

Summary
As an organism that has evolved to live in environments ranging from soil to the cytosol of mammalian cells, Listeria monocytogenes must regulate the secretion and activity of protein products that promote survival within these habitats. The post-translocation chaperone PrsA2 has been adapted to assist in the folding and activity of L. monocytogenes secreted proteins required for bacterial replication within host cells. Here we present the first structure/function investigation of the contributions of PrsA2 to protein secretion and activity as well as to bacterial virulence. Domain swap experiments with the closely related L. monocytogenes PrsA1 protein combined with targeted mutagenesis indicate distinct functional roles for the PrsA2 peptidyl-prolyl isomerase (PPIase) and the N- and C-terminal domains in pathogenesis. In contrast to other PrsA-like proteins described thus far in the literature, an absolute in vivo requirement for PrsA2 PPIase activity is evident in mouse infection models. This work illustrates the diversity of function associated with L. monocytogenes PrsA2 that serves to promote bacterial life within the infected host.
Keywords: PrsA, peptidyl-prolyl isomerase (PPIase), chaperone, SurA, LLO
Introduction
Bacterial pathogens depend upon the activity of secreted protein products to establish infection within a susceptible host. Secreted virulence factors establish the first contact with host cells and aide in bacterial attachment, invasion, and/or subversion of host immune defenses. Bacteria have thus developed a variety of secretion systems and mechanisms to fold, stabilize, and deliver active proteins across the bacterial membrane. For Gram-negative bacteria, complex secretion machineries have evolved that facilitate transit of proteins across the double membrane and, in some cases, directly into host cells (Cascales, 2008; Desvaux et al., 2009; Donnenberg, 2000; Gerlach and Hensel, 2007; Marlovits and Stebbins, 2010). In Gram-positive bacteria, the majority of proteins are thought to be secreted in an unfolded state across the single bacterial cell membrane to enter the Gram-positive periplasmic space that exists between the membrane and the cell wall (Matias and Beveridge, 2005, 2006, 2008; Sarvas et al., 2004; Simonen and Palva, 1993; van Wely et al., 2001) The periplasm of Gram-positive bacteria presents a challenging environment for protein folding and function as a result of its characteristic high cation concentration, low pH, and high density of negative charge (Sarvas et al., 2004). Optimal protein folding, activity, and localization under such conditions likely requires dedicated protein chaperones as well as proteases to ensure secreted protein function and to maintain optimal secretion homeostasis.
Listeria monocytogenes is a Gram-positive bacterium that transitions between life in the outside environment and life within the cytosol of infected mammalian host cells (Dussurget et al., 2004; Freitag, 2006; Freitag et al., 2009; Scortti et al., 2007). L. monocytogenes adaptation to life within a mammalian host is accompanied by large increases in the number and amount of secreted proteins and by the regulated release of factors that facilitate intracellular survival (Mueller and Freitag, 2005; Port and Freitag, 2007; Shetron-Rama et al., 2003; Alonzo and Freitag, 2010; Port and Freitag, 2007). Many of the physiological changes that enable bacterial replication within the cytosol initiate at the transcriptional level, mediated primarily by activation of the virulence regulator PrfA, but also occur at the translational and post-translational levels (Desvaux et al., 2006; Desvaux and Hebraud, 2006; Desvaux et al., 2009; Desvaux et al., 2010; Freitag et al., 2009; Johansson et al., 2002; Loh et al., 2009). Secreted virulence factors such as the cytolysin listeriolysin O (LLO) and the broad specificity phospholipase PC-PLC, both of which mediate phagosomal membrane lysis, are produced at increased levels within the host but are sequestered in a folded and functional state at the bacterial surface until environmental conditions trigger their release (Geoffroy et al., 1987; Glomski et al., 2002; Marquis and Hager, 2000; Portnoy et al., 1992; Schnupf and Portnoy, 2007). Secreted virulence proteins are thus not only synthesized by L. monocytogenes in greater abundance during host cell infection but they may also be sequestered at the bacterial surface. Proper folding is required to prevent the accumulation of inactive proteins at the membrane-cell wall interface and the triggering of a membrane stress response; incorrectly folded proteins are rapidly degraded by quality-control proteases (Hyyrylainen et al., 2001; Hyyrylainen et al., 2005; Sarvas et al., 2004).
Recent studies have suggested that the ability of L. monocytogenes to regulate secreted protein stability, function, and localization during replication within host cells depends upon the activity of a chaperone known as PrsA2 (Alonzo and Freitag, 2010). PrsA2 is one of two secreted chaperones in L. monocytogenes predicted to function as a peptidyl-prolyl isomerase (PPIase) within the Gram-positive periplasm (Alonzo et al., 2009; Alonzo and Freitag, 2010; Zemansky et al., 2009). L. monocytogenes mutants lacking PrsA2 are severely attenuated for virulence such that bacterial burdens in the livers and spleens of infected animals are reduced by more than 100,000-fold; and the protein appears to be directly involved in maintaining secreted virulence factor stability and activity (Alonzo et al., 2009; Zemansky et al., 2009). Functioning in conjunction with the secreted heat shock protease/chaperone HtrA, PrsA2 has been hypothesized to play a critical role in countering secretion stress resulting from the activation of PrfA within the host cell cytosol and the subsequent increase in protein secretion (Alonzo and Freitag, 2010). While PrsA2 has been demonstrated to be essential for pathogenesis, no functional role has yet to be ascribed to the second highly similar L. monocytogenes secreted PPIase/chaperone PrsA1 (Alonzo et al., 2009; Alonzo and Freitag, 2010).
The PrsA protein of Bacillus subtilis shares a high degree of sequence similarity with PrsA2 and has been extensively studied (Hyyrylainen et al., 2001; Hyyrylainen et al., 2010; Jacobs et al., 1993; Kontinen et al., 1991; Kontinen and Sarvas, 1993; Vitikainen et al., 2001; Vitikainen et al., 2004; Vitikainen et al., 2005; Wahlstrom et al., 2003). In contrast to L. monocytogenes PrsA1 and PrsA2, B. subtilis PrsA is an essential protein that is directly involved in the proper folding of a diverse repertoire of secreted proteins (Kontinen et al., 1991; Kontinen and Sarvas, 1993; Vitikainen et al., 2005). Recently the essential role for PrsA in B. subtilis viability was associated with gross cell wall structural defects imparted by a loss of Penicillin Binding Protein (PBP) stability and/or activity upon PrsA depletion (Hyyrylainen et al., 2010). Both PrsA1 and PrsA2 of L. monocytogenes are anticipated to have the same fold as B. subtilis PrsA with helical N and C-terminal domains surrounding a central PPIase domain (Alonzo et al., 2009; Tossavainen et al., 2006; Vitikainen et al., 2004). The PPIase domain of B. subtilis PrsA has been demonstrated to be functional in vitro, and while the domain itself is indispensable, its enzymatic activity may be dispensable for protein function in vivo (Tossavainen et al., 2006; Vitikainen et al., 2004). In other Gram-positive organisms, including Lactococcus lactis (where the PrsA protein completely lacks PPIase activity), the PPIase domain is not required for chaperone activity (Drouault et al., 2002; Missiakas et al., 1996; Rouviere and Gross, 1996). Thus far, only limited information exists regarding structural and functional aspects of most PrsA-like proteins, despite their pivotal contributions to Gram-positive protein secretion.
In this study, we investigated the functional contributions of the PrsA2 N- and C-terminus and PPIase domains to protein secretion and activity as well as bacterial virulence. Our findings indicate distinct functional roles for the PPIase and the N and C-terminal domains that significantly impact L. monocytogenes pathogenesis. In contrast to PrsA-like proteins described thus far in the literature, we have identified an in vivo requirement for the PPIase domain of L. monocytogenes PrsA2 through the use of mouse infection models. This work illustrates the functional diversity of L. monocytogenes PrsA2 that enables bacterial life within the infected host.
Results
Predicted structural organization of L. monocytogenes PrsA2 and PrsA1
L. monocytogenes PrsA2 shares a significant degree of amino acid similarity with PrsA of Bacillus subtilis (45% identity, and 65% similarity) [Fig 1A and B and (Adler et al., 2009)]. B. subtilis PrsA is a chaperone comprised of N and C-terminal domains that are moderately conserved among other PrsA homologues, as well as a highly conserved central parvulin-type PPIase domain [Fig 1B and (Tossavainen et al., 2006; Vitikainen et al., 2004)]. Chemical cross-linking studies indicate that B. subtilis PrsA forms dimers and possibly multimers in solution (Hyyrylainen et al., 2010). The predicted modular organization of L. monocytogenes PrsA2 and PrsA1 is similar to that of B. subtilis PrsA with N and C-terminal domains flanking a central PPIase domain, and heat stable PrsA2 dimers are visible when the purified protein is subjected to SDS-PAGE with and without chemical crosslinking (Fig. 1C). The PPIase domains of the three proteins share identical putative active site residues (Fig. 1B).
Figure 1. L. monocytogenes PrsA2 domain organization and construction of PrsA1/PrsA2 domain swap mutants.
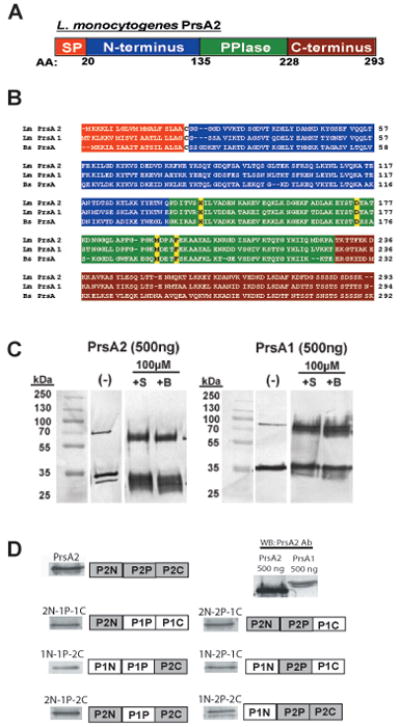
(A) Illustration of the predicted domain architecture of PrsA2. Included are an N-terminal signal peptide (orange), an N-terminal domain of 135 amino acids (blue), a central PPIase domain of 93 amino acids (green), and a C-terminal domain of 66 amino acids (red). (B) Chemical crosslinking of purified PrsA1 and PrsA2. 500ng of purified PrsA1 and PrsA2 proteins were chemically crosslinked with 10μM of sulfo-ethylene glycol bis[succinimidylsuccinate] (+S) or Bis[sulfosuccinimidyl] suberate (+B) for 1 hour at room-temperature followed by SDS-PAGE and western blotting for detection of PrsA dimers. All data is representative of at least three independent experiments. (−), no crosslinker added. (C) Amino acid sequence alignment of L. monocytogenes PrsA2 and PrsA1 and B. subtilis PrsA. Each domain is colored as in (A). The four amino acids known to be critical for PPIase activity in B. subtilis PrsA are highlighted in yellow. (D) PrsA1/PrsA2 chimeric domain swap mutants introduced into ΔprsA1 ΔprsA2 mutant strains using the site-specific integration vector pPL2. Each construct is under the expression control of the prsA2 promoter. The six constructs generated are shown along with respective Western blots using PrsA2 polyclonal antibody. Because all of the constructs generated contain a substantial portion of the PrsA1 protein, a Western blot was also performed on equivalent amounts (500 ng) of PrsA1 and PrsA2 using polyclonal PrsA2 antibody. Based on differences in antibody recognition of PrsA1 and PrsA2, approximately equivalent amounts of chimera proteins were expressed in L. monocytogenes (see also Supplementary Fig. S1).
Construction of PrsA1/PrsA2 domain swap mutants reveals specific functional contributions of the PrsA2 N and C-termini
Previous studies have indicated that PrsA1 has no apparent functional overlap with PrsA2 for virulence-associated activities (hemolytic activity, cell-to-cell spread, and phospholipase activity) or for bacterial virulence in mice despite sharing a high degree of amino acid sequence similarity (58% identity and 75% similarity) [(Alonzo et al., 2009; Alonzo and Freitag, 2010) and Fig. 1B]. To gain a better understanding of PrsA2 structure/function and to further explore the apparent lack of functional redundancy between PrsA2 and PrsA1, mutant proteins were constructed containing different domain combinations of the PrsA1 and PrsA2 N-terminal, C-terminal, or PPIase domains. Six mutants comprising different domain combinations were constructed (P1N-P1P-P2C, P2N-P1P-P1C, P1N-P2N-P1C, P2N-P2P-P1C, P1N-P2P-P2C, and P2N-P1P-P2C) and introduced into a ΔprsA1 ΔprsA2 mutant strain using the integrative plasmid vector pPL2 (Lauer et al., 2002) (Fig. 1D). Each mutant protein was confirmed to be stably expressed, secreted, and localized at the cell surface as demonstrated by Western blot analysis of bacterial surface preparations and in comparison to wild type PrsA2 expressed in ΔprsA1 ΔprsA2 + pPL2-prsA2 complement strains (Fig. 1D). Antibodies directed against PrsA2 recognized PrsA1 albeit with reduced affinity (Fig. 1D), however approximately equivalent amounts of the chimeric mutant protein were determined to be expressed in L. monocytogenes when the apparent abundance of detectable protein was corrected based on the affinity of antibody recognition (Figs. 1D and Fig. S1).
Strains containing each of the six domain swap chimera proteins were tested for complementation of PrsA2-associated phenotypes as reflected by hemolytic activity, phospholipase activity, and the ability to form zones of clearing or plaques in monolayers of fibroblast cells (Alonzo et al., 2009) (Table 1). All constructs that contained the PrsA2 N-terminal domain restored nearly all of the defective phenotypes of ΔprsA1 ΔprsA2 strains with the exception of the P2N-P1P-P1C construct which did not restore hemolytic activity. In contrast, two chimeric constructs (P1N-P1P-P2C and P1N-P2P-P2C) fully complemented hemolytic activity but not phospholipase activity or plaque formation (Table 1); these chimeras shared the PrsA2 C-terminus. Chimeras containing the PrsA2 PPIase domain in conjunction with either one of the PrsA2 N or C terminal domains demonstrated increased complementation of PrsA2-associated activities over corresponding chimeras with PrsA1 PPIase (see P1N-P2P-P1C, P2N-P2P-P1C and P1N-P2P-P2C) (Table 1). The PrsA2 PPIase domain therefore appears to enhance protein activity for chimeras lacking either the PrsA2 N terminal or the C terminal domain, however when both PrsA2 N and C terminal domains were present the protein was fully functional regardless of the source of the PPIase domain (see P2N-P1P-P2C, Table 1). Taken together, the data from PrsA1 - PrsA2 protein chimera experiments point to a critical role for the PrsA2 N-terminal domain in chaperone function as well as to a distinct role for the C-terminal domain relating to LLO-dependent hemolytic activity. The PrsA2 PPIase domain enhances protein activity in PrsA2-PrsA1 chimeras that lack either the PrsA2 N or C terminal domains, suggesting that the conserved PPIase differs in function between PrsA2 and PrsA1.
Table 1.
Plaque formation, and hemolytic and phospholipase activities of domain swap mutants.
| PrsA2 Chimeric Construct | Plaque Size (% WT) | Hemolytic Units | Phospholipase Activity |
|---|---|---|---|
| PrsA2 (Full length) | 100 +/− 3.16 | 100 +/− 9.5 | + |
| PrsA1 (Full length) | 20.2 +/− 0.98*** | 31.8 +/− 4/4*** | − |
| ΔprsA1ΔprsA2 | 20.0 +/− 2.10*** | 37.5 +/− 3.5*** | − |
| P2N-P1P-P1C | 102.8 +/− 2.80 | 44.0 +/− 5.8*** | + |
| P1N-P1P-P2C | 23.3 +/− 3.40*** | 88.0 +/− 8.0 | − |
| P1N-P2P-P1C | 72.5 +/− 1.30*** | 40.8 +/− 4.2*** | − |
| P2N-P2P-P1C | 100.1 +/− 8.60 | 80.0 +/− 13.6 | + |
| P1N-P2P-P2C | 61.6 +/− 1.60*** | 66.7 +/− 6.7 | − |
| P2N-P1P-P2C | 101.7 +/− 8.60 | 100 +/− 6.2 | + |
Statistical significance was calculated using a 1-way ANOVA with Tukey’s multiple comparison test. Statistically significant difference compared to PrsA2 (Full Length) (ΔprsA2 + pPL2- prsA2) are indicated (***, P< .0001)
Targeted mutagenesis within the N and C terminal domains of PrsA2 suggests that multiple contact sites are required for protein function
Although limited structural information exists for PrsA-like proteins from Gram-positive bacteria, the X-ray crystal structure of a related molecule, the E. coli periplasmic chaperone/PPIase SurA, has been determined (Bitto and McKay, 2002, 2003; Xu et al., 2007). PSI-BLAST searches reveal that L. monocytogenes PrsA2 shares significant sequence identity in its N and C terminal domains with the corresponding regions of E. coli SurA (J. Whisstock, unpublished). The SurA N-terminal domain features a loose cluster of helices followed by 2 distinct PPIase domains that can be deleted without loss of chaperone function (unlike the central PPIase domain of B subtilis) (Behrens et al., 2001; Bitto and McKay, 2002, 2003; Rouviere and Gross, 1996; Xu et al., 2007). The SurA C-terminal helices are folded back onto the N-terminal domain. Together the N-and C-terminal regions form a cleft that most likely is central to chaperone function and overall SurA activity (Bitto and McKay, 2003). Based on sequence alignments with SurA, it is suggested that L. monocytogenes PrsA2 similarly has a loose N terminal helical domain followed by a single central PPIase domain and a C-terminal helix. Using SurA as a template for the structure of PrsA2, it is suggested that the N and C terminal domains of PrsA2 are predicted to come together to form a substrate binding cleft (Fig 2A). We further suggest that two small areas within the N terminus are predicted to contribute to the chaperone binding cleft (#1 and #2 in Figs. 2A and 2B). Additionally, two regions within the C terminal domain that vary between PrsA2 and PrsA1 are predicted to form the base of the cleft (#3 and #4 in Figs. 2A and 2B).
Figure 2. The structure of E. coli SurA indicating the position of mutations introduced in this study.

(A) The structure of SurA was used as a guide to design mutations in PrsA2. By analogy with SurA, PrsA2 is predicted to act as a dimer with contributions from both the N and C-terminus of each monomer. PPIase domain (red), N-terminus of monomer 1 (green), N-terminus of monomer 2 (light blue), C terminus of monomer 2 (yellow and blue). Purple regions represent predicted contact regions for substrate binding in the C-terminus of monomer 2 and the N-terminus of monomer 1. (B) Amino acid sequence alignment of PrsA2 and PrsA1. Black shading indicates identical amino acids between PrsA2 and PrsA1; gray shading indicates similar amino acids; and red highlighting represents the four initial regions of the putative substrate binding cleft targeted for mutagenesis. Western blot of the nine mutants is shown (C).
To test whether individual residues within the predicted SurA-like chaperone binding cleft contribute to PrsA2 chaperone activity, selected mutations were introduced in the cleft as depicted in Fig. 2A. Six targeted mutations were introduced into the N-terminal portion of PrsA2 (F77A, Q105A, F77A + F102A, F102A + Q105A, F77A + Q105A, and F77A + F102A + Q105A) and three larger PrsA1 for PrsA2 domain swaps within the C-terminal base of the cleft (DKPATKTTFEKDKKA → VKKTEKGTYAKEKAN, QKTLKKEYK → TAALKKELK, or both swaps combined) (Fig. 2B). Mutant gene constructs were introduced into the L. monocytogenes ΔprsA1 ΔprsA2 double deletion strain on the integrative plasmid pPL2, and Western blot analysis of the strains indicated that the mutant proteins were stably expressed on the bacterial surface (Fig 2C). Surprisingly, nearly all of the mutant constructs fully restored hemolytic and phospholipase activities as well as plaque formation (Table 2). Strains containing the N terminal prsA2 F77A + F102A and prsA2 F77A + F102A + Q105A mutations were modestly compromised for plaque formation, and prsA2 F77A + F102A for hemolytic activity, whereas none of the C terminal substitution mutants were affected for activity (Table 2). Thus, while the substitution of large domains of PrsA2 with PrsA1 resulted in significant alterations in PrsA2 activity, the introduction of more targeted mutations within these regions had little effect. These results suggest at least one of two possibilities: (1) despite the predicted structural similarity, PrsA2 differs significantly from SurA in its structural organization, and/or (2) multiple substrate contact sites contribute to PrsA2 chaperone function such that the loss of contacts in select regions can be functionally compensated by contacts in other regions of the protein.
Table 2.
Plaque formation, and hemolytic and phospholipase activities of targeted mutants.
| PrsA2 Targeted Construct | Plaque Size (% WT) | Hemolytic Units | Phospholipase Activity |
|---|---|---|---|
| PrsA2 (Full length) | 100 +/− 3.16 | 100 +/− 9.5 | + |
| ΔprsA1ΔprsA2 | 20 +/− 2.10*** | 37.5 +/− 3.5*** | − |
| F77A | 97.9 +/− 1.40 | 75 +/− 8.3 | + |
| Q105A | 97.5 +/− 1.71 | 72.5 +/− 13.6 | + |
| F77A + Q105A | 91.45 +/− 1.42*** | 78.3 +/− 9.3 | + |
| F77A + F102A | 78.5 +/− 1.10*** | 54.4 +/− 1.8** | + |
| F102A + Q105A | 95.16 +/− 2.06 | 71.7 +/− 10.2 | + |
| F77A + F102A + Q105A | 81.3 +/− 1.41*** | 84.2 +/− 11.1 | + |
| P1→P2 #3 | 103.2 +/− 1.90 | 78 +/− 6.9 | + |
| P1→P2 #4 | 100.4 +/− 1.70 | 78 +/− 6.9 | + |
| P1→P2 #3 + P1→ P2 #4 | 97.2 +/− 1.81 | 88.9 +/− 7.3 | + |
Statistical significance was calculated using a 1-way ANOVA with Tukey’s multiple comparison test. Statistically significant differences compared to PrsA2 (Full Length) (ΔprsA2 + pPL2- prsA2) are indicated (***, P< .0001, **,P<.005).
Functional investigation of the PrsA2 PPIase domain
The PrsA1/PrsA2 domain swap experiments suggested that the PrsA2 PPIase domain contributes to PrsA2 activity based on its ability to enhance the activity of constructs lacking either the N or C terminal domain of PrsA2 (Table 3). To further explore the functional contributions of the PrsA2 PPIase domain while also investigating potential modularity in function for the N and C- terminal domains (Alonzo et al., 2009; Alonzo and Freitag, 2010), we constructed a PrsA2 N+C terminal fusion protein that lacked the entire PPIase domain. The PrsA2 N+C terminal fusion construct comprises amino acids 1–135 of the N-terminus of the protein and 229–293 of the C-terminus (Fig 3A). In addition, a second mutant was constructed that contained an amino acid substitution (D174A) within the predicted active site of the PrsA2 PPIase domain; the construction of this mutant was based on a similar mutant described for B. subtilis PrsA (Tossavainen et al., 2006). The PrsA2 N+C terminal fusion, as well as PrsA2 D174A, were confirmed to be stably expressed and secreted by ΔprsA2 strains (Fig. 3A and B).
Table 3.
Proteins identified in wild type 2D-gels, but not ΔprsA2 + pPL2-prsA2 (N+C)
| Functional Class | Gene No. | Protein Name | Protein Description | Peptide Match | Coverage (%) | Protein Score | Reference |
|---|---|---|---|---|---|---|---|
| Cell Surface and Cell Wall Metabolism | lmo1438 | Penicillin binding protein (putative) | 27 | 47 | 1932 | (Guinane et al., 2006) | |
| lmo2039 | PbpB | Penicillin binding protein B | 15 | 24 | 608 | (Guinane et al., 2006) | |
| lmo2229 | Pbp4 | Penicillin binding protein (putative) | 3 | 9 | 50 | (Guinane et al., 2006; Zawadzka -Skomial et al., 2006) | |
| lmo2505 | P45 | Peptidoglycan Lytic Protein | 2 | 7 | 522 | (Schubert et al., 2000) | |
| lmo0927 | LtaS | Lipoteichoic acid synthase | 1 | 2.6 | 55 | (Webb et al., 2009) | |
| Flagellar apparatus proteins | lmo0706 | FlgL | Flagellar hook associated protein | 9 | 18 | 373 | |
| Specific Metabolic Pathways | lmo1670 | PykA | Pyruvate kinase | 7 | 16 | 268 | |
| Transformation/Competence | lmo0275 | ComEC | Competence Protein | 3 | 5 | 87 | |
| Amino-acyl t-RNA Synthetases | lmo2416 | ThrS | Threonyl t-RNA synthetase | 4 | 23 | 81 |
Figure 3. The central domain of PrsA2 has PPIase activity.

(A) Upper panel: construction of a PrsA2 N+C mutant lacking the entire central PPIase domain. Lower panels: PrsA2 Western blots of ΔprsA2 + pPL2- prsA2, ΔprsA2 + pPL2-prsA2 N+C, andΔprsA2 + pPL2-prsA2 D174A surface proteins to confirm synthesis and localization of PrsA2 mutant constructs. (B) SDS-PAGE gel of purified recombinant PrsA2 (N+C), PrsA2, PrsA1, and PrsA2 (D174A) (3 μg loaded). (C–F) Purified PrsA2, PrsA2 (N+C), PrsA2 (D174A), and PrsA1 proteins were used in a protease coupled assay for peptidyl prolyl isomerase activity. (C) PrsA2 has PPIase activity. 6.0μM PrsA2 (◆) accelerates the rate of cleavage of the tetrapeptide substrate Suc-Ala-Phe-Pro-Phe-pNA compared to the negative controls [no PPIase (X) and Cyclophilin + cyclosporine A (□)]. Cyclophilin (0.1 μM) (○) is shown as a positive control for PPIase activity. (D) PrsA2 exhibits a dose-dependent increase in activity. PrsA2 at the following concentrations [0.1 μM, 1 μM, 3 μM, 6 μM, and 10 μM (all ◆)] was used in the same PPIase assay as (C). The rate of substrate cleavage was measured at increasing concentrations of PrsA2 protein. (E) PPIase activity resides in the central parvulin-like domain and Aspartate 174 maintains optimal activity. A PrsA2 (N+C) (■) terminal contruct lacking its PPIase domain is no longer capable of catalyzing the cis → trans isomerization of the tetrapeptide substrate, while PrsA2 (D174A) (△) displays a modest decrease in activity compared to wild type PrsA2 (◆). (F) PrsA1 is less active than PrsA2 for PPIase activity. 6 μM and 10 μM (both ▽) PrsA1 are far less efficient and promoting cis→trans isomerization than 6 μM PrsA2. In C, D, E, and F; X, No PPIase added.
Because the PPIase activity of PrsA-like proteins in a number of organisms appears to be either dispensable or completely non-functional (Drouault et al., 2002; Missiakas et al., 1996; Rouviere and Gross, 1996; Vitikainen et al., 2004; Weininger et al., 2010), we first determined whether PrsA2 and PrsA1 displayed any PPIase activity in vitro. C-terminal his-tagged fusion proteins of PrsA2, PrsA1, PrsA2 D174A, and PrsA2 N+C were purified (Fig. 3B) and used in a standard protease-coupled assay developed for the detection of PPIase activity (Fischer et al., 1984; Fischer et al., 1989; Fischer et al., 1992; Harding et al., 1989; Missiakas et al., 1996; Rouviere and Gross, 1996; Takahashi et al., 1989; Tossavainen et al., 2006; Vitikainen et al., 2004). The assay measures the efficiency of PPIase-catalyzed cis → trans isomerization of a commercially available tetrapeptide substrate that, following cis conversion to the trans isomer, is recognized and cleaved by chymotrypsin to result in yellow color formation. The rate of tetrapeptide cleavage was observed to increase substantially upon the addition of 6 μM PrsA2 (Fig. 3C) and to increase in a concentration dependent manner (Fig. 3D). Although the PrsA2 activity on this tetrapeptide substrate was weak in comparison to the positive control bovine cyclophilin (0.1 μM) (Fig. 3C), it was similar to that previously reported for B. subtilis PrsA and E. coli SurA (Missiakas et al., 1996; Rouviere and Gross, 1996; Tossavainen et al., 2006; Vitikainen et al., 2004).
To confirm that the PrsA2 PPIase domain was responsible for the observed PPIase activity, the same assay was used to detect activity for the PrsA2 N+C fusion and the PrsA2 D174A active site mutant (predicted to have ~50% activity based on a similar B. subtilis PrsA substitution mutant) (Tossavainen et al., 2006). As anticipated, the PrsA2 N+C mutant was unable to catalyze the cis → trans isomerization of the tetrapeptide substrate, displaying a rate of isomerization that was similar to that of tetrapeptide substrate alone (Fig. 3E). PrsA2 D174A catalyzed cis → trans isomerization but at a reduced rate in comparison to wild type PrsA2 (Fig. 3E). Lastly, PrsA1 was found to exhibit only weak PPIase activity based upon its modest acceleration of tetrapeptide cleavage in comparison to PrsA2 (Fig. 3F). Taken together these results confirm that both PrsA2 and PrsA1 have bona fide PPIase activity.
PrsA2 PPIase activity is not required for the restoration of hemolytic or phospholipase activity nor for intracellular growth or cell-to-cell spread
In B. subtilis, as well as in other bacteria, the parvulin-like PPIase activity of PrsA appears dispensable for many if not all of its associated functions (Drouault et al., 2002; Missiakas et al., 1996; Rouviere and Gross, 1996; Vitikainen et al., 2004; Weininger et al., 2010). To investigate whether PrsA2 PPIase activity makes a detectable contribution to LLO or PC-PLC activity or to bacterial growth and spread within infected cell monolayers, we examined the ability of the prsA2 N+C construct lacking the PPIase domain to complement ΔprsA2 mutant strain defects. The prsA2 N+C construct restored hemolytic activity of a ΔprsA2 mutant to 85% of wild type levels (Fig. 4A), and appeared to fully restore phospholipase activity based on zones of opacity on egg yolk agar plates (Fig. 4B). Most strikingly, prsA2 N+C fully complemented the severe cell-to-cell spread defect of the ΔprsA2 mutant (Fig. 4C). These data indicate that the PPIase domain of PrsA2 is dispensable for PrsA2 functions relating to LLO and PC-PLC activity as well as bacterial growth and spread within cell monolayers.
Figure 4. The PPIase domain of PrsA2 is not required for restoration of in vitro defects associated with a ΔprsA2 mutant.
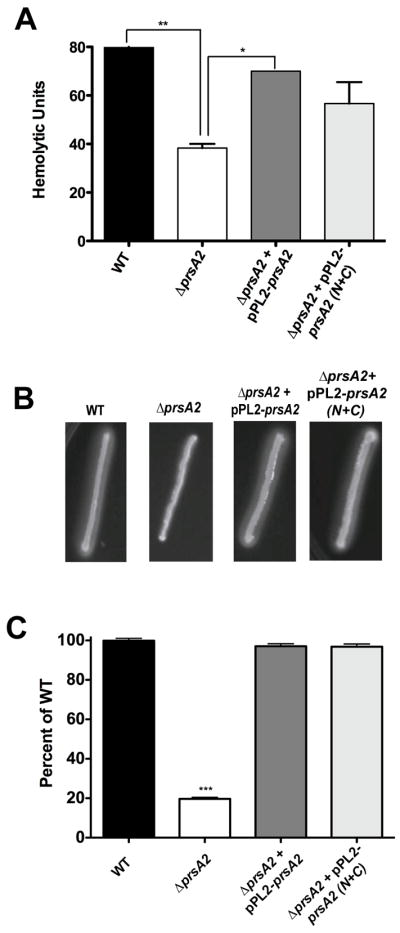
ΔprsA2 + pPL2- prsA2, and ΔprsA2 + pPL2-prsA2 N+C mutants were used to assay restoration of hemolytic activity, phospholipase activity, and plaque formation compared to that of wild type and a ΔprsA2 mutant. (A) Hemolytic activity was determined by measuring the ability of bacterial culture supernatants to lyse sheep’s red blood cyles. Hemolytic units are described as the reciprocal of the dilution resulting in 50% lysis of RBCs. (B) Examination of PC-PLC based phospholipase acticity. Phospholipase activity was measured by streaking strains onto egg yolk agar followed by incubation at 37°C for 24 hours and examining the zone of opacity that is formed. (C) L2 plaque formation assays to measure cell-to-cell spread. L2 cells were infected for 1 hour with each strain followed by the addition of gentamicin in an agarose overlay. Plaques were measured after 72 hours and are reflected as % of wild type.
PrsA2 PPIase activity is required for full virulence in animals
To assess a potential requirement for the PrsA2 PPIase activity during animal infection, mice were infected withΔprsA2 strains containing a pPL2 plasmid integrated copy of either wild type, prsA2 N+C, or prsA2 D174A active site mutant. As previously described, a ΔprsA2 mutant is severely attenuated for virulence in mice based on the bacterial burdens recovered from the liver and spleen at 72 hours post-infection (100,000-fold and 10,000-fold fewer bacteria in livers and spleen respectively in comparison to mice infected with wild type L. monocytogenes) (Fig. 5). In contrast, complementation with wild type prsA2 restored virulence to wild type levels. L. monocytogenes strains containing prsA2 N+C partially restored virulence, however the bacterial burdens recovered from liver and spleens were still 136- and 167-fold lower than those recovered from mice infected with wild type bacteria (Fig. 5). Consistent with the loss of PPIase activity resulting in a virulence defect, the prsA2 D174A partial activity mutant restored bacterial virulence to a greater degree than the PPIase deficient N+C mutant, but to a lesser degree than PrsA2 containing a fully functional PPIase domain (Fig. 5). These experiments indicate a role for PrsA2 PPIase activity in bacterial virulence within infected animals.
Figure 5. The PPIase domain is critical for restoration of virulence in animal models of infection.

Mice were intravenously infected with 2 × 104 CFU of wild type (●), ΔprsA2 (□),ΔprsA2 + pPL2- prsA2 (X), ΔprsA2 + pPL2-prsA2 N+C (▽), and ΔprsA2 + pPL2-prsA2 D174A (■). At 72 hours post-infection livers and spleens were isolated, homogenized, and plated for bacterial CFU on solid BHI agar plates. CFU from the livers and spleens of a minimum of five mice are shown as scatter plots. Solid lines indicated the median value for each group. Statistical significance was calculated using a 1-way ANOVA with Tukey’s multiple comparison test. Statistically significant difference compared to WT and ΔprsA2 + pPL2- prsA2 are indicated (***, P < .0001).
Analysis of bacterial supernatant proteins reveals putative PrsA2 PPIase-dependent substrates
To identify proteins whose stability and/or localization may be dependent upon PrsA2 PPIase activity, we isolated supernatant proteins from wild type, ΔprsA2, and prsA2 N+C strains and compared the protein profiles (Alonzo and Freitag, 2010). Interestingly, pPL2-prsA2 N+C nearly fully restored the secreted protein profiles associated with ΔprsA2 mutants to those observed for wild type based on 2D-gel patterns (F. Alonzo, unpublished data). Nine proteins were identified in wild type supernatants that were not present in those derived from the ΔprsA2 + pPL2-prsA2 N+C strain, suggesting a possible role for PPIase activity for stability and/or localization of these proteins (Table 3). Most striking from the list of nine proteins was that three were predicted penicillin binding proteins (Lmo1438, Lmo2039, and Lmo2229), while two had functions associated with modification of the bacterial cell surface (Lmo2505, and Lmo0927). Additional proteins identified included a flagellar hook-associated protein (Lmo0706), and three proteins of predicted cytosolic function (PykA, ComEC, and a threonyl tRNA synthetase). These results indicate that while the secreted protein profiles of a ΔprsA2 mutant can be largely restored by the introduction of prsA2 N+C, a subset of proteins consisting primarily of PBP’s and cell surface modifying enzymes require the presence of the PrsA2 PPIase domain for stability and/or correct localization.
PrsA2 PPIase is required for optimal resistance to β-lactam antibiotics
Given that the PrsA2 PPIase domain may be important for L. monocytogenes PBP folding and/or activity, we assessed whether bacterial strains lacking PrsA2 PPIase exhibited any change in sensitivity to β-lactam antibiotics. Minimal inhibitory concentrations (MICs) for bacterial growth in the presence of either a β-lactam antibiotic (Penicillin G), or ribosomally targeted antibiotic (gentamicin) were determined for mutant and wild type strains. Wild type, ΔprsA2, ΔprsA2 + pPL2-prsA2, and ΔprsA2 + pPL2-prsA2 N+C exhibited identical MICs (0.32 μg/ml) with gentamicin (Fig. 6A), however the ΔprsA2 and ΔprsA2 + pPL2-prsA2 N+C strains displayed increased sensitivities to Penicillin G (0.02 and 0.04 μg/ml respectively) in comparison to wild type and the ΔprsA2 + pPL2-prsA2 complemented strain (both had an MIC of 0.08 μg/ml) (Fig. 6B). These data support a role for PrsA2 PPIase activity in the folding and/or activity of L. monocytogenes PBPs.
Figure 6. The PPIase domain of PrsA2 is required to maintain wild type levels of resistance to β-lactam antibiotics.

The minimum inhibitory concentration required to prevent growth of wild type, ΔprsA2,ΔprsA2 + pPL2- prsA2, and ΔprsA2 + pPL2-prsA2 N+C was determined by inoculation of each strain into broth culture containing serial dilutions of either gentamicin (A) or penicillinG (B). Statistical significance was calculated using a 1-way ANOVA with Tukey’s multiple comparison test. Statistically significant difference compared to WT and ΔprsA2 + pPL2- prsA2 are indicated (***, P < .0001).
Discussion
As an organism that has evolved to live in environments ranging from soil to the cytosol of mammalian cells, L. monocytogenes must adjust and regulate the expression and activity of protein products that enable survival and replication within these diverse and specific habitats. The secreted chaperone PrsA2 has been adapted to facilitate the folding, stability, and activity of L. monocytogenes secreted proteins required for bacterial survival within host cells (Alonzo et al., 2009; Alonzo and Freitag, 2010; Zemansky et al., 2009). PrsA2 is required for bacterial fitness under conditions of PrfA activation (such as within the cytosol) where the abundance of L. monocytogenes secreted protein products dramatically increases from the levels observed for bacteria in broth culture (Alonzo and Freitag, 2010). In this work we have initiated the first structure/function analysis of this critical virulence chaperone and have identified distinct functional roles for its active PPIase domain and for the N and C terminal domains in L. monocytogenes pathogenesis and drug resistance. The existence of two functionally distinct and non-essential PrsA-like proteins in L. monocytogenes has provided an exceptional opportunity to define and compare novel mechanistic aspects and substrate specificities for two members of an important class of Gram-positive post-translocation secretion chaperones.
As PrsA1 and PrsA2 share significant homology and yet share no apparent functional overlap (Alonzo et al., 2009; Alonzo and Freitag, 2010), domain swapping experiments yielded a ready means for examining and defining functional regions of PrsA2. PrsA1 has yet to be associated with any secreted protein substrate or phenotype, thus the domain swap constructs also provided a method to test whether portions of the protein were indeed functional. Comparisons of PrsA1 and PrsA2 chimeric mutants indicated that the PrsA2 N-terminus plays an important role in mediating PrsA2 substrate interactions, as the introduction of this domain alone in place of the PrsA1 N terminus resulted in the near full complementation of ΔprsA2 associated phenotypes with the exception of hemolytic activity (Table 1). In comparison, experiments in B. subtilis using pentapeptide insertional analysis of PrsA also suggested that the N-terminus of the protein has a number of residues critical for chaperone activity. Similarly, even partial truncations and at least two pentapeptide insertions within the C-terminus were found to abrogate function (Vitikainen et al., 2004). In contrast to the PrsA2 N terminal domain, the C terminal domain restored at least partial hemolytic activity whenever it was present, but failed to restore either phospholipase activity or plaque formation. These results indicate distinct functional roles for the PrsA2 N and C termini, while also demonstrating that PrsA1 domains are at least partially functional when combined with PrsA2. The PrsA2 PPIase domain appeared to enhance PrsA2 function, and was sufficient to increase L. monocytogenes plaque formation in cell monolayers when provided together with the PrsA1 N and C terminal domains (Table 1). PrsA1 and PrsA2 thus share both structural and sequence homology while maintaining distinct functional substrate specificities.
Predicted structural similarities between E. coli SurA and L. monocytogenes PrsA2 suggested contact regions that could contribute to the formation and specificity of the PrsA2 chaperone binding cleft (Bitto and McKay, 2002, 2003; Xu et al., 2007) (Fig. 2). Consistent with this predicted model, L. monocytogenes PrsA2 forms heat resistant dimers in solution (Fig. 1B). However, targeted mutagenesis of the predicted contact sites within the proposed substrate binding cleft region had at best modest effects on PrsA2-associated activities (Table 2). These modest effects could either indicate that the actual structure of PrsA2 differs significantly from that of SurA, or alternatively that multiple amino acid contact sites make up the substrate binding pocket, such that loss of one or more amino acids does not prevent substrate binding. Given that PrsA2 potentially interacts with a significant number of substrate proteins (Alonzo and Freitag, 2010), it seems feasible that multiple residues may contribute to low affinity or transient chaperone interactions.
The enhanced rate of cleavage of the tetrapeptide substrate (Suc-Ala-Phe-Pro-Phe-pNitroanilide) in the presence of chymotrypsin confirmed that PrsA2 is a bone fide prolyl cis → trans isomerase (Fig. 3). Removal of the PPIase domain or the introduction of a point mutation within the active site either abolished or diminished PPIase activity, confirming the central domain of PrsA2 as the source of PPIase activity. While functional, PrsA1 had far less PPIase activity than PrsA2 in the presence of the same tetrapeptide substrate (Fig. 3F). The two proteins may either have altered substrate specificities or differ in their rates of catalysis as a result of amino acid variations within the conserved PPIase domain. B. subtilis PrsA has also been shown to have PPIase activity associated with its central domain (Tossavainen et al., 2006; Vitikainen et al., 2004) and it would be interesting to compare functional similarities between B. subtilis PrsA and L. monocytogenes PrsA2 versus PrsA1. The activity of bacterial parvulins toward the commercial tetrapeptide substrates used in this work is low in relation to PPIases of other organisms (compare the activity of bovine cyclophilin to PrsA2 in Fig. 3) (Fischer et al., 1984; Fischer et al., 1989; Fischer et al., 1992; Harding et al., 1989; Missiakas et al., 1996; Rouviere and Gross, 1996; Takahashi et al., 1989; Tossavainen et al., 2006; Vitikainen et al., 2004). Both B. subtilis PrsA and E. coli SurA also display weak activities using the same tetrapeptide substrates (Missiakas et al., 1996; Rouviere and Gross, 1996; Tossavainen et al., 2006; Vitikainen et al., 2004), suggesting that this substrate may not be optimal for this class of PPIases.
PrsA2 is unique thus far from other PrsA-like proteins in that a functional requirement for the PrsA2 PPIase activity can be demonstrated in vivo. Evidence in other organisms has suggested that the PPIase activity of PrsA-like proteins is dispensable, with some PrsA-like molecules lacking any apparent activity associated with the domain. In B. subtilis the PPIase domain itself is indispensable for function, though PPIase enzymatic activity may be dispensable (Drouault et al., 2002; Missiakas et al., 1996; Rouviere and Gross, 1996; Vitikainen et al., 2004). With PrsA2, the PPIase domain as well as its enzymatic activity were found to be required for the restoration of full virulence to a prsA2 deletion strain in animal infection models (Fig. 5). While the N and C-terminal PrsA2 fusion protein was sufficient for the restoration of a number of in vitro defects (hemolytic activity, phospholipase activity, and plaque formation), the absence of the PPIase domain restored only partial virulence in animals (Fig. 4 and 5). Consistent with a requirement for PPIase activity, the PrsA2 partial function mutant (prsA2 D174A) complemented the virulence defect of a prsA2 deletion mutant more fully than the prsA2 N+C mutant, but still did not reach wild type levels (Fig. 5). Proteomic analysis of supernatant proteins from wild type and the prsA2 N+C complemented deletion strain identified at least nine proteins whose localization was altered in the absence of the PrsA2 PPIase domain (Table 3). Five of the nine proteins had predicted functions relating to cell wall biosynthesis or remodeling; these included three PBPs (Lmo1438, Lmo2039, and Lmo2229), a cell wall hydrolase (Lmo2505, or P45), and Lmo0927, a lipoteichoic acid synthase.(Schubert et al., 2000; Webb et al., 2009). The association of PrsA2 PPIase activity with cell wall synthesis and remodeling proteins is notable in light of work in B. subiltis, where a critical role for PrsA in promoting PBP folding/activity and bacterial surface integrity was recently described (Hyyrylainen et al., 2010). Proteomic analyses of prsA2 deletion mutants in L. monocytogenes has previously revealed a substantial number of mislocalized PBPs, as well as cell wall remodeling enzymes (Alonzo and Freitag, 2010). The association of PBP activity with PrsA2 PPIase activity was further established biologically in that the prsA2 N+C construct was unable to complement increased bacterial sensitivity to Penicillin G exhibited by ΔprsA2 mutants. Alterations in cell wall structure and/or stability may account for the residual virulence defect observed for strains containing prsA2 N+C.
The study of PrsA2 functional activity in L. monocytogenes provides an opportunity to gain a better understanding of the role of these enzymes in Gram-positive protein secretion and physiology. PrsA-like proteins have been targeted for study based on their potential ability to facilitate the folding and stability of heterologous proteins with commercial value (Kim et al., 2005; Lindholm et al., 2006; Vitikainen et al., 2005). PrsA-like proteins have been implicated in the pathogenesis of other Gram-positive bacteria (Ma et al., 2006; Williams et al., 2003), but little remains known regarding the substrate specificities of these proteins or their mechanisms of action. Based on our findings, we speculate that the PrsA2 PPIase domain and N and C-terminal domains each contribute to the recognition of distinct substrates, but may share functional overlap in terms of chaperone activity. Future studies focused on the investigation of PrsA2 with individual substrate proteins will provide additional insight into the fundamental processes underlying post-secretion protein folding and regulation of secreted virulence factor activity in L. monocytogenes.
Experimental Procedures
Bacterial strains, media and culture conditions
The bacterial strains used in this study are listed in Table 4. L. monocytogenes 10403S containing either a deletion of prsA2 (NF-L1651), or a deletion of both prsA2 as well as prsA1 (NF-L1631) was used to assess the pPL2-based complementation of the PrsA2 N+C terminal fusion and PrsA1/PrsA2 domain swap chimeras respectively (Alonzo et al., 2009; Alonzo and Freitag, 2010). Escherichia coli strains TOP10, DH5α I/q, and SM10 were used for construction and propagation of recombinant plasmids. All bacterial strains were grown overnight at 37°C with shaking in brain heart infusion broth (BHI) (Difco Laboratories, Detroit, MI) or Luria broth (LB) (Invitrogen, Carlsbad, CA) unless otherwise described. Antibiotic concentrations used were as follows: chloramphenicol, 15 μg/ml (E. coli) and 5 μg/ml (L. monocytogenes); ampicillin, 100μg/ml; and streptomycin, 200 μg/ml. In all cases, the integration plasmid pPL2 was used for complementation studies (Lauer et al., 2002).
Table 4.
Bacterial strains used in this work
| Strain | Genotype | Designation | Reference |
|---|---|---|---|
| TOP10 | E. coli host strain for recombinant pPL2 plasmids | ||
| SM10 | E. coli conjugation strain for recombinant pPL2 plasmids | ||
| DH5α I/q | E. coli host strain for recombinant PrsA1, PrsA2, N+C, D174A expression vectors | ||
| NF-L100 | L. monocytogenes 10403S | wild type | |
| NF-L1651 | L. monocytogenes 10403S ΔprsA2 | ΔprsA2 | (Alonzo et al., 2009) |
| NF-L1631 | L. monocytogenes 10403S ΔprsA1ΔprsA2 | ΔprsA1ΔprsA2 | (Alonzo and Freitag, 2010) |
| NF-L1656 | NF-L1651 + pPL2- prsA2 | ΔprsA2 + pPL2-prsA2 | (Alonzo et al., 2009) |
| NF-L1924 | NF-L1631 + pPL2-prsA2 | ΔprsA1ΔprsA2 + pPL2-prsA2 | (Alonzo and Freitag, 2010) |
| NF-L1674 | NF-L1651 + pPL2-prsA2 (N+C) | ΔprsA2 + pPL2-prsA2 (N+C) | This work |
| NF-L1672 | NF-L1651 + pPL2-prsA2 (D174A) | ΔprsA2 + pPL2-prsA2 (D174A) | This work |
| NF-L1885 | NF-L1631 + pPL2-(p2n-p1p-p1c) | P2N-P1P-P1C | This work |
| NF-L1887 | NF-L1631 + pPL2-(p1n-p1p-p2c) | P1N-P1P-P2C | This work |
| NF-L1991 | NF-L1631 + pPL2-(p1n-p2p-p1c) | P1N-P2P-P1C | This work |
| NF-L1990 | NF-L1631 + pPL2-(p2n-p2p-p1c) | P2N-P2P-P1C | This work |
| NF-L2021 | NF-L1631 + pPL2-(p1n-p2p-p2c) | P1N-P2P-P2C | This work |
| NF-L1892 | NF-L1631 + pPL2-(p2n-p1p-p2c) | P2N-P1P-P2C | This work |
| NF-L2024 | NF-L1631 + pPL2-prsA2 (F77A) | ΔprsA1 ΔprsA2 + pPL2-prsA2 (F77A) | This work |
| NF-L2026 | NF-L1631 + pPL2-prsA2 (F77A + F102A) | ΔprsA1 ΔprsA2 + pPL2-prsA2 (F77A + F102A) | This work |
| NF-L3003 | NF-L1631 + pPL2-prsA2 (Q105A) | ΔprsA1 ΔprsA2 + pPL2-prsA2 (Q105A) | This work |
| NF-L3014 | NF-L1631 + pPL2-prsA2 (F77A + Q105A) | ΔprsA1 ΔprsA2 + pPL2-prsA2 (F77A + Q105A) | This work |
| NF-L3012 | NF-L1631 + pPL2-prsA2 (F102A + F105A) | ΔprsA1 ΔprsA2 + pPL2-prsA2 (F77A + F102A) | This work |
| NF-L3016 | NF-L1631 + pPL2-prsA2 (F77A + F102A + Q105A) | ΔprsA1 ΔprsA2 + pPL2-prsA2 (F77A + F102A + Q105A) | This work |
| NF-L2027 | NF-L1631 + pPL2-prsA2 (P1→P2 #3) | ΔprsA1 ΔprsA2 + pPL2-prsA2 (P1→P2 #3) | This work |
| NF-L2028 | NF-L1631 + pPL2-prsA2 (P1→P2 #4) | ΔprsA1 ΔprsA2 + pPL2-prsA2 (P1→P2 #4) | This work |
| NF-L3001 | NF-L1631 + pPL2-prsA2 (P1→P2 #3 + P1→P2 #4) | ΔprsA1 ΔprsA2 + pPL2-prsA2 (P1→P2 #3 + P1→P2 #4) | This work |
| NF-E1942 | DH5α I/q + pQE60 - prsA2(N+C) | PrsA2 (N+C)-6his | This work |
| NF-E1965 | DH5α I/q + pQE60 - prsA1 | PrsA1-6his | This work |
| NF-E1764 | DH5α I/q + pQE60 - prsA2 | PrsA2-6his | This work |
| NF-E1969 | DH5α I/q + pQE60 – prsA2(D174A) | PrsA2 (D174A)-6his | This work |
Construction of PrsA2 (N+C) and complementation studies
A B. subtilis PrsA (N+C) mutant was previously described and used as a model for construction of L. monocytogenes PrsA2 (N+C) (Vitikainen et al., 2004). Briefly, splicing by overlap extension polymerase chain reaction (SOEing PCR) was used to fuse bases 1–405 of the prsA2 coding sequence (corresponding to amino acids 1–135) with bases 687–882 (corresponding to amino acids 229–293). Initially, two DNA fragments were generated by PCR using primer pairs NC-a/NC-b and NC-c/NC-d (all oligonucleotides are listed in Table 3). The two fragments (~1060 base pairs and ~200 base pairs) were purified and used in a second PCR reaction to generate a fragment of ~1260 base pairs containing both KpnI and SacI restriction sites. The fragment was digested and subcloned into appropriately digested pPL2, introduced into E. coli SM10 by electroporation, and subsequently introduced into L. monocytogenes ΔprsA2 (NF-L1651) by conjugation (Freitag, 2000; Lauer et al., 2002). The resulting strain expressing a truncated PrsA2 (200 amino acids) lacking its putative PPIase domain (93 amino acids) was designated NF-L1674.
Construction of PrsA1/PrsA2 domain swap chimeras
Construction of domain swap mutants between PrsA1 and PrsA2 was carried out using SOEing PCR. Briefly, six constructs were generated, each containing various combinations either of the PrsA1 or PrsA2 N-terminus, PPIase domain, or C-terminus. The constructs are indicated as follows: P2N-P1P-P1C, P1N-P1P-P2C, P1N-P2P-P1C, P2N-P2P-P1C, P1N-P2P-P2C, and P2N-P1P-P2C (Fig. 1C). These variants allowed for assessment of each domain’s contribution to PrsA1 and/or PrsA2 function. The oligonucleotides used for construction of each mutant PrsA protein are shown in Supplemental Table 1 and described briefly below. An initial series of PCRs were performed to generate fragments corresponding to each domain of interest (PrsA1 or PrsA2). The total number of initial PCRs required was based on the overall number of swaps for that particular construct. For example, to generate the DNA fragment corresponding to P2N-P1P-P1C only two initial PCRs were needed (one fragment corresponding to the prsA2 promoter and N-terminus together, and a second fragment corresponding to the coding sequence of the PrsA1 PPIase and C-terminal domains together). In contrast, a more complex domain swap, such as P1N-P2P-P1C required four initial PCRs corresponding to the prsA2 promoter (all constructs were designed to be under the control of the prsA2 promoter region), PrsA1 N-terminus, PrsA2 PPIase, and PrsA1 C-terminus respectively. The resultant fragments generated by the initial PCRs for each of the constructs described were combined and used in a second SOEing PCR reaction to generate a fragment of approximately 1.5kb (corresponding to the full length chimera of the swapped prsA1 and prsA2 sequences). The fragments (designed to contain KpnI and SacI restriction sites) were subcloned into appropriately digested pPL2 and sequenced to confirm the presence of the expected domains and validate correct fusion junctions prior to conjugation into L. monocytogenes. Upon confirmation of each prsA chimera, the recombinant plasmids were introduced into SM10 E. coli by electroporation, followed by introduction into L. monocytogenes ΔprsA1 ΔprsA2 (NF-L1631) via conjugation. DNA sequencing was again used to confirm each domain swap construct in L. monocytogenes. Strain designations are indicated in Table 4.
Construction of PrsA2 targeted mutations
Targeted PrsA2 mutations were constructed based on either predicted substrate contact sites (as determined by molecular modeling of the E. coli SurA chaperone binding cleft as well as similarities to B. subtilis PrsA) or variation between the PrsA1 and PrsA2 amino acid sequences (Alonzo et al., 2009; Bitto and McKay, 2002, 2003; Xu et al., 2007). Ten PrsA2 mutants were generated by either site-directed mutagenesis (F77A, F77A + F102A, Q105A, F77A + Q105A, F102A + Q105A, F77A + F102A + Q105A, and D174A), or SOEing PCR [a swap of the coding regions for amino acids 225–239 (#3) or 254–262 (#4) or both (#3 + #4) of prsA2 for prsA1] and used in subsequent complementation studies. Site-directed mutagenesis was carried out using a kit provided by USB (Cleveland, OH). Briefly, recombinant pPL2 containing full-length prsA2 (pNF1255) was purified and used in a PCR reaction containing mutagenic primer (77-FtoA, 77-AtoF, 102-FtoA, 105-QtoA, 174-DtoA) and a non-mutagenic common primer in pPL2 (pPL2common). After PCR, the parent plasmid (not mutated) was digested by incubation in the presence of DpnI for one hour. The PCR mixture was subsequently transformed into E. coli TOP10 and transformants screened for the presence or absence of the desired mutation by PCR and DNA sequencing. For small amino acid swaps between PrsA1 and PrsA2, SOEing PCR was used as described above using the primers listed in Supplemental Table 1. Upon confirmation of each prsA2 mutant, the recombinant plasmids were introduced into SM10 E. coli by electroporation, followed by introduction into L. monocytogenes ΔprsA1 ΔprsA2 (NF-L1631) via conjugation. DNA sequencing was again used to confirm each mutant construct in L. monocytogenes. Strain designations are designated in Table 4.
Expression and purification of PrsA1, PrsA2, PrsA2 (D174A), and PrsA2 (N+C) 6X-his-tagged proteins
The DNA sequence corresponding to the mature form of the PrsA2 enzyme lacking its N-terminal signal peptide [amino acids 22–294 (PrsA1) and 21–293 (PrsA2)] was amplified from L. monocytogenes 10403S genomic DNA (for wild type prsA1 and prsA2), or pPL2 vectors containing prsA2 (N+C) or prsA2 (D174A) using primer pairs PrsA1-6hisA/PrsA1-6hisB and PrsA2-6hisA/PrsA26hisB to generate an ~1.5 kb PCR product containing NcoI and BglII restriction sites. The product was digested with NcoI and BglII and subcloned into appropriately digested pQE60 C-terminal 6X-His expression vector (Qiagen, Valencia, CA) followed by transformation into DH5α I/q E. coli. The resulting strains were designated NF-E1764 (prsA2-6his), NF-E1965 (prsA1-6his), NF-E1942 [prsA2 (N+C)-6his], and NF-E1969 [prsA2 (D174A)-6his].
Expression and purification of recombinant PrsA2 proteins was carried out using the methods described by the supplier of the pQE60 vector (Qiagen, Valencia, CA). Briefly, a 4 ml overnight culture of each strain was inoculated at a 1:50 dilution into 100 mls of LB containing ampicillin (100 μg/ml). The culture was grown with shaking at 37°C until an approximate OD600 nm of 0.5, at which point IPTG was added at a final concentration of 0.8 mM. Cultures were allowed to continue growing for another 5 hours followed by centrifugation at 8500 RPM for 15 minutes and subsequent freezing of the bacterial pellet at −80 °C overnight. Bacterial cell lysates were prepared by resuspending the bacterial pellet in 20 ml of Wash Buffer (500 mM NaPO4, 300 mM NaCl, 10 mM imidazole, pH 7.4) followed by the addition of Lysozome (0.3 mg/ml) (Sigma, St. Louis, MO) and protease inhibitor cocktail (Pierce, Pittspurgh, PA). The cell suspension was subsequently sonicated 10X on ice (10 sec. On, 30 sec. Off) followed by the addition of Triton X-100 (final concentration 1%) and incubation at room temperature for 30 minutes. Lysates were clarified by centrifugation at 11,000 RPM for 30 minutes followed by passage through a 0.22 μm sterile filter. Purification of PrsA2/PrsA1 from bacterial lysates was carried out by metal affinity chromatography using Cobalt resin as described by the supplier (Pierce, Pittsburgh, PA). The eluted protein fraction was dialyzed overnight against final storage buffer (20 mM HEPES, 140 mM NaCl, 10% glycerol, 1mM DTT, pH 7.4), aliquoted, and frozen at −80 °C. Final protein concentration and purity was determined using the Bicinchoninic Acid Assay Kit (Pierce, Pittsburgh, PA) and SDS-PAGE analysis.
Protease-coupled assay for peptidyl-prolyl isomerase activity
An assay measuring the ability of peptidyl-prolyl isomerases to catalyze the cis-trans interconversion of small tetrapeptide substrates covalently linked to p-nitroanilide has been previously described (Fischer et al., 1984; Fischer et al., 1989; Fischer et al., 1992; Harding et al., 1989; Missiakas et al., 1996; Rouviere and Gross, 1996; Takahashi et al., 1989; Tossavainen et al., 2006; Vitikainen et al., 2004). Purified PrsA proteins were added to 1ml of assay buffer (20 mM HEPES, 140 mM NaCl, 1 mM DTT, pH 7.4) on ice at the final concentrations specified in the text. After allowing the protein to equilibrate in the assay buffer for ~5 minutes on ice, the tetrapeptide substrate (Suc-Ala-Phe-Pro-Phe-pNitroanilide) was added at a final concentration of 37.5 μM and mixed by pipetting up and down. The protein + peptide solution was immediately added to a cuvette containing a 10 μl solution of chymotrypsin (20 μg/μl) for a final chymotrypsin concentration of 0.2 mg/ml. Upon addition of chymotrypsin, the entire solution was mixed by pipetting up and down three times, followed immediately by measurement in a spectrophotometer at 390 nm. This was the “zero” time point. Beginning at 10 seconds and every 10 seconds thereafter (for a maximum of six minutes) measurements were read to follow color formation over time (cleavage of the trans form of the tetrapeptide substrate by chymotrypsin). Assays were carried out a minimum of three times for each PrsA protein/concentration tested. Cleavage of the tetrapeptide substrate in the absence of PrsA was used as a negative control and addition of CyclophilinA from calf thymus (Sigma, St. Louis, MO) was used as a positive control for PPIase activity. Where described PPIase inhibitors were added (both to test the purity of purified protein samples and confirm the inhibition of Cyclophilin by CyclosporinA) at the following concentrations: CyclosporinA (5.0 μM), FK-506 (5.0 μM), and Juglone (7.0 μM) (Vitikainen et al., 2004).
PrsA1 and PrsA2 chemical crosslinking
Chemical crosslinking was done as previously described by Miner et. al (2008) for PrfA with minor modifications. After separation of purified protein samples by SDS-PAGE, proteins were transferred to PVDF membranes, and PrsA1 and PrsA2 were detected using a 1:2500 dilution of a monoclonal antibody directed against the 6His tag in 1X PBST (Phosphate buffered saline solution plus 0.05% Tween-20) followed by incubation with a 1:2500 dilution of a polyclonal Goat-anti Mouse secondary antibody conjugated to alkaline-phosphatase (SouthernBiotech, Birmingham, AL). Bands were visualized colorimetrically with the addition of 10ml of a BCIP/NBT Plus solution (SouthernBiotech, Birmingham, AL)
Measurement of hemolytic activity
The hemolytic activity for all strains tested was measured as previously described (Alonzo et al., 2009; Alonzo and Freitag, 2010; Camilli et al., 1989). Bacterial cultures were grown for five hours from a 1:10 dilution of overnight culture in LB. Supernatants were collected and normalized based on Optical Density (OD) at 600 nm. Bacterial cultures at higher OD were appropriately diluted into fresh LB to match that of the bacterial culture with the lowest overall OD. Serial dilutions of the culture supernatant were added to phosphate buffered saline (PBS) (pH 5.0) containing 1mM DTT and incubated for 30 minutes at 37°C, after which 100 μl of washed sheep’s red blood cells (RBCs) were added followed by additional incubation for 30 minutes at 37°C. Bacterial supernatant/RBC mixtures were pelleted by centrifugation in a microcentrifuge for 1 minute at maximum speed, and the supernatant dilution resulting in 50% lysis of blood cells was determined based on visual inspection of the pellet. Hemolytic units are described as the reciprocal of the dilution resulting in 50% lysis of RBCs. All hemolysin assays were conducted a minimum of 5 times and data shown is an average of all experiments.
Measurement of bacterial phospholipase activity
The measurement of bacterial secreted phospholipase activity on solid egg yolk agar plates was conducted as previously described (Mueller and Freitag, 2005). Briefly, single colonies of each strain tested were struck onto solid egg yolk agar containing activated charcoal (0.2%) and glucose-6-phosphate (25mM) to enhance the prfA dependent expression of plcB (Yeung et al., 2005). Plates were incubated for 24 hours at 37°C followed by visual inspection of the zone of opacity (indicative of phospholipase activity) surrounding the bacterial streaks. Results were obtained from a minimum of three independent experiments.
L2 plaque assays
Plaque assays were conducted as previously described (Sun et al., 1990). Monolayers of L2 fibroblasts in six-well culture dishes were infected at a multiplicity of infection (MOI) of 30 bacteria to 1 fibroblast for 1 hour. Infected monolayers were subsequently washed three times with PBS followed by addition of a DMEM/Agarose overlay containing gentamicin (10 μg/ml) to kill extracellular bacteria. After 72 hours, plaques were measured using a micrometer. Data shown are an average of the plaques measured from a minimum of three independent experiments with wild type plaque size set to 100%.
Animal infections
All animal procedures were approved by the University of Illinois at Chicago (UIC) Animal Care Committee (ACC) and were conducted in the Biological Resources Laboratory (BRL). 2 × 104 colony forming units of each of the strains tested was injected via tail into 6–8 week old female Swiss Webster mice (Harlan, Madison, WI) as previously described (Alonzo et al., 2009). After 72 hours, mice were sacrificed, and livers and spleens were isolated, homogenized and plated on solid media for enumeration of bacterial burden to each organ.
Preparation of bacterial surface proteins for Westen blot and supernatant proteins for two-dimensional polyacrylamide gel electrophoresis (2D-PAGE)
Bacterial surface and supernatant protein isolation was conducted as previously described (Alonzo et al., 2009; Alonzo and Freitag, 2010; Port and Freitag, 2007). To isolate surface proteins, 25 ml of bacterial culture (grown for 5 hours in BHI and normalized to OD600nm 1.4) was collected by spinning in a floor centrifuge at 9000 RPM for 30 minutes. The resultant bacterial pellet was resuspended in 200 μl of 2% SDS boiling buffer (2% SDS, 5% β-mercaptoethanol, 10% glycerol, 60 mM Tris, pH 6.8), boiled for 5 minutes, and clarified by centrifugation at 11,000 RPM for 30 minutes. For supernatant proteins used in 2D-PAGE studies, 200 ml of bacterial culture was collected after 5 hours of growth in BHI, and supernatants were collected and precipitated with trichloro-acetic acid as previously described (Alonzo and Freitag, 2010; Port and Freitag, 2007). Precipitated proteins were resuspended in 5% SDS boiling buffer (5% SDS, 5% β-mercaptoethanol, 10% glycerol, 60 mM Tris, pH 6.8) and sent to Kendrick Labs for electrophoresis. All supernatant samples were prepared on three independent occasions and used for 2D-PAGE resulting in a minimum of two biological replicates for all gels. Samples from biological replicates were each analyzed a minimum of three times resulting in three technical replicates to validate consistency between samples.
2D-PAGE analysis of bacterial secreted proteins
Two-dimensional gel electrophoresis of supernatant proteins from wild type or ΔprsA2 + pPL2-prsA2(N+C) strains was conducted by Kendrick Labs using the carrier ampholine method of isoelectric focusing as previously described and indicated below (O’Farrell, 1975). Isolelectric focusing was carried out in a glass tube of inner diameter 2.0 mm using 2% pH 4–8 mix ampholines (GE Healthcare, Piscataway, NJ and Serva, Heidelberg, Germany) for 9600 volt-hrs. One μg of an IEF internal standard, tropomyosin, was added to the sample. This protein migrated as a doublet with lower polypeptide spot of MW 33,000 and pI 5.2. A tube gel pH gradient plot was determined with a surface pH electrode. After isoelectric focusing and equilibration for 10 minutes in Buffer O (10% glycerol, 50mM dithiothreitol, 2.3% SDS and 0.0625 M Tris, pH 6.8), each tube gel was sealed to the top of a stacking gel that overlaid a 10% acrylamide slab gel (0.75mm thick). SDS slab gel electrophoresis was carried out for about 4 hrs at 15 mA/gel. The following proteins were used as molecular weight standards: myosin (220,000), phosphorylase A (94,000), catalase (60,000), actin (43,000), carbonic anhydrase (29,000) and lysozyme (14,000) (Sigma-Aldrich, St. Louis, MO). These standards appear along the basic edge of the Coomassie blue stained 10% acrylamide slab gel. The Coomassie blue stained gels were dried between sheets of cellophane with the acid edge to the left.
Protein identification by LC-MS-MS tandem mass spectrometry
To identify protein spots whose abundance was altered between wild type and ΔprsA2 + pPL2-prsA2 N+C, gels were placed on a light box and visually inspected for spots clearly present in one gel but entirely absent from the other (Alonzo and Freitag, 2010). Identified spots were cut from the gel using a clean scalpel blade, and placed in 200 μl of deionized water for further processing. The remainder of the protein spot digestion and mass spectrometry analyses were carried out by the Proteomics Core Facility, part of the Research Resources Center and the University of Illinois at Chicago, as previously described and indicated below (Alonzo and Freitag, 2010). Spots were cut into 1 mm cubes with a scalpel followed by washing the spots with 100 mM ammonium bicarbonate, reduction with dithiothreitol in ammonium bicarbonate, and alkylation with iodoacetamide in the dark. Samples were digested overnight with Promega Modified Sequencing Grade Trypsin in ammonium bicarbonate (Promega, Madison, WI). Peptides were liberated using three consecutive extractions with ammonium bicarbonate at 37°C followed by sample concentration using a speedvac.
LC/MS/MS was carried out using a Thermo Instruments LTQ-FT equipped with a Dionex Ultimate 3000 two-dimensional microcapillary HPLC system (Thermo Fisher Scientific, Waltham, MA). Peptides were separated on a C18 column eluting with a gradient. Generated peak lists were extracted from the resulting chromatograms as MGF files (Mascot Generic Format) using ReAdW (Institute for Systems Biology, Seattle, WA) and in-house software, then searched using a Mascot 2.2 search engine (Matrix Science, Boston, MA) against the List_monocyt Listeria monocytogenes NCBI database (53458 sequences; 14892194 residues) using a peptide tolerance of 10 ppm and carbamidomethylation of cysteine and oxidation of methionine as variable modifications. Scaffold 2.4 (Proteome Software, Portland, OR) software was used to merge and display only the results with a 95% confidence and 2 or more unique peptide matches. The average false discovery rate was between 3%–5% as estimated by Mascot using automated decoy database searching. Mascot scores >46 were considered statistically significant P < 0.05.
SDS-PAGE and Western Blot analyses
All protein samples were run on 10% SDS-PAGE gels for 1.5 hours at 180V. Prior to staining, gels were fixed in a 50% methanol 7% acetic acid solution followed by washing 3X for 15 minutes in deionized water. Gels were subsequently stained for 1 hour with Bios-safe Coomassie R-250 (Bio-Rad, Hercules, CA), followed by destaining overnight in 1 L water prior to image acquisition. For Western blots, proteins were transferred to poly-vinylidine di-fluoride membranes at 30V for 1 hour, followed by blocking in PBS + 0.05% Tween (PBST) containing 5% milk for an additional hour. 20 mls PBST containing primary polyclonal antibody against PrsA2 (1:2,500 dilution) was then incubated with the membranes for 1.5 hours, washed three times for 10 minutes each with PBST, and incubated for an additional hour with a 1:2,500 dilution of secondary polyclonal antibody conjugated to alkaline phosphatase (Southern Biotech, Birmingham, AL). Blots were developed via the addition of BCIP-NBT-Plus (5-bromo-4-chloro-3-indolylphosphate- nitroblue tetrazolium) (Southern Biotech, Birmingham, AL) for approximately 5 minutes. All images were acquired using an Alpha Imager 2200 (Alpha Innotech, San Leandro, CA).
Determination of antibiotic minimum inhibitory concentrations (MICs)
Serial dilutions of either gentamicin or PencillinG were made into brain heart infusion media in 4 ml polypropylene test tubes. Each tube was subsequently inoculated with 2 μl of overnight culture of the following strains (wild type, ΔprsA2, ΔprsA2 + pPL2-prsA2, and ΔprsA2 + pPL2-prsA2 (N+C). Inoculated cultures were allowed to incubate at 37°C for 16 hours followed by determination of the minimal antibiotic concentration required for complete inhibition of bacterial growth. MICs were repeated a minimum of three times in duplicate.
Supplementary Material
Acknowledgments
We thank Brad Winters for help with the prsA2 site directed mutagenesis experiments and members of the Freitag lab for helpful discussions. This work was supported by Public Health Service grants AI41816 and AI083241 (N.E.F) from NIAID and an American Heart Association Predoctoral Fellowship 0910080G (F.A.), and by funding from the UIC Center for Clinical and Translational Science (UL1RR029879). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the funding sources.
References
- Adler A, Fimbres A, Marcinak J, Johnson A, Zheng X, Hasegawa S, Shulman ST. Inflammatory pseudotumor of the heart caused by Listeria monocytogenes infection. J Infect. 2009;58:161–163. doi: 10.1016/j.jinf.2008.12.007. [DOI] [PubMed] [Google Scholar]
- Alonzo F, 3rd, Port GC, Cao M, Freitag NE. The posttranslocation chaperone PrsA2 contributes to multiple facets of Listeria monocytogenes pathogenesis. Infect Immun. 2009;77:2612–2623. doi: 10.1128/IAI.00280-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonzo F, 3rd, Freitag NE. Listeria monocytogenes PrsA2 is required for virulence factor secretion and bacterial viability within the host cell cytosol. Infect Immun. 2010;7:4944–4967. doi: 10.1128/IAI.00532-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens S, Maier R, de Cock H, Schmid FX, Gross CA. The SurA periplasmic PPIase lacking its parvulin domains functions in vivo and has chaperone activity. Embo J. 2001;20:285–294. doi: 10.1093/emboj/20.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitto E, McKay DB. Crystallographic structure of SurA, a molecular chaperone that facilitates folding of outer membrane porins. Structure. 2002;10:1489–1498. doi: 10.1016/s0969-2126(02)00877-8. [DOI] [PubMed] [Google Scholar]
- Bitto E, McKay DB. The periplasmic molecular chaperone protein SurA binds a peptide motif that is characteristic of integral outer membrane proteins. J Biol Chem. 2003;278:49316–49322. doi: 10.1074/jbc.M308853200. [DOI] [PubMed] [Google Scholar]
- Camilli A, Paynton CR, Portnoy DA. Intracellular methicillin selection of Listeria monocytogenes mutants unable to replicate in a macrophage cell line. Proc Natl Acad Sci USA. 1989;86:5522–5526. doi: 10.1073/pnas.86.14.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascales E. The type VI secretion toolkit. EMBO Rep. 2008;9:735–741. doi: 10.1038/embor.2008.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desvaux M, Dumas E, Chafsey I, Hebraud M. Protein cell surface display in Gram-positive bacteria: from single protein to macromolecular protein structure. FEMS Microbiol Lett. 2006;256:1–15. doi: 10.1111/j.1574-6968.2006.00122.x. [DOI] [PubMed] [Google Scholar]
- Desvaux M, Hebraud M. The protein secretion systems in Listeria: inside out bacterial virulence. FEMS Microbiol Rev. 2006;30:774–805. doi: 10.1111/j.1574-6976.2006.00035.x. [DOI] [PubMed] [Google Scholar]
- Desvaux M, Hebraud M, Talon R, Henderson IR. Secretion and subcellular localizations of bacterial proteins: a semantic awareness issue. Trends Microbiol. 2009;17:139–145. doi: 10.1016/j.tim.2009.01.004. [DOI] [PubMed] [Google Scholar]
- Desvaux M, Dumas E, Charsey I, Chambon C, Hebraud M. Comprehensive appraisal of the extracellular proteins from a monoderm bacterium: theoretical and empirical exoproteomics of Listeria monocytogenes EGD-e by secretomics. J Proteome Res. 2010;9:5076–5092. doi: 10.1021/pr1003642. [DOI] [PubMed] [Google Scholar]
- Donnenberg MS. Pathogenic strategies of enteric bacteria. Nature. 2000;406:768–774. doi: 10.1038/35021212. [DOI] [PubMed] [Google Scholar]
- Drouault S, Anba J, Bonneau S, Bolotin A, Ehrlich SD, Renault P. The peptidyl-prolyl isomerase motif is lacking in PmpA, the PrsA-like protein involved in the secretion machinery of Lactococcus lactis. Appl Environ Microbiol. 2002;68:3932–3942. doi: 10.1128/AEM.68.8.3932-3942.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dussurget O, Pizarro-Cerda J, Cossart P. Molecular determinants of Listeria monocytogenes virulence. Annu Rev Microbiol. 2004;58:587–610. doi: 10.1146/annurev.micro.57.030502.090934. [DOI] [PubMed] [Google Scholar]
- Fischer G, Bang H, Mech C. Determination of enzymatic catalysis for the cis-trans-isomerization of peptide binding in proline-containing peptides. Biomed Biochim Acta. 1984;43:1101–1111. [PubMed] [Google Scholar]
- Fischer G, Wittmann-Liebold B, Lang K, Kiefhaber T, Schmid FX. Cyclophilin and peptidyl-prolyl cis-trans isomerase are probably identical proteins. Nature. 1989;337:476–478. doi: 10.1038/337476a0. [DOI] [PubMed] [Google Scholar]
- Fischer G, Bang H, Ludwig B, Mann K, Hacker J. Mip protein of Legionella pneumophila exhibits peptidyl-prolyl-cis/trans isomerase (PPlase) activity. Mol Microbiol. 1992;6:1375–1383. doi: 10.1111/j.1365-2958.1992.tb00858.x. [DOI] [PubMed] [Google Scholar]
- Freitag NE. Genetic tools for use with Listeria monocytogenes. In: Fischetti VA, Novick RP, Ferretti JJ, Portnoy DA, Rood JI, editors. Gram-postive pathogens. Washington D.C: ASM Press; 2000. pp. 488–498. [Google Scholar]
- Freitag NE. From hot dogs to host cells: how the bacterial pathogen Listeria monocytogenes regulates virulence gene expression. Future Microbiol. 2006;1:89–101. doi: 10.2217/17460913.1.1.89. [DOI] [PubMed] [Google Scholar]
- Freitag NE, Port GC, Miner MD. Listeria monocytogenes - from saprophyte to intracellular pathogen. Nat Rev Microbiol. 2009;7:623–628. doi: 10.1038/nrmicro2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoffroy CLGJ, Alouf JE, Berche P. Purification, characterization, and toxicity of the sulfhydryl-activated hemolysin listeriolysin O from Listeria monocytogenes. Infect Immun. 1987;55:1641–1646. doi: 10.1128/iai.55.7.1641-1646.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlach RG, Hensel M. Protein secretion systems and adhesins: the molecular armory of Gram-negative pathogens. Int J Med Microbiol. 2007;297:401–415. doi: 10.1016/j.ijmm.2007.03.017. [DOI] [PubMed] [Google Scholar]
- Glomski IJ, Gedde MM, Tsang AW, Swanson JA, Portnoy DA. The Listeria monocytogenes hemolysin has an acidic pH optimum to compartmentalize activity and prevent damage to infected host cells. J Cell Biol. 2002;156:1029–1038. doi: 10.1083/jcb.200201081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinane CM, Cotter PD, Ross RP, Hill C. Contribution of penicillin-binding protein homologs to antibiotic resistance, cell morphology, and virulence of Listeria monocytogenes EGDe. Antimicrob Agents Chemother. 2006;50:2824–2828. doi: 10.1128/AAC.00167-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding MW, Galat A, Uehling DE, Schreiber SL. A receptor for the immunosuppressant FK506 is a cis-trans peptidyl-prolyl isomerase. Nature. 1989;341:758–760. doi: 10.1038/341758a0. [DOI] [PubMed] [Google Scholar]
- Hyyrylainen HL, Bolhuis A, Darmon E, Muukkonen L, Koski P, Vitikainen M, Sarvas M, Pragai Z, Bron S, van Dijl JM, Kontinen VP. A novel two-component regulatory system in Bacillus subtilis for the survival of severe secretion stress. Mol Microbiol. 2001;41:1159–1172. doi: 10.1046/j.1365-2958.2001.02576.x. [DOI] [PubMed] [Google Scholar]
- Hyyrylainen HL, Sarvas M, Kontinen VP. Transcriptome analysis of the secretion stress response of Bacillus subtilis. Appl Microbiol Biotechnol. 2005;67:389–396. doi: 10.1007/s00253-005-1898-1. [DOI] [PubMed] [Google Scholar]
- Hyyrylainen HL, Marciniak BC, Dahncke K, Pietiainen M, Courtin P, Vitikainen M, Seppala R, Otto A, Becher D, Chapot-Chartier MP, Kuipers OP, Kontinen VP. Penicillin-binding protein folding is dependent on the PrsA peptidyl-prolyl cis-trans isomerase in Bacillus subtilis. Mol Microbiol. 2010 doi: 10.1111/j.1365-2958.2010.07188.x. [DOI] [PubMed] [Google Scholar]
- Jacobs M, Andersen JB, Kontinen V, Sarvas M. Bacillus subtilis PrsA is required in vivo as an extracytoplasmic chaperone for secretion of active enzymes synthesized either with or without pro-sequences. Mol Microbiol. 1993;8:957–966. doi: 10.1111/j.1365-2958.1993.tb01640.x. [DOI] [PubMed] [Google Scholar]
- Johansson J, Mandin P, Renzoni A, Chiaruttini C, Springer M, Cossart P. An RNA thermosensor controls expression of virulence genes in Listeria monocytogenes. Cell. 2002;110:551–561. doi: 10.1016/s0092-8674(02)00905-4. [DOI] [PubMed] [Google Scholar]
- Kim JH, Park IS, Kim BG. Development and characterization of membrane surface display system using molecular chaperon, prsA, of Bacillus subtilis. Biochem Biophys Res Commun. 2005;334:1248–1253. doi: 10.1016/j.bbrc.2005.07.024. [DOI] [PubMed] [Google Scholar]
- Kontinen VP, Saris P, Sarvas M. A gene (prsA) of Bacillus subtilis involved in a novel, late stage of protein export. Mol Microbiol. 1991;5:1273–1283. doi: 10.1111/j.1365-2958.1991.tb01901.x. [DOI] [PubMed] [Google Scholar]
- Kontinen VP, Sarvas M. The PrsA lipoprotein is essential for protein secretion in Bacillus subtilis and sets a limit for high-level secretion. Mol Microbiol. 1993;8:727–737. doi: 10.1111/j.1365-2958.1993.tb01616.x. [DOI] [PubMed] [Google Scholar]
- Lauer P, Chow MY, Loessner MJ, Portnoy DA, Calendar R. Construction, Characterization, and Use of Two Listeria monocytogenes Site-Specific Phage Integration Vectors. J Bacteriol. 2002;184:4177–4186. doi: 10.1128/JB.184.15.4177-4186.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindholm A, Ellmen U, Tolonen-Martikainen M, Palva A. Heterologous protein secretion in Lactococcus lactis is enhanced by the Bacillus subtilis chaperone-like protein PrsA. Appl Microbiol Biotechnol. 2006;73:904–914. doi: 10.1007/s00253-006-0551-y. [DOI] [PubMed] [Google Scholar]
- Loh E, Dussurget O, Gripenland J, Vaitkevicius K, Tiensuu T, Mandin P, Repoila F, Buchrieser C, Cossart P, Johansson J. A trans-acting riboswitch controls expression of the virulence regulator PrfA in Listeria monocytogenes. Cell. 2009;139:770–779. doi: 10.1016/j.cell.2009.08.046. [DOI] [PubMed] [Google Scholar]
- Ma Y, Bryant AE, Salmi DB, Hayes-Schroer SM, McIndoo E, Aldape MJ, Stevens DL. Identification and characterization of bicistronic speB and prsA gene expression in the group A Streptococcus. J Bacteriol. 2006;188:7626–7634. doi: 10.1128/JB.01059-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marlovits TC, Stebbins CE. Type III secretion systems shape up as they ship out. Curr Opin Microbiol. 2010;13:47–52. doi: 10.1016/j.mib.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquis H, Hager EJ. pH-regulated activation and release of a bacteria-associated phospholipase C during intracellular infection by Listeria monocytogenes. Mol Microbiol. 2000;35:289–298. doi: 10.1046/j.1365-2958.2000.01708.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matias VR, Beveridge TJ. Cryo-electron microscopy reveals native polymeric cell wall structure in Bacillus subtilis 168 and the existence of a periplasmic space. Mol Microbiol. 2005;56:240–251. doi: 10.1111/j.1365-2958.2005.04535.x. [DOI] [PubMed] [Google Scholar]
- Matias VR, Beveridge TJ. Native cell wall organization shown by cryo-electron microscopy confirms the existence of a periplasmic space in Staphylococcus aureus. J Bacteriol. 2006;188:1011–1021. doi: 10.1128/JB.188.3.1011-1021.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matias VR, Beveridge TJ. Lipoteichoic acid is a major component of the Bacillus subtilis periplasm. J Bacteriol. 2008;190:7414–7418. doi: 10.1128/JB.00581-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner MD, Port GC, Freitag NE. Functional impact of mutational activation on the Listeria monocytogenes central virulence regulator PrfA. Microbiol. 2008;154:3579–3598. doi: 10.1099/mic.0.2008/021063-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missiakas D, Betton JM, Raina S. New components of protein folding in extracytoplasmic compartments of Escherichia coli SurA, FkpA and Skp/OmpH. Mol Microbiol. 1996;21:871–884. doi: 10.1046/j.1365-2958.1996.561412.x. [DOI] [PubMed] [Google Scholar]
- Mueller KJ, Freitag NE. Pleiotropic enhancement of bacterial pathogenesis resulting from the constitutive activation of the Listeria monocytogenes regulatory factor PrfA. Infect Immun. 2005;73:1917–1926. doi: 10.1128/IAI.73.4.1917-1926.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Farrell PH. High resolution two-dimensional electrophoresis of proteins. J Biol Chem. 1975;250:4007–4021. [PMC free article] [PubMed] [Google Scholar]
- Port GC, Freitag NE. Identification of novel Listeria monocytogenes secreted virulence factors following mutational activation of the central virulence regulator, PrfA. Infect Immun. 2007;75:5886–5897. doi: 10.1128/IAI.00845-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portnoy DA, Tweten RK, Kehoe M, Bielecki J. Capacity of listeriolysin O, streptolysin O, and perfringolysin O to mediate growth of Bacillus subtilis within mammalian cells. Infect Immun. 1992;60:2710–2717. doi: 10.1128/iai.60.7.2710-2717.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouviere PE, Gross CA. SurA, a periplasmic protein with peptidyl-prolyl isomerase activity, participates in the assembly of outer membrane porins. Genes Dev. 1996;10:3170–3182. doi: 10.1101/gad.10.24.3170. [DOI] [PubMed] [Google Scholar]
- Sarvas M, Harwood CR, Bron S, van Dijl JM. Post-translocational folding of secretory proteins in Gram-positive bacteria. Biochim Biophys Acta. 2004;1694:311–327. doi: 10.1016/j.bbamcr.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Schnupf P, Portnoy DA. Listeriolysin O: a phagosome-specific lysin. Microbes Infect. 2007;9:1176–1187. doi: 10.1016/j.micinf.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Schubert K, Bichlmaier AM, Mager E, Wolff K, Ruhland G, Fiedler F. P45, an extracellular 45 kDa protein of Listeria monocytogenes with similarity to protein p60 and exhibiting peptidoglycan lytic activity. Arch Microbiol. 2000;173:21–28. doi: 10.1007/s002030050003. [DOI] [PubMed] [Google Scholar]
- Scortti M, Monzo HJ, Lacharme-Lora L, Lewis DA, Vazquez-Boland JA. The PrfA virulence regulon. Microbes Infect. 2007;9:1196–1207. doi: 10.1016/j.micinf.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Shetron-Rama LM, Mueller K, Bravo JM, Bouwer HG, Way SS, Freitag NE. Isolation of Listeria monocytogenes mutants with high-level in vitro expression of host cytosol-induced gene products. Mol Microbiol. 2003;48:1537–1551. doi: 10.1046/j.1365-2958.2003.03534.x. [DOI] [PubMed] [Google Scholar]
- Simonen M, Palva I. Protein secretion in Bacillus species. Microbiol Rev. 1993;57:109–137. doi: 10.1128/mr.57.1.109-137.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun AN, Camilli A, Portnoy DA. Isolation of Listeria monocytogenes small-plaque mutants defective for intracellular growth and cell-to-cell spread. Infect Immun. 1990;58:3770–3778. doi: 10.1128/iai.58.11.3770-3778.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Hayano T, Suzuki M. Peptidyl-prolyl cis-trans isomerase is the cyclosporin A-binding protein cyclophilin. Nature. 1989;337:473–475. doi: 10.1038/337473a0. [DOI] [PubMed] [Google Scholar]
- Tossavainen H, Permi P, Purhonen SL, Sarvas M, Kilpelainen I, Seppala R. NMR solution structure and characterization of substrate binding site of the PPIase domain of PrsA protein from Bacillus subtilis. FEBS Lett. 2006;580:1822–1826. doi: 10.1016/j.febslet.2006.02.042. [DOI] [PubMed] [Google Scholar]
- van Wely KH, Swaving J, Freudl R, Driessen AJ. Translocation of proteins across the cell envelope of Gram-positive bacteria. FEMS Microbiol Rev. 2001;25:437–454. doi: 10.1111/j.1574-6976.2001.tb00586.x. [DOI] [PubMed] [Google Scholar]
- Vitikainen M, Pummi T, Airaksinen U, Wahlstrom E, Wu H, Sarvas M, Kontinen VP. Quantitation of the capacity of the secretion apparatus and requirement for PrsA in growth and secretion of alpha-amylase in Bacillus subtilis. J Bacteriol. 2001;183:1881–1890. doi: 10.1128/JB.183.6.1881-1890.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitikainen M, Lappalainen I, Seppala R, Antelmann H, Boer H, Taira S, Savilahti H, Hecker M, Vihinen M, Sarvas M, Kontinen VP. Structure-function analysis of PrsA reveals roles for the parvulin-like and flanking N- and C-terminal domains in protein folding and secretion in Bacillus subtilis. J Biol Chem. 2004;279:19302–19314. doi: 10.1074/jbc.M400861200. [DOI] [PubMed] [Google Scholar]
- Vitikainen M, Hyyrylainen HL, Kivimaki A, Kontinen VP, Sarvas M. Secretion of heterologous proteins in Bacillus subtilis can be improved by engineering cell components affecting post-translocational protein folding and degradation. J Appl Microbiol. 2005;99:363–375. doi: 10.1111/j.1365-2672.2005.02572.x. [DOI] [PubMed] [Google Scholar]
- Wahlstrom E, Vitikainen M, Kontinen VP, Sarvas M. The extracytoplasmic folding factor PrsA is required for protein secretion only in the presence of the cell wall in Bacillus subtilis. Microbiology. 2003;149:569–577. doi: 10.1099/mic.0.25511-0. [DOI] [PubMed] [Google Scholar]
- Webb AJ, Karatsa-Dodgson M, Grundling A. Two-enzyme systems for glycolipid and polyglycerolphosphate lipoteichoic acid synthesis in Listeria monocytogenes. Mol Microbiol. 2009;74:299–314. doi: 10.1111/j.1365-2958.2009.06829.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weininger U, Jakob RP, Kovermann M, Balbach J, Schmid FX. The prolyl isomerase domain of PpiD from Escherichia coli shows a parvulin fold but is devoid of catalytic activity. Protein Sci. 2010;19:6–18. doi: 10.1002/pro.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RC, Rees ML, Jacobs MF, Pragai Z, Thwaite JE, Baillie LW, Emmerson PT, Harwood CR. Production of Bacillus anthracis protective antigen is dependent on the extracellular chaperone, PrsA. J Biol Chem. 2003;278:18056–18062. doi: 10.1074/jbc.M301244200. [DOI] [PubMed] [Google Scholar]
- Xu X, Wang S, Hu YX, McKay DB. The periplasmic bacterial molecular chaperone SurA adapts its structure to bind peptides in different conformations to assert a sequence preference for aromatic residues. J Mol Biol. 2007;373:367–381. doi: 10.1016/j.jmb.2007.07.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung PS, Zagorski N, Marquis H. The metalloprotease of Listeria monocytogenes controls cell wall translocation of the broad-range phospholipase C. J Bacteriol. 2005;187:2601–2608. doi: 10.1128/JB.187.8.2601-2608.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawadzka-Skomial J, Markiewicz Z, Nguyen-Disteche M, Devreese B, Frere JM, Terrak M. Characterization of the bifunctional glycosyltransferase/acyltransferase penicillin-binding protein 4 of Listeria monocytogenes. J Bacteriol. 2006;188:1875–1881. doi: 10.1128/JB.188.5.1875-1881.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemansky J, Kline BC, Woodward JJ, Leber JH, Marquis H, Portnoy DA. Development of a mariner-based transposon and identification of Listeria monocytogenes determinants, including the peptidyl-prolyl isomerase PrsA2, that contribute to its hemolytic phenotype. J Bacteriol. 2009;191:3950–3964. doi: 10.1128/JB.00016-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.