

In the last several years Mo and W imido alkylidene complexes that have the formula M(NR)(CHR’)(OR”)(Pyr), where Pyr is a pyrrolide or a substituted pyrrolide and OR” usually is an aryloxide, have been prepared and explored.1 These MonoAlkoxidePyrrolide (MAP) species have many new features of fundamental interest, one of the most important being a stereogenic metal center.2 Ring-opening cross-metatheses with 1 were found to be highly Z-selective (as well as enantioselective); the reason is that a “large” aryloxide in combination with a relatively “small” (adamantyl) imido ligand prevents formation of a TBP metallacyclobutane intermediate in which a metallacycle substituent points toward the axial aryloxide (away from the axial imido ligand).3 ROMP of substituted norbornadienes with 2 as an initiator led to >99% cis and >99% syndiotactic polymers4 while 3 promoted homocoupling of neat terminal olefins to Z olefins at 80-120 °C.5

Since tungsten appears to be superior to molybdenum for Z-selective homocoupling with compounds of type 3,5 it would be desirable to employ an imido group smaller than those shown in 3 in order to increase the rate of homocoupling and perhaps at the same time increase the efficiency for forming Z product. We turned to the synthesis of 3,5-dimethylphenyl imido complexes of tungsten since adamantylimido complexes of tungsten are unknown6 and since 3,5-dimethylphenylimido alkylidene complexes of molybdenum have been prepared.7
We found that W(NAr”)(CHCMe2Ph)(OTf)2(dme) (Ar” = 3,5-dimethylphenyl) can be prepared using a procedure analogous to that employed for similar tungsten species, as shown in Scheme 1. Since W(NAr”)2Cl2(dme) (4) was obtained only as a red oil it was used in the next step without purification. W(NAr”)2(CH2CMe2Ph)2 (5) was obtained as a yellow solid in an overall yield of 30% and converted to W(NAr”)(CHCMe2Ph)(OTf)2(dme) (6) in 67% yield.
Scheme 1.
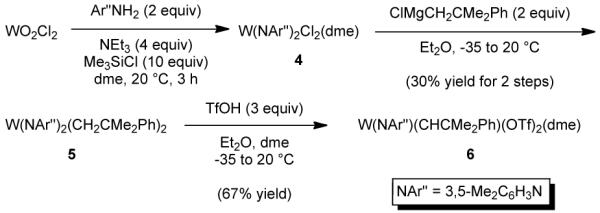
The synthesis of W(NAr”)(CHCMe2Ph)(OTf)2(dme) (6).
A variety of bispyrrolide species (7a,b,c) could be prepared from 6, as shown in Scheme 2. Tungsten MAP species (8a and 8b) are generated in good yields, while two species of type 8 were turned into the more crystalline and readily isolated metallacyclobutane complexes (9) by exposing a solution of 8 to an ethylene atmosphere.
Scheme 2.
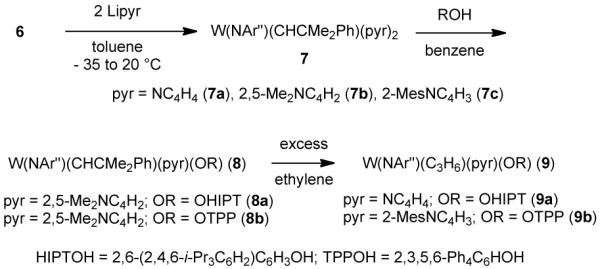
The synthesis of MAP species.
X-ray structural studies of 9a (Figure 1) and 9b (Figure 2) show each to have the expected TBP geometry with the imido and aryloxide ligands in apical positions. Bond distances and angles are similar to those found for W(NAr)(C3H6)(pyr)(OHIPT) (Ar = 2,6-i-Pr2C6H3).4a The W-O-Cipso angle is relatively large (160.80(13)°) in 9a, consistent with significant steric demands for the HIPTO ligand. The W-O-Cipso angle in 9b is slightly smaller (157.73(14)°), as one might expect for the less sterically demanding OTPP ligand. In the solid state, the 3,5-dimethylphenyl and mesityl rings in 9b show an interdigitated π -stacking; the C(61)…C(81) distance is 3.405 Å and the W(1)-N(2)-C(61) angle (164.32(16)°) is reduced from what it is in 9a (176.83(16)°) in response to the π-stacking interaction.
Figure 1.
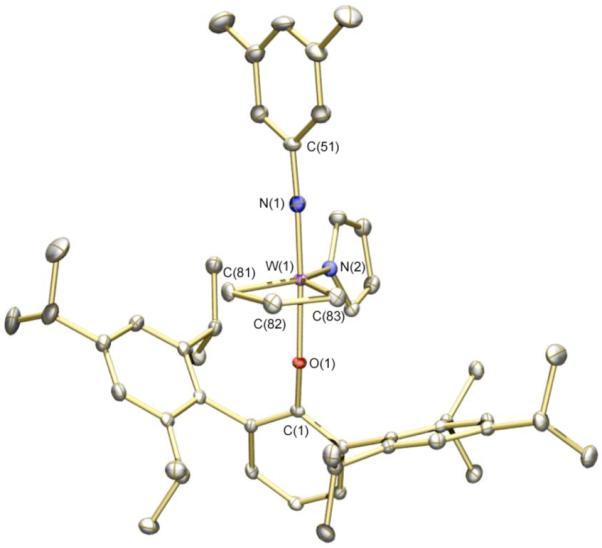
Thermal ellipsoid drawing of 9a. Ellipsoids are displayed at 50% probability level. Hydrogen atoms are omitted. Selected distances (Å) and angles (deg): W(1)-N(1) = 1.7617(18) Å, W(1)-N(2) = 2.0536(16) Å, W(1)-O(1) = 1.9713(13) Å, W(1)-C(81) = 2.0697(19) Å, W(1)-C(82) = 2.353(2) Å, W(1)-C(83) = 2.0428(19) Å, W(1)-N(1)-C(51) = 176.83(16)°, W(1)-O(1)-C(1) = 160.80(13)°, N(1)-W(1)-C(81) = 91.28(8)°, N(1)-W(1)-C(83) = 94.32(8)°, C(81)-W(1)-C(83) = 83.91(8)°.
Figure 2.
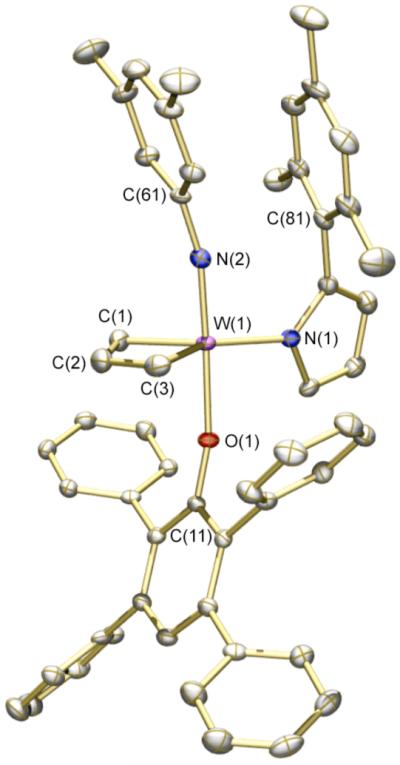
Thermal ellipsoid drawing of 9b. Ellipsoids are displayed at 50% probability level. Hydrogen atoms are omitted. Selected distances (Å) and angles (deg): W(1)-N(1) = 2.0540(18) Å, W(1)-N(2) = 1.7571(19) Å, W(1)-O(1) = 1.9797(14) Å, W(1)-C(1) = 2.049(2) Å, W(1)-C(3) = 2.077(2) Å, C(61)-C(81) = 3.405 Å, W(1)-N(2)-C(61) = 164.32(16)°, N(2)-W(1)-O(1) = 173.29(7)°, W(1)-O(1)-C(11) = 157.73(14)°.
Results for the homocoupling of 1-hexene in benzene are shown in Table 1. W(NAr”)(C3H6)(pyr)(OHIPT) (9a) catalyzes the homocoupling of 1-hexene to >99% Z-5-decene at room temperature (entry 1). (Reactions were monitored until no further change was observed.) When 8a was employed (entry 2), a slight decrease in the Z content was observed. Conversion is limited by bimolecular decomposition of intermediate methylidene species to give an ethylene complex, a type of reaction that has been documented in other circumstances.8 For example, treatment of a sample of 8a with ethylene leads to formation of W(NAr”)(CH2)(2,5-Me2pyr)(OHIPT) (10), according to proton NMR spectra. (See Supporting Information.) Crystals of the metallacyclopentane species, W(NAr”)(C4H8)(2,5-Me2pyr)(OHIPT) (11) were isolated from an NMR sample of 10 after standing the sample for two months at −35 °C . An X-ray structural study (see Supporting Information) shows 11 to be approximately a square pyramid with the imido group in the apical position; this stucture is analogous to that of a related tungstacyclopentane complex in the literature.9 Facile bimolecular decomposition is likely to be a consequence of the relatively small size of the 3,5-dimethylimido group. There is little to no Z-selectivity for the homocoupling of 1-hexene with 9b at room temperature (entry 3) or at 0 °C (entry 4) as a consequence of a lower steric demand of the OTPP ligand compared to the OHIPT ligand.
Table 1.
Catalyst screening for the homocoupling of 1-hexene
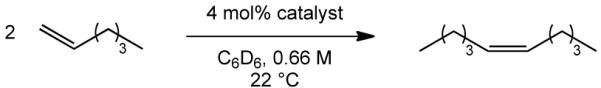 | ||||
|---|---|---|---|---|
| Entry | Catalyst | time | %conv. | %Z |
| 1 | W(3,5-Me2C6H3N)(pyr)(OHIPT)(C3H6) (9a) | 16 h | 32 | >99 |
| 2 | W(3,5-Me2C6H3N)(2,5-Me2pyr)(OHIPT)(CHCMe2Ph) (8a) | 3 d | 48 | 94 |
| 3 | W(3,5-Me2C6H3N)(2-Mespyr)(OTPP)(C3H6) (9b) | 2 h | 54 | 20 |
| 4 | W(3,5-Me2C6H3N)(2-Mespyr)(OTPP)(C3H6) (9b), 0 °C | 24 h | 40 | ~30 |
Speed and conversion of homocoupling can be improved by performing the reaction in neat substrate (Table 2). 1-Hexene, 1-octene, and methyl-10-undecenoate are homocoupled in 45-89% yield with Z-selectivities of >99%. We propose that the substrates listed in entries 4, 5, and 6 yield product with a lower Z content as a consequence of the greater steric hindrance that is required in the α,β disubstituted molybdacyclobutane intermediates. All homocouplings performed at elevated temperatures led to relatively low yields of product as a consequence of catalyst decomposition.
Table 2.
Z-Selective homocoupling of various terminal olefins in the presence of 9a
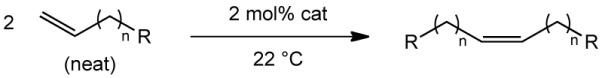 | |||||
|---|---|---|---|---|---|
| Entry | n | R | time | %conv. | %cis |
| 1 | 3 | Me | 4 h | 74 | >99 |
| 2 | 5 | Me | 4 d | 89 | >99 |
| 3 | 8 | CO2Me | 2 d | 45 | >99 |
| 4 | 1 | Ph | 24 h | 35 | 97 |
| 5 | 1 | SiMe3 | 4 h | 45 | 81 |
| 6 | 1 |
|
2.5 h | 61 | 92 |
Resistance of the Z double bond in the homocoupled products toward isomerization to E for the first three substrates in Table 2 suggests that it should be possible to homocouple terminal olefins in the presence of an internal E C=C bond. This is shown to be the case for the substrate shown in equation 1; the triolefin product is obtained in 60% yield (eq 1) as a single E,Z,E isomer. Formation of high % Z homocoupled products in the presence of E olefins elsewhere in the molecule, has never been observed, to the best of our knowledge.
 |
It is clear that the success of highly Z selective homocoupling depends upon the juxtaposition of a “large” aryloxide and a “small” imido group in the intermediate metallacyclobutane, and no secondary isomerization of Z to E. Activity and Z efficiency are high when the 3,5-Me2C6H3N/OHIPT combination is employed, but at the expense of catalyst stability. In general, we expect that catalysts will have to be finely tuned even further in order to optimize the yield and Z-selectivity for a given substrate.
Supplementary Material
Acknowledgment
We are grateful to the National Science Foundation (CHE-0554734 to R.R.S.) and to the National Institutes of Health (Grant GM-59426 to R.R.S. and A.H.H.) for financial support.
Footnotes
Supporting Information Available. Experimental details for the synthesis of all compounds and metathesis reactions. Supporting Information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Schrock RR. Chem. Rev. 2009;109:3211. doi: 10.1021/cr800502p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Malcolmson SJ, Meek SJ, Sattely ES, Schrock RR, Hoveyda AH. Nature. 2008;456:933. doi: 10.1038/nature07594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ibrahem I, Yu M, Schrock RR, Hoveyda AH. J. Am. Chem. Soc. 2009;131:3844. doi: 10.1021/ja900097n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Flook MM, Jiang AJ, Schrock RR, Müller P, Hoveyda AH. J. Am. Chem. Soc. 2009;131:7962. doi: 10.1021/ja902738u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Flook MM, Gerber LCH, Debelouchina GT, Schrock RR. Macromolecules. 2010;43:7515. doi: 10.1021/ma101375v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Flook MM, Ng VWL, Schrock RR. J. Am. Chem. Soc. 2011;132:1784. doi: 10.1021/ja110949f. [DOI] [PubMed] [Google Scholar]
- 5.Jiang AJ, Zhao Y, Schrock RR, Hoveyda AH. J. Am. Chem. Soc. 2009;131:16630. doi: 10.1021/ja908098t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pilyugina T. Massachusetts Institute of Technology thesis. [Google Scholar]
- 7.Oskam JH, Fox HH, Yap KB, McConville DH, O’Dell R, Lichtenstein BJ, Schrock RR. J. Organomet. Chem. 1993;459:185. [Google Scholar]
- 8.Marinescu SC, King AJ, Schrock RR, Singh S, Müller P, Takase MK. Organometallics. 2010;29:6816. [Google Scholar]
- 9.Arndt S, Schrock RR, Müller P. Organometallics. 2007;26:1279. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.