact with the 120’s loop from the CPD.
Figure 1.
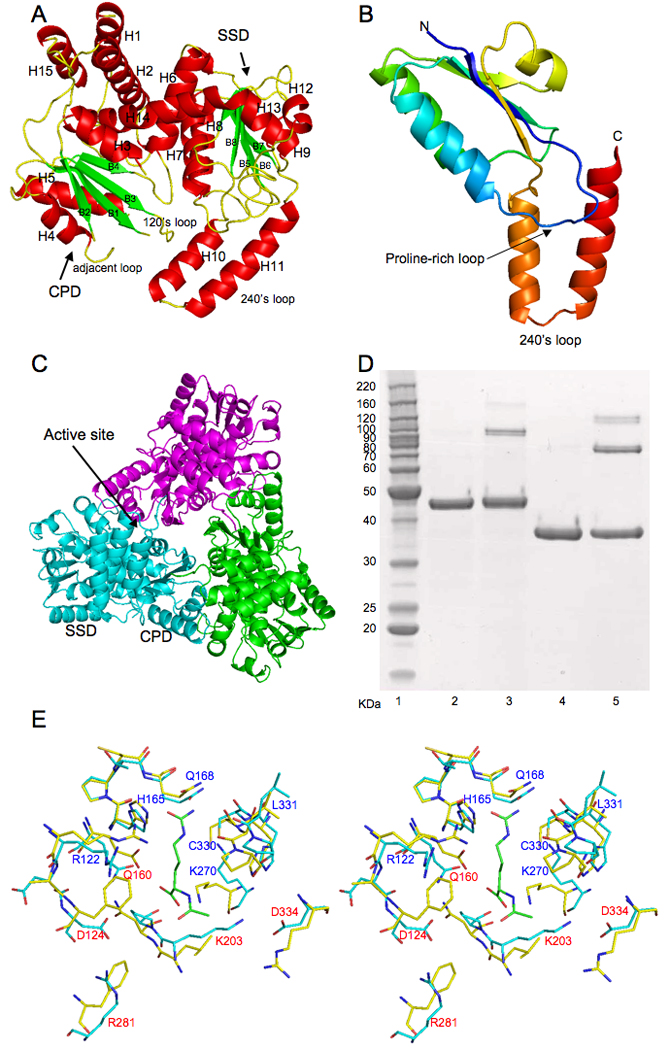
A, Ribbon diagram of UTCase subunit. Green arrows indicate the direction of strands in β-sheet, α helices are in red. B, Ribbon diagram of the knotted region of E. coli UTCase (residues 208–326). Color changes continuously from blue (first residue in the region) to red (last residue in the region). The proline-rich loop (from dark blue to light blue) seems to be critical for the knot formation. The 240’s loop (from brown to red) was threaded through the proline-rich loop to form a 31 trefoil knot. C, Ribbon diagram of a trimer. Subunits are colored pink, green, and light blue. D, Cross-linking of E. coli UTCase. Lane 1: Molecular marker; Lane 2: E. coli UTCase without cross-linking agent; Lane 3: E. coli UTCase with cross-linking agent; Lane 4: E. coli OTCase without cross-linking agent as control; Lane 5: E. coli OTCase with cross-linking agent as control. E, Stereo view of the proposed substrate binding site of UTCase (shown in light blue sticks) in comparison to AOTCase (shown in light yellow sticks, PDB accession number 3KZK). Bound N-acetyl-L-citrulline for AOTCase is shown in green sticks. Conserved residues are labeled as blue. Different residues are label as red.
The molecular functional unit for UTCase is a trimer, which is assembled by a three-fold crystallographic symmetry in the crystal structure, similar to the catalytic trimer in other transcarbamylases (Fig. 1C). Cross-linking indicates that the trimer is also present in solution (Fig. 1D). The molecular weight calculated from gel filtration chromatography is 148 KDa, in agreement with a trimeric structure. UTCase seems to have more similar interface interactions to AOTCase and SOTCase than to OTCase. The interface is extensive with the buried surface area of 1388 Å2, calculated with a probe radius of 1.4 Å and program AREAIMOL at CCP4 suite33.
The putative active site is located at the interface between two domains and shared by adjacent subunits as in other transcarbamylases (Fig. 1C). Although the crystals were grown from a buffer containing 30 mM S-allantoin, no electron density consistent with allantoin was identified in the active site.
Residues involved in binding CP could be reliably identified by aligning the current structure and sequence with other transcarbamylases (Supporting Information Fig. S2), even though the loop adjacent to the CP binding site (residue 92–102), termed the 80’s loop in E. coli ATCase and OTCase structures, which usually contributes one or two residues to binding substrates, and the STRTR (Ser71-Thr72-Arg73-Thr74-Arg75) CP binding motif were not modeled in the current structure. Specifically, Ser71, Arg73, Thr74, Arg122 and Gln98 from the adjacent subunit would be expected to bind the phosphate moiety of CP. The disorder in the adjacent loop is not surprising, since it is often disordered in the absence of the first substrate, CP22,41. However, the disorder of the STRTR binding motif was unexpected, since the STRTR motif is ordered in all known transcarbamylase structures in both the absence and presence of CP.
The side-chains of residues His165 and Gln168 and the carbonyl oxygen atoms of Cys330 and Leu331 involved in binding the carbamyl moiety of CP or the ureido group of the second substrate are well defined. Lys270, whose equivalent Lys252 in AOTCase interacts with the carboxyl group of acetylornithine, is conserved, but forms a hydrogen bond with Cys330. The side chain of Gln160 occupies a similar position to Glu144 in AOTCase, which interacts with the carboxyl group of acetylornithine. Essential hydrophobic residues, Leu184 and Phe114 in AOTCase which form the acetylornithine binding site, are replaced by Lys203 and Asp124 in UTCase, increasing the size and hydrophilic environment of the second binding site (Fig. 1E). The positive charged residues, Arg298 and His180, which hydrogen bond to Glu92 in AOTCase or the carboxyl group of succinyl moiety in SOTCase, are replaced by Asp334 and Ser198 in UTCase, respectively. As a result of these substitutions, the second substrate binding site of UTCase has a negative electrophilic surface.
A search with PDBeFold servers42 confirmed that UTCase is a member of the transcarbamylase family (http://www.ebi.ac.uk/msd-srv/ssm/) (Supporting Information Table S1). Transcarbamylases with known structures include OTCase, ATCase, AOTCase and SOTCase. AOTCase appears to be most closely related to UTCase based on Q score and root mean square deviation (RMSD). However, the P and Z scores indicate that OTCase is UTCase’s closest relative. This discrepancy may reflect differences in the degree of sequence and structural similarity of different regions of pairs of transcarbamylases. Although SOTCase also has a knotted fold, it is not as closely related to UTCase as AOTCase. As expected, ATCase is the most distantly related transcarbamylase.
A deep 31 trefoil knot similar to the knots in AOTCase and SOTCase was identified using the protein knot detection server43,44. The knot involves residues 194–305, close to the proposed second substrate binding site (Fig. 1B). Because of the presence of two extra helices (H10 and H11) in the 240’s loop and one extra helix (H15) at the C-terminus, UTCase needs to thread through approximately 124 residues through the proline-rich loop (residues 195–207) during protein folding, in comparison to 85 and 70 residues in AOTCase and SOTCase, respectively. Although knots in protein structures are very rare45,46, the knot is usually preserved across species and even across kingdoms. However, a deep trefoil knot which is present in UTCase, AOTCase and SOTCase, is absent in ATCase and OTCase47,48. Sequence and structural alignments suggest that a proline-rich loop is a pre-requisite for forming a knot21,49. UTCase, which has both a proline-rich loop and a knot, is consistent with this hypothesis (Supporting Information Fig. S2). Phylogenetic tree analysis indicates that a knotted member usually occupies a primary sub-branch of the phylogenetic tree close to the root that does not include unknotted members of the family49–51. Recent studies of thermally and mechanically induced unfolding processes suggest that the presence of a knot increases the intrinsic stability of the protein52. However, more importantly, the presence of the knot restructures the active site to allow the protein to recognize different substrates.
Sequence and structure comparisons of UTCase with other transcarbamylases indicate that UTCase incorporates features of several of other transcarbamylases. Four loop regions, the adjacent loop (or 80’s loop in E. coli OTCase and ATCase), the 120’s loop, the proline-rich loop, and the 240’s loop (or SMG loop termed in human OTCase)21 have the most sequence diversity and define the specificity of the second substrate binding. UTCase has an 80’s loop similar to that of E. coli OTCase, but much shorter than those of AOTCase and SOTCase. It had an extended 120’s loop similar to those in ATCase and SOTCase. The proline-rich loop is similar to those of AOTCase and SOTCase, but one residue longer. The 240’s loop is the most diverse region among transcarbamylases and is the key second substrate recognition site for unknotted transcarbamylase. The 240’s loop in UTCase is 18–20 residues longer than those in other transcarbamylases. The extra loop with an extra α-helix (H13), which is more common in catabolic OTCases, is also present in UTCase in the 340’s region.
UTCase appears to be an ancestral transcarbamylase that has structural and sequence features in different parts of its structure that are both similar and different compared to other transcarbamylases. During evolution, the disappearance of the proline-rich loop and shortening of the 240’s loop seems to convert the knotted form of transcarbamylase to an unknotted form49. More rapid evolution of disordered and loop regions than ordered regions53 seems to have enabled members of the transcarbamylase family to develop different binding specificities.
The ygeW gene encoded protein is likely to be a catabolic transcarbamylase
A BLAST search of protein sequences in the NBCI database identified 52 species (sub-strains were excluded) with ygeW-like genes (Supporting Information Table S2). A sequence alignment for UTCases from all species is provided in the Supporting Information Fig. S3. Pair-wise comparisons demonstrated that ygeW encoded protein sequences are highly conserved with more than 75% sequence similarity and 65% sequence identity. The largest protein, from Oribacterium sinus F0268, has 415 residues and the smallest one, from Shigella sonnei Ss046, has 372 residues. Most sequence variants are in the N-terminal region, implying that this region is not functionally significant. In the database, only one species, Spirochaeta smaragdinae DSM 11293, has two ygeW genes. The sequence identity between these two ygeW encoded proteins is 79%. All species, except Trichomonas vaginalis G3, are bacterial. No species from archaea, fungi, plants and animals have an ygeW-like gene. The ygeW-like gene (TVAG_038539) of Trichomonas vaginalis G3 is of particular interest since this organism is the most common sexually transmitted protozoan in the world and its resistance to metronidazole is increasing. If the yegW gene of T, vaginalis is essential, it might provide a potential therapeutic target.
To find clues about possible function, genes adjacent to ygeW were examined in all 52 sequenced genomes that contain it. The ygeW gene is close to the ygeA gene (separated by 3 or less genes) that encodes CK in 43 genomes (Supporting Information Table S2). In 9 genomes, the ygeW gene was not near ygeA. Thus, in approximately 83% of the genomes examined, ygeW and ygeA were in close proximity.
Anabolic transcarbamylases catalyze the forward reaction of transferring the carbamyl group of CP to the amino group of the second substrate, while catabolic transcarbamylase catalyzes the back reaction to produce CP. Thermodynamically the forward reaction is favored. However, the back reaction is favored when CK converts CP and ADP to ATP27. Among transcarbamylases, ATCase plays an anabolic role only in the pyrimidine biosynthetic pathway while OTCase can play both anabolic and catabolic roles in arginine biosynthetic and degradation, respectively. However, anabolic and catabolic OTCase reactions are usually catalyzed by two different enzymes with genes located in different regions of the genome. For example, the anabolic OTCase gene, argF, is located in the arginine biosynthetic gene cluster, while the catabolic OTCase gene, arcB, is located next to the CK gene, arcC. Putrescine transcarbamylase (PTCase) plays only a catabolic role in the agmatine degradation pathway. Its gene, ptcA (termed aguB in some literatures), is close to the agmatine deiminase and CK gene50,51,54. Gene context analysis seemed to suggest that the ygeW encoded protein in most bacteria has a catabolic role and produces CP, which is used by CK to produce ATP. However, in some bacteria the possibility of the ygeW encoded protein playing an anabolic role cannot be ruled out. Specifically, since two ygeW genes were found in Spirochaeta smaragdinae DSM 11293, it is not clear whether both encoded proteins play a catabolic role, or whether one of them plays an anabolic role.
The structure of E. coli UTCase is also consistent with a catabolic role. In both ATCase and anabolic OTCase, substrate binding is ordered, with CP binding first55,56, and binding of the second substrate causing the 240’s loop to swing in and form part of the active site. The disorder of the STRTR CP binding motif in the current UTCase structure is unique, since all other transcarbamylase structures have an ordered STRTR CP binding site even in the absence of any substrates41,57. The disorder of the STRTR motif in UTCase suggests that it may catalyze the catabolic reaction with a ureido-containing substrate binding first and phosphate binding next to order the STRTR binding motif and facilitate the formation of CP. This structural feature is in contrast to those in AOTCase and SOTCase, which are knotted transcarbamylases and play an anabolic role in the arginine biosynthetic pathway22, in which the equivalent SM(L)RTR binding motifs for CP are well ordered even in the absence of substrates.
Biological function of ygeW encoded protein remains unknown
In the genome sequence of E. coli, the only gene that could encode a novel transcarbamylase is the ygeW gene. However, no transcarbamylase activity can be detected for the recombinant protein with allantoin, or twenty five other related compounds as potential substrate including 20 naturally occurring amino acids, N-acetyl-L-ornithine, N-succinyl-L-ornithine, L-ornithine, oxamate, β-alanine and putrescine. Since OHCU, allantoate, ureidoglycolate and ureidoglycine, are not available, we cannot test the possibility that the enzyme uses these ureido-containing intermediates in the purine degradation pathway as substrate. Our structural study further ruled out that enzyme used allantoin as substrate because allantoin did not bind to the crystalline enzyme and suggested that the natural substrate is bulkier than either oxalurate or allantoin, and is either positive charged itself or binds positively charged metal ions around to compensate the negatively charged environment of the binding site. Thus, even though extensive effort has been made to elucidate the biological function of UTCase, it remains unknown.
Supplementary Material
Acknowledgments
We thank David Davies and Fred Dyda for facilitating use of the diffraction equipment in the Molecular Structure Section of the National Institute of Health. This work was supported by Public Health Service grant DK-47870 (to M. T.) and DK-067935 (to D. S.) from the National Institute of Diabetes, Digestive and Kidney Diseases. This work was also supported in part by the U.S. Department of Energy under contract W-31-109-Eng-38.
Abbreviations
- AOTCase
N-acetyl-L-ornithine transcarbamylase
- CK
carbamate kinase
- CP
carbamyl phosphate
- CPD
carbamyl phosphate domain
- HIU
5-hydroxyisourate
- OHCU
2-oxo-4-hydroxy-4-carboxy-5-ureidoimidazoline
- OTCase
ornithine transcarbamylase
- OXTCase
oxamate transcarbamylase
- PDB
Protein Data Bank
- PTCase
putrescine transcarbamylase
- RMSD
root mean square deviation
- SOTCase
N-succinyl-L-ornithine transcarbamylase
- SSD
second substrate domain
- UTCase
transcarbamylase with unknown function
REFERENCES
- 1.Vogels GD, Van der Drift C. Degradation of purines and pyrimidines by microorganisms. Bacteriological reviews. 1976;40(2):403–468. doi: 10.1128/br.40.2.403-468.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hall AP, Barry PE, Dawber TR, McNamara PM. Epidemiology of gout and hyperuricemia. A long-term population study. The American journal of medicine. 1967;42(1):27–37. doi: 10.1016/0002-9343(67)90004-6. [DOI] [PubMed] [Google Scholar]
- 3.Ames BN, Cathcart R, Schwiers E, Hochstein P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: a hypothesis. Proceedings of the National Academy of Sciences of the United States of America. 1981;78(11):6858–6862. doi: 10.1073/pnas.78.11.6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chou HC, Lee CZ, Ma LC, Fang CT, Chang SC, Wang JT. Isolation of a chromosomal region of Klebsiella pneumoniae associated with allantoin metabolism and liver infection. Infection and immunity. 2004;72(7):3783–3792. doi: 10.1128/IAI.72.7.3783-3792.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramazzina I, Folli C, Secchi A, Berni R, Percudani R. Completing the uric acid degradation pathway through phylogenetic comparison of whole genomes. Nature chemical biology. 2006;2(3):144–148. doi: 10.1038/nchembio768. [DOI] [PubMed] [Google Scholar]
- 6.Bomalaski JS, Clark MA. Serum uric acid-lowering therapies: where are we heading in management of hyperuricemia and the potential role of uricase. Current rheumatology reports. 2004;6(3):240–247. doi: 10.1007/s11926-004-0075-3. [DOI] [PubMed] [Google Scholar]
- 7.Werner AK, Romeis T, Witte CP. Ureide catabolism in Arabidopsis thaliana and Escherichia coli. Nature chemical biology. 2010;6(1):19–21. doi: 10.1038/nchembio.265. [DOI] [PubMed] [Google Scholar]
- 8.Barker HA. Streptococcus allantoicus and the Fermentation of Allantoin. Journal of bacteriology. 1943;46(3):251–259. doi: 10.1128/jb.46.3.251-259.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bojanowski R, Gaudy E, Valentine RC, Wolfe RS. Oxamic Transcarbamylase of Streptococcus Allantoicus. Journal of bacteriology. 1964;87:75–80. doi: 10.1128/jb.87.1.75-80.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tigier H, Grisolia S. Induction of Carbamyl-P Specific Oxamate Transcarbamylase by Parabanic Acid in a Streptococcus. Biochemical and biophysical research communications. 1965;19:209–214. doi: 10.1016/0006-291x(65)90506-1. [DOI] [PubMed] [Google Scholar]
- 11.Valentine RC, Bojanowski R, Gaudy E, Wolfe RS. Mechanism of the allantoin fermentation. The Journal of biological chemistry. 1962;237:2271–2277. [PubMed] [Google Scholar]
- 12.Valentine RC, Wolfe RS. Phosphorolysis of carbamyl oxamic acid. Biochimica et biophysica acta. 1960;45:389–391. doi: 10.1016/0006-3002(60)91467-0. [DOI] [PubMed] [Google Scholar]
- 13.Valentine RC, Wolfe RS. Biosynthesis of carbamyl oxamic acid. Biochemical and biophysical research communications. 1960;2(6):384–387. [Google Scholar]
- 14.Valentine RC, Wolfe RS. Phosphate-dependent degradation of urea. Nature. 1961;191:925–926. doi: 10.1038/191925b0. [DOI] [PubMed] [Google Scholar]
- 15.Xi H, Schneider BL, Reitzer L. Purine catabolism in Escherichia coli and function of xanthine dehydrogenase in purine salvage. Journal of bacteriology. 2000;182(19):5332–5341. doi: 10.1128/jb.182.19.5332-5341.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cusa E, Obradors N, Baldoma L, Badia J, Aguilar J. Genetic analysis of a chromosomal region containing genes required for assimilation of allantoin nitrogen and linked glyoxylate metabolism in Escherichia coli. Journal of bacteriology. 1999;181(24):7479–7484. doi: 10.1128/jb.181.24.7479-7484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rintoul MR, Cusa E, Baldoma L, Badia J, Reitzer L, Aguilar J. Regulation of the Escherichia coli allantoin regulon: coordinated function of the repressor AllR and the activator AllS. Journal of molecular biology. 2002;324(4):599–610. doi: 10.1016/s0022-2836(02)01134-8. [DOI] [PubMed] [Google Scholar]
- 18.Walker JR, Altamentova S, Ezersky A, Lorca G, Skarina T, Kudritska M, Ball LJ, Bochkarev A, Savchenko A. Structural and biochemical study of effector molecule recognition by the E.coli glyoxylate and allantoin utilization regulatory protein AllR. Journal of molecular biology. 2006;358(3):810–828. doi: 10.1016/j.jmb.2006.02.034. [DOI] [PubMed] [Google Scholar]
- 19.Lespinet O, Labedan B. ORENZA: a web resource for studying ORphan ENZyme activities. BMC bioinformatics. 2006;7:436. doi: 10.1186/1471-2105-7-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi D, Morizono H, Cabrera-Luque J, Yu X, Roth L, Malamy MH, Allewell NM, Tuchman M. Structure and catalytic mechanism of a novel N-succinyl-L-ornithine transcarbamylase in arginine biosynthesis of Bacteroides fragilis. The Journal of biological chemistry. 2006b;281(29):20623–20631. doi: 10.1074/jbc.M601229200. [DOI] [PubMed] [Google Scholar]
- 21.Shi D, Yu X, Cabrera-Luque J, Chen TY, Roth L, Morizono H, Allewell NM, Tuchman M. A single mutation in the active site swaps the substrate specificity of N-acetyl-L-ornithine transcarbamylase and N-succinyl-L-ornithine transcarbamylase. Protein Sci. 2007;16(8):1689–1699. doi: 10.1110/ps.072919907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi D, Yu X, Roth L, Morizono H, Hathout Y, Allewell NM, Tuchman M. Expression, purification, crystallization and preliminary X-ray crystallographic studies of a novel acetylcitrulline deacetylase from Xanthomonas campestris. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2005;61(Pt 7):676–679. doi: 10.1107/S1744309105018051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical biochemistry. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 24.Morizono H, Cabrera-Luque J, Shi D, Gallegos R, Yamaguchi S, Yu X, Allewell NM, Malamy MH, Tuchman M. Acetylornithine transcarbamylase: a novel enzyme in arginine biosynthesis. Journal of bacteriology. 2006;188(8):2974–2982. doi: 10.1128/JB.188.8.2974-2982.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pastra-Landis SC, Foote J, Kantrowitz ER. An improved colorimetric assay for aspartate and ornithine transcarbamylases. Analytical biochemistry. 1981;118(2):358–363. doi: 10.1016/0003-2697(81)90594-7. [DOI] [PubMed] [Google Scholar]
- 26.Morizono H, Tuchman M, Rajagopal BS, McCann MT, Listrom CD, Yuan X, Venugopal D, Barany G, Allewell NM. Expression, purification and kinetic characterization of wild-type human ornithine transcarbamylase and a recurrent mutant that produces 'late onset' hyperammonaemia. The Biochemical journal. 1997;322(Pt 2):625–631. doi: 10.1042/bj3220625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sainz G, Tricot C, Foray MF, Marion D, Dideberg O, Stalon V. Kinetic studies of allosteric catabolic ornithine carbamoyltransferase from Pseudomonas aeruginosa. European journal of biochemistry / FEBS. 1998;251(1–2):528–533. doi: 10.1046/j.1432-1327.1998.2510528.x. [DOI] [PubMed] [Google Scholar]
- 28.Shi D, Sagar V, Jin Z, Yu X, Caldovic L, Morizono H, Allewell NM, Tuchman M. The crystal structure of N-acetyl-L-glutamate synthase from Neisseria gonorrhoeae provides insights into mechanisms of catalysis and regulation. The Journal of biological chemistry. 2008;283(11):7176–7184. doi: 10.1074/jbc.M707678200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kabsch W. Xds. Acta crystallographica. 2010;66(Pt 2):125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Read RJ. Pushing the boundaries of molecular replacement with maximum likelihood. Acta crystallographica. 2001;57(Pt 10):1373–1382. doi: 10.1107/s0907444901012471. [DOI] [PubMed] [Google Scholar]
- 31.Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta crystallographica. 2004;60(Pt 3):432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 32.Panjikar S, Parthasarathy V, Lamzin VS, Weiss MS, Tucker PA. Auto-Rickshaw: an automated crystal structure determination platform as an efficient tool for the validation of an X-ray diffraction experiment. Acta crystallographica. 2005;61(Pt 4):449–457. doi: 10.1107/S0907444905001307. [DOI] [PubMed] [Google Scholar]
- 33.The CCP4 suite: programs for protein crystallography. Acta crystallographica. 1994;50(Pt 5):760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 34.Sheldrick GM. A short history of SHELX. Acta Crystallogr A. 2008;64(Pt 1):112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 35.Hao Q. ABS: a program to determine absolute configuration and evaluate anomalous scatterer substructure. Journal of Applied Crystallography. 2004;Volume 37:498–499. [Google Scholar]
- 36.Morris RJ, Zwart PH, Cohen S, Fernandez FJ, Kakaris M, Kirillova O, Vonrhein C, Perrakis A, Lamzin VS. Breaking good resolutions with ARP/wARP. Journal of synchrotron radiation. 2004;11(Pt 1):56–59. doi: 10.1107/s090904950302394x. [DOI] [PubMed] [Google Scholar]
- 37.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nature structural biology. 1999;6(5):458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 38.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta crystallographica. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 39.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta crystallographica. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. Journal of Applied Crystallography. 1993;Volume 26:283–291. [Google Scholar]
- 41.Jin L, Seaton BA, Head JF. Crystal structure at 2.8 A resolution of anabolic ornithine transcarbamylase from Escherichia coli. Nature structural biology. 1997;4(8):622–625. doi: 10.1038/nsb0897-622. [DOI] [PubMed] [Google Scholar]
- 42.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta crystallographica. 2004;60(Pt 12 Pt 1):2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 43.Kolesov G, Virnau P, Kardar M, Mirny LA. Protein knot server: detection of knots in protein structures. Nucleic acids research. 2007;35(Web Server issue):W425–W428. doi: 10.1093/nar/gkm312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lai YL, Yen SC, Yu SH, Hwang JK. pKNOT: the protein KNOT web server. Nucleic acids research. 2007;35(Web Server issue):W420–W424. doi: 10.1093/nar/gkm304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taylor WR. A deeply knotted protein structure and how it might fold. Nature. 2000;406(6798):916–919. doi: 10.1038/35022623. [DOI] [PubMed] [Google Scholar]
- 46.Taylor WR, Lin K. Protein knots: A tangled problem. Nature. 2003;421(6918):25. doi: 10.1038/421025a. [DOI] [PubMed] [Google Scholar]
- 47.Virnau P, Mirny LA, Kardar M. Intricate knots in proteins: Function and evolution. PLoS computational biology. 2006;2(9):e122. doi: 10.1371/journal.pcbi.0020122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lua RC, Grosberg AY. Statistics of knots, geometry of conformations, and evolution of proteins. PLoS computational biology. 2006;2(5):e45. doi: 10.1371/journal.pcbi.0020045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Potestio R, Micheletti C, Orland H. Knotted vs. unknotted proteins: evidence of knot-promoting loops. PLoS computational biology. 2010;6(7) doi: 10.1371/journal.pcbi.1000864. e1000864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Naumoff DG, Xu Y, Stalon V, Glansdorff N, Labedan B. The difficulty of annotating genes: the case of putrescine carbamoyltransferase. Microbiology (Reading, England) 2004;150(Pt 12):3908–3911. doi: 10.1099/mic.0.27640-0. [DOI] [PubMed] [Google Scholar]
- 51.Naumoff DG, Xu Y, Glansdorff N, Labedan B. Retrieving sequences of enzymes experimentally characterized but erroneously annotated : the case of the putrescine carbamoyltransferase. BMC genomics. 2004;5(1):52. doi: 10.1186/1471-2164-5-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sulkowska JI, Sulkowski P, Szymczak P, Cieplak M. Stabilizing effect of knots on proteins. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(50):19714–19719. doi: 10.1073/pnas.0805468105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brown CJ, Takayama S, Campen AM, Vise P, Marshall TW, Oldfield CJ, Williams CJ, Dunker AK. Evolutionary rate heterogeneity in proteins with long disordered regions. Journal of molecular evolution. 2002;55(1):104–110. doi: 10.1007/s00239-001-2309-6. [DOI] [PubMed] [Google Scholar]
- 54.Griswold AR, Chen YY, Burne RA. Analysis of an agmatine deiminase gene cluster in Streptococcus mutans UA159. Journal of bacteriology. 2004;186(6):1902–1904. doi: 10.1128/JB.186.6.1902-1904.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shi D, Morizono H, Yu X, Tong L, Allewell NM, Tuchman M. Human ornithine transcarbamylase: crystallographic insights into substrate recognition and conformational changes. The Biochemical journal. 2001;354(Pt 3):501–509. doi: 10.1042/0264-6021:3540501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang J, Stieglitz KA, Cardia JP, Kantrowitz ER. Structural basis for ordered substrate binding and cooperativity in aspartate transcarbamoylase. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(25):8881–8886. doi: 10.1073/pnas.0503742102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vitali J, Colaneri MJ, Kantrowitz E. Crystal structure of the catalytic trimer of Methanococcus jannaschii aspartate transcarbamoylase. Proteins. 2008;71(3):1324–1334. doi: 10.1002/prot.21667. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.