

Abstract
Optimized DNA vectors were constructed comprising the proteome of SIV including the structural, enzymatic, regulatory, and accessory proteins. In addition to native antigens as produced by the virus, fusion proteins and modified antigens with altered secretion, cellular localization and stability characteristics were generated. The DNA vectors were tested for expression upon transfection in human cells. In addition, the vectors were tested either alone or in combinations in mice and macaques, which provided an opportunity to compare immune responses in two animal models. DNA only immunization using intramuscular injection in the absence or presence of in vivo electroporation did not alter the phenotype of the induced T cell responses in mice. Although several fusion proteins induced immune responses to all the components of a polyprotein, we noted fusion proteins that abrogated immune response to some of the components. Since the expression levels of such fusion proteins were not affected, these data suggest that the immune recognition of certain components was altered by the fusion. Testing different DNA vectors in mice and macaques revealed that a combination of DNAs producing different forms of the same antigen generated more balanced immune responses, a desirable feature for an optimal AIDS vaccine.
Keywords: DNA vaccine, electroporation, immune response, mice, macaque, SIV, HIV, antibody, cellular immune responses
1. Introduction
DNA vaccination has great potential due to its safety, versatility, and scalability. DNA vaccines can be administered repeatedly without generating immunity against the vector itself. DNA vaccines for veterinary applications are in use, and several human candidate DNA vaccines are in clinical trials [1]. A key problem in using HIV DNA vaccines has been the relative low immunogenicity in humans [1-5]. This has been in part a result of low dose of antigen due to suboptimal DNA plasmids and to poor DNA delivery methods. A significant advance in the field was the application of expression-optimized SIV/HIV antigens, which led to great increases in expression and immunogenicity of the encoded proteins [6-12]. The codon changes introduced in these genes alter the stability and transport of the encoded mRNA resulting in increased expression [6-9]. In addition to the use in DNA vaccines, RNA-optimized genes are necessary for the efficient expression of antigens in viral vectors such as Adenovirus and Herpes-based vectors (for reviews see [13-16]), which use the nuclear machinery of the infected cells for virus expression. In contrast, poxvirus vectors (MVA, ALVAC), which do not encounter nuclear events and are strictly limited to the cytoplasm (for review see [17-19]), are less sensitive to RNA optimization for increased gene expression. Due to the great enhancement of expression achieved by optimized vectors, this technology is considered critical for the generation of effective recombinant vaccines, as well as optimizing the results of gene transfer.
Another step advancing the use of DNA as vaccine modality has been the introduction of in vivo electroporation (EP) as DNA delivery method. Studies by several groups, including ours, have demonstrated that DNA delivery by in vivo electroporation enhances uptake and immunogenicity of DNA vaccines [20-32].
The power and ease of DNA manipulation has allowed the generation of a number of variant antigens, and the evaluation of such antigens for the selection of optimal vaccines requires rapid and efficient techniques. Here we describe procedures for the sequential evaluation and selection of optimal vectors in vitro and in vivo using the mouse and macaque animal models. The expression vectors were first evaluated in transient transfection experiments to characterize the stability and secretion of the produced SIV antigens. Selected vectors were then used in in vivo studies. We found that combinations of DNAs producing different forms of SIV antigens induce more balanced cellular and humoral immune responses both in mice and macaques.
2. Materials and methods
2.1. Plasmids
We designed RNA/codon-optimized SIV genes by removing RNA processing and instability sequences in the mRNA via multiple codon replacements without altering the encoded protein according to our previously described methodology [6-9]. The genes were chemically synthesized, confirmed by nucleotide sequencing (GeneArt Inc, Regensburg, Germany), and cloned into the eukaryotic expression vector pCMV.kan [33] between the human CMV promoter and the bovine growth hormone polyadenylation signal. No splice sites were included in the vector and predicted splice sites (score >0.4) were eliminated from all coding sequences by appropriate codon changes, to minimize the possibility of splicing. Several versions of optimized genes encoding the same protein were generated and analyzed.
The RNA-optimized SIVmac239 p57gag gene in plasmid 1S (also called gagDX [33]) has 85 nt changes of 1533 nt of gag (95% identity wild type gag gene), resulting in a small increase of gag GC-content (49.7% compared to 46.1% of wild type gag). Another p57gag gene (plasmid 206S) has 279 nucleotide changes (82% identity wild type gag gene) introduced throughout the gene, resulting in an increase in GC-content to 67.5%. p57gag/pVAX contains the gag gene from plasmid 206S including AUG and surrounding sequence inserted into the commercially available expression vector pVAX1 (Invitrogen, Carlsbad, CA). Plasmid 10S produces the non-myristoylated form of p57gag. The p39gag (plasmid 208S) spans aa 1-364. Fusions of human MCP-3 to p57gag (plasmid 214S) and to p39gag (plasmid 209S) and the fusion of the murine MCP-3 to p39gag (plasmid 213S) were generated. The fusion of a protein-destabilizing b-catenin (CATE) peptide to p57gag (plasmid 2S) has been described [33].
The RNA-optimized SIVmac239 env genes in plasmid 64S has 552 nt of the 2640 nt changed (79% sequence identity to the wild type env gene). The env gene in plasmid 99S has an additional 181 nt changes. Both env genes have similar GC-content of 61.7% and 61.6%, respectively, which is higher than the wild type env (43.1%). Additional expression vectors derived from plasmid 64S having Env protein sequence modifications were produced: The Env signal peptide (aa 1-22) was replaced by the human MCP-3 (plasmid 73S), the murine MCP-3 (plasmid 115S) or the human tissue-type plasminogen activator (tPA) signal sequence (plasmid 78S).
An inactive SIVmac239 Pol protein (plasmid 88S) was generated similar to the previously reported inactive HIV-1 NL4-3 Pol (plasmid 133H) [29]. The following mutations were introduced into SIV Pol: deletion 26-DTG-28 in Protease [34], 183-YMDD-186 in Reverse Transcriptase [35], 473E in RNaseH [36], and D64, D113 and E150 in Integrase [37]. The following Pol protein modifications were generated: fusion to LAMP (plasmid 103S) using pLAMP/CMV.kan 6L and fusions to the murine or human MCP-3. The following signal peptides of the murine MCP3-Pol fusion protein were used: murine GM-CSF (plasmid 195S), murine IP-10 (plasmid 196S), native MCP-3 (plasmid 197S), IgE (plasmid 193S), IL-4 (plasmid 194S). The human MCP3-Pol fusion protein contains the murine IP-10 signal peptide (plasmid 216S).
Plasmid NefTatVif (NTV, plasmid 84S), generated by analogy to the previously reported HIV-1 Nef-Tat-Vif [29], expresses a fusion protein comprising the SIVmac239 Nef (aa 4-260), a GA-linker, Tat (aa 2-130), a GS-linker, and Vif (aa 2-213). The NefTat fusion (NT, plasmid 122S) was generated by deleting vif from NTV. NTV and NT were inserted into the LAMP/CMV.kan plasmid 7L, generating LAMP-NTV (plasmid 147S) and LAMP-NT (plasmid 148S), respectively. In SIV RevNefTat (RNT, plasmid 166S) the complete Rev (aa 1-107) was fused via a linker (SGAPGGS) to the N-terminus of NT.
The Pol-NTV protein (plasmid 35S) contains the inactive SIVmac239 Pol (from plasmid 88S) fused to NTV (from plasmid 84S). The b-catenin signal [33] was added at N-terminus of Pol (plasmid 44S). The GagPol polyprotein (plasmid 82S) lacks the Gag myristoylation signal, the frameshifting signal and the pseudoknot, as well as the 63 C-terminal aa of Gag. Gag (aa 1-438) was fused via an SGGAH-linker to the inactive pol gene and includes 12 aa preceding the PRT start. Fusion proteins containing Nef either at C-terminus (plasmid 179S) or N-terminus (plasmid 185S) were generated. The HIV Gag-Nef fusion protein (plasmid 113H) contains HXB2 Gag (aa 1-500) linked via an ASAAA linker to the Nef core (aa 65-150).
Several LAMP expression vectors (6L, 7L, 8L, 9L) were generated, containing the LAMP gene inserted into pCMV.kan [38] vector and differ in the multi-cloning sites inserted between the LAMP luminal domain and the C-terminal portion spanning the transmembrane domain and the cytoplasmic tail to facilitate in-frame cloning of the gene of interest.
2.2. In vitro expression of DNA plasmids
The expression levels of all the proteins were evaluated upon transient transfection in human 293 cells using the calcium phosphate coprecipitation technique. Briefly, 1×106 cells in complete DMEM plus 10% fetal bovine serum (FBS) were plated onto 60 mm tissue culture dishes and allowed to adhere overnight. The following day, the cells were transfected with a DNA mixture containing typically 100 ng of each SIV plasmid, 100 ng of the GFP expressing plasmid pFRED143 [39] as transfection control and bluescript DNA up to 7 mg. Forty-eight hours post transfection, culture supernatants (3 ml) and cells (1 ml extract) were harvested either in 0.5× RIPA or 1% Triton-Tris. Protein expression was analyzed by Western immunoblots. The proteins were resolved by electrophoresis on 10 or 12% sodium dodecyl sulfate polyacrylamide gels (SDS-PAGE) (NuPAGE, Invitrogen) and transferred onto nitrocellulose membranes. SIV Gag, Pol and Env were detected upon probing the membranes with pooled plasma from SIVmac251 infected Indian rhesus macaques (dilution 1:5000 [33]) followed by goat anti-monkey Horseradish Peroxidase-conjugated serum (dilution 1:10,000 Fitzgerald, Concord, MA). SIV Nef was detected using a mouse anti-Nef antibody (1:5,000 dilution of SIVmac251 Nef Monoclonal Antibody 17.2; Cat# 2659; National Institutes of Health, AIDS Research and Reference Reagent Program, Bethesda, MD), followed incubation with sheep anti-mouse IgG Horseradish Peroxidase (dilution 1:5,000; GE Healthcare, UK). The proteins were visualized by enhanced chemiluminescence (ECL plus Western blotting Detection System, GE HealthCare, Piscataway, NJ) and autography.
The production of SIV Gag was quantified using the SIV p27gag antigen capture assay (ZeptoMetrix Corporation, Buffalo, NY). Of note, the SIV p27gag antigen capture assay measures p57gag, p39gag and Gag fusion proteins with different efficiencies. Therefore, this assay does not allow precise comparisons of differently processed forms of Gag and was used to compare the levels of a given Gag protein in different compartments.
2.3. Immunogenicity in DNA vaccinated mice
The immunogenicity of the SIV antigens was evaluated in 6 to 8 weeks old female Balb/c mice obtained from Charles River Laboratories, Inc. (Frederick, MD). Mice were housed at the National Cancer Institute, Frederick, MD, in a temperature-controlled, light-cycled facility, and were cared for under the guidelines of the National Institutes of Health. For DNA vaccination, typically 100 μg of total DNA in 100 μl of PBS (Invitrogen) was injected intramuscularly with needle and syringe in the left and right quadriceps (50 μg/50 μl per dose). The mice were immunized at weeks 0 and 4 or at weeks 0, 3 and 6. Some mice were also immunized using electroporation as DNA delivery method using Elgen1000 pulse generator (Biomedical Corp., Blue Bell, PA USA). The splenocytes and blood were collected two weeks after the last vaccination. Cellular immune responses were evaluated either by ELISPOT or by intracellular cytokine staining (ICS), and plasma endpoint antibody titers for Gag and Env were measured by ELISA (Advanced BioScience Laboratories, Inc., Kensington, MD).
2.4. Immunogenicity in DNA vaccinated rhesus macaques
The Indian rhesus macaques (Macaca mulatta) were housed and handled in accordance with the standards of the Association for the Assessment and Accreditation of Laboratory Animal Care International. Naïve macaques (N=8) were immunized by intramuscular injection of 1 ml of a water solution containing 2 mg of a mixture of plasmids expressing the SIV antigens in either native or modified forms followed by in vivo electroporation (EP) using the Elgen device (Inovio Pharmaceuticals, Blue Bell, PA USA). These animals have been previously described [31]. Cellular immune responses in PBMC were measured by intracellular cytokine staining. Humoral immune responses (endpoint titers) in plasma were measured by ELISA (Advanced Bioscience Laboratory, Kensington, MD).
Another group of macaques (N=4), infected with SIVmac251 for 1.8 years, was subjected to anti-retroviral treatment (ART) to control viremia for a period of 22 weeks. The ART protocol [40] was modified to the following regimen: 20 mg/kg ((R)-9-(2-phosphonylmethoxypropyl)adenine (PMPA), and 50 mg/kg FTC, both injected subcutaneously once daily, and 5 mg/kg Didanosine (ddI), injected intravenously once daily. After 19 weeks, ddI was discontinued, and 100 mg integrase inhibitor was given orally twice per day (Integrase inhibitor L-000870812, Merck). At week 6 and 14 after ART start, the animals were vaccinated intramuscularly with 1 mg (700 ul in water) of a mixture of plasmids expressing the combination of both native and modified antigens, followed by in vivo electroporation (EP) using the Elgen device (Inovio Pharmaceuticals, Blue Bell, PA USA). Cellular immune responses were measured at the day of EP1 and 2 weeks post EP2.
2.5. Analysis of cellular immune responses by ELISPOT assay
ELISPOT assays were performed using the mouse IFNg ELISPOT Antibody Pair (Millipore, Bedford, MA) following the manufacturer's instruction with minor modifications (BD Biosciences, San Diego, CA). Briefly, splenocytes were seeded in triplicate at 4 × 105/well in RPMI 1640 containing 10% FCS alone or with SIV-specific peptide pools (1 μg/ml for each peptide). The SIV peptide pools used in this assay consist of 15-mer peptides with an overlap of 11 amino acids (Infinity Inc. Biotech Research and Resource, Aston, PA) spanning the complete Gag (105 peptides), Env (211 peptides), Pol (262 peptides), Nef (63 peptides), Tat (29 peptides), Vif (50 peptides), and Rev (23 peptides) proteins. The plates were incubated for 18 hrs at 37°C in 5% CO2 and subsequently the splenocytes were treated with ice-cold water, and washed. Biotinylated rabbit polyclonal anti-IFN-g (BD Biosciences) at 0.5 μg/ml in PBS containing 10% FBS was added to the plates and incubated for 2 hrs at room temperature, washed, and incubated for 1 hr at room temperature with streptavidin-alkaline phosphatase (Southern Biotechnology, Birmingham, AL) in PBS containing 5% FBS and 0.002% Tween. Following washes, the plates were developed with one-step Nitro Blue Tetrazolium/5-bromo-4-chloro-3-indolylphospate (KPL, Gaithersburg, MD). The spots were counted on a C.T.L. ELISPOT reader (Cellular Technology Ltd., BD Biosciences, San Diego, CA) and analyzed using ImmunoSpot software version 2.06. The cut-off was defined as the average spots obtained in the negative control wells (triplicates) plus 2 standard deviations. Specific spots to a given peptide pool were calculated by subtracting the cut-off value and adjusted to the number of spot-forming cells per million splenocytes.
2.6. Flow cytometry
To determine the number and phenotype of SIV-specific T cells, isolated mouse splenocytes or macaque PBMC were incubated at a density of 2×106 cells/ml and 1.5×106 cells/ml, respectively, in the presence of SIV peptide pools (15-mer, 11 aa overlap; 1ug/ml each peptide) and monensin to prevent cytokine secretion. Immunostaining and flow cytometric analysis of rhesus PBMC was performed as described [32, 33, 40]. Immunostaining and flow cytometric analysis of mouse splenocytes was performed using the following antibody cocktail for cell surface staining: CD3-allophycocyanin-Cy7, CD4-PerCP, CD8-PECy7 (BD Biosciences). The cells were washed twice with PBS containing 0.5% FBS and fixed and permeabilized with the Cytofix/Cytoperm kit (BD Biosciences). Intracellular cytokine staining was performed using IFN-g-FITC antibody (BD Biosciences). After intracellular staining, the cells were washed twice with perm/wash buffer (BD Biosciences) and the samples were analyzed on LSRII flow cytometer (BD Biosciences). Data analysis was performed using the FlowJo platform (Tree Star, Inc., Ashland, OR) and all antigen specific responses were reported after subtracting values obtained from incubating samples with medium alone.
2.7. Humoral immune responses
Humoral immune responses to SIV Gag, Env, Nef in vaccinated mice and macaques and to HIV Gag in mice were assessed by ELISA. Serial 4-fold dilutions of the plasma samples were assessed and the optical absorbance at 450 nm was determined (Advanced BioScience Laboratory, Kensington, MD). The binding titers are reported as the reciprocal of the highest dilution scoring positive having a value higher than the average plus 3 standard deviations obtained with control plasma from naïve mice or macaques.
Semi-quantitative assessment of humoral immune responses to SIV Nef, Rev, Tat, and Pol in vaccinated macaques was performed by Western immunoblots. Cell extracts (16 μl of 1 ml) of human 293 cells transfected with 1 μg of the respective expression vectors were separated on 12% SDS-PAGE (NuPAGE Bis-Tris, Invitrogen) and probed with plasma samples (1:200 dilution) of individual immunized macaques from a previously described study [31].
3. Results
3.1. Comparison of SIV gag expression vectors
High level expression of the myristoylated SIV p57gag protein has been obtained upon expression optimization (RNA or codon optimization) of the SIVmac239 gag gene as we reported previously [33, 41, 42] (Fig. 1A). In the present work, we used several vectors to compare expression and immunogenicity in mice. In plasmid gagDX (plasmid 1S) a limited amount of nucleotide changes (85 nt of the 1533 nt spanning gag gene; see Materials and methods) were introduced in the wild type gag gene. These changes targeted predicted RNA processing and instability sequences in the areas shown experimentally to decrease RNA stability and expression. Another SIV p57gag expression vector (plasmid 206S) has more nucleotide changes throughout the gag gene (354 nt; see Materials and methods). Despite the substantial RNA sequence changes and significant higher GC-content of the gag gene in plasmid 206S, we noted only a small ∼1.5× increase in Gag production compared to the levels obtained by the first generation optimized plasmid 1S (Fig. 1A). These findings are in agreement with our previously reported RNA-optimization of HIV-1 gag [7-9], where we identified that limited changes within the HIV gag sequence were sufficient to overcome the effect of the negative-acting RNA sequences present in the wild type transcript, which are responsible for the poor expression in the absence of the viral posttranscriptional Rev regulation.
Fig. 1.
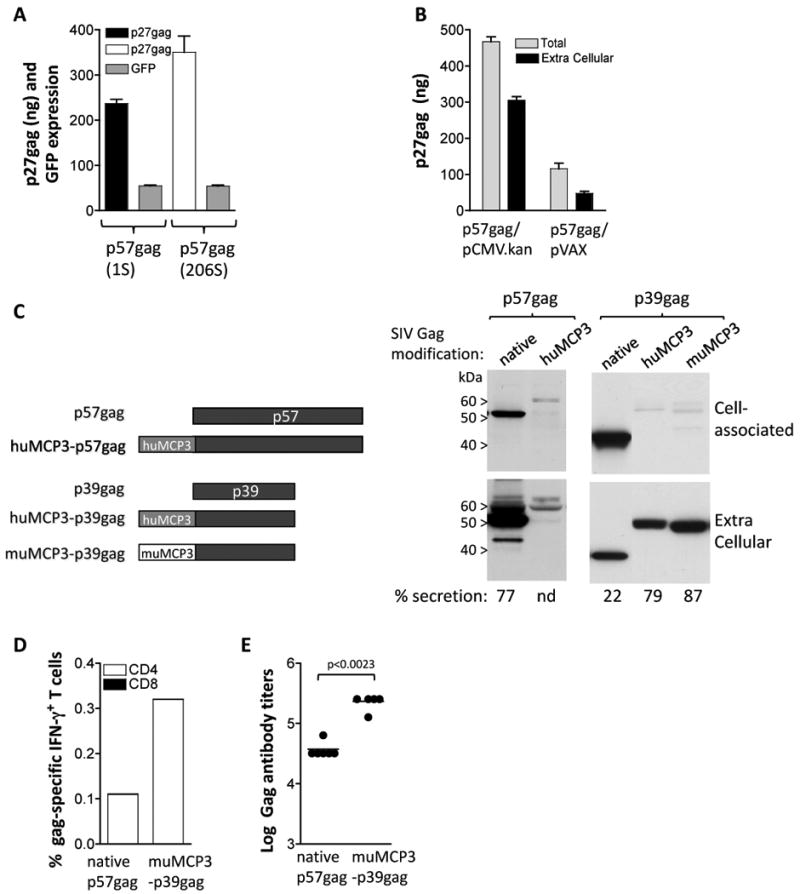
Optimization of SIV Gag expression and immunogenicity in mice. (A) Human 293 cells were transfected in triplicate with 50 ng of gag expression plasmids 1S and 206S and 100 ng of the GFP expression plasmid FRED143. Two days later the cells were harvested and the Gag and GFP levels were measured. Total p27gag (cell-associated and supernatant) and relative GFP values (×100) and SEM are shown. Expression of p57gag from plasmid 206S resulted in 1.5× higher Gag production compared to the levels obtained by plasmid 1S. (B) Comparison of expression of p57gag from the CMV.kan (plasmid 206S) and pVAX plasmids. The two plasmids contain the same p57gag gene including the optimized AUG initiator sequence. HEK 293 cells were transfected with 50 ng of the indicated plasmids and 2 days later Gag expression was measured by antigen capture assay. The mean and SD of triplicate transfection are shown. The mean GFP values (×100) obtained from the cotransfected pFRED143 plasmid are 657 and 740 for p57gag/CMV.kan and p57gag/pVAX, respectively. (C) The cartoon depicts the native p57gag and p39gag and modifications thereof. The fusion proteins comprise the human MCP-3 and p57gag or the p39gag; the murine MCP-3 and p39gag. Plasmids expressing the different forms of Gag were transfected into human 293 cells. The expression vectors producing the native p57gag (plasmid 1S), the fusion of MCP-3 with p57gag (plasmid 214S), p39gag (plasmid 208S), the fusions of human MCP-3 (plasmid 209S) and the murine MCP-3 (plasmid 213S) with p39gag are shown. Human 293 cells were transfected with the p57gag plasmid series or the p39gag plasmid series, respectively, in different experiments. Two days later, the cells were harvested. The Gag proteins from the supernatants (1/250 of sample) and the cell extracts (1/250 of sample) were separated on SDS-PAGE and were visualized on Western immunoblot using pooled antiserum from SIVmac251 infected macaques. Transfection efficiencies were controlled by co-transfection of GFP encoding plasmid pFRED143. The relative GFP values (×100) were 260 and 150 for the p57 plasmid series, and 220, 170 and 160 for the p39 plasmid series respectively. (D) Groups of Balb/c mice (N=6) were immunized with DNA plasmids producing the native p57gag (plasmid 1S) or the murine MCP3-p39gag (plasmid 171S) at day 0 and week 4 and were sacrificed 2 weeks later. Gag-specific cellular immune responses were analyzed in pooled splenocytes by flow cytometric analysis. (E) The endpoint titers of the Gag-specific binding antibody from the mice that received the plasmids described in panel B were determined by ELISA from individual serial 4-fold diluted plasma samples. Statistical analysis was performed using two-tailed Mann Whitney t test.
The RNA-optimized coding sequences were inserted into the vector pCMV.kan comprising the human CMV promoter, an optimal surrounding of the AUG initiator codon (derived from HIV-1 tat) to prevent translation initiation from internal AUGs [43], the bovine growth hormone (BGH) polyadenylation site, the kanamycin resistance gene and a plasmid backbone optimized for growth in bacteria [33]. Expression of SIV p57gag from pCMV.kan was compared to that using the commercially available plasmid backbone (pVAX1, Invitrogen), used in many DNA vaccine projects. Side-by-side comparison of the two vectors carrying the same gag gene (from plasmid 206S) showed ∼5× higher expression using pCMV.kan plasmid backbone (Fig. 1B). We conclude that optimization of both, the cDNA and the plasmid backbone, are important to obtain the most efficiently expressed plasmids.
Expression of the p57gag plasmid showed that ∼80% of Gag is found in the extracellular compartment (Fig. 1C), mostly in immature particles as expected (data not shown). In an effort to improve immunogenicity of Gag, we replaced the myristoylation signal with the complete MCP-3 chemokine region (MCP3) anticipating the attraction of antigen presenting cells by this fusion protein (Fig. 1C). Of note, we have generated fusions to both the murine and the human MCP-3 with similar results for in vitro expression, and we used the matching chemokine in mouse and macaque studies, respectively. Fusion of MCP-3 to p57gag generated an unstable protein that accumulated poorly. For this reason, we tested the fusion of MCP-3 to p39gag, which comprises the p19gag matrix and p27gag capsid parts of Gag. The p39gag protein did not form particles and was poorly secreted (∼20%) compared to p57gag (secretion of ∼80%). The MCP3-p39gag fusion protein was efficiently produced and was found primarily in the extracellular compartment (∼80-90% of the fusion protein) as reported previously [33]. Side-by-side comparison of the p57gag and the p39gag plasmid showed similar levels of expression by the Western immunoblot assay (data not shown). We also reported that fusion to a b-catenin (CATE) peptide providing a strong ubiquination signal renders the protein intracellular [33]. In addition to the fusion with SIVgag (plasmid 2S), the b-catenin sequence has been used to generate several SIV fusion proteins (see below).
We tested the immunogenicity of the different forms of SIV Gag (Fig. 1 D-E) upon injection of the respective DNAs in mice using needle and syringe as delivery method. Cellular immune responses were evaluated in pooled splenocytes using intracellular cytokine staining. Fig. 1D shows that vaccination with the plasmid expressing MCP3-p39gag induced higher cellular immune responses compared to the native p57gag antigen. Both forms of Gag induced almost exclusively antigen-specific CD4+ T cells. Mice vaccinated by the CATE-p57gag showed similar levels of gag-specific CD4+ T cell responses like those obtained by vaccination with the plasmid expressing the native gag (data not shown). We also tested the immunogenicity of MCP3-gag using in vivo electroporation (EP) as DNA delivery method in mice. The Gag-specific cellular immune responses were also dominated by CD4+ T cells using the EP DNA delivery method (data not shown). Similar to the mouse data, we have reported that SIV p57Gag induces primarily CD4 responses in macaques [28, 31, 32, 38].
Humoral immune responses were determined by endpoint ELISA measurements from individual vaccinated mice (Fig. 1E). The MCP3-p39gag fusion protein induced significantly higher levels of binding antibodies than the native p57gag. It is postulated that the fusion protein targets antigen-presenting cells promoting induction of increased immune responses. Thus, modification of Gag affected both localization and immunogenicity of the protein, and resulted in increased antibody response.
3.2. Comparison of Env expression vectors
Using a similar approach as outlined above for Gag, we generated expression plasmids producing different modified forms of SIV Env proteins (Fig. 2). Two variants of RNA-optimized env genes (plasmid 64S and 99S) showed similar levels of expression in human 293 cells (Fig. 2A, lanes 1 and 2) and both plasmids were used in animal studies. Plasmids producing Env having the native signal peptide replaced by the tPA signal peptide (lane 4) or having Env being fused to the human (lane 3) or murine (lane 5) MCP-3 chemokine were also generated. High levels of Env were produced from all the env expression plasmids (lanes 1 to 5), although we noted that the MCP3-env levels were reproducibly slightly lower. Analyzing the extracellular levels of Env, we noted that fusion to MCP-3 delayed gp120-gp41 processing (lanes 3 and 5), and that the presence of the human tissue-type plasminogen activator (tPA) signal peptide (lane 4) also delayed processing but to a lesser extent. It is likely that the structure and intracellular trafficking of these hybrid Env proteins is altered by the N-terminal modifications, affecting the processing of the precursor gp160. The HIV-1 env 6101 (clade B) expression plasmids producing the native and modified forms were previously described [44]. Similarly, we found that upon fusion of the tPA signal peptide to HIVenv 6101 less gp120 was produced and that the MCP-3 env fusion showed very poor gp120 production (data not shown), in agreement with the SIV Env results (see above). From the constructs tested, we conclude that Env is processed best when it contains its native leader peptide.
Fig. 2.
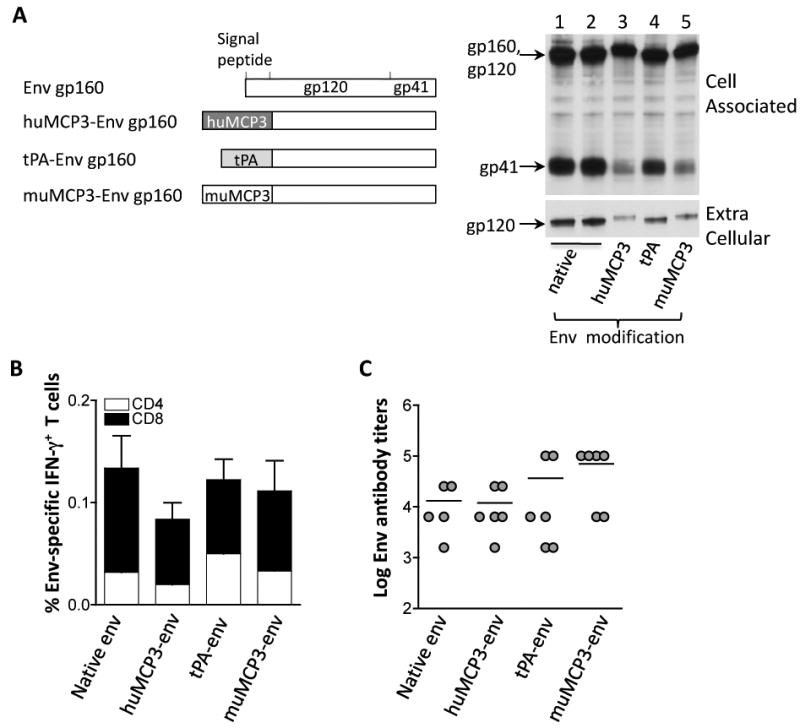
Immunogenicity of native and modified SIV Env proteins in mice. (A) The cartoon depicts the native gp160env as well as the modified gp160env proteins which have the native signal peptide replaced by the human or murine MCP-3 chemokine or the tPA signal peptide. Human 293 cells were transfected with 100 ng of the plasmids expressing the native gp160env (plasmid 64S, lane 1; plasmid 99S, lane 2); the modified gp160env proteins with the native signal peptide replaced by the human (plasmid 73S, lane 3) or murine MCP-3 (plasmid 115S, lane 5) chemokine or the tPA signal peptide (plasmid 78S; lane 4). Two days later, the cells were harvested and 1/250 of the supernatants and cell extracts, respectively, were visualized by Western immunoblot analysis using pooled antiserum from SIVmac251 infected macaques. Transfection efficiency was controlled by co-transfection of GFP encoding plasmid pFRED143. The relative GFP values (× 100) obtained from lanes 1-5 were: 30, 40, 110, 100 and 50, respectively. (B) Immunogenicity of Env produced from the indicated plasmids was assessed. Groups of Balb/C mice (N=5-6) received the plasmids expressing the native (plasmid 64S), the human MCP3-env (plasmid 73S) or murine MCP3-env (plasmid 115S) or the tPA-env (plasmid 78S). The mice were vaccinated 3 times (day 0, week 3 and week 6) and were sacrificed 2 weeks later. Env-specific cellular immune responses were analyzed from splenocytes of individual mice and provided as mean and standard error (SEM) of IFN-γ-producing CD4+ and CD8+T cells. (C) The endpoint titers of the Env-specific binding antibody from the individual mice described in panel B were determined by ELISA from serial 4-fold dilutions of individual plasma samples. The mean titers are indicated.
Immune responses to the different Env proteins were evaluated upon intramuscular injection of mice with the various plasmids. Analysis of cellular immune responses showed the induction of overall similar levels of Env-specific T cell responses (Fig. 2B). We previously reported that different forms of HIV env 6101 (native, fusion to tPA or MCP3) induced similar levels of cellular responses [44], corroborating our finding of the SIV Env described here. All forms of SIV Env induced both CD4+ and CD8+ IFN-g-producing T cells, with predominant CD8 responses. In agreement with the results shown above, mice receiving the Env plasmids via in vivo electroporation also showed predominant Env-specific CD8+ T cell responses (data not shown).
Analysis of humoral immune responses showed potent responses induced by the different Env proteins, especially by the muMCP3-env fusion protein (Fig. 2C). Despite the lower levels of extracellular Env produced from the MCP3-env fusion protein, we found a trend for higher levels of anti-Env antibody responses.
3.3. Comparison of immunogenicity upon Gag and Env co-expression in mice and macaques
Next, we tested DNA vaccine mixtures that consisted of plasmids expressing the antigens in either their native form (Gag, Env) or the modified forms (CATE-Gag, MCP3-Gag, MCP3-Env) or a combination of plasmids expressing the native Env and modified forms of Gag antigens in Balb/c mice (Fig. 3A-B). We compared the immune response in mice to that obtained in rhesus macaques immunized with the same plasmids (Fig. 3C-E). Analysis of the splenocytes from immunized mice (Fig. 3A) showed that the group receiving the plasmids expressing the native antigens induced higher responses to Env, whereas the group receiving the plasmids expressing the modified antigens showed predominant Gag responses. Interestingly, the combination of native Env and modified Gag antigens showed the most balanced responses to both Gag and Env (Fig. 3A). The humoral immune responses reflected the cellular responses. The native Env and the modified Gag showed higher responses and the combination of these native and modified antigens induced more balanced responses (Fig. 3B). We noted also that coimmunization of plasmids expressing the native Gag and Env results in proportionally lower humoral immune responses to Gag whereas the responses to Env are increased (compare to Fig. 1E and 2C). This data suggest that Env might form a better immunogen when associated with Gag in virus like-particles, which mimic the functional Env complex on the surface of virions. In contrast, the Gag protein may be less accessible as part of such particles.
Fig. 3.

Immunogenicity of different mixtures of plasmids expressing Gag and Env in mice and macaques. (A) Groups of Balb/c mice (N=6) were immunized with a mixture of plasmids (50 μg each) expressing the native forms of p57gag (plasmid 1S) and gp160env (plasmid 99S) or a mixture of plasmids expressing the modified forms of Gag (25 μg of the CATE-p57gag [plasmid 2S] and 25 μg MCP3-p39gag [plasmid 21S]) and 50 μg MCP3-env (plasmid 73S). A third group of mice received a combination of the DNA plasmids (“combined antigens”) expressing the modified Gag (25 μg of each gag plasmid 2S and 21S) and native gp160env (50 μg of the env plasmid 99S). The mice were immunized at day 0 and week 4 and were sacrificed 2 weeks later. Gag- and Env-specific cellular immune responses were measured by ELISPOT in splenocytes from individual mice. The mean and SEM are shown. (B) Gag- and Env-specific endpoint titers of binding antibodies from pooled plasma samples from the mice shown in panel A were determined by ELISA and the log of antibody titers are shown and the mean is shown. (C) Indian rhesus macaques (N=8) were vaccinated with a mixture of plasmids expressing native or modified SIV antigens using IM delivery followed by in vivo electroporation. The data of the group receiving the native and modified forms of the antigens are modified from Rosati et al. [31] and show the data from week 4 post EP4. The mean and SEM are shown. (D) A group SIV-infected and ART-treated macaques (N=4) received a mixture of DNAs expressing thecombination of the native and modified antigens using IM delivery followed by in vivo electroporation. The cellular responses (mean and SEM) from the time of EP1 and 2 weeks post EP2 are shown. (E) Endpoint Env and Gag antibody titers were determined by ELISA from the animals (shown in panel C) by ELISA using individual plasma of macaques immunized with DNA expressing the native (left panel) and modified (right panel) antigens. Mean and SEM are shown.
We further used these plasmid mixtures (described in Fig. 3A & B) to immunize Indian rhesus macaques using intramuscular DNA injection followed by in vivo electroporation (Fig. 3C-E). Figure 3C shows that immunization with plasmids expressing the native antigens induced predominant immune responses to Env, whereas the modified antigens induced predominant Gag responses (data modified from [33]). Accordingly, we also found that the humoral immune responses parallel the cellular responses in that the native Env and the modified Gag are most potent (Fig. 3E). We used a combination of DNAs producing the native Env and modified Gag antigens in another group of macaques (Fig. 3D). These animals were previously infected by SIVmac251 but control viremia due to multi-component ART treatment (see Material and methods). Vaccination of these animals using the DNA mixture expressing a combination of the antigens also showed induction of more balanced cellular immune responses. It is noteworthy, that the testing of these DNA vaccine mixtures in mice and macaques led to similar conclusions and showed that the mouse model provided an excellent guide to identify the most immunogenic vaccine mixture. In conclusion, the mixture of plasmids expressing native Env and modified SIV Gag induced balanced cellular and humoral immune responses, and is proposed for further studies.
3.4. Generation of Pol expression vectors
We also generated and studied a series of pol expression vectors. Pol was produced from an RNA-optimized gene that contains several known [34-37] inactivating mutations in protease, reverse transcriptase, RNAseH and integrase (Fig. 4A). Modifications of the SIV Pol protein were generated to test whether expression and antigenicity could be further improved. First, we inserted pol into LAMP, generating a LAMP-Pol fusion protein (Fig. 4A). Transient transfection experiments showed that the LAMP-Pol fusion protein (lane 2) is more stable than Pol alone (lane 1) resulting in increased protein accumulation at steady-state. Similarly, we found that fusion of inactive HIV Pol to LAMP stabilized Pol (data not shown). We further generated a series Pol fusion proteins that contain the murine MCP-3 (Fig 4A). In contrast to MCP3-gag, the muMCP3-pol was not secreted (not shown). We tested several signal peptides i.e. murine GM-CSF (lane 3), murine IP-10 (lane 4) as well as the IgE, IL-4 or the native MCP-3 signal (data not shown), which did not further improve accumulation or localization of Pol. Similar levels of Pol were produced upon fusion to the human MCP-3 chemokine with the murine IP-10 signal peptide (lane 5).
Fig. 4.
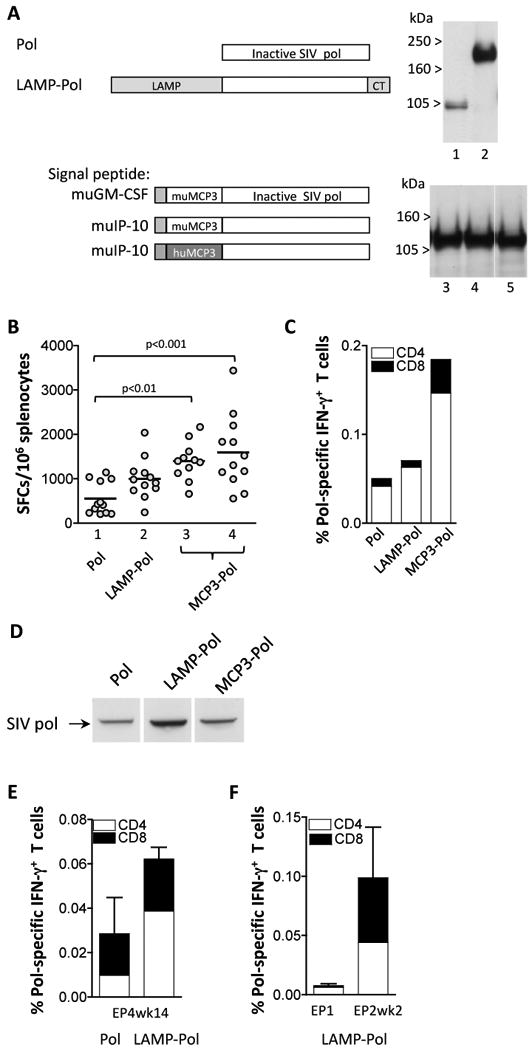
Increased immunogenicity by the modified SIV-pol. (A) The cartoon depicts the Pol protein that contains mutations inactivating the enzymatic activities in protease, reverse transcriptase, RNaseH, and integrase (plasmid 88S, lane 1). Fusions of Pol with LAMP (plasmid 103S, lane 2) or with murine MCP-3 that has the murine GM-CSF (plasmid 195S, lane 3) or murine IP-10 (plasmid 196S, lane 4) signal peptide are shown. The fusion of Pol to human MCP-3 with the murine IP-10 signal peptide is shown (plasmid 216S, lane 5). Human 293 cells were cotransfected with 100 ng of the indicated plasmids and 100 ng of GFP expressing plasmid, pFRED143. Gag protein expression was analyzed 2 days later by Western immunoblot using pooled plasma from SIVmac251 infected macaques. Data shown in lanes 1 and 2 and in lanes 3-5 are from two independent experiments. The relative GFP values (×100) were: lanes 1 and 2: 340, 220; lanes 3-5: 140, 160, and 140. (B) Balb/c mice (N=12) were immunized with different pol expression plasmids at day 0 and week 4 and sacrificed 2 weeks later. Splenocytes of individual animals were analyzed by ELIspot assay. Lane 1, pol (plasmid 88S); lane 2, LAMP-pol (plasmid 103S); lane3, muMCP3-pol with the murine GM-CSF leader (plasmid 195S); lane 4, muMCP3-pol with the murine IP-10 signal peptide (plasmid 196S). Statistical analysis was performed using one-way ANOVA, Dunn's Multiple comparison test. The mean values are indicated. (C) Pooled splenocytes were analyzed by flow cytometric analysis and the levels of Pol-specific IFN-g+ CD4+ and CD8+ T cells were measured. (D) Pooled plasma from the mice immunized with Pol (panel B, lane 1), LAMP-pol (panel B, lane 2) and muMCP3-pol (panel B, lane 4) were used in a Western immunoblot assay to detect Pol proteins produced upon transient transfection of human 293 cells with 1 μg of the Pol plasmid (plasmid 88S). Two days later cell extracts were harvested in 0.5X RIPA and loaded on to a 12% NuPAGE Bis-Tris gel. The blot was probed using a 1:200 dilution of pooled plasma samples from the 3 vaccinated groups of mice. (E) The rhesus macaques described in Fig. 3C were coimmunized with a plasmid expressing the native or the modified Pol proteins as indicated. The immune responses determined by flow cytometry were measured at 14 weeks post EP4. The percent of pol-specific IFN-g producing CD4+ and CD8+ T cells are shown (mean and SEM). (F) The rhesus macaques described in Fig. 3D were co-immunized with the plasmid expressing LAMP-Pol. The immune responses determined by flow cytometry were measured at day of EP1 and 2 weeks post EP2. The percent of pol-specific IFN-g producing CD4+ and CD8+ T cells are shown (mean and SEM) as described for panel E.
The immunogenicity of selected plasmids was tested in Balb/c mice after 2 vaccinations via the IM route. Fig. 4B shows significant higher levels of cellular immune responses in mice vaccinated with plasmid expressing the MCP3-Pol proteins, whereas the LAMP-Pol showed a trend of higher responses. The Pol responses induced by either of the Pol proteins were mainly mediated by CD4+ T cells (Fig. 4C). Pooled plasma samples from the immunized mice were analyzed for the presence of anti-Pol antibody. Fig. 4D shows that all three plasma samples contained anti-Pol antibodies as determined by Western immunoblot assay with the following ranking: LAMP-pol>MCP3-Pol>Pol.
We further compared the immunogenicity of Pol and LAMP-Pol upon vaccination of naïve rhesus macaques using the respective plasmids delivered via IM using electroporation (modified from[31]). Fig. 4E shows higher responses in the group of vaccinated macaques that received the plasmid producing LAMP-Pol. The induced cellular responses were mediated by both CD4+ and CD8+ T cells. The LAMP-Pol plasmid was also shown to induce high levels of recall responses upon DNA immunization of ART-treated SIV-infected rhesus macaques (Fig. 4F).
We analyzed the plasma from individual vaccinated macaques (from Fig. 4E) for the presence of anti-Pol antibody by the Western immunoblot assays (as described for Fig. 4D). We found that 0/6 animals from the group that received the native Pol and 2/8 animals that received the LAMP-Pol showed anti-Pol reactivity. Therefore, modification of Pol led to increased expression and immunogenicity, as also observed for Gag, in both mice and macaques.
3.5. Generation of expression vectors combining accessory proteins
To produce accessory proteins efficiently, we generated several DNA plasmids expressing fusion proteins consisting of Rev, Tat and Nef or of Nef, Tat and Vif (Fig. 5). Fig. 5A shows that both Rev-Nef-Tat (RNT) and Rev-Tat-Nef (RTN) as well as a CATE-RNT fusion proteins are well expressed. The immunogenicity study in mice showed induction of overall similar levels of cellular immune responses by these fusion proteins (Fig. 5B) as well as similar levels of responses to the individual antigens.
Fig. 5.
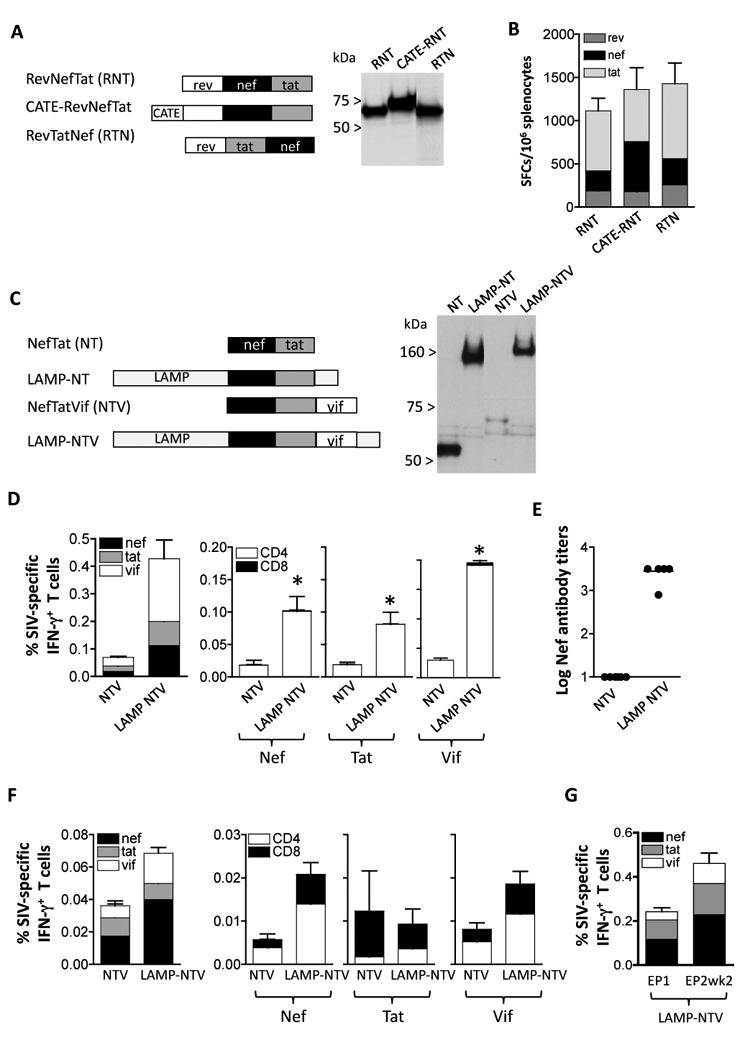
Immunogenicity to accessory proteins. (A) Cartoon depicts fusion proteins consisting of Rev (aa 1-107), Nef (aa 4-260) and Tat (aa 2-130). Two configurations changing the position of Nef (plasmid RevNefTat [RNT, 166S] and RevTatNef [RTN, plasmid 168S]; and the b-Catenin-RevNefTat (CATE-RNT; RNT [plasmid 174S] are shown. Expression of the proteins from 293 cells transfected with 100 ng of the respective DNAs was visualized on Western immunoblots probed with anti-Nef antibody. The relative GFP relative values (×100) of lanes 1-3 were: 300, 270, and 300, respectively. (B) Immunogenicity in Balb/c mice upon IM vaccination using plasmids expressing the fusion proteins shown in panel A. Mice were immunized at day 0 and week 4 with 100 μg of the indicated plasmids and sacrificed 2 weeks later. The levels of Rev-, Nef- and Tat-specific cellular immune responses were determined by ELISPOT assay. The mean and SEM are shown. (C) Cartoon depicts fusions of NefTat with LAMP and Vif Expression of the proteins from 293 cells transfected with 200 ng of DNA was visualized on Western immunoblots probed with anti-Nef antibody. The relative GFP values (×100) of the transfection in lanes 1-4: 240, 250, 270, and 260, respectively. (D) Immunogenicity of the NTV and LAMP-NTV fusion proteins shown in panel C. Balb/C mice (N=6) were immunized at day 0 week 3 and week 6 with 100 μg of the indicated plasmids and sacrificed 2 weeks later. Cellular immune responses to Nef, Tat and Vif were determined from splenocytes from individual mice (left panel) and antigen-specific CD4+ and CD8+ T cells were measured (right panels). Statistical analysis was performed using two-tailed Mann Whitney t test, with p values of 0.043 (Nef), 0.043 (Tat) and 0.0022 (Vif). The mean and SEM are shown. (E) The humoral immune responses to Nef were analyzed from plasma samples from individual mice (right panel) as endpoint ELISA titers. The mean is shown. (F) Immunogenicity of NTV and LAMP-NTV in rhesus macaques. Naïve macaques (see Figure 3C) were vaccinated with NTV and LAMP-NTV plasmids as described [31]. The levels of antigen-specific IFN-g producing total (left panel) and CD4+ and CD8+ T cells (right panels) were measured by flow cytometry at 14 weeks post EP4. Mean and SEM are shown. (G) The ART-treated macaques were co-immunized with the LAMP-pol expression vector. The immune responses determined by flow cytometry were measured at the day of EP1 and 2 weeks post EP2. The percent of antigen-specific IFN- g producing CD4+ and CD8+ T cells are shown (Mean and SEM).
We generated a second series of plasmids consisting of a Nef-Tat fusion protein having Rev replaced by Vif (Fig. 5C). We noted that inclusion of Vif in the Nef-Tat fusion led to great reduction in Nef-Tat-Vif levels. Vif has been reported to be an unstable protein and we confirmed that Vif is subject to degradation through the proteasome [46]. To counterbalance the destabilizing effect of Vif, we generate a LAMP-NTV fusion protein. Transient transfection experiments confirmed that the LAMP-NTV levels were significantly higher than those produced by the NTV plasmid and are similar to those obtained by LAMP-NT plasmid.
The immunogenicity of plasmids expressing NTV and LAMP-NTV was tested in Balb/c mice. Fig. 5D shows that LAMP-NTV fusion protein induced significantly higher levels of cellular immune responses than NTV. Both NTV and the LAMP-NTV induced primarily CD4 mediated responses to all the components Nef, Tat and Vif Plasma of the vaccinated mice were monitored for anti-Nef binding antibodies by ELISA. We found significantly higher anti-Nef antibody responses in mice vaccinated with LAMP-NTV expressing plasmid (Fig. 5E).
Analysis of macaques vaccinated by in vivo electroporation with plasmids producing NTV and LAMP-NTV (Fig. 5F) showed higher levels of cellular responses induced by LAMP-NTV (modified from [31]). Analysis of the individual antigens showed that responses to Nef and Vif were mediated mainly by CD4+ T cells, although significant levels of CD8+ T cell responses were present (Fig. 5F). The Tat responses were overall low and mediated predominantly by CD8+ T cells. Upon DNA immunization of ART-treated SIV-infected rhesus macaques using the LAMP-NTV DNA plasmid, substantial recall responses to all the components Nef, Tat and Vif were detected (Fig. 5G). The plasma samples of the NTV and LAMP-NTV DNA-vaccinated macaques (from Fig. 5F) were also screened for the presence of anti-Nef, -Vif and -Tat antibodies by Western immunoblots. Evaluation of the responses in the group receiving the native versus the modified antigens (positive animals/total macaques) revealed: 0/6 versus 2/8 for anti-Vif responses; 2/6 versus 6/8 for anti-Tat responses; 4/6 versus 8/8 for anti-Nef responses. Therefore, the LAMP-NTV fusion is a more potent vaccine plasmid, inducing higher cellular and humoral responses in macaques, similar to the results obtained in mice.
3.6. Nef immunogenicity is affected by partners in fusion proteins
We further explored the Nef immunogenicity using Pol (Fig. 6A) or GagPol (Fig. 6C) fusion proteins that contain the NTV fusion or Nef alone. Fig. 6A shows that Pol-NTV or CATE-pol-NTV plasmids were well expressed. Despite the high level of expression, the analysis of the immunogenicity (Fig. 6B) of these fusion proteins from vaccinated Balb/c mice showed only responses to Pol but not to Nef (or Tat and Vif).
Fig. 6.
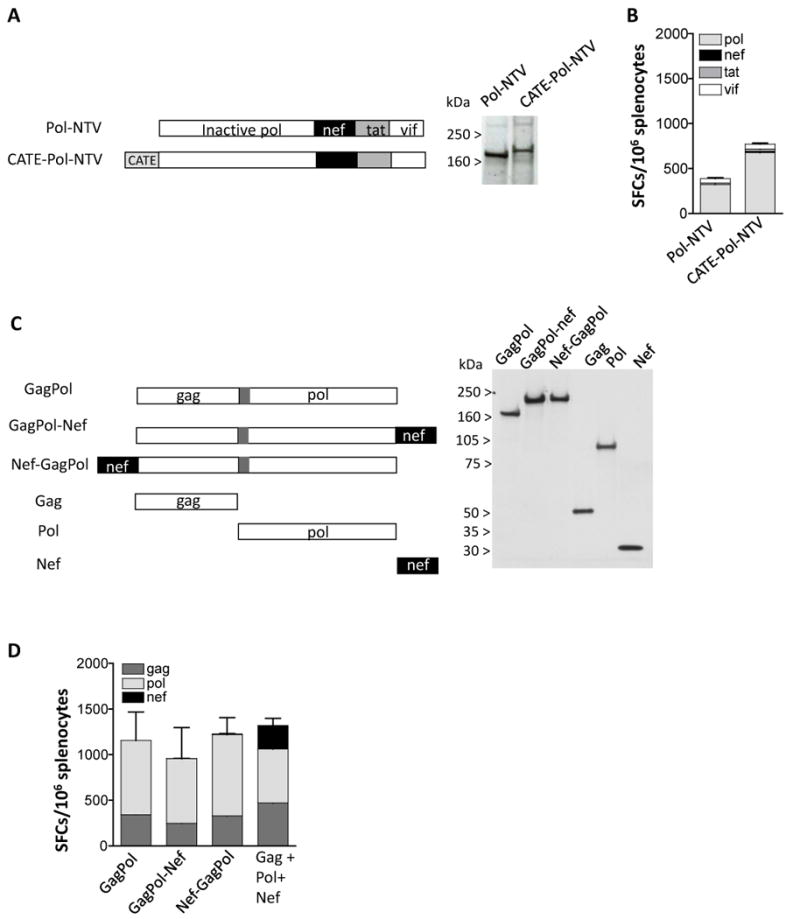
Loss of Nef Immunogenicity when Nef is expressed as part of GagPol or Pol fusion proteins. (A) The Pol-NTV (plasmid 35S) and the CATE-Pol-NTV (plasmid 44S) fusion plasmids were expressed in human 293 cells upon transfection of 100 ng of DNA. Two days later the cell extracts were analyzed on Western immunoblot using pooled plasma from SIVmac251 infected macaques. The relative GFP relative values (× 100) shown in lanes 1-2 were 570 and 570, respectively. (B) The immunogenicity of the pol-NTV fusion proteins. Balb/c mice (N=6) were vaccinated twice with 100 μg of DNA at day 0 and week 4, and splenocytes were prepared 2 weeks later. Cellular immune responses to pol, Nef, Tat, and Vif peptide pools were analyzed by ELISPOT. The mean and SEM are shown. (C) The GagPol fusion protein contains Gag (aa 1-439) artificially linked (via 13 aa of the ORF preceding Pol) to Pol (aa 1-951) (plasmid 82S). Nef was fused to the C-term (plasmid 179S) or the N-term (plasmid 185S) of GagPol. The plasmids expressing the fusion proteins were expressed in human 293 cells and analyzed on Western immunoblot probed with pooled plasma from SIVmac251 infected macaques. Plasmids expressing only Gag (plasmid 10S; non-myristoylated gag), Pol (plasmid 88S) and Nef (plasmid 180S) are shown. The relative GFP values (× 100) of the transfections shown in lanes 1-6 were: 140, 150, 150, 150, 150 and 140. (D) The immunogenicity of the indicated GagPol fusion proteins was tested. Balb/c mice (N=6) were vaccinated with 100 μg twice at day 0 and week 4, and splenocytes were prepared 2 weeks later. Cellular immune responses to Gag, Pol, and Nef peptide pools were analyzed by ELISPOT. The mean and SEM are shown.
Figure 6C shows that the addition of Nef either at the C terminus or at the N-terminus of the GagPol fusion protein did not affect the expression level of the fusion proteins. Interestingly, analysis of mice vaccinated with these plasmids showed responses only to Gag and Pol, but not to Nef (Fig. 6D). Vaccination of mice in the same study with a mixture of 3 plasmids producing Gag, Pol and Nef separately showed responses to all antigens (Fig. 6D). Analysis of immunogenicity of plasmids producing HIV-1 Gag alone or a Gag-Nef fusion protein in mice also showed similar levels of immune responses to Gag but a total lack of immune responses to Nef (data not shown). Thus, we conclude that careful evaluation of the immunogenicity of all the components of a polyprotein needs to be performed. The combination of Nef with either SIV Pol, SIV Gag-pol or HIV-1 Gag abolished cellular responses to Nef. In contrast, Nef is able to induce immune responses when combined with Tat and Vif or Rev and Tat as shown in Fig. 5. It is possible that the inclusion of a proteolytic cleavage site could improve the Nef immunogenicity but this concept has not been tested. Thus, careful evaluation of antigens when presented in combination as fusion proteins is critical not only for levels of expression but importantly for immunogenicity of the individual components.
4. Discussion
Optimal immunogen selection is essential for the development of an effective AIDS vaccine. Poor immunogens may not induce potent responses or may generate responses by diverting the immune system to focus on decoy or highly variable regions [47-52]. Model systems to assess immunogens in a comprehensive manner will be important to accelerate vaccine design. DNA is an important future vaccine modality and also a great tool for the development of new immunogen designs through the power of genetic engineering. Rapid development of variant immunogens and testing using a DNA platform yields rapid results. It is also important to assess the predictability of DNA vaccine results in model systems. The mouse model for testing DNA vaccine immunogenicity has not been used extensively for SIV/HIV antigens because these genes are poorly expressed in mice. The development of RNA/codon optimized expression vectors allowed for efficient expression in mice, and opened the possibility to study the same vectors in comparative studies in mice and macaques. The improved delivery technology by in vivo electroporation resulted in high and consistent results in both systems and allows for comparison of the same vectors giving strong immune responses in both systems. A limitation of DNA vaccination in non-human primates compared to mice is that macaques receive a lower vaccine dose per body weight. The lower levels of total circulating antigens in macaques could explain the slower rate of development of immune responses after DNA vaccination. In particular, typically 3-4 vaccinations are necessary to induce high levels of immune responses in macaques, whereas 2 vaccinations are sufficient in mice.
In this report, we have used comparisons in mice and macaques to rank our immunogens in both systems in an effort to predict immunogen performance before testing in humans. Our analysis suggests that several vectors rank similarly in terms of cellular immune responses. In addition, several vectors gave similar biases for generation of CD4 or CD8 responses, i.e. SIV Gag induces CD4 responses and SIV Env induces primarily CD8 responses. Similarly, in DNA vaccinated macaques, we reported the induction of predominant Gag-specific CD4 responses [31, 32]) and Env-specific CD8 responses [31] in blood.
Several of the SIV plasmids expressing the native antigens have been used in DNA-only vaccination studies using either needle and syringe or in vivo electroporation [29, 31] [41, 42, 53-55] or in prime-boost vaccination studies [45, 56, 57]. In an effort to increase the immunogenicity of SIV and HIV antigens when provided as DNA vaccines, we tested several modified version of each antigen, which affected secretion, stability, or intracellular trafficking of the antigen in a predictable manner. As expected from previous results [28, 31-33]), we found that several modifications had strong effects on the immune response generated by a given antigen. The MCP3-gag, the LAMP-pol and LAMP-NTV fusions proteins in mice (this report) and in a comparative study in macaques [31] showed greater induction of immune responses than the native antigens. We also found that combinations of Gag modified to enter the secretory pathway (MCP3-Gag) and particle-forming native Gag showed broader immune responses (Fig. 3) both in mice and macaques. We further reported that macaques immunized by in vivo electroporation using plasmids expressing modified antigens developed long-lasting (> 2 years) cellular and humoral immune responses and furthermore that these immune responses were disseminated to the mucosal tissues [32], and we attribute this to both the improved immunogens and the improved DNA delivery via in vivo electroporation.
Many vaccine projects involve the generation of artificial antigens fusing two or more pathogen proteins or fragments of proteins [58-60]. Other approaches involve the selective inclusion of regions from several proteins that are predicted to be highly immunogenic [47-52]. It is generally assumed that the immune system will recognize such protein portions in a manner similar to the full protein, but this depends on complex steps of antigen processing. It is therefore prudent to study antigen processing and immunogenicity when generating artificial proteins or fusions. For CD8 responses restricted by MHC class I, great progress has been made in understanding the rules allowing antigen processing and presentation [61-64]. Examining several fusion vectors including the Nef protein, we found, unexpectedly, that Nef immunogenicity was suppressed by the fusions to Pol, Gag-Pol and Gag. This did not depend on the position of Nef within the fusion, or due to fusion protein instability, because the effect was selective for Nef. Further work is needed to identify the specific Nef responses suppressed and the differences in processing that preclude induction of Nef responses. These results demonstrate the importance of appropriate antigen design and testing for maximal effects.
In conclusion, to generate a practical method of vaccination able to induce potent, broad, and durable cellular and humoral responses, we tested combination of vectors expressing different modifications of the same antigen. It is interesting that in both mice and macaques the results support the notion that the combination of different forms of the antigens gave broader immune responses than the individual forms alone, which supports the idea to combine vectors for optimal results. Thus, antigen expression levels alone while being critical may not sufficient to induce the best immune responses. In summary, we demonstrated that in both mice and macaques, optimization of expression together with modifications of antigens provide an additional improvement over the powerful in vivo electroporation as delivery method to obtain high levels of long-lasting cellular and humoral immune responses.
Acknowledgments
We are grateful to D. Weiss, J. Treece, I. Kalisz, V. Kalyanaraman, S. Orndorff, P. Markham and staff at Advanced BioScience Laboratories, Inc., Kensington, for their expert help. We thank the AIDS Research and Reagent Program (NIH) for antibodies, K. Nagashima for electron microscopy, D. Hazuda (Merck) for the integrase inhibitor, J. Bear for technical assistance, and T. Jones for editorial assistance. This research was supported by the Intramural Research Program of NIH, National Cancer Institute, Center for Cancer Research. Part of this work was supported by the grant RR-00169 from the National Center for Research Resources, NIH, to the California National Primate Research Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kutzler MA, Weiner DB. DNA vaccines: ready for prime time? Nat Rev Genet. 2008;9:776–88. doi: 10.1038/nrg2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vasan S, Schlesinger SJ, Huang Y, Hurley A, Lombardo A, Chen Z, et al. Phase 1 safety and immunogenicity evaluation of ADVAX, a multigenic, DNA-based clade C/B′ HIV-1 candidate vaccine. PLoS One. 2010;5:e8617. doi: 10.1371/journal.pone.0008617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Catanzaro AT, Roederer M, Koup RA, Bailer RT, Enama ME, Nason MC, et al. Phase I clinical evaluation of a six-plasmid multiclade HIV-1 DNA candidate vaccine. Vaccine. 2007 May 16;25:4085–92. doi: 10.1016/j.vaccine.2007.02.050. [DOI] [PubMed] [Google Scholar]
- 4.Tavel JA, Martin JE, Kelly GG, Enama ME, Shen JM, Gomez PL, et al. Safety and immunogenicity of a Gag-Pol candidate HIV-1 DNA vaccine administered by a needle-free device in HIV-1-seronegative subjects. J Acquir Immune Defic Syndr. 2007;44:601–5. doi: 10.1097/QAI.0b013e3180417cb6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.MacGregor RR, Boyer JD, Ugen KE, Lacy KE, Gluckman SJ, Bagarazzi ML, et al. First human trial of a DNA-based vaccine for treatment of human immunodeficiency virus type 1 infection: safety and host response. J Infect Dis. 1998;178:92–100. doi: 10.1086/515613. [DOI] [PubMed] [Google Scholar]
- 6.Nasioulas G, Zolotukhin AS, Tabernero C, Solomin L, Cunningham CP, Pavlakis GN, et al. Elements distinct from human immunodeficiency virus type 1 splice sites are responsible for the Rev dependence of env mRNA. J Virol. 1994;68:2986–93. doi: 10.1128/jvi.68.5.2986-2993.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schneider R, Campbell M, Nasioulas G, Felber BK, Pavlakis GN. Inactivation of the human immunodeficiency virus type 1 inhibitory elements allows Rev-independent expression of Gag and Gag/protease and particle formation. J Virol. 1997;71:4892–903. doi: 10.1128/jvi.71.7.4892-4903.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwartz S, Campbell M, Nasioulas G, Harrison J, Felber BK, Pavlakis GN. Mutational inactivation of an inhibitory sequence in human immunodeficiency virus type 1 results in Rev-independent gag expression. J Virol. 1992;66:7176–82. doi: 10.1128/jvi.66.12.7176-7182.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwartz S, Felber BK, Pavlakis GN. Distinct RNA sequences in the gag region of human immunodeficiency virus type 1 decrease RNA stability and inhibit expression in the absence of Rev protein. J Virol. 1992;66:150–9. doi: 10.1128/jvi.66.1.150-159.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andre S, Seed B, Eberle J, Schraut W, Bultmann A, Haas J. Increased immune response elicited by DNA vaccination with a synthetic gp120 sequence with optimized codon usage. J Virol. 1998;72:1497–503. doi: 10.1128/jvi.72.2.1497-1503.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wagner R, Graf M, Bieler K, Wolf H, Grunwald T, Foley P, et al. Rev-independent expression of synthetic gag-pol genes of human immunodeficiency virus type 1 and simian immunodeficiency virus: implications for the safety of lentiviral vectors. Hum Gene Ther. 2000;11:2403–13. doi: 10.1089/104303400750038507. [DOI] [PubMed] [Google Scholar]
- 12.Graf M, Deml L, Wagner R. Codon-optimized genes that enable increased heterologous expression in mammalian cells and elicit efficient immune responses in mice after vaccination of naked DNA. Methods Mol Med. 2004;94:197–210. doi: 10.1385/1-59259-679-7:197. [DOI] [PubMed] [Google Scholar]
- 13.Taylor TJ, Brockman MA, McNamee EE, Knipe DM. Herpes simplex virus. Front Biosci. 2002;7:d752–64. doi: 10.2741/taylor. [DOI] [PubMed] [Google Scholar]
- 14.Dudek T, Knipe DM. Replication-defective viruses as vaccines and vaccine vectors. Virology. 2006;344:230–9. doi: 10.1016/j.virol.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 15.Roizman B, Sears AE. Herpes simplex viruses and their replication. In: Fields BN, Knipe DM, Chanock RM, Hirsch MS, Melnick JL, Monath TP, Roizman B, editors. Virology. Second. New York: Raven Press; 1990. pp. 1795–842. [Google Scholar]
- 16.Horwitz MS. Adenoviridae and their replication. In: Fields BN, Knipe DM, Chanock RM, Hirsch MS, Melnick JL, Monath TP, Roizman B, editors. Virology. Second. New York: Raven Press; 1990. pp. 1679–722. [Google Scholar]
- 17.Moss B, Carroll MW, Wyatt LS, Bennink JR, Hirsch VM, Goldstein S, et al. Host range restricted, non-replicating vaccinia virus vectors as vaccine candidates. Adv Exp Med Biol. 1996;397:7–13. doi: 10.1007/978-1-4899-1382-1_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Franchini G, Gurunathan S, Baglyos L, Plotkin S, Tartaglia J. Poxvirus-based vaccine candidates for HIV: two decades of experience with special emphasis on canarypox vectors. Expert Rev Vaccines. 2004;3:S75–88. doi: 10.1586/14760584.3.4.s75. [DOI] [PubMed] [Google Scholar]
- 19.Moss B. Poxviridae and their replication. In: Fields BN, Knipe DM, Chanock RM, Hirsch MS, Melnick JL, Monath TP, Roizman B, editors. Virology. New York: Raven Press; 1990. pp. 2079–112. [Google Scholar]
- 20.Aihara H, Miyazaki J. Gene transfer into muscle by electroporation in vivo. Nat Biotechnol. 1998;16:867–70. doi: 10.1038/nbt0998-867. [DOI] [PubMed] [Google Scholar]
- 21.Mathiesen I. Electropermeabilization of skeletal muscle enhances gene transfer in vivo. Gene Ther. 1999;6:508–14. doi: 10.1038/sj.gt.3300847. [DOI] [PubMed] [Google Scholar]
- 22.Prud'homme GJ, Glinka Y, Khan AS, Draghia-Akli R. Electroporation-enhanced nonviral gene transfer for the prevention or treatment of immunological, endocrine and neoplastic diseases. Curr Gene Ther. 2006;6:243–73. doi: 10.2174/156652306776359504. [DOI] [PubMed] [Google Scholar]
- 23.Rizzuto G, Cappelletti M, Maione D, Savino R, Lazzaro D, Costa P, et al. Efficient and regulated erythropoietin production by naked DNA injection and muscle electroporation. Proc Natl Acad Sci U S A. 1999;96:6417–22. doi: 10.1073/pnas.96.11.6417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Z, Troilo PJ, Wang X, Griffiths TG, Pacchione SJ, Barnum AB, et al. Detection of integration of plasmid DNA into host genomic DNA following intramuscular injection and electroporation. Gene Ther. 2004;11:711–21. doi: 10.1038/sj.gt.3302213. [DOI] [PubMed] [Google Scholar]
- 25.Widera G, Austin M, Rabussay D, Goldbeck C, Barnett SW, Chen M, et al. Increased DNA vaccine delivery and immunogenicity by electroporation in vivo. J Immunol. 2000;164:4635–40. doi: 10.4049/jimmunol.164.9.4635. [DOI] [PubMed] [Google Scholar]
- 26.Otten G, Schaefer M, Doe B, Liu H, Srivastava I, zur Megede J, et al. Enhancement of DNA vaccine potency in rhesus macaques by electroporation. Vaccine. 2004;22:2489–93. doi: 10.1016/j.vaccine.2003.11.073. [DOI] [PubMed] [Google Scholar]
- 27.Otten GR, Schaefer M, Doe B, Liu H, Megede JZ, Donnelly J, et al. Potent immunogenicity of an HIV-1 gag-pol fusion DNA vaccine delivered by in vivo electroporation. Vaccine. 2006;24:4503–9. doi: 10.1016/j.vaccine.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 28.Rosati M, Valentin A, Jalah R, Patel V, von Gegerfelt A, Bergamaschi C, et al. Increased immune responses in rhesus macaques by DNA vaccination combined with electroporation. Vaccine. 2008;26:5223–9. doi: 10.1016/j.vaccine.2008.03.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luckay A, Sidhu MK, Kjeken R, Megati S, Chong SY, Roopchand V, et al. Effect of plasmid DNA vaccine design and in vivo electroporation on the resulting vaccine-specific immune responses in rhesus macaques. J Virol. 2007;81:5257–69. doi: 10.1128/JVI.00055-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hirao LA, Wu L, Khan AS, Hokey DA, Yan J, Dai A, et al. Combined effects of IL-12 and electroporation enhances the potency of DNA vaccination in macaques. Vaccine. 2008;26:3112–20. doi: 10.1016/j.vaccine.2008.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosati M, Bergamaschi C, Valentin A, Kulkarni V, Jalah R, Patel V, et al. DNA vaccination in rhesus macaques induces potent immune responses and decreases acute and chronic viremia after SIVmac251 challenge. Proc Natl Acad Sci U S A. 2009;06:15831–6. doi: 10.1073/pnas.0902628106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patel V, Valentin A, Kulkarni V, Rosati M, Bergamaschi C, Jalah R, et al. Induction of Mucosal SIV-Specific Immune Responses Upon Intramuscular In Vivo Electroporation Of Optimized DNA Vaccines In Rhesus Macaques. Vaccine. 2010;28:4827–36. [Google Scholar]
- 33.Rosati M, von Gegerfelt A, Roth P, Alicea C, Valentin A, Robert-Guroff M, et al. DNA vaccines expressing different forms of simian immunodeficiency virus antigens decrease viremia upon SIVmac251 challenge. J Virol. 2005;79:8480–92. doi: 10.1128/JVI.79.13.8480-8492.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Loeb DD, Swanstrom R, Everitt L, Manchester M, Stamper SE, Hutchison CA., 3rd Complete mutagenesis of the HIV-1 protease. Nature. 1989;340:397–400. doi: 10.1038/340397a0. [DOI] [PubMed] [Google Scholar]
- 35.Larder BA, Purifoy DJ, Powell KL, Darby G. Site-specific mutagenesis of AIDS virus reverse transcriptase. Nature. 1987;327:716–7. doi: 10.1038/327716a0. [DOI] [PubMed] [Google Scholar]
- 36.Schatz O, Cromme FV, Gruninger-Leitch F, Le Grice SF. Point mutations in conserved amino acid residues within the C-terminal domain of HIV-1 reverse transcriptase specifically repress RNase H function. FEBS Lett. 1989;257:311–4. doi: 10.1016/0014-5793(89)81559-5. [DOI] [PubMed] [Google Scholar]
- 37.Leavitt AD, Shiue L, Varmus HE. Site-directed mutagenesis of HIV-1 integrase demonstrates differential effects on integrase functions in vitro. J Biol Chem. 1993;268:2113–9. [PubMed] [Google Scholar]
- 38.Valentin A, Chikhlikar P, Patel V, Rosati M, Maciel M, Chang KH, et al. Comparison of DNA vaccines producing HIV-1 Gag and LAMP/Gag chimera in rhesus macaques reveals antigen-specific T-cell responses with distinct phenotypes. Vaccine. 2009;27:4840–9. doi: 10.1016/j.vaccine.2009.05.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stauber RH, Horie K, Carney P, Hudson EA, Tarasova NI, Gaitanaris GA, et al. Development and applications of enhanced green fluorescent protein mutants. Biotechniques. 1998;24:462–6. 8–71. doi: 10.2144/98243rr01. [DOI] [PubMed] [Google Scholar]
- 40.Valentin A, von Gegerfelt A, Rosati M, Miteloudis G, Alicea C, Bergamaschi C, et al. Repeated DNA Therapeutic Vaccination of Chronically SIV-Infected Macaques Provides Additional Virological Benefit. Vaccine. 2010;28:1962–74. doi: 10.1016/j.vaccine.2009.10.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schadeck EB, Sidhu M, Egan MA, Chong SY, Piacente P, Masood A, et al. Plasmid encoded IL-12 functions as a DNA vaccine adjuvant and augments SIVgag-specific cell-mediated and humoral immune responses in Rhesus macaques. Vaccine. 2006;24:4677–87. doi: 10.1016/j.vaccine.2005.10.035. [DOI] [PubMed] [Google Scholar]
- 42.Schadeck EB, Sidhu M, Egan MA, Chong SY, Piacente P, Masood A, et al. A dose sparing effect by plasmid encoded IL-12 adjuvant on a SIVgag-plasmid DNA vaccine in rhesus macaques. Vaccine. 2006;24:4677–87. doi: 10.1016/j.vaccine.2005.10.035. [DOI] [PubMed] [Google Scholar]
- 43.Schwartz S, Felber BK, Pavlakis GN. Mechanism of translation of monocistronic and multicistronic human immunodeficiency virus type 1 mRNAs. Mol Cell Biol. 1992;12:207–19. doi: 10.1128/mcb.12.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Megati S, Garcia-Hand D, Cappello S, Roopchand V, Masood A, Xu R, et al. Modifying the HIV-1 env gp160 gene to improve pDNA vaccine-elicited cell-mediated immune responses. Vaccine. 2008;26:5083–94. doi: 10.1016/j.vaccine.2008.03.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaur A, Sanford HB, Garry D, Lang S, Klumpp SA, Watanabe D, et al. Ability of herpes simplex virus vectors to boost immune responses to DNA vectors and to protect against challenge by simian immunodeficiency virus. Virology. 2007;357:199–214. doi: 10.1016/j.virol.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fujita M, Akari H, Sakurai A, Yoshida A, Chiba T, Tanaka K, et al. Expression of HIV-1 accessory protein Vif is controlled uniquely to be low and optimal by proteasome degradation. Microbes Infect. 2004;6:791–8. doi: 10.1016/j.micinf.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 47.Barouch DH, O'Brien KL, Simmons NL, King SL, Abbink P, Maxfield LF, et al. Mosaic HIV-1 vaccines expand the breadth and depth of cellular immune responses in rhesus monkeys. Nat Med. 16:319–23. doi: 10.1038/nm.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fischer W, Liao HX, Haynes BF, Letvin NL, Korber B. Coping with viral diversity in HIV vaccine design: a response to Nickle et al. PLoS Comput Biol. 2008;4:e15. doi: 10.1371/journal.pcbi.0040015. author reply e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fischer W, Perkins S, Theiler J, Bhattacharya T, Yusim K, Funkhouser R, et al. Polyvalent vaccines for optimal coverage of potential T-cell epitopes in global HIV-1 variants. Nat Med. 2007;13:100–6. doi: 10.1038/nm1461. [DOI] [PubMed] [Google Scholar]
- 50.Santra S, Liao HX, Zhang R, Muldoon M, Watson S, Fischer W, et al. Mosaic vaccines elicit CD8+ T lymphocyte responses that confer enhanced immune coverage of diverse HIV strains in monkeys. Nat Med. 16:324–8. doi: 10.1038/nm.2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nickle DC, Jojic N, Heckerman D, Jojic V, Kirovski D, Rolland M, et al. Comparison of immunogen designs that optimize peptide coverage: reply to Fischer et al. PLoS Comput Biol. 2008;4:e25. doi: 10.1371/journal.pcbi.0040025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nickle DC, Rolland M, Jensen MA, Pond SL, Deng W, Seligman M, et al. Coping with viral diversity in HIV vaccine design. PLoS Comput Biol. 2007;3:e75. doi: 10.1371/journal.pcbi.0030075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boyer JD, Robinson TM, Kutzler MA, Parkinson R, Calarota SA, Sidhu MK, et al. SIV DNA vaccine co-administered with IL-12 expression plasmid enhances CD8 SIV cellular immune responses in cynomolgus macaques. J Med Primatol. 2005;34:262–70. doi: 10.1111/j.1600-0684.2005.00124.x. [DOI] [PubMed] [Google Scholar]
- 54.Boyer JD, Robinson TM, Kutzler MA, Vansant G, Hokey DA, Kumar S, et al. Protection against simian/human immunodeficiency virus (SHIV) 89.6P in macaques after coimmunization with SHIV antigen and IL-15 plasmid. Proc Natl Acad Sci U S A. 2007;104:18648–53. doi: 10.1073/pnas.0709198104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Halwani R, Boyer JD, Yassine-Diab B, Haddad EK, Robinson TM, Kumar S, et al. Therapeutic vaccination with simian immunodeficiency virus (SIV)-DNA+IL-12 or IL-15 induces distinct CD8 memory subsets in SIV-infected macaques. J Immunol. 2008;180:7969–79. doi: 10.4049/jimmunol.180.12.7969. [DOI] [PubMed] [Google Scholar]
- 56.Hel Z, Nacsa J, Tryniszewska E, Tsai WP, Parks RW, Montefiori DC, et al. Containment of simian immunodeficiency virus infection in vaccinated macaques: correlation with the magnitude of virus-specific pre- and postchallenge CD4+ and CD8+ T cell responses. J Immunol. 2002;169:4778–87. doi: 10.4049/jimmunol.169.9.4778. [DOI] [PubMed] [Google Scholar]
- 57.Vaccari M, Boasso A, Ma ZM, Cecchinato V, Venzon D, Doster MN, et al. CD4+ T-cell loss and delayed expression of modulators of immune responses at mucosal sites of vaccinated macaques following SIV(mac251) infection. Mucosal Immunol. 2008;1:497–507. doi: 10.1038/mi.2008.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hanke T, McMichael A. Pre-clinical development of a multi-CTL epitope-based DNA prime MVA boost vaccine for AIDS. Immunol Lett. 1999;66:177–81. doi: 10.1016/s0165-2478(98)00164-3. [DOI] [PubMed] [Google Scholar]
- 59.Hanke T, Samuel RV, Blanchard TJ, Neumann VC, Allen TM, Boyson JE, et al. Effective induction of simian immunodeficiency virus-specific cytotoxic T lymphocytes in macaques by using a multiepitope gene and DNA prime-modified vaccinia virus Ankara boost vaccination regimen. J Virol. 1999;73:7524–32. doi: 10.1128/jvi.73.9.7524-7532.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McMichael A, Mwau M, Hanke T. Design and tests of an HIV vaccine. Br Med Bull. 2002;62:87–98. doi: 10.1093/bmb/62.1.87. [DOI] [PubMed] [Google Scholar]
- 61.Lazaro E, Godfrey SB, Stamegna P, Ogbechie T, Kerrigan C, Zhang M, et al. Differential HIV epitope processing in monocytes and CD4 T cells affects cytotoxic T lymphocyte recognition. J Infect Dis. 2009;200:236–43. doi: 10.1086/599837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Troyer RM, McNevin J, Liu Y, Zhang SC, Krizan RW, Abraha A, et al. Variable fitness impact of HIV-1 escape mutations to cytotoxic T lymphocyte (CTL) response. PLoS Pathog. 2009;5:e1000365. doi: 10.1371/journal.ppat.1000365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Le Gall S, Stamegna P, Walker BD. Portable flanking sequences modulate CTL epitope processing. J Clin Invest. 2007;117:3563–75. doi: 10.1172/JCI32047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tenzer S, Wee E, Burgevin A, Stewart-Jones G, Friis L, Lamberth K, et al. Antigen processing influences HIV-specific cytotoxic T lymphocyte immunodominance. Nat Immunol. 2009;10:636–46. doi: 10.1038/ni.1728. [DOI] [PubMed] [Google Scholar]