

Abstract
The symptoms of schizophrenia involve profound dysfunction of the prefrontal cortex (PFC). PFC networks create our “mental sketch pad”, and PFC dysfunction contributes to symptoms such as cognitive deficits, thought disorder, delusions and hallucinations. Neuropathological studies of schizophrenia have shown marked loss of dendritic spines in deep layer III, the sublayer where PFC microcircuits reside. The microcircuits consist of recurrent excitatory pyramidal cell networks that interconnect on spines, and excite each other via NMDA receptor signaling. The pyramidal cell persistent firing is sculpted by lateral inhibition from GABAergic basket and chandelier cells, thus creating tuned, persistent firing needed for accurate representational knowledge (i.e. working memory). The strength of pyramidal cell network connections is markedly and flexibly altered by intracellular signaling pathways in dendritic spines, a process called Dynamic Network Connectivity (DNC). DNC proteins such as HCN channels are concentrated on dendritic spines in deep layer III. Under optimal conditions, network inputs to pyramidal cells are strengthened by noradrenergic alpha-2A inhibition of cAMP-HCN channel signaling, and sculpted by dopamine D1-cAMP-HCN channel weakening of inappropriate inputs. However, with stress exposure, high levels of cAMP-HCN channel signaling produces a collapse in network firing. With chronic stress exposure, spines reduce in size and are lost, and this process involves increased PKC signaling. Importantly, molecules that normally strengthen PFC networks connections and/or reverse the stress response, are often genetically altered in schizophrenia. As exposure to stress is a key factor in the precipitation of schizophrenic symptoms, these dysregulated signaling pathways in deep layer III may interact with already vulnerable circuitry to cause spine loss and the descent into illness.
Overview
Schizophrenia is associated with altered prefrontal cortical (PFC) circuits, arising from both developmental insults in utero (e.g. as discussed in this issue), and continuing in the mature brain, e.g. with waves of gray matter loss in late adolescence and adulthood (Vidal et al., 2006; Sun et al., 2008) (Figure 1A). Exposure to stress is a key factor in the precipitation of symptoms in adolescence and in subsequent exacerbation of symptoms (Breier et al., 1991; Nuechterlein et al., 1992; Miller et al., 2001; Dawson et al., 2010), suggesting that the environment can interact with already vulnerable circuitry to aggravate cortical deterioration. The current review focuses on mechanisms that may contribute to PFC network weakening in young adulthood, and the signaling pathways that contribute to dendritic spine loss in response to stress.
Figure 1.
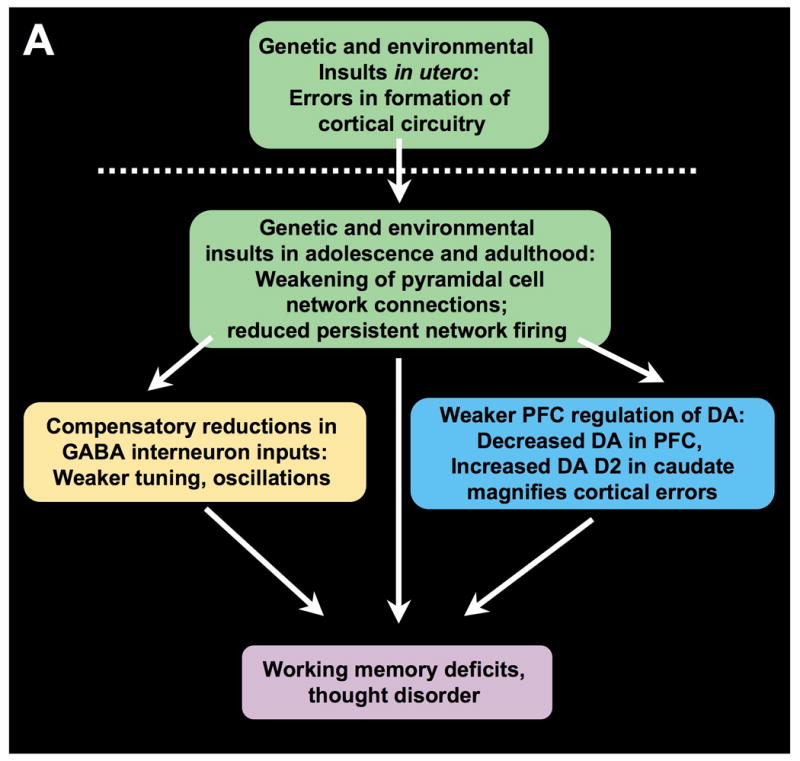
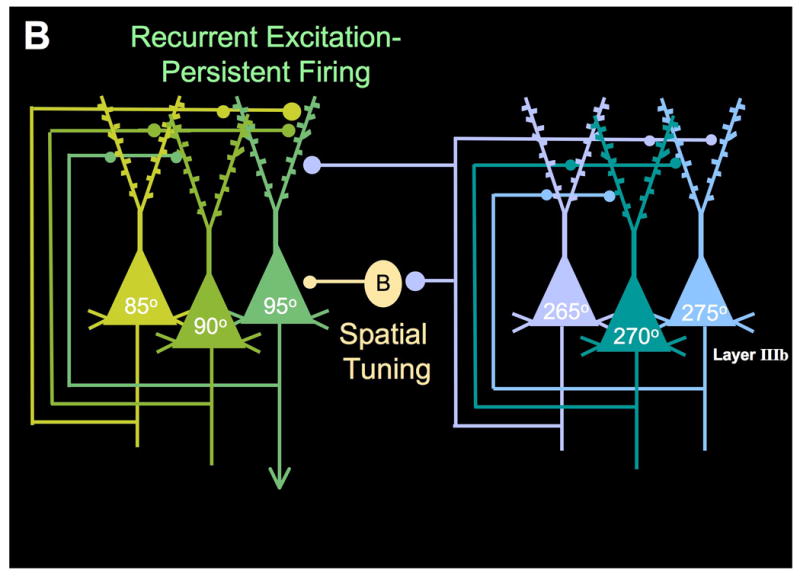
Prefrontal cortical circuitry and schizophrenia. A. An overview of a possible progression of brain changes in schizophrenia. Genetic and/or environmental insults alter the precise formation of cortical pyramidal cell microcircuits in utero. These insults continue to weaken NMDA-mediated, pyramidal cell connections in the maturing brain, particularly during adolescence when there are normal rearrangements of cortical circuitry. Layer III pyramidal cell recurrent microcircuits may be particularly vulnerable, as this layer is the focus of spine loss in schizophrenia. Loss of NMDA microcircuit activity leads to a variety of compensatory changes, including reduction in GABAergic actions, reduced DA inputs to PFC, and increased DA inputs to caudate. Reductions in GABA may contribute to weaker oscillatory activity in brain. Reductions in both GABA and D1 receptor stimulation in PFC would erode information processing in PFC, increasing inappropriate network inputs. Increased DA inputs in caudate magnify cortical errors to contribute to thought disorder, hallucinations and delusions. Adapted from Lewis and Gonzalez-Burgos, 2006, with emphasis on vulnerability of pyramidal cell recurrent connections. B. The layer III PFC microcircuits that subserve spatial working memory in primate dorsolateral PFC. Based on the work of Goldman-Rakic and colleagues (Goldman-Rakic, 1995).
We have discovered powerful intracellular signaling pathways that rapidly alter the strength of PFC network connections, a process termed Dynamic Network Connectivity (DNC) (Arnsten et al., 2010). Sustained activation of protein kinase C (PKC) and cAMP signaling pathways during chronic stress exposure contributes to dendritic spine loss from layer III pyramidal cells, where DNC proteins are concentrated (Arnsten, 2009). Deep layer III is also the site of PFC pyramidal cell recurrent connections (Kritzer and Goldman-Rakic, 1995; González-Burgos et al., 2000), and the sublayer where spine loss is particularly evident in schizophrenia (Glantz and Lewis, 2000). We posit that recurrent pyramidal cell network synapses are particularly fragile, and that genetic insults-although expressed globally- may have greater ramifications at this vulnerable site. Understanding the processes that modulate these synaptic connections may provide the opportunity to protect PFC gray matter and prevent the descent into severe illness.
Prefrontal cortical dysfunction in schizophrenia
The PFC guides behavior, thought and emotion using representational knowledge, a process often referred to as working memory (Goldman-Rakic, 1995). This ability to represent, maintain and manipulate information that is no longer in the environment is thought to be the foundation of abstract thought, and the neural basis of our “mental sketch pad”. As eloquently described by Fuster (Fuster, 2008), persistent firing by PFC neurons is able to “wed the past to the future” (so-called cross-temporal mediation; Fuster, 2000), allowing us to plan ahead for the end of a sentence or the start of a career. The PFC is critical for flexible, high order decision-making (Lee et al., 2007) and top-down regulation of attention (Knight et al., 1995; Buschman and Miller, 2007), including the suppression of interference (Thompson-Schill et al., 2002), set-shifting (Robbins, 2007), and the ability to sustain attention over a long delay (Wilkins et al., 1987). Studies of human subjects also have discovered remarkable roles for PFC circuits in metacognition and insight, including feedback about what is real (Simons et al., 2008), what we know (Jurado et al., 1998), and whether we are making errors (Modirrousta and Fellows, 2008).
Patients with schizophrenia exhibit profound deficits in PFC function that are fundamental components of this illness (Weinberger et al., 1986; Barch, 2005; Keedy et al., 2006; Keefe et al., 2006). For example, reduced activity in the right PFC during a challenging working memory task highly correlates with symptoms of thought disorder (Perlstein et al., 2001). A patient with severe limits in working memory abilities would be unable to hold the initial goal of a sentence or thought long enough to sustain its conclusion through the distraction of competing associations. Foresight also has been positively associated with gray matter volume in the right PFC of patients with schizophrenia (Eack et al., 2008). Patients sometimes describe a lack of foresight as an agonizing experience of being trapped in an eternal present, being unable to envision even the near future, much like a small child who finds it intolerable to wait (“Are we there yet?”). This impairment in cross-temporal mediation (Fuster, 2000) may be present in both young children with immature PFC function, and in patients, where PFC has become dysfunctional.
The so-called “positive” symptoms of schizophrenia also involve PFC dysfunction. Reduced activity of the right PFC is associated with delusional thinking (Corlett et al., 2007). There is also evidence that auditory hallucinations involve impaired communication between the left PFC and Wernicke's area (Ford et al., 2002). Normally, the PFC emits a signal, termed efference copy (or corollary discharge) to tag a response as internally generated. This suppresses activity in Wernicke's area during inner speech, and assigns the voice as self-generated (Ford et al., 2002). Aberrations in corollary discharge may interfere with this process and misassign voices to an external source. Thus, there are likely many ways in which dysfunctional PFC circuits contribute to symptoms of schizophrenia.
Recurrent excitatory networks: the cellular basis of working memory
Extensive research has identified the PFC microcircuits that underlie spatial working memory in monkeys (Figure 1B). The ability to represent a visuospatial position arises from recurrent excitation between pyramidal cells in deep layer III of area 46 (Kritzer and Goldman-Rakic, 1995), the dorsolateral PFC surrounding the caudal principal sulcus (Goldman-Rakic, 1995). This PFC region receives highly processed visuospatial information from the parietal association cortex, and even small lesions to this area produce permanent deficits in spatial working memory abilities (Funahashi et al., 1993). The recurrent excitatory circuits interconnect on dendritic spines (e.g. Figure 2), which have recently been shown to depend on NMDA receptor stimulation (Arnsten et al., 2010). Thus, pyramidal cells with similar spatial characteristics excite each other to keep information “in mind” over a delay period when there is no information available in the environment.
Figure 2.
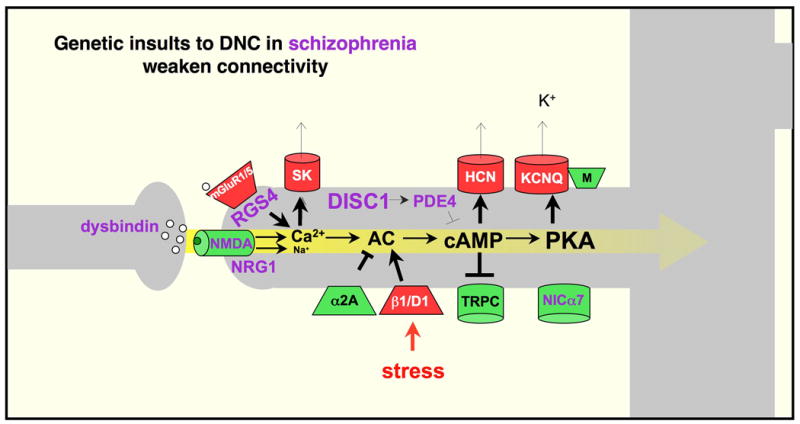
Some of the DNC signaling pathways that regulate PFC network strength, and their relationship to schizophrenia; mechanisms that strengthen connectivity are shown in green; those that weaken connectivity are shown in red. Genetic alterations in patients with schizophrenia frequently involve DNC proteins that normally serve to strengthen pyramidal cell connections, and to inhibit stress signaling pathways (shown in purple). Thus, these genetic insults would weaken PFC network connections and increase vulnerability to stress exposure. Adapted from (Arnsten et al., 2010).
Representation of a visuospatial position also requires spatial tuning, such that the neuron fires to the memory of one region of visuospace but not to others. This is accomplished in part through lateral inhibition by GABAergic interneurons (Rao et al., 1999; Rao et al., 2000). In particular, parvalbumin-containing basket cells and interneurons provide spatial tuning, as shown in Figure 1B. Thus, inhibition of GABAa receptors leads to noisy neuronal response to all spatial directions (Rao et al., 1999; Rao et al., 2000).
Dendritic spine loss in layer III of patients with schizophrenia
Neuropathological studies of dorsolateral PFC from patients with schizophrenia have shown reduced neuropil (Selemon et al., 1998) and loss of dendritic spines (Glantz and Lewis, 2000), particularly in deep layer III where PFC recurrent microcircuits reside. Lewis and Gonzalez-Burgos (Lewis and Gonzalez-Burgos, 2006) have described a cascade of primary genetic insults to PFC pyramidal cell circuits, followed by compensatory events that may combine to produce the symptoms of schizophrenia (a variation on this scheme is presented in Figure 1A):
Neurodevelopmental errors caused by genetic and/or environmental insults as the cortex develops in utero, leading to the formation of abnormal PFC circuitry, followed by
progressive PFC spine loss in adolescence and continuing into adulthood, leading to greatly reduced pyramidal cell network excitation. The weakening of pyramidal cell network activity would then lead to a number of compensatory changes:
weakening of specific (parvalbumin-containing) GABA synapses due to loss of excitatory drive from pyramidal cell networks. The expression of the synthetic enzyme for GABA, GAD67, is dependent on NMDA stimulation from pyramidal cell networks (Kinney et al., 2006), and GAD67 is markedly reduced in the PFC of patients with schizophrenia (Volk et al., 2000). The reduction in GABA would in turn reduce neuronal network tuning, and thus make information noisier;
reduced cortical drive on DA neurons projecting to PFC. As described below, this would further erode spatial tuning, and alter the timing of corollary discharge. A compensatory increase in D1 receptors in PFC may also magnify the stress response and contribute to spine loss, also decribed below; and
increased DA release in caudate, which would magnify cortical inputs and PFC network errors.
It is also possible that the primary genetic insult in some families could afflict GABA interneurons (Gonzalez-Burgos et al., 2010), leading to compensatory reductions in NMDA signaling and the emergence of a similar phenotype. Indeed, a major challenge for current research is to determine how so many different genetic insults can all lead to similar neuropathology and symptoms. In this context, it is remarkable that a large number of already-identified genetic insults in schizophrenia are DNC proteins that regulate the synaptic strength of recurrent PFC network connections (Arnsten et al., 2010). Specifically, schizophrenia is often associated with alterations in molecules that normally serve to strengthen connections, and to protect PFC networks from the detrimental effects of stress.
Molecular regulation of PFC excitatory networks: Dynamic Network Connectivity
Recent research has revealed that the physiological strength of PFC network connections dynamically changes in coordination with arousal state. Molecular signaling events in the spines of PFC pyramidal dendrites can open or close ion channels near synaptic connections to rapidly and reversibly alter the strength of network inputs (Wang et al., 2007). Importantly, these DNC signaling proteins are concentrated in deep layer III (Arnsten et al., 2010), the site of PFC microcircuits (Kritzer and Goldman-Rakic, 1995), and of the greatest spine loss in schizophrenia (Glantz and Lewis, 2000). We propose that these molecular events, in combination with the challenge of sustaining neural activity in the absence of “bottom-up” environmental stimulation, make layer III PFC recurrent connections the “weakest link” in higher cognitive functioning (Arnsten et al., 2010).
A schematic illustration of DNC mechanisms is presented in Figure 2, whereby specific network connections onto spines can be weakened via calcium and/or cAMP-PKA increasing the open state of potassium channels localized on spines near excitatory, NMDA synapses. The ability to rapidly and reversibly weaken a network connection may serve several purposes. Under optimal arousal conditions, a specific subset of network inputs can be weakened (e.g. as with dopamine D1 receptor stimulation described below), to dynamically alter the breadth of network inputs based on current cognitive demands. Global weakening of network inputs may also serve key roles in 1) providing negative feedback to constrain over-excitability in recurrent excitatory circuits, 2) conserving energy under conditions of fatigue, and 3) rapidly taking PFC “offline”, in response to danger, to switch control of behavior to more primitive brain regions that mediate instinctive reactions. For example, high levels of catecholamine release during stress exposure drives the production of cAMP (Fig 2), which disconnects PFC networks but strengthens the emotional and habitual responses of the amygdala and basal ganglia (Arnsten, 2009). The loss of PFC cognitive function with even mild, uncontrollable stress has now been observed in both animals and humans, and likely contributes to a broad range of mental illnesses (reviewed in (Arnsten, 2009).
There are also DNC mechanisms that inhibit the stress response and strengthen network connections. PFC connections are strengthened when cAMP signaling is inhibited, potassium channels close, and other, depolarizing channels (e.g. TRPC) are opened. For example, noradrenergic stimulation of alpha-2A receptors strengthens network firing during the delay by inhibiting cAMP-HCN channel signaling in spines, preferentially increasing the neuron's response to its preferred direction (Wang et al., 2007). This specific pattern of response is thought to arise from selective localization of alpha2A receptors on spines receiving inputs from pyramidal cells with shared tuning properties (schematically diagrammed in Figure 3). Similarly, DISC1 (Disrupted In Schizophrenia) is found in spines in the primate PFC (Kirkpatrick et al., 2006), and is known to regulate cAMP through interactions with the phosphodiesterases, enzymes that catabolize cAMP (Millar et al., 2007; Murdoch et al., 2007). The ability to precisely control the amount and location of cAMP actions within a PFC pyramidal cell may define the quality and extent of its network connections, and thus the cellular basis of cognitive content.
Figure 3.
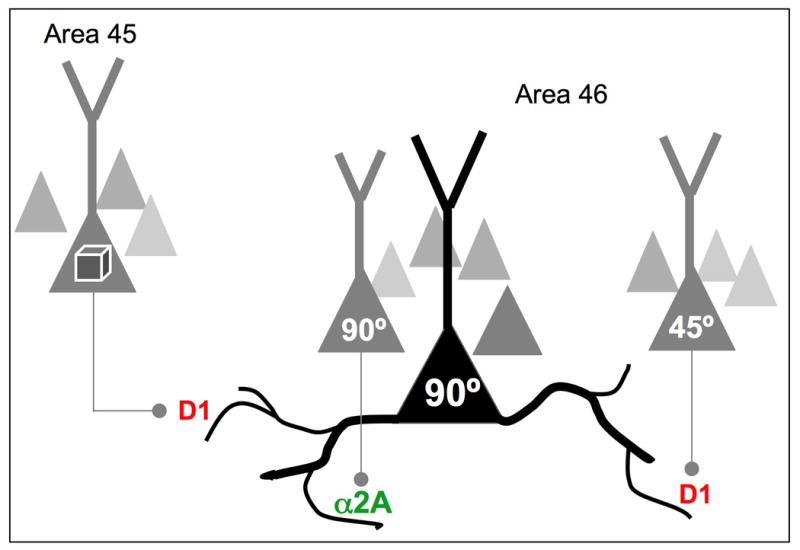
Recordings from PFC neurons indicate that network inputs from neurons with similar vs. dissimilar tuning characteristics are modulated differently. Noradrenergic stimulation of α2A-AR strengthens delay-related firing for a neuron's preferred direction, suggesting that these receptors modulate network inputs from neurons with shared stimulus properties (e.g. as shown in this example, strengthening inputs from other 90° neurons). In contrast, dopamine D1 signaling can weaken network connections from neurons with dissimilar characteristics, (e.g. weakening inputs from a 45° neuron). We have proposed that D1 receptor stimulation may also weaken network inputs from other cortical areas, (e.g. visual feature information arriving from area 45), thus determining the breadth of network inputs. Based on (Arnsten et al., 2009).
Dopamine D1 Receptor Stimulation: The Double-edged Sword
Dopamine D1 receptors are localized on dendritic spines near excitatory network inputs in the primate dorsolateral PFC (Smiley et al., 1994). For over 20 years, it has been appreciated that D1 receptor stimulation produces an inverted U dose response, whereby either too little or too much impairs working memory performance (Arnsten and Goldman-Rakic, 1990; Arnsten et al., 2009). Total blockade of D1 receptors appears to reduce all neuronal firing, likely due to the loss of fundamental excitatory influences (reviewed in Arnsten et al., 2009). However, D1 receptors also have powerful effects on PFC network inputs. Recent studies show that DA D1 receptor stimulation weakens network inputs via increased cAMP signaling (Vijayraghavan et al., 2007). Under optimal conditions during a spatial working memory task, D1 receptor stimulation weakens the neuron's response to nonpreferred spatial directions, and thus reduces “noise” and sharpens spatial tuning (Vijayraghavan et al., 2007). In vitro recordings from PFC slices in both rat (Gorelova et al., 2002) and monkey (Kroner et al., 2007) indicate that sculpting actions may also arise from D1 activation of fast-spiking interneurons, and possible reductions in presynaptic glutamate release (Gao et al., 2001). All of these mechanisms may combine to restrict neuronal firing. Thus, inadequate D1 receptor stimulation leads to noisy neuronal firing, with PFC neurons firing during the delay to the memory of all spatial directions (Vijayraghavan et al., 2007). However, under conditions of very high levels of D1 receptor stimulation- as occurs during stress exposure (Murphy et al., 1996)- all PFC networks are disconnected, and all memory-related firing is suppressed (Vijayraghavan et al., 2007). Neuronal firing patterns are restored via inhibition of cAMP signaling (ibid). These powerful D1 actions may contribute to the rapid loss of dorsolateral PFC functions with exposure to even mild, uncontrollable stressors.
Although spatial working memory for a small region of visuospace benefits from D1 sculpting actions, other PFC cognitive operations that rely on broad network inputs can actually be harmed by dopamine D1 receptor actions (Arnsten et al., 2009). For example, attentional set-shifting likely requires broad inputs, and this ability is impaired by D1 receptor actions in PFC (Granon et al., 2000). These data indicate that dopamine D1 sculpting actions must be dynamically regulated according to momentary cognitive demands to provide optimal modulation, e.g. slightly higher dopamine release for tasks requiring narrow tuning (e.g remembering 90°, as shown in Figure 3), and less dopamine release for cognitive challenges needing wider network inputs (combining 90° and 45°, or combining spatial and feature information, as shown in Figure 3). These data may also explain why insightful, “out-of-the box” solutions are more likely to arise when a subject has no pressure to perform (Subramaniam et al., 2008; Arnsten et al., 2009). Altogether, the dopamine D1 receptor findings highlight the need for precise regulation of network connections, and thus the need for precise regulation of D1-cAMP signaling.
Chronic stress: Sustained weakening of PFC network connections leads to spine loss
While acute stress induces a transient, physiological weakening of PFC network connections, exposure to chronic stress additionally induces architectural changes in PFC pyramidal cells. Chronic stress causes dendritic retraction and loss of spines, and these structural changes are associated with impaired PFC function (Radley et al., 2008), including severe working memory loss (Hains et al., 2009). Weakened PFC connectivity has even been seen in humans undergoing a mild, sustained stress (Liston et al., 2009). Spine loss can also be induced in PFC through chronic exposure to corticosterone, and is associated with reduced NMDA receptors and BDNF levels (Gourley et al., 2009), suggesting that some aspects of the stress response may be mediated through this hormone. In normal subjects, these architectural changes in animals and humans reverse once the stressor is removed (Liston et al., 2009). However, environmental and genetic insults can reduce resilience to stress exposure. For example, in studies of rodent PFC, architectural changes have been observed in the adult offspring of mothers who were stressed during pregnancy (Murmu et al., 2006).
The signaling events that underlie stress-induced spine loss are beginning to be explored. Sustained elevation of protein kinase C (PKC) signaling may contribute, as increased PKC activity has been observed in PFC in response to stress exposure (Birnbaum et al., 2004). In vitro studies of hippocampal pyramidal cells have shown that sustained activation of PKC induces spine loss through phosphorylation of MARCKS, unanchoring MARCKS from the membrane, which in turn leads to collapse of the spine's actin cytoskeleton (Calabrese and Halpain, 2005) (schematically illustrated in Figure 4). Similar results were observed in vivo in the rat PFC, where inhibition of PKC signaling prevented spine loss and protected working memory performance during chronic stress exposure (Hains et al., 2009). Sustained elevations in PKC signaling also induce actin collapse in growth cones (Torreano et al., 2005) and microspikes (Yokoyama et al., 1998), the latter via pMARCKS as well.
Figure 4.
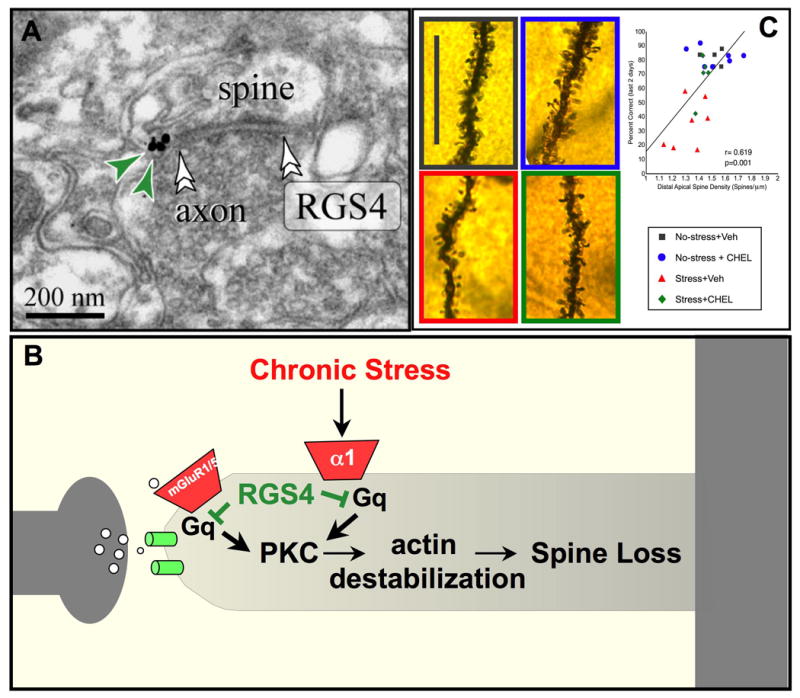
RGS4 inhibition of PKC signaling may protect PFC gray matter from the detrimental effects of chronic stress exposure. A. RGS4 is abundant in PFC neurons, and is localized near synapses in dendritic spines in monkey dorsolateral PFC (Paspalas et al., 2009). B. A possible signaling mechanism whereby RGS4 inhibits Gq-PKC signaling in spines. Sustained increases in PKC signaling are known to cause collapse of the actin cytoskeleton and spine loss via phosphorylation of MARCKS, which in turn unanchors actin from the membrane (see text). C. Exposure to chronic stress induces working memory deficits and spine loss from layer III PFC pyramidal cells in rats. Inhibition of PKC signaling during stress exposure protects working memory and dendritic spines, and there is a significant correlation between these measures. Thus, loss of RGS4 in schizophrenia may leave patients more vulnerable to spine loss. Black= control conditions; Red= saline treatment during chronic stress exposure; Blue = PKC inhibition under control conditions, Green = PKC inhibition during chronic stress exposure. Adapted from (Hains et al., 2009).
Preliminary evidence indicates that inhibition of cAMP signaling may also protect PFC structure and function from chronic stress (Arnsten, unpublished). It is not known how sustained elevation of cAMP signaling leads to spine loss rather than enlargement in PFC. Like PKC, increased cAMP-PKA signaling can defibrillize actin in microspikes (Fleming et al., 2004), perhaps through unanchoring actin via FAK (Serrels et al., 2010). The findings that elevated PKC and cAMP signaling may cause defibrillation of actin in both microspikes and PFC spines during stress exposure suggests these very different cellular processes may share some essential regulatory processes (although PKA reduces actin filaments in microspikes by inhibiting RhoA signaling (Fleming et al., 2004), while activation of RhoA signaling causes spine loss in hippocampal neurons (Sfakianos et al., 2007). Interestingly, microspikes formation relies on CDC42 signaling, a molecule that is reduced in layers III-VI in the dorsolateral PFC of patients with schizophrenia (Hill et al., 2006; Ide and Lewis, 2010). It is possible that reductions in CDC42 interact with DNC stress pathways in layer III to contribute to preferential spine loss in layer III in patients.
Spine loss may also occur through reductions in NMDA receptor actions. Stresslike neurochemical conditions markedly suppress neuronal firing in the monkey PFC (Birnbaum et al., 2004; Vijayraghavan et al., 2007). If these conditions are sustained in chronic stress, large reductions in network firing would lead to reduced glutamate stimulation of NMDA receptors. Recent studies have shown that decreases in NMDA signaling lead to spine shrinkage via DISC1 sequestration of Kalirin-7 in the synapse and inhibition of rac1 signaling (Hayashi-Takagi et al., 2010). It is likely that multiple, interacting pathways lead to spine loss during stress, and understanding these molecular events will be key to rescuing gray matter loss in patients with schizophrenia.
Genetic insults to “molecular brakes” on stress pathways in schizophrenia
A variety of signaling events can strengthen PFC network connections and/or provide brakes on the stress response to restore PFC network connectivity. A remarkable number of these molecules have been associated with genetic insults in schizophrenia (shown in purple in Figure 2), which would lead to weakened network connections and dysregulation of the stress response. Although not all of these genes have been confirmed by GWAS, the working model indicates how even rare genetic alterations may produce a similar phenotype if they also weaken key PFC networks:
NMDA receptor signaling- PFC network firing during working memory depends on glutamate actions at NMDA receptors on pyramidal cell spines (Arnsten et al., 2010) and Wang and Arnsten, unpublished). The reduction in NMDA receptor signaling in schizophrenia is widely discussed, and may arise from a number of factors, including genetic insults to such NMDA regulatory proteins as NRG1 and dysbindin (reviewed in Krystal et al., 2003; Ross et al., 2006; Javitt, 2010; Kristiansen et al., 2010). Reduced NMDA receptor signaling can lead to spine shrinkage and loss (Hayashi-Takagi et al., 2010), suggesting that both functional and architectural dysfunction can arise from these insults (see above). NMDA receptors have also been reported on GABAergic interneurons, although this may only occur during adolescence (Wang and Gao, 2009). Thus, the impact of altered NMDA signaling on GABAergic interneuronal function may only contribute during a discrete but important time period (Belforte et al., 2010).
Nicotinic α7 receptors- Nicotinic α7 receptors are depolarizing, cation channels that have been localized on dendritic spines in the rat PFC, in addition to their traditional presynaptic location (Duffy et al., 2009). Behavioral studies have shown that stimulation of α7 receptors strengthens the working memory and attention functions of the PFC e.g. (Levin et al., 2006; Boess et al., 2007; Chan et al., 2007), including reversal of cognitive deficits induced by NMDA receptor blockade (Buccafusco and Terry, 2009). Our recent data show that stimulation of α7 receptors in PFC strengthens memory-related firing, while blockade of this receptor markedly reduces PFC network firing in monkeys performing a working memory task (Yang et al., 2010). Genetic alterations of the α7 receptor have been linked to attentional gating deficits in patients with schizophrenia for many years (Leonard and Freedman, 2006; Martin and Freedman, 2007; Severance and Yolken, 2008). It is possible that the high prevalence of smoking in patients with schizophrenia arises from their need to increase nicotine stimulation of α7 receptors to strengthen PFC network connections. Nicotinic a7 agonists are currently being developed as potential cognitive enhancers that may be especially useful in patients with schizophrenia (Hajós and Rogers, 2010; Thomsen et al., 2010).
RGS4 and PI/PKC signaling- RGS4 inhibits Gq signaling, and thus can reduce a number of cellular actions, including IP3 calcium release (Fig 2) and PKC signaling (Fig 4). RGS4 protein and mRNA are greatly reduced in the PFC of patients with schizophrenia (Mirnics et al., 2001; Erdely et al., 2006; Volk et al., 2010), and several genetic studies have found links between RGS4 and mental illness (Chowdari et al., 2002; Morris et al., 2004; Fallin et al., 2005). Loss of RGS4 would disinhibit Gq signaling, potentially amplifying the effects of alpha-1, 5HT2 and mGluR1/Gq signaling in the spine and reducing PFC network activity (Figs. 2B, D). Recent studies have also observed an increase in mGluR1a mRNA in the PFC of patients with schizophrenia (Volk et al., 2010), further driving Gq signaling and reducing neuronal firing. As shown in Figure 4, loss of RGS4 would also disinhibit PKC signaling, which could aggravate spine loss (Hains et al., 2009). In support of this hypothesis, polymorphisms in RGS4 are associated with PFC gray matter loss, reduced connectivity and reduced BOLD signals in patients with schizophrenia (Prasad et al., 2005; Buckholtz et al., 2007).
DISC1 and cAMP signaling- A translocation in the gene encoding for DISC1 has been associated with high rates of mental illness in a large Scottish pedigree (Millar et al., 2000; Millar et al., 2005). Genetic insults to DISC1 likely contribute to altered cortical development (Ishizuka et al., 2006), and may also lead to weakened network connectivity. DISC1 is localized in dendritic spines in the primate PFC (Kirkpatrick et al., 2006), and interacts with the cAMP-catabolizing enzyme, PDE4 (Millar et al., 2007; Murdoch et al., 2007). DISC1 is thought to activate PDE4s under conditions of high cAMP production to provide negative feedback. Thus, loss of DISC1 function should lead to increased cAMP in spines, as shown in Figure 2. As described above, increased cAMP signaling in suppresses PFC neuronal firing, and may also contribute to spine loss during chronic stress. Preliminary data from our lab show that knockdown of DISC1 in the rat medial PFC lowers the threshold for stress-induced PFC dysfunction (Gamo et al., 2010). In this regard it is particularly interesting that a DISC1 polymorphism is associated with increased psychosis, cognitive dysfunction, and PFC gray matter loss in patients with schizophrenia (Cannon et al., 2005; Szeszko et al., 2008).
Interestingly, significant associations have been observed between reduced PFC BOLD activity and single nucleotide polymorphisms in genes encoding for DISC1, NMDA, α7 nicotinic and α2A adrenergic receptors in an fMRI study of patients with schizophrenia performing an oddball task (Liu et al., 2009). Thus, this nonbiased study uncovered a set of DNC-associated proteins needed for strong PFC function.
These data may explain why such a wide variety of genetic insults can lead to a similar phenotype of weakened PFC network firing. Although it is unlikely that treatments will be able to correct abnormalities in PFC circuit formation that have occurred during development, there is hope that we may be able to ameliorate some of the genetic errors in DNC proteins that weaken PFC circuits with targeted medications. The appropriate treatment at key time periods, e.g. in adolescence, may slow the progression of PFC network demise and the manifestation of schizophrenia symptoms.
Dopamine actions in schizophrenia
Dopamine is dysregulated in schizophrenia. A long and consistent history has shown excessive DA stimulation of D2 receptors in caudate (Wong et al., 1986; Seeman et al., 1989; Laruelle et al., 1996; Kegeles et al., 2010), and more recent work suggests reduced DA in PFC post-mortem, at least at later ages (Akil et al., 1999). As there is likely increased DA innervation of layer III in the PFC during adolescence (Rosenberg and Lewis, 1995), there may be a hyperDA state during this key time period, which rapidly erodes with advancing age. As shown in Figure 1 A, the dysregulation of DA in PFC and caudate is likely secondary to insults in PFC circuits that normally control DA activity (Lewis and Gonzalez-Burgos, 2006). Dopamine dysregulation would likely have multiple consequences:
D1 receptor signaling in PFC: PET imaging studies have shown increased D1 receptor binding in PFC in schizoprenia that correlates with cognitive deficits (Abi-Dargham et al., 2002). The upregulation in D1 receptors is thought to be a compensatory response to reduced DA stimulation (Guo et al., 2003). Given that optimal DA D1 receptor stimulation in PFC requires rapid, dynamic changes based on cognitive demands, it is likely that D1 receptor stimulation is often suboptimal in patients with schizophrenia, with
inadequate D1 receptor stimulation under basal conditions, leading to noisy firing and inappropriate network connections, perhaps contributing to loose associations, and
excessive D1 receptor stimulation suppressing all firing under conditions such as mild stress when there is increased DA release onto upregulated D1 receptors (Vijayraghavan et al., 2007); Fig 1A).
These data suggest that patients with schizophrenia would have narrowed inverted U's with a limited range of optimal modulation, and thus be especially vulnerable to stress (Arnsten et al., 2009). One can speculate that this would be further aggravated in patients with genetic insults in DISC1 and/or PDE4 signaling with dysregulation of cAMP signaling (Arnsten et al., 2009). Exaggerated D1 receptor signaling also might contribute to spine loss during stress exposure.
D2 receptor signaling in PFC: In contrast to D1 receptor modulation of delay-related firing, D2 receptors modulate response-related firing (Wang et al., 2004). Interestingly, some of the responses of PFC neurons occur during the motor response, and are thought to represent corollary discharge. D2 receptor stimulation increases response-related firing, and speeds the response, while reduced D2 receptor stimulation slows and reduces response-related firing (Wang et al., 2004). Alterations in either the timing and/or the amplitude of corollary discharge may interfere with the “mental tag” that denotes that a response is self-generated (Arnsten et al., 2009). In humans, D2 alterations in PFC may interfere with corollary discharge emanating from Broca's area, contributing to auditory hallucinations (Ford et al., 2002).
D2 receptor signaling in caudate: As described above, extensive evidence documents increased DA D2 receptor stimulation in the caudate of patients with schizophrenia. Dopamine D2 receptor signaling normally serves to disinhibit the indirect pathways through the basal ganglia, thus increasing motor, cognitive and affective responses (Steiner and Gerfen, 1998). The caudate is the striatal structure mediating cognitive responses, and receives extensive inputs from the association cortices (Alexander et al., 1986). Thus, excessive D2 receptor stimulation in caudate would magnify incoming cortical errors (Fig 1 A).
Increased DA release during stress exposure could thus lead to a constellation of detrimental actions: disconnection of PFC networks, inaccurate timing of corollary discharge from PFC, and magnification of cortical errors in caudate. These actions may contribute to worsening of symptoms with stress exposure.
Ideas for future research
Research on the regulation of PFC connections is in its infancy, and thus many questions remain. This arena is particularly challenging to study, as it requires studies of cognitively-engaged animals. Furthermore, it is likely that there are important differences between medial PFC and dorsolateral PFC in this domain, which limits some aspects of the research to primates. Examples of future research include determination of whether DNC proteins are in the same spines or in distinct patterns. For example, if DISC1 and α2A receptors are in the same spines, α2A receptor stimulation might substitute for some aspects of lost DISC1 function. Conversely, if they are in different sets of spines, this strategy would likely not succeed. Another interesting study would be to see whether certain genetic changes increase vulnerability to stress-induced PFC dysfunction, as predicted by animal data. For example, the findings suggest that individuals with DISC1 translocation would be especially vulnerable to stress.
Summary
In summary, stress exposure leads to weakened PFC networks and PFC dendritic spine loss. A remarkable number of genetic insults in families with schizophrenia afflict molecules that normally serve to inhibit the stress response in PFC. As the symptoms of schizophrenia often have their onset in response to stress, these stress mechanisms may be key to the descent into illness. Understanding these molecular actions may give rise to treatments that can protect PFC gray matter and retard the onset of symptoms.
Acknowledgments
The research in this article was supported by Public Health Service grants PO1 AG030004, MERIT Award AG06036, and 1RL1AA017536 within U54RR024350, and a NARSAD Distinguished Investigator Award to AFTA.
Footnotes
Disclosures- AFTA and Yale University receive royalties from Shire Pharmaceuticals for the sale of Intuniv (extended release guanfacine) for the treatment of ADHD and related disorders. AFTA receives funds from Shire for research related to α2A noradrenergic signaling mechanisms, and performs consulting and speaking engagements with Shire. AFTA also has previously received research funds from Marinus Pharmaceuticals for the development of the PKC inhibitor, chelerythrine, for the treatment of mental illness.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abi-Dargham A, Mawlawi O, Lombardo I, Gil R, Martinez D, Huang Y, Hwang DR, Keilp J, Kochan L, Van Heertum R, Gorman JM, Laruelle M. Prefrontal dopamine D1 receptors and working memory in schizophrenia. J Neurosci. 2002;22:3708–3719. doi: 10.1523/JNEUROSCI.22-09-03708.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akil M, Pierri JN, Whitehead RE, Edgar CL, Mohila C, Sampson AR, Lewis DA. Lamina-specific alterations in the dopamine innervation of the prefrontal cortex in schizophrenic subjects. Am J Psychiatry. 1999;156:1580–1589. doi: 10.1176/ajp.156.10.1580. [DOI] [PubMed] [Google Scholar]
- Alexander GE, DeLong MR, Strick PL. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Ann Rev Neurosci. 1986;9:357–381. doi: 10.1146/annurev.ne.09.030186.002041. [DOI] [PubMed] [Google Scholar]
- Arnsten AFT. Stress signaling pathways that impair prefrontal cortex structure and function. Nature Reviews Neuroscience. 2009;32:267–287. doi: 10.1038/nrn2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AFT, Goldman-Rakic PS. Stress impairs prefrontal cortex cognitive function in monkeys: role of dopamine. Soc Neurosci Abstr. 1990;16:164. [Google Scholar]
- Arnsten AFT, Vijayraghavan S, Wang M, Gamo NJ, Paspalas CD. Dopamine's influence on prefrontal cortical cognition: Actions and circuits in behaving primates. In: Bjorklund A, Dunnett S, Iversen L, Iversen S, editors. Dopamine Handbook. Oxford, UK: Oxford University Press; 2009. pp. 230–249. [Google Scholar]
- Arnsten AFT, Paspalas CD, Gamo NJ, Y Y, Wang M. Dynamic Network Connectivity: A new form of neuroplasticity. Trends Cog Sci. 2010;14:365–375. doi: 10.1016/j.tics.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barch DM. The cognitive neuroscience of schizophrenia. Annu Rev Clin Psychol. 2005;1:321–353. doi: 10.1146/annurev.clinpsy.1.102803.143959. [DOI] [PubMed] [Google Scholar]
- Belforte JE, Zsiros V, Sklar ER, Jiang Z, Yu G, Li Y, Quinlan EM, Nakazawa K. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat Neurosci. 2010;13:76–83. doi: 10.1038/nn.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum SB, Yuan P, Wang M, Vijayraghavan S, Bloom A, Davis D, Gobeske K, Sweatt D, Manji HK, Arnsten AFT. Protein kinase C overactivity impairs prefrontal cortical regulation of working memory. Science. 2004;306:882–884. doi: 10.1126/science.1100021. [DOI] [PubMed] [Google Scholar]
- Boess FG, De Vry J, Erb C, Flessner T, Hendrix M, Luithle J, Methfessel C, Riedl B, Schnizler K, van der Staay FJ, van Kampen M, Wiese WB, Koenig G. The novel alpha7 nicotinic acetylcholine receptor agonist N-[(3R)-1-azabicyclo[2.2.2]oct-3-yl]-7-[2-(methoxy)phenyl]-1-benzofuran-2-carboxamide improves working and recognition memory in rodents. J Pharmacol Exp Ther. 2007;321:716–725. doi: 10.1124/jpet.106.118976. [DOI] [PubMed] [Google Scholar]
- Breier A, Wolkowitz O, Pickar D. Stress and schizophrenia: Advances in neuropsychiatry and psychopharmacology. In: Tamminga C, Schult S, editors. Schizophrenia Research. New York: Raven Press, Ltd.; 1991. [Google Scholar]
- Buccafusco JJ, Terry AVJ. A reversible model of the cognitive impairment associated with schizophrenia in monkeys: potential therapeutic effects of two nicotinic acetylcholine receptor agonists. Biochem Pharmacol. 2009;78:852–862. doi: 10.1016/j.bcp.2009.06.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckholtz JW, Meyer-Lindenberg A, Honea RA, Straub RE, Pezawas L, Egan MF, Vakkalanka R, Kolachana B, Verchinski BA, Sust S, Mattay VS, Weinberger DR, Callicott JH. Allelic variation in RGS4 impacts functional and structural connectivity in the human brain. J Neurosci. 2007;27:1584–1593. doi: 10.1523/JNEUROSCI.5112-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschman TJ, Miller EK. Top-down versus bottom-up control of attention in the prefrontal and posterior parietal cortices. Science. 2007;315:1860–1862. doi: 10.1126/science.1138071. [DOI] [PubMed] [Google Scholar]
- Calabrese B, Halpain S. Essential role for the PKC target MARCKS in maintaining dendritic spine morphology. Neuron. 2005;48:77–90. doi: 10.1016/j.neuron.2005.08.027. [DOI] [PubMed] [Google Scholar]
- Cannon TD, Hennah W, van Erp TG, Thompson PM, Lonnqvist J, Huttunen M, Gasperoni T, Tuulio-Henriksson A, Pirkola T, Toga AW, Kaprio J, Mazziotta J, Peltonen L. Association of DISC1/TRAX haplotypes with schizophrenia, reduced prefrontal gray matter, and impaired short-and long-term memory. Arch Gen Psychiatry. 2005;62:1205–1213. doi: 10.1001/archpsyc.62.11.1205. [DOI] [PubMed] [Google Scholar]
- Chan WK, Wong PT, Sheu FS. Frontal cortical alpha7 and alpha4beta2 nicotinic acetylcholine receptors in working and reference memory. Neuropharmacology. 2007;2007:1641–1649. doi: 10.1016/j.neuropharm.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Chowdari KV, Mirnics K, Semwal P, Wood J, Lawrence E, Bhatia T, Deshpande SN, Ferrell RE, Middleton FA, Devlin B, Levitt P, Lewis DA, Nimgaonkar VL. Association and linkage analyses of RGS4 polymorphisms in schizophrenia. Hum Mol Genet. 2002;11:1373–1380. doi: 10.1093/hmg/11.12.1373. [DOI] [PubMed] [Google Scholar]
- Corlett PR, Murray GK, Honey GD, Aitken MR, Shanks DR, Robbins TW, Bullmore ET, Dickinson A, Fletcher PC. Disrupted prediction-error signal in psychosis: evidence for an associative account of delusions. Brain. 2007;130(Pt 9):2387–2400. doi: 10.1093/brain/awm173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson ME, Schell AM, Rissling A, Ventura J, Subotnik KL, Nuechterlein KH. Psychophysiological prodromal signs of schizophrenic relapse: A pilot study. Schizophr Res. 2010 Aug 17; doi: 10.1016/j.schres.2010.07.029. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy AM, Zhou P, Milner TA, Pickel VM. Spatial and intracellular relationships between the alpha7 nicotinic acetylcholine receptor and the vesicular acetylcholine transporter in the prefrontal cortex of rat and mouse. Neuroscience. 2009;161:1091–1103. doi: 10.1016/j.neuroscience.2009.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eack SM, George MM, Prasad KM, Keshavan MS. Neuroanatomical substrates of foresight in schizophrenia. Schizophr Res. 2008;103:62–70. doi: 10.1016/j.schres.2008.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdely HA, Tamminga CA, Roberts RC, Vogel MW. Regional alterations in RGS4 protein in schizophrenia. Synapse. 2006;59:472–479. doi: 10.1002/syn.20265. [DOI] [PubMed] [Google Scholar]
- Fallin MD, Lasseter VK, Avramopoulos D, Nicodemus KK, Wolyniec PS, McGrath JA, Steel G, Nestadt G, Liang KY, Huganir RL, Valle D, Pulver AE. Bipolar I disorder and chizophrenia: a 440-single-nucleotide polymorphism screen of 64 candidate genes among Ashkenazi Jewish case-parent trios. Am J Hum Genet. 2005;77:918–936. doi: 10.1086/497703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming YM, Frame MC, Houslay MD. PDE4-regulated cAMP degradation controls the assembly of integrin-dependent actin adhesion structures and REF52 cell migration. J Cell Sci. 2004;117:2377–2388. doi: 10.1242/jcs.01096. [DOI] [PubMed] [Google Scholar]
- Ford JM, Mathalon DH, Whitfield S, Faustman WO, Roth WT. Reduced communication between frontal and temporal lobes during talking in schizophrenia. Biol Psychiatry. 2002;51:485–492. doi: 10.1016/s0006-3223(01)01335-x. [DOI] [PubMed] [Google Scholar]
- Funahashi S, Bruce CJ, Goldman-Rakic PS. Dorsolateral prefrontal lesions and oculomotor delayed-response performance: evidence for mnemonic “scotomas”. J Neurosci. 1993;13:1479–1497. doi: 10.1523/JNEUROSCI.13-04-01479.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster JM. Executive frontal functions. Exp Brain Res. 2000;133:66–70. doi: 10.1007/s002210000401. [DOI] [PubMed] [Google Scholar]
- Fuster JM. The Prefrontal Cortex. 4th. Academic Press; 2008. [Google Scholar]
- Gamo NJ, Kata A, Boven L, Bryan C, Lo T, Anighoro K, Bermudez L, Peng K, Annor A, Taylor S, Patel K, Duque A, Simen AA, A AFT. Knockdown of Disrupted in Schizophrenia 1 (DISC1) in the rat prefrontal cortex lowers the threshold for stress-induced cognitive dysfunction. Society for Neuroscience Abstracts. 2010 in press. [Google Scholar]
- Gao WJ, Krimer LS, Goldman-Rakic PS. Presynaptic regulation of recurrent excitation by D1 receptors in prefrontal circuits. Proc Natl Acad Sci USA. 2001;98:295–300. doi: 10.1073/pnas.011524298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Archives General Psychiatry. 2000;57:65–73. doi: 10.1001/archpsyc.57.1.65. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS. Cellular basis of working memory. Neuron. 1995;14:477–485. doi: 10.1016/0896-6273(95)90304-6. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Burgos G, Hashimoto T, Lewis DA. Alterations of cortical GABA neurons and network oscillations in schizophrenia. Curr Psychiatry Rep. 2010;12:335–344. doi: 10.1007/s11920-010-0124-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Burgos G, Barrionuevo G, Lewis DA. Horizontal synaptic connections in monkey prefrontal cortex: an in vitro electrophysiological study. Cereb Cortex. 2000;10:82–92. doi: 10.1093/cercor/10.1.82. [DOI] [PubMed] [Google Scholar]
- Gorelova NA, Seamans JK, Yang CR. Mechanisms of dopamine activation of fast-spiking interneurons that exert inhibition in rat prefrontal cortex. J Neurophysiol. 2002;88:150–166. doi: 10.1152/jn.00335.2002. [DOI] [PubMed] [Google Scholar]
- Gourley SL, Kedves AT, Olausson P, Taylor JR. A history of corticosterone exposure regulates fear extinction and cortical NR2B, GluR2/3, and BDNF. Neuropsychopharmacology. 2009;34:707–716. doi: 10.1038/npp.2008.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granon S, Passetti F, Thomas KL, Dalley JW, Everitt BJ, Robbins TW. Enhanced and impaired attentional performance after infusion of D1 dopaminergic receptor agents into rat prefrontal cortex. J Neurosci. 2000;20:1208–1215. doi: 10.1523/JNEUROSCI.20-03-01208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo N, Hwang DR, Lo ES, Huang YY, Laruelle M, Abi-Dargham A. Dopamine depletion and in vivo binding of PET D1 receptor radioligands: implications for imaging studies in schizophrenia. Neuropsychopharmacology. 2003;28:1703–1711. doi: 10.1038/sj.npp.1300224. [DOI] [PubMed] [Google Scholar]
- Hains AB, Vu MA, Maciejewski PK, van Dyck CH, Gottron M, Arnsten AF. Inhibition of protein kinase C signaling protects prefrontal cortex dendritic spines and cognition from the effects of chronic stress. Proc Natl Acad Sci U S A. 2009;106:17957–17962. doi: 10.1073/pnas.0908563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajós M, Rogers BN. Targeting alpha7 nicotinic acetylcholine receptors in the treatment of schizophrenia. Curr Pharm Des. 2010;16:538–554. doi: 10.2174/138161210790361434. [DOI] [PubMed] [Google Scholar]
- Hayashi-Takagi A, Takaki M, Graziane N, Seshadri S, Murdoch H, Dunlop AJ, Makino Y, Seshadri AJ, Ishizuka K, Srivastava DP, Xie Z, Baraban JM, Houslay MD, Tomoda T, Brandon NJ, Kamiya A, Yan Z, Penzes P, Sawa A. Disrupted-in-Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat Neurosci. 2010;13:327–332. doi: 10.1038/nn.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JJ, Hashimoto T, Lewis DA. Molecular mechanisms contributing to dendritic spine alterations in the prefrontal cortex of subjects with schizophrenia. Mol Psychiatry. 2006;11:557–566. doi: 10.1038/sj.mp.4001792. [DOI] [PubMed] [Google Scholar]
- Ide M, Lewis DA. Altered cortical CDC42 signaling pathways in schizophrenia: implications for dendritic spine deficits. Biol Psychiatry. 2010;68:25–32. doi: 10.1016/j.biopsych.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka K, Paek M, Kamiya A, Sawa A. A review of Disrupted-ln-Schizophrenia-1 (DISC1): neurodevelopment, cognition, and mental conditions. Biol Psychiatry. 2006;59:1189–1197. doi: 10.1016/j.biopsych.2006.03.065. [DOI] [PubMed] [Google Scholar]
- Javitt DC. Glutamatergic theories of schizophrenia. Isr J Psychiatry Relat Sci. 2010;47:4–16. [PubMed] [Google Scholar]
- Jurado MA, Junqué C, Vendrell P, Treserras P, Grafman J. Overestimation and unreliability in “feeling-of-doing” judgements about temporal ordering performance: impaired self-awareness following frontal lobe damage. J Clin Exp Neuropsychol. 1998;20:353–364. doi: 10.1076/jcen.20.3.353.816. [DOI] [PubMed] [Google Scholar]
- Keedy SK, Ebens CL, Keshavan MS, Sweeney JA. Functional magnetic resonance imaging studies of eye movements in first episode schizophrenia: smooth pursuit, visually guided saccades and the oculomotor delayed response task. Psychiatry Res. 2006;146:199–211. doi: 10.1016/j.pscychresns.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Keefe RS, Perkins DO, Gu H, Zipursky RB, Christensen BK, Lieberman JA. A longitudinal study of neurocognitive function in individuals at-risk for psychosis. Schizophr Res. 2006;88:26–35. doi: 10.1016/j.schres.2006.06.041. [DOI] [PubMed] [Google Scholar]
- Kegeles LS, Abi-Dargham A, Frankle WG, Gil R, Cooper TB, Slifstein M, Hwang DR, Huang Y, Haber SN, Laruelle M. Increased synaptic dopamine function in associative regions of the striatum in schizophrenia. Arch Gen Psychiatry. 2010;67:231–239. doi: 10.1001/archgenpsychiatry.2010.10. [DOI] [PubMed] [Google Scholar]
- Kinney JW, Davis CN, Tabarean I, Conti B, Bartfai T, Behrens MM. A specific role for NR2A-containing NMDA receptors in the maintenance of parvalbumin and GAD67 immunoreactivity in cultured interneurons. J Neurosci. 2006;26:1604–1615. doi: 10.1523/JNEUROSCI.4722-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick B, Xu L, Cascella N, Ozeki Y, Sawa A, Roberts RC. DISC1 immunoreactivity at the light and ultrastructural level in the human neocortex. J Comp Neurol. 2006;497:436–450. doi: 10.1002/cne.21007. [DOI] [PubMed] [Google Scholar]
- Knight RT, Grabowecky MF, Scabini D. Role of human prefrontal cortex in attention control. Adv Neurol. 1995;66:21–34. [PubMed] [Google Scholar]
- Kristiansen LV, Patel SA, Haroutunian VH, Meador-Woodruff JH. Expression of the NR2B-NMDA receptor subunit and its Tbr-1/CINAP regulatory proteins in postmortem brain suggest altered receptor processing in schizophrenia. Synapse. 2010;64:495–502. doi: 10.1002/syn.20754. [DOI] [PubMed] [Google Scholar]
- Kritzer MF, Goldman-Rakic PS. Intrinsic circuit organization of the major layers and sublayers of the dorsolateral prefrontal cortex in the rhesus monkey. J Comp Neurol. 1995;359:131–143. doi: 10.1002/cne.903590109. [DOI] [PubMed] [Google Scholar]
- Kroner S, Krimer LS, Lewis DA, Barrionuevo G. Dopamine increases inhibition in the monkey dorsolateral prefrontal cortex through cell type-specific modulation of interneurons. Cereb Cortex. 2007;17:1020–1032. doi: 10.1093/cercor/bhl012. [DOI] [PubMed] [Google Scholar]
- Krystal JH, D'Souza DC, Mathalon D, Perry E, Belger A, Hoffman R. NMDA receptor antagonist effects, cortical glutamatergic function, and schizophrenia: toward a paradigm shift in medication development. Psychopharmacology. 2003;169:215–233. doi: 10.1007/s00213-003-1582-z. [DOI] [PubMed] [Google Scholar]
- Laruelle M, Abi-Dargham A, van Dyck CH, Gil R, D'Souza CD, Erdos J, McCance E, Rosenblatt W, Fingado C, Zoghbi SS, Baldwin RM, Seibyl JP, Krystal JH, Charney DS, Innis RB. Single photon emission computerized tomography imaging of amphetamine-induced dopamine release in drug-free schizophrenic subjects. Proc Natl Acad Sci U S A. 1996;93:9235–9240. doi: 10.1073/pnas.93.17.9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Rushworth MF, Walton ME, Watanabe M, Sakagami M. Functional specialization of the primate frontal cortex during decision making. J Neurosci. 2007;27:8170–8173. doi: 10.1523/JNEUROSCI.1561-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard S, Freedman R. Genetics of chromosome 15q13-q14 in schizophrenia. Biol Psychiatry. 2006;60:115–122. doi: 10.1016/j.biopsych.2006.03.054. [DOI] [PubMed] [Google Scholar]
- Levin ED, McClernon FJ, Rezvani AH. Nicotinic effects on cognitive function: behavioral characterization, pharmacological specification, and anatomic localization. Psychopharmacology. 2006;184:523–539. doi: 10.1007/s00213-005-0164-7. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Gonzalez-Burgos GR. Pathophysiologically based treatment interventions in schizophrenia. Nature Medicine. 2006;12:1016–1022. doi: 10.1038/nm1478. [DOI] [PubMed] [Google Scholar]
- Liston C, McEwen BS, Casey BJ. Psychosocial stress reversibly disrupts prefrontal processing and attentional control. Proc Nat Acad Sci USA. 2009;106:912–917. doi: 10.1073/pnas.0807041106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Pearlson G, Windemuth A, Ruano G, Perrone-Bizzozero NI, Calhoun V. Combining fMRI and SNP data to investigate connections between brain function and genetics using parallel ICA. Hum Brain Mapp. 2009;30:241–255. doi: 10.1002/hbm.20508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LF, Freedman R. Schizophrenia and the alpha7 nicotinic acetylcholine receptor. Int Rev Neurobiol. 2007;78:225–246. doi: 10.1016/S0074-7742(06)78008-4. [DOI] [PubMed] [Google Scholar]
- Millar JK, Wilson-Annan JC, Anderson SL, Christie S, Taylor MS, Semple CA, Devon RS, Clair DM, Muir WJ, Blackwood DH, Porteous DJ. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet. 2000;9:1415–1423. doi: 10.1093/hmg/9.9.1415. [DOI] [PubMed] [Google Scholar]
- Millar JK, Mackie S, Clapcote SJ, Murdoch H, Pickard BS, Christie S, Muir WJ, Blackwood DH, Roder JC, Houslay MD, Porteous DJ. Disrupted in schizophrenia 1 and hosphodiesterase 4B: towards an understanding of psychiatric illness. J Physiol. 2007;584:401–405. doi: 10.1113/jphysiol.2007.140210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar JK, Pickard BS, Mackie S, James RS, Christie S, Buchanan SR, Malloy MP, Chubb JE, Huston E, Baillie GS, Thomson PA, Hill EV, Brandon NJ, Rain JC, Camargo LM, Whiting PJ, Houslay MD, Blackwood DH, Muir WJ, Porteous DJ. DISC1 and PDE4B are interacting genetic factors in schizophrenia that regulate cAMP signaling. Science. 2005;310:1187–1191. doi: 10.1126/science.1112915. [DOI] [PubMed] [Google Scholar]
- Miller PL, Lawrie SM, Hodges A, Clafferty R, Cosway R, Johnstone EC. Genetic liability, illicit drug use, life stress and psychotic symptoms: preliminary findings from the Edinburgh study of people at high risk for schizophrenia. Soc Psychiatry Psychiatr Epidemiol. 2001;36:338–342. doi: 10.1007/s001270170038. [DOI] [PubMed] [Google Scholar]
- Mirnics K, Middleton FA, Stanwood GD, Lewis DA, Levitt P. Disease-specific changes in regulator of G-protein signaling 4 (RGS4) expression in schizophrenia. Mol Psychiatry. 2001;6:293–301. doi: 10.1038/sj.mp.4000866. [DOI] [PubMed] [Google Scholar]
- Modirrousta M, Fellows LK. Dorsal medial prefrontal cortex plays a necessary role in rapid error prediction in humans. J Neurosci. 2008;28:14000–14005. doi: 10.1523/JNEUROSCI.4450-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris DW, Rodgers A, McGhee KA, Schwaiger S, Scully P, Quinn J, Meagher D, Waddington JL, Gill M, Corvin AP. Confirming RGS4 as a susceptibility gene for schizophrenia. Am J Med Genet. 2004;125B:50–53. doi: 10.1002/ajmg.b.20109. [DOI] [PubMed] [Google Scholar]
- Murdoch H, Mackie S, Collins DM, Hill EV, Bolger GB, Klussmann E, Porteous DJ, Millar JK, Houslay MD. Isoform-selective susceptibility of DISC1/phosphodiesterase-4 complexes to dissociation by elevated intracellular cAMP levels. J Neurosci. 2007;27:9513–9524. doi: 10.1523/JNEUROSCI.1493-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murmu MS, Salomon S, Biala Y, Weinstock M, Braun K, Bock J. Changes of spine density and dendritic complexity in the prefrontal cortex in offspring of mothers exposed to stress during pregnancy. Eur J Neurosci. 2006;24:1477–1487. doi: 10.1111/j.1460-9568.2006.05024.x. [DOI] [PubMed] [Google Scholar]
- Murphy BL, Arnsten AFT, Goldman-Rakic PS, Roth RH. Increased dopamine turnover in the prefrontal cortex impairs spatial working memory performance in rats and monkeys. Proc Nat Acad Sci USA. 1996;93:1325–1329. doi: 10.1073/pnas.93.3.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuechterlein KH, Dawson ME, Gitlin M, Ventura J, Goldstein MJ, Snyder KS, Yee CM, Mintz J. Developmental processes in schizophrenic disorders: longitudinal studies of vulnerability and stress. Schizophr Bull. 1992;18:387–425. doi: 10.1093/schbul/18.3.387. [DOI] [PubMed] [Google Scholar]
- Paspalas CD, Selemon LD, Arnsten AF. Mapping the Regulator of G Protein Signaling 4 (RGS4): Presynaptic and postsynaptic substrates for neuroregulation in prefrontal cortex. Cereb Cortex. 2009;19:2145–2155. doi: 10.1093/cercor/bhn235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlstein WM, Carter CS, Noll DC, Cohen JD. Relation of prefrontal cortex dysfunction to working memory and symptoms in schizophrenia. Am J Psychiatry. 2001;158:1105–1113. doi: 10.1176/appi.ajp.158.7.1105. [DOI] [PubMed] [Google Scholar]
- Prasad KM, Chowdari KV, Nimgaonkar VL, Talkowski ME, Lewis DA, Keshavan MS. Genetic polymorphisms of the RGS4 and dorsolateral prefrontal cortex morphometry among first episode schizophrenia patients. Mol Psychiatry. 2005;10:213–219. doi: 10.1038/sj.mp.4001562. [DOI] [PubMed] [Google Scholar]
- Radley JJ, Rocher AB, Rodriguez A, Ehlenberger DB, Dammann M, McEwen BS, Morrison JH, Wearne SL, Hof PR. Repeated stress alters dendritic spine morphology in the rat medial prefrontal cortex. J Comp Neurol. 2008;507:1141–1150. doi: 10.1002/cne.21588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SG, Williams GV, Goldman-Rakic PS. Isodirectional tuning of adjacent interneurons and pyramidal cells during working memory: evidence for microcolumnar organization in PFC. J Neurophysiol. 1999;81:1903–1916. doi: 10.1152/jn.1999.81.4.1903. [DOI] [PubMed] [Google Scholar]
- Rao SG, Williams GV, Goldman-Rakic PS. Destruction and creation of spatial tuning by disinhibition: GABA(A) blockade of prefrontal cortical neurons engaged by working memory. J Neurosci. 2000;20:485–494. doi: 10.1523/JNEUROSCI.20-01-00485.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins TW. Shifting and stopping: fronto-striatal substrates, neurochemical modulation and clinical implications. Philos Trans R Soc Lond B Biol Sci. 2007;362:917–932. doi: 10.1098/rstb.2007.2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg DR, Lewis DA. Postnatal maturation of the dopaminergic innervation of monkey prefrontal cortices: A tyrosine hydroxylase immunohistochemical analysis. J Comp Neurol. 1995;358:383–400. doi: 10.1002/cne.903580306. [DOI] [PubMed] [Google Scholar]
- Ross CA, Margolis RL, Reading SA, Pletnikov M, Coyle JT. Neurobiology of schizophrenia. Neuron. 2006;52:139–153. doi: 10.1016/j.neuron.2006.09.015. [DOI] [PubMed] [Google Scholar]
- Seeman P, Niznik HB, Guan HC. Elevation of dopamine D2 receptors in schizophrenia is underestimated by radioactive raclopride. Arch Gen Psych. 1989;47:1170–1172. doi: 10.1001/archpsyc.1990.01810240090014. [DOI] [PubMed] [Google Scholar]
- Selemon LD, Rajkowska G, Goldman-Rakic PS. Elevated neuronal density in prefrontal area 46 in brains from schizophrenic patients: application of a three-dimensional, stereologic counting method. J Comp Neurol. 1998;392:402–412. [PubMed] [Google Scholar]
- Serrels B, Sandilands E, Serrels A, Baillie GS, Houslay MD, Brunton VG, Canel M, Machesky LM, Anderson Kl, Frame MC. A complex between FAK, RACK1, and PDE4D5 controls spreading initiation and cancer cell polarity. Curr Biol. 2010;20:1086–1092. doi: 10.1016/j.cub.2010.04.042. [DOI] [PubMed] [Google Scholar]
- Severance EG, Yolken RH. Novel alpha7 nicotinic receptor isoforms and deficient cholinergic transcription in schizophrenia. Genes Brain Behav. 2008;7:37–45. doi: 10.1111/j.1601-183X.2007.00323.x. [DOI] [PubMed] [Google Scholar]
- Sfakianos MK, Eisman A, Gourley SL, Bradley WD, Scheetz AJ, Settleman J, Taylor JR, Greer CA, Williamson A, Koleske AJ. Inhibition of Rho via Arg and p190RhoGAP in the postnatal mouse hippocampus regulates dendritic spine maturation, synapse and dendrite stability, and behavior. J Neurosci. 2007;27:10982–10992. doi: 10.1523/JNEUROSCI.0793-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons JS, Henson RN, Gilbert SJ, Fletcher PC. Separable forms of reality monitoring supported by the anterior prefrontal cortex. J Cogn Neurosci. 2008;20:447–457. doi: 10.1162/jocn.2008.20036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smiley JF, Levey AI, Ciliax BJ, Goldman-Rakic PS. D1 dopamine receptor immunoreactivity in human and monkey cerebral cortex: predominant and extrasynaptic localization in dendritic spines. Proc Natl Acad Sci U S A. 1994;91:5720–5724. doi: 10.1073/pnas.91.12.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner H, Gerfen CR. Role of dynorphin and enkephalin in the regulation of striatal output pathways and behavior. Exp Brain Res. 1998;123:60–76. doi: 10.1007/s002210050545. [DOI] [PubMed] [Google Scholar]
- Subramaniam K, Kounios J, Parrish TB, Jung-Beeman M. A brain mechanism for facilitation of insight by positive affect. J Cogn Neurosci. 2008 Jun 25; doi: 10.1162/jocn.2009.21057. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Sun D, Phillips L, Velakoulis D, Yung A, McGorry PD, Wood SJ, van Erp TG, Thompson PM, Toga AW, Cannon TD, Pantelis C. Progressive brain structural changes mapped as psychosis develops in ‘at risk’ individuals. Schizophr Res. 2008 Feb 8; doi: 10.1016/j.schres.2008.11.026. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeszko PR, Hodgkinson CA, Robinson DG, Derosse P, Bilder RM, Lencz T, Burdick KE, Napolitano B, Betensky JD, Kane JM, Goldman D, Malhotra AK. DISC1 is associated with prefrontal cortical gray matter and positive symptoms in schizophrenia. Biol Psychol. 2008;79:103–110. doi: 10.1016/j.biopsycho.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson-Schill SL, Jonides J, Marshuetz C, Smith EE, D'Esposito M, Kan IP, Knight RT, Swick D. Effects of frontal lobe damage on interference effects in working memory. Cogn Affect Behav Neurosci. 2002;2:109–120. doi: 10.3758/cabn.2.2.109. [DOI] [PubMed] [Google Scholar]
- Thomsen MS, Hansen HH, Timmerman DB, Mikkelsen JD. Cognitive improvement by activation of alpha7 nicotinic acetylcholine receptors: from animal models to human pathophysiology. Curr Pharm Des. 2010;16:323–343. doi: 10.2174/138161210790170094. [DOI] [PubMed] [Google Scholar]
- Torreano PJ, Waterman-Storer CM, Cohan CS. The effects of collapsing factors on F-actin content and microtubule distribution of Helisoma growth cone. Cell Motil Cytoskeleton. 2005;60:166–179. doi: 10.1002/cm.20051. [DOI] [PubMed] [Google Scholar]
- Vidal CN, Rapoport JL, Hayashi KM, Geaga JA, Sui Y, McLemore LE, Alaghband Y, Giedd JN, Gochman P, Blumenthal JD, Gogtay N, Nicolson R, Toga AW, Thompson PM. Dynamically spreading frontal and cingulate deficits mapped in adolescents with schizophrenia. Arch Gen Psychiatry. 2006;63:25–34. doi: 10.1001/archpsyc.63.1.25. [DOI] [PubMed] [Google Scholar]
- Vijayraghavan S, Wang M, Birnbaum SG, Bruce CJ, Williams GV, Arnsten AFT. Inverted-U dopamine D1 receptor actions on prefrontal neurons engaged in working memory. Nature Neuroscience. 2007;10:376–384. doi: 10.1038/nn1846. [DOI] [PubMed] [Google Scholar]
- Volk DW, Eggan SM, Lewis DA. Alterations in metabotropic glutamate receptor 1α and Regulator of G Protein Signaling 4 in the prefrontal cortex in schizophrenia. Am J Psychiatry. 2010 doi: 10.1176/appi.ajp.2010.10030318. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk DW, Austin MC, Pierri JN, Sampson AR, Lewis DA. Decreased glutamic acid decarboxylase67 messenger RNA expression in a subset of prefrontal cortical gamma-aminobutyric acid neurons in subjects with schizophrenia. Arch Gen Psychiatry. 2000;57:237–245. doi: 10.1001/archpsyc.57.3.237. [DOI] [PubMed] [Google Scholar]
- Wang HX, Gao WJ. Cell type-specific development of NMDA receptors in the interneurons of rat prefrontal cortex. Neuropsychopharmacology. 2009;34:2028–2040. doi: 10.1038/npp.2009.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Vijayraghavan S, Goldman-Rakic PS. Selective D2 receptor actions on the functional circuitry of working memory. Science. 2004;303:853–856. doi: 10.1126/science.1091162. [DOI] [PubMed] [Google Scholar]
- Wang M, Ramos B, Paspalas C, Shu Y, Simen A, Duque A, Vijayraghavan S, Brennan A, Dudley AG, Nou E, Mazer JA, McCormick DA, Arnsten AFT. Alpha2A-adrenoceptor stimulation strengthens working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell. 2007;129:397–410. doi: 10.1016/j.cell.2007.03.015. [DOI] [PubMed] [Google Scholar]
- Weinberger DR, Berman KF, Zee RF. Physiologic dysfunction of dorsolateral prefrontal cortex in schizophrenia. I. Regional cerebral blood flow evidence. Archives General Psychiatry. 1986;43:114–124. doi: 10.1001/archpsyc.1986.01800020020004. [DOI] [PubMed] [Google Scholar]
- Wilkins AJ, Shallice T, McCarthy R. Frontal lesions and sustained attention. Neuropsychologia. 1987;25:359–365. doi: 10.1016/0028-3932(87)90024-8. [DOI] [PubMed] [Google Scholar]
- Wong DF, Wagner HNJ, Tune LE, Dannals RF, Pearlson GD, Links JM, Tamminga CA, Broussolle EP, Ravert HT, Wilson AA, Toung JK, Malat J, Williams JA, O'Tuama LA, Snyder SH, Kuhar MJ, Gjedde A. Positron emission tomography reveals elevated D2 dopamine receptors in drug-naive schizophrenics. Science. 1986;234:1558–1563. doi: 10.1126/science.2878495. [DOI] [PubMed] [Google Scholar]
- Yang Y, Gamo NJ, Arnsten AFT, Wang M. Alpha7 nicotinic receptor influences on prefrontal cortex network physiology. Society for Neuroscience Abstracts. 2010 In press. [Google Scholar]
- Yokoyama Y, Ito T, Hanson V, Schwartz GK, Aderem AA, Holland JF, Tamaya T, Ohnuma T. PMA-induced reduction in invasiveness is associated with hyperphosphorylation of MARCKS and talin in invasive bladder cancer cells. Int J Cancer. 1998;75:774–779. doi: 10.1002/(sici)1097-0215(19980302)75:5<774::aid-ijc18>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]