

Abstract
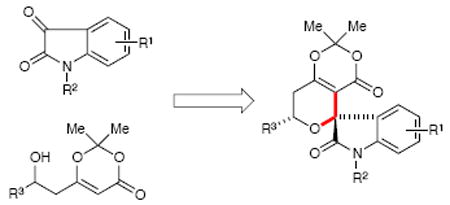
A Brønsted acid-catalyzed Prins-type cyclization sequence to construct spirooxindole pyrans in high yields and excellent diastereoselectivity has been developed. The combination of a β-hydroxy dioxinone fragment and isatin dimethyl acetal generate oxa-spirooxindoles efficiently. These compounds are diversifiable scaffolds that tap into the rich chemistry of dioxinones.
The spirooxindole alkaloids are a class of natural products containing a privileged bicyclic architecture.1 Given their broad and interesting biological activities, convergent and stereoselective strategies to access this highly substituted system continue to fuel ongoing investigations of their therapeutic potential.2 Toward this goal, a variety of synthetic strategies have been developed for the pyrrolidine-indole and spirooxindole core, including Mannich reactions, oxidative rearrangements, MgI2-promoted ring expansions 1,3-dipolar cycloadditions, radical cyclizations, and intramolecular Heck reactions.3,4
A potentially promising subset of these natural products are the oxa-spirooxindoles which are an intriguing fusion of two priviledged motifs, the tetrahydropyranone and pyrrolidinyl spirooxindole substructures (Scheme 1). They are characterized by a spiro ring fusion at the 3-position of the oxindole core with varied substitution about the pyran and/or oxindole ring.5 General approaches to this target are considerably more limited6 and include a stereoselective vinylogous aldol reaction of vinyl malononitrile to isatin derivatives under mild conditions.7 In 2009, Porco disclosed Prins cyclizations with isatin ketals.8 Most recently, Shintani and Hiyashi have reported a palladium- catalyzed decarboxylative cyclization of γ-methylidene-δ-valerolactones with isatins.9 We envisioned that a catalytic approach that a) was modular with respect to the indole and pyran unit, and b) delivered oxa-spirooxindoles with high levels of selectivity (including control over absolute stereochemistry) would permit rapid construction of this bioactive motif.
Scheme 1.
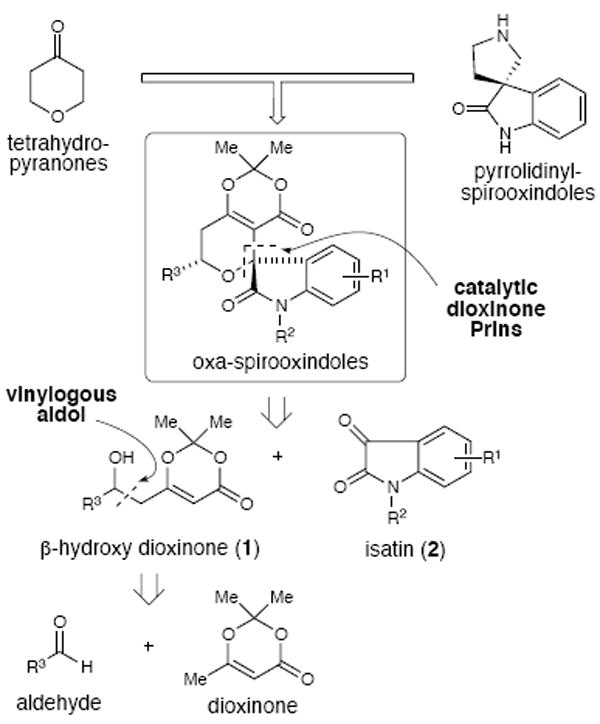
A modular strategy to the spirooxindole-pyrans
In 2005, we reported the Lewis acid-catalyzed condensation of β-hydroxy-dioxinones with aldehydes to produce 2,6-cis-disubstituted tetrahydropyran-4-ones.10 The β-hydroxy-dioxinones are useful building blocks and straightforward to prepare from aldehydes and dioxinone by known catalytic enantioselective vinylogous aldol reactions.11 In addition to the established reactions with dioxinones such as acylations,12 photochemistry13 and alkylations,14 this moiety possesses a deactivated enoltype alkene and provides a versatile synthetic handle in the form of a protected β-ketoester. The use of β-hydroxy dioxinone fragments in Prins-type chemistry is a component of our larger platform aimed at developing new methods toward the construction of oxygen-containing heterocycles2 and their subsequent application in total synthesis/chemical biology activities.15
During investigations to engage additional electrophiles beyond aldehydes for our tetrahydropyranone method, we uncovered that a β-hydroxy dioxinone underwent addition to acetone in the presence of a strong Lewis acid (BF3•OEt2) in a Prins-type process. Based on this observation, we initiated an examination of of isatins as potential electrophiles16 which could in turn provide facile access to oxa-spirooxindole structures.
At the onset, our overall strategy focused on a sequential (not tandem) combination of a general aldehyde, a three-carbon acetoacetate fragment (dioxinone) through a vinylous aldol followed by a Prins reaction with the resulting β-hydroxy dioxinone and isatin (2, Scheme 1). The three separate inputs in this reaction design would make it amenable to diversity-oriented synthesis.17 We first explored the reactivity of N-methylated isatin 3a with β-hydroxy-dioxinone 1a under catalytic scandium(III) triflate conditions, which resulted in no reaction (entry 1, Table 1).18 When utilizing a more robust Lewis acid such as BF3•OEt2, even stirring at 23°C for 12 hours only afforded a 4% yield of the desired tricyclic product (entry 2). We then decided to increase the reactivity of the N-methyl isatin substrate by converting it into the N-methyl isatin dimethyl ketal 3b, which would presumably generate a reactive oxocarbenium ion in situ under the acidic conditions. Unfortunately, the reaction with this new isatin derivative was still not promoted by scandium(III) triflate (entry 3), but BF3•OEt2 did afford the desired product 4a in 26% yield with excellent diastereoselectivity (entry 4). With this encouraging result, we performed a thorough Lewis acid screen19 and identified Sn(IV) or silyl triflates as good promoters of this reaction, but reducing the amounts below one equivalent for this reaction provided significantly reduced yields of the desired products.
Table 1.
Acid Optimization
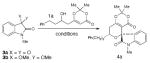 | ||||
|---|---|---|---|---|
| entry | 3 | conditions[a] | dr (%)[b] | yield (%)[c] |
| 1 | 3a | 20 mol % Sc(OTf)3/CaSO4, 0 °C, 4 h; 20 °C, 12 h | – | 0 |
| 2 | 3a | BF3•OEt2, 0 °C, 4 h; 23 °C, 12 h | 20:1 | 4 |
| 3 | 3b | 20 mol % Sc(OTf)3/CaSO4, 0 °C, 4 h; 20 °C, 12 h | – | 0 |
| 4 | 3b | BF3•OEt2, 0 °C, 4 h; 23 °C, 12 h | 20:1 | 26 |
| 5 | 3b | 5 mol % TfOH | 20:1 | 32 |
| 6 | 3b | 20 mol % TfOH | 20:1 | 52 |
| 7 | 3b | 20 mol % TfOH, 5 Å MS | 20:1 | 70 |
| 8 | 3b | 20 mol % TfOH, 5 Å MS[d] | 20:1 | 93 |
Reaction conditions (entries 1-6): 1a (1.0 equiv), 3a or 3b (1.0 equiv), CH2Cl2 (0.05 M), temperature and time as indicated; (entries 7-16): 1a (1.0 equiv), 3b (1.0 equiv), 0.05 M in CH2Cl2, 0 °C, 30 min; warm to 20 °C, 1 h;
Diastereoselectivity was determined from 1HNMR spectra of unpurified material.
Yield of isolated tricyclic product.
1.5 equivalents of dioxinone 1a.
We then turned our attention to employing catalytic amounts of Brønsted acids to facilitate this process. The reaction was not promoted by H3PO4, p-TSA•H2O, TFA or Tf2NH;20 however, 50 mol % of TfOH, H2SO4 and MeSO3H all catalyzed the reaction (data not shown). Our best result was achieved with 20 mol % of TfOH, 1 equivalent of isatin diketal 3b and 1.5 equivalents of dioxinone 1a, affording desired product 4a in a 93% yield with excellent levels of diastereoselectivity favoring the 2,6-cis isomer (entry 8). It is worth noting that addition of flame dried 5 Å molecular sieves to the reaction mixture was crucial to achieve the high yields for this process.
With these catalytic Brønsted conditions, we surveyed isatin ketal substrate scope and found that both electron-donating and withdrawing substituents are tolerated at either the 5 or 6 position when the nitrogen of the isatin was methyl or benzyl group-protected (Table 2, entries 1-7). The relative stereochemistry of these tricyclic products was determined by X-ray crystallographic analysis.21 In addition, the unprotected isatin substrate was compatible with the reaction conditions (entry 7), but no reaction was observed with more sterically demanding substrates, such as 4-bromo-isatin (data not shown). We then explored the substrate scope of the dioxinones and found that with alkyl (linear and branched) or aromatic substitution gave moderate to high yields (51-88%) with no adverse impact on diastereoselectivity of the resulting spiro-tricyclic product 4 (entries 8-15).
Table 2.
Substrate Scope
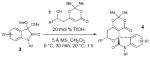 | |||||||
|---|---|---|---|---|---|---|---|
| entry | 3 | R1 | R2 | 1 | R3 | dr | yield (%) |
| 1 | 3b | H | Me | 1a | (CH2)2Ph | 20:1 | 93 (4a) |
| 2 | 3c | 5-OMe | Me | 1a | (CH2)2Ph | 20:1 | 89 (4b) |
| 3 | 3d | 6-Br | Me | 1a | (CH2)2Ph | 20:1 | 80 (4c) |
| 4 | 3e | H | Bn | 1a | (CH2)2Ph | 20:1 | 71 (4d) |
| 5 | 3f | 5-OMe | Bn | 1a | (CH2)2Ph | 20:1 | 94 (4e) |
| 6 | 3g | 6-Br | Bn | 1a | (CH2)2Ph | 20:1 | 81 (4f) |
| 7 | 3h | H | H | 1a | (CH2)2Ph | 20:1 | 72 (4g) |
| 8 | 3b | H | Me | 1b | Ph | 20:1 | 81 (4h) |
| 9 | 3b | H | Me | 1c | 3,5-(CH3O)2C6H3 | 20:1 | 63 (4i) |
| 10 | 3b | H | Me | 1d | cyclohexyl | 20:1 | 88 (4j) |
| 11 | 3b | H | Me | 1e | cyclopropyl | 20:1 | 66 (4k) |
| 12 | 3b | H | Me | 1f | CH2CO2CH3 | 20:1 | 69 (4l) |
| 13 | 3b | H | Me | 1g | (CH3)2CCH2OBn | 20:1 | 66 (4m) |
| 14 | 3b | H | Me | 1h | (CH2)2OBn | 20:1 | 51 (4n) |
| 15 | 3b | H | Me | 1i | (CH2)5N3 | 20:1 | 57 (4o) |
To shed light on the reaction mechanism of this diastereoselective process, we isolated the minor diastereomer of 4g from the reaction under TMSOTf conditions and submitted this diastereomer to the optimized catalytic acid conditions in Table 2. No conversion to major diastereomer 4g was observed, which supports that this Prins cyclization is a kinetically controlled process. The current rationale for the high diastereoselectivity observed in this reaction is shown in Scheme 2. The combination of the isatin ketal and β-hydroxy dioxinone leads to initial formation of the oxocarbenium ion intermediate. The aryl substituent of the oxindole moiety adopts a pseudo-equatorial position, syn to the R substituent of the dioxinone. There may also exist additional stabilization resulting from O (lone pair)–π interaction between the carbonyl of the dioxinone and the aryl ring of the isatin.22 The intervention of a 2-oxonia Cope [3,3]-sigmatropic process during Prins reaction could potentially epimerize the stereochemistry of the dioxinone under the reaction conditions.23 However, complete transfer of enantiomeric excess was observed (60% ee 1a with isatin 3b provides 4a in 60% ee).
Scheme 2.
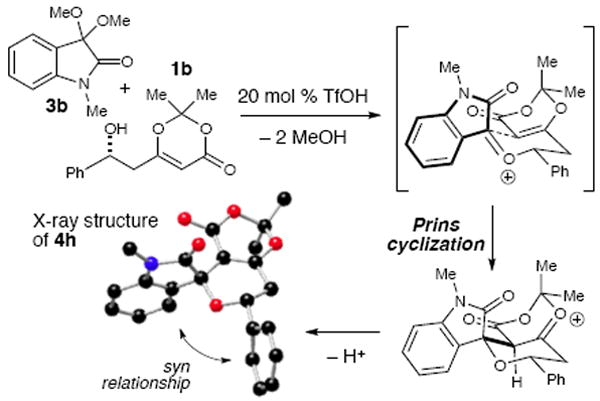
Proposed cyclization mechanism
By tapping into the embedded β-keto ester functional group of the new tricyclic dioxinone oxa-spirooxindoles, a variety of different structures can be accessed quickly. A thermally-initiated retro [4+2] cycloaddition allows access to a reactive acyl ketene intermediate (Scheme 3). When the tricyclic dioxinone 4h is heated in wet DMSO, a standard decarboxylation of the in situ generated β-keto acid affords tetrahydropyranone 5. Under similar thermal parameters, the dioxinone functional group of 4h can be exchanged for a thiouracil 6 by heating with thiourea (42%).24 Lastly, a simple acylation with methanol produces the β- keto ester (7). When methyl hyrdrazine is added after this esterification, the tricyclic pyrrolidinone (8) can be isolated in excellent yield for the two steps (81%).
Scheme 3.
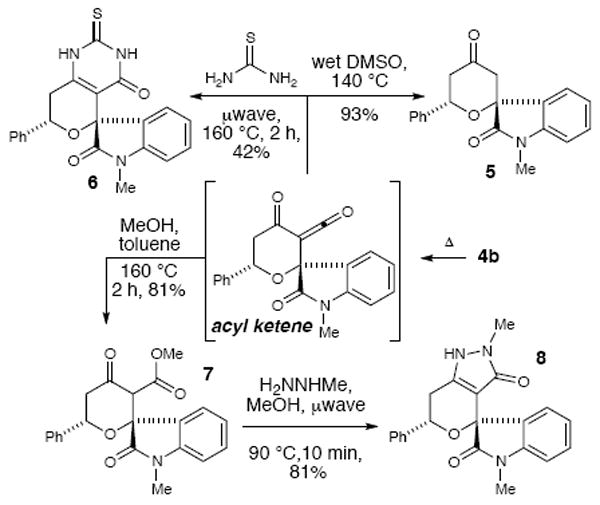
Diversification of tricyclic spirooxindole.
We have also explored different ketal structures beyond isatins (Scheme 4). For example, the combination of a β-hydroxy dioxinone and the dimethyl ketal of methyl benzyl formate under the Brønsted acid conditions produced trisubstituted tetrahydropyran 10a. In addition, we were able to obtain the pentacyclic product 10b by adding to the dimethyl ketal of 1,2-naphthylenedione. However, ketal substrates such as the dimethyl ketal of phenyl acetone or benzophenone are not suitable substrates. This lack of reactivity may be due to stabilization of the corresponding oxocarbenium intermediate generated with the adjacent phenyl (or bisphenyl) groups. The lack of an adjacent, destabilizing electron withdrawing group as with the isatins may impede C–C bond formation to close the THP ring. Further exploration of the α-keto substrate scope is currently underway and will provide more insight into the expanded potential of this method.
Scheme 4.
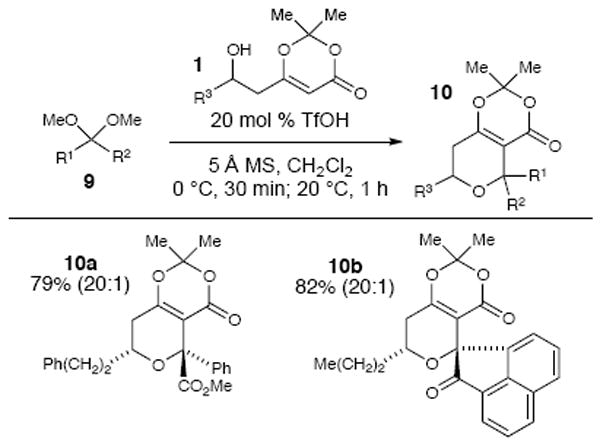
Additional substrates.
In summary, we have developed a modular and highly stereoselective approach to construct spirooxindole pyrans in high yield with catalytic Brønsted acid. The overall combination of an aldehyde, an acetoacetate fragment (dioxinone) and isatin provides an efficient and diastereoselective route for diversified products with complete transfer of stereochemistry from the β-hydroxy intermediate to the products. The high levels of 2,6-cis selectivity observed for this catalytic process is attributed to the equatorial preference of the oxindole moiety. This study sets the stage for the generation of focused libraries for screening efforts geared toward leveraging small compound synthesis for advances in biomedical research. Efforts to realize this potential and advance dioxinone-driven Prins methods in new directions are ongoing in our laboratory.
Supplementary Material
Acknowledgments
Support for this work was provided by the National Institutes of General Medical Sciences (NIGMS P50 GM086145-01) and the National Cancer Institute (R01 CA126827), Amgen, GlaxoSmithKline and AstraZeneca. E.A.C. thanks the Malkin Scholars Program for support (Robert H. Lurie Comprehensive Cancer Center at Northwestern University).
Footnotes
Supporting Information Available Experimental procedures and spectral data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Mander L, Liu H-W. Natural Products Structural Diversity II. Vol. 2 Elsevier; 2010. [Google Scholar]
- 2.(a) Marti C, Carreira EM. Eur J Org Chem. 2003:2209. [Google Scholar]; (b) Galliford CV, Scheidt KA. Angew Chem Int Ed. 2007;46:8748. doi: 10.1002/anie.200701342. [DOI] [PubMed] [Google Scholar]; (c) Trost BM, Brennan MK. Synthesis. 2009:3003. [Google Scholar]
- 3.Alper PB, Meyers C, Lerchner A, Siegel DR, Carreira EM. Angew Chem Int Ed. 1999;38:3186.Galliford CV, Martenson JS, Stern C, Scheidt KA. Chem Commun. 2007;631 doi: 10.1039/b609155e.Nair V, Mathai S, Augustine A, Viji S, Radhakrishnan KV. Synthesis. 2004:2617.Zhang Y, Wang L, Zhang M, Fun HK, Xu JH. Org Lett. 2004;6:4893. doi: 10.1021/ol048028t.Alcaide B, Almendros P, Rodriguez-Acebes R. J Org Chem. 2006;71:2346. doi: 10.1021/jo0525027.Overman L, Watson D. J Org Chem. 2006;71:2587. doi: 10.1021/jo052335a. and references therein.
- 4.(a) Chen T, Xu X-P, Ji S-J. J Comb Chem. 2010;12:659. doi: 10.1021/cc100064c. [DOI] [PubMed] [Google Scholar]; (b) Chen X, Wei Q, Luo S, Xiao H, Gong L. J Am Chem Soc. 2009;131:13819. doi: 10.1021/ja905302f. [DOI] [PubMed] [Google Scholar]; (c) Companyó X, Zea A, Alba A-NR, Mazzanti A, Moyano A, Rios R. Chem Commun. 2010;46:6953. doi: 10.1039/c0cc01522a. [DOI] [PubMed] [Google Scholar]; (d) Sridhar R, Srinivas B, Madhav B, Reddy V, Nageswar Y, Rao K. Can J Chem. 2009;87:1704. [Google Scholar]; (e) Helan V, Mills A, Drewry D, Grant D. J Org Chem. 2010;75:6693. doi: 10.1021/jo101077g. [DOI] [PubMed] [Google Scholar]
- 5.Nair V, Mathai S, Mathew SC, Rath NP. Tetrahedron. 2005;61:2849. [Google Scholar]
- 6.Basavaiah D, Rao JS, Reddy RJ, Rao AJ. Chem Commun. 2005;2621 doi: 10.1039/b500224a. [DOI] [PubMed] [Google Scholar]
- 7.Babu TH, Karthik K, Perumal PT. Synlett. 2010;1128 [Google Scholar]
- 8.Castaldi MP, Troast DM, Porco JA. Org Lett. 2009;11:3362. doi: 10.1021/ol901201k. For a related formal [5+2] transformation with isatin ketals, see, Zhang Y, Panek JS. Org Lett. 2009;11:3366. doi: 10.1021/ol901202t.
- 9.(a) Shintani R, Hayashi S, Murakami M, Takeda M, Hayashi T. Org Lett. 2009;11:3754. doi: 10.1021/ol901348f. [DOI] [PubMed] [Google Scholar]; (b) Shintani R, Tsuji T, Park S, Hayashi T. J Am Chem Soc. 2010;132:7508. doi: 10.1021/ja1023223. [DOI] [PubMed] [Google Scholar]
- 10.Morris WJ, Custar DW, Scheidt KA. Org Lett. 2005;7:1113. doi: 10.1021/ol050093v. [DOI] [PubMed] [Google Scholar]
- 11.(a) Singer RA, Carreira EM. J Am Chem Soc. 1995;117:12360. [Google Scholar]; (c) Villano R, Acocella MR, De Rosa M, Soriente A, Scettri A. Tetrahedron: Asymmetry. 2004;15:2421. [Google Scholar]
- 12.(a) Boeckman RK, Pruitt JR. J Am Chem Soc. 1989;111:8286. [Google Scholar]; (b) Boeckman RK, Shao PC, Wrobleski ST, Boehmler DJ, Heintzelman GR, Barbosa AJ. J Am Chem Soc. 2006;128:10572. doi: 10.1021/ja0581346. [DOI] [PubMed] [Google Scholar]; (c) Calo F, Richardson J, Barrett AGM. Org Lett. 2009;11:4910. doi: 10.1021/ol901979x. [DOI] [PubMed] [Google Scholar]
- 13.(a) Dritz JH, Carreira EM. Tetrahedron Lett. 1997;38:5579. [Google Scholar]; (b) Winkler JD, Bowen CM, Liotta F. Chem Rev. 1995;95:2003. [Google Scholar]
- 14.Seebach D, Misslitz U, Uhlmann P. Chem Ber. 1991;124:1845. [Google Scholar]
- 15.(a) Crane EA, Scheidt KA. Angew Chem Int Ed. 2010;49:8316. doi: 10.1002/anie.201002809. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Custar DW, Zabawa TP, Hines J, Crews CM, Scheidt KA. J Am Chem Soc. 2009;131:12406. doi: 10.1021/ja904604x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Custar DW, Zabawa TP, Scheidt KA. J Am Chem Soc. 2008;130:804. doi: 10.1021/ja710080q. [DOI] [PubMed] [Google Scholar]; (d) Farmer RL, Biddle MM, Nibbs AE, Huang X, Bergan RC, Scheidt KA. ACS Med Chem Lett. 2010;1:400. doi: 10.1021/ml100110x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) da Silva JFM, Garden SJ, Pinto AC. J Braz Chem Soc. 2001;12:273. [Google Scholar]; (b) Vyas DJ, Frohlich R, Oestreich M. J Org Chem. 2010;75:6720. doi: 10.1021/jo101420e. [DOI] [PubMed] [Google Scholar]; (c) Lai H, Huang Z, Wu Q, Qin Y. J Org Chem. 2008;74:283. doi: 10.1021/jo802036m. [DOI] [PubMed] [Google Scholar]; (d) Toullec PY, Jagt RBC, de Vries JG, Feringa BL, Minnaard AJ. Org Lett. 2006;8:2715. doi: 10.1021/ol0608101. [DOI] [PubMed] [Google Scholar]; (e) Tomita D, Yamatsugu K, Kanai M, Shibasaki M. J Am Chem Soc. 2009;131:6946. doi: 10.1021/ja901995a. [DOI] [PubMed] [Google Scholar]; (f) Nair V, Rajesh C, Dhanya R, Rath NP. Tetrahedron Lett. 2002;43:5349. doi: 10.1021/ol025503j. [DOI] [PubMed] [Google Scholar]; (g) Smet M, Van Oosterwijck C, Van Hecke K, Van Meervelt L, Vandendriessche A, Dehaen W. Synlett. 2004:2388. [Google Scholar]; (h) Liang B, Kalidindi S, Porco JA, Stephenson CRJ. Org Lett. 2010;12:572. doi: 10.1021/ol902764k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burke MD, Berger EM, Schreiber SL. J Am Chem Soc. 2004;126:14095. doi: 10.1021/ja0457415. and references therein.
- 18.1a and related compounds can be prepared in high ee for some cases, see: Singer RA, Carreira EM. J Am Chem Soc. 1995;117:12360.De Rosa M, Acocelli MR, Villano R, Soriente A, Scettri A. Tetrahedron: Asymmetry. 2003;14:2499.Custar DW, Zabawa TP, Hines J, Crews CM, Scheidt KA. J Am Chem Soc. 2009;131:12406. doi: 10.1021/ja904604x. However, for this study, we employed racemic material. For a report of scandium(III)-catalyzed additions to isatins, see: Hanhan NV, Sahin AH, Chang TW, Fettinger JC, Franz AK. Angew Chem Int Ed. 2010;49:744. doi: 10.1002/anie.200904393.
- 19.In addition to the Lewis and Bronsted acids shown Table 1, the following were examined and provided inferior results: TiCl4, SnCl4, ZnCl2, Zn(OTf)2, Mg(OTf)2, Sm(OTf)3, La(OTf)3, TMSOTf, TMSCl, and HNTf2.
- 20.(a) Ishihara K, Hiraiwa Y, Yamamoto H. Synlett. 2001:1851. [Google Scholar]; (b) Yamamoto H, Futatsugi K. Angew Chem Int Ed. 2005;44:1924. doi: 10.1002/anie.200460394. [DOI] [PubMed] [Google Scholar]
- 21.See Supporting Information for details and CCDC numbers.
- 22.Gung BW, Xue X, Reich HJ. J Org Chem. 2005;70:7232. doi: 10.1021/jo050874+. [DOI] [PubMed] [Google Scholar]
- 23.Roush WR, Dilley GJ. Synlett. 2001:955.Jaber JJ, Mitsui K, Rychnovsky SD. J Org Chem. 2001;66:4679. doi: 10.1021/jo010232w. Racemization was not observed in Porco’s recent Prins reactions with isatins, see reference 8a.
- 24.We thank Dr. James Leahy (Exelixis) for this suggestion.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.