

Abstract
Background
Catecholamines increase heart rate by augmenting the cAMP responsive HCN4 ‘pacemaker current’ (If) and/or by promoting inward Na+/Ca2+ exchanger current (INCX), by a ‘Ca2+ clock’ mechanism in sinoatrial nodal cells (SANCs). The importance, identity and function of signals that connect If and Ca2+ clock mechanisms are uncertain and controversial, but the multifunctional Ca2+ and calmodulin-dependent protein kinase II (CaMKII) is required for physiological heart rate responses to β-adrenergic receptor (β-AR) stimulation. The aim of this stuy is to measure the contribution of the Ca2+ clock and CaMKII to cardiac pacing independent of β-AR agonist stimulation.
Methods and Results
We used the L-type Ca2+ channel agonist BayK 8644 (BayK) to activate the SANC Ca2+ clock. BayK and isoproterenol were similarly effective in increasing rates in SANCs and Langendorff-perfused hearts from WT control mice. In contrast, SANCs and isolated hearts from mice with CaMKII inhibition by transgenic expression of an inhibitory peptide (AC3-I) were resistant to rate increases by BayK. BayK only activated CaMKII in control SANCs, but increased ICa equally in all SANCs, indicating that increasing ICa was insufficient and suggesting CaMKII activation was required for heart rate increases by BayK. BayK did not increase If or protein kinase A (PKA)-dependent phosphorylation of phospholamban (at Ser16), indicating that increased SANC Ca2+ by BayK did not augment cAMP/PKA signaling at these targets. Late diastolic intracellular Ca2+ release and INCX were significantly reduced in AC3-I SANCs and the response to BayK was eliminated by ryanodine in all groups.
Conclusions
The Ca2+ clock is capable of supporting physiological fight or flight responses, independent of β-AR stimulation or If increases. Complete Ca2+ clock and β-AR stimulation responses require CaMKII.
Keywords: Ca2+/calmodulin-dependent protein kinase (CaMKII), sinoatrial node cells, L-type Ca2+ channels, pacemaker current, sarcoplasmic reticulum
Introduction
The fight or flight response to increase heart rate is a fundamental aspect of cardiovascular physiology, but recent findings have unsettled a core concept that increases in heart rate are exclusively due to a cyclic adenosine monophosphate (cAMP)-activated pacemaker current (If). Accumulating data support a view that sinoatrial node (SAN) pacemaker automaticity is an outcome of cell membrane depolarization triggered by an inward If, conducted primarily by HCN4, and inward Na+/Ca2+ exchange current (INCX), activated by spontaneous Ca2+ release from the sarcoplasmic reticulum (SR).1-4 Increases in If and INCX steepen the slope of phase 4 diastolic depolarization (DD) leading to faster SAN cell (SANC) action potential triggering.4-5 Catecholamines activate the cAMP and protein kinase A (PKA) signaling pathway by agonist actions at β adrenergic receptors (β-AR), and so have the capability of increasing If, SR Ca2+ release and INCX.6 However, recent work suggests that SANCs also utilize a β-AR independent mechanism for promoting cAMP activity by Ca2+-dependent activation of adenylyl cyclase.7 Despite the potential interconnections between β-AR-initiated and Ca2+ responsive signaling pathways for augmenting If and INCX to increase heart rate, we are unaware of any studies examining the signaling mechanisms engaged by increasing intracellular Ca2+, independent of β-AR agonist stimulation, in SANCs. Isoproterenol, a β-AR agonist, increases intracellular Ca2+ in SANCs and activates the Ca2+/calmodulin-dependent protein kinase II (CaMKII).8 We recently reported that CaMKII was required for the physiological heart rate response to isoproterenol,9 but it is unknown if CaMKII actions require β-AR activation or if CaMKII activation in the absence of β-AR activation is sufficient to increase heart rate. We designed our study to distinguish the contributions of Ca2+ and β-AR signaling to SAN automaticity and to determine if increases in intracellular Ca2+ and CaMKII were capable of increasing SAN rates to a similar extent as β-AR stimulation.
Here we increased SANC intracellular Ca2+ directly, independently of β-AR stimulation, by increasing L-type Ca2+ current (ICa) with BayK 8644 (BayK). BayK increased mouse and dog SANC automaticity over a dynamic range that was similar to increases evoked by isoproterenol, suggesting that a Ca2+ dependent mechanism is robust and capable of supporting the majority of heart rate increases required for the physiological fight or flight response in small and large animal models.10-11 BayK increased heart rate and SANC automaticity significantly less in Langendorff-perfused isolated mouse hearts and in isolated SANCs with CaMKII inhibition due to transgenic myocardial expression of a CaMKII inhibitory peptide (AC3-I) compared to WT littermates or transgenic control mice with myocardial expression of an inactive scrambled form of AC3-I (AC3-C). These findings suggested that heart rate responses to increased Ca2+ relied on activation of CaMKII. BayK increased ICa equally in AC3-I, AC3-C and WT SANCs, showing that reduced BayK response in AC3-I SANCs was due to actions ‘downstream’ to ICa. The increases in rate by BayK were independent of If because If was not increased by BayK and was equivalent in AC3-I and control SANCs, suggesting that increasing SANC Ca2+ with BayK did not activate a cAMP pathway with access to HCN4. Furthermore, BayK did not increase phosphorylation of a bona fide PKA phosphorylation target site on phospholamban (Ser16) in SANCs, suggesting that increasing SANC Ca2+ does not lead to a generalized increase in PKA activity. BayK increases in SANC automaticity required SR Ca2+ release, because they were eliminated by ryanodine and because AC3-I SANCs failed to show increased frequency of late diastolic SR Ca2+ release events or enhanced INCX in response to BayK, compared to controls. These data show that CaMKII activation, independent of β-AR stimulation, can support heart rate increases over a dynamic range that is similar to the classical β-AR agonist stimulated fight or flight response. CaMKII is required for increasing heart rate by β-AR and Ca2+-based mechanisms in SANCs.
Methods
Methodological details are included in the supplemental materials (available online at http://circep.ahajournals.org/). Single SANCs from mouse and dog were isolated.9, 12 We recorded APs, If and INCX with perforated patch access, ICa was recorded with conventional whole celll mode. All voltage and current clamp studies were performed at 36°C. Intracellular Ca2+ transients and sparks were measured with a confocal microscope (LSM510, Carl Zeiss MicroImaging) using the line scan mode, as described.12-13 Western analyses were performed to measure the phosphorylated (Ser16 or Thr17) and total phospholamban expression in SAN tissue. The data are presented as means ± the standard error of the mean (S.E.M,). The statistical significance was evaluated with one-way, two-way ANOVA or Student’s t-test as appropriate, Student-Newman-Keuls was used for post hoc analysis. P<0.05 is considered statistically significant.
Results
Equivalent SAN rate acceleration by BayK and β-AR stimulation
We used a selective L-type Ca2+ channel agonist, BayK,14 to examine if increasing ICa affected the spontaneous activity of SANCs. The SANCs were perfused for at least 5 mins with different concentrations of BayK. BayK significantly and dose-dependently increased the pacemaking activity of SANCs isolated from WT mice (Fig 1A and B). Interestingly, the rate-dose response relationship to BayK was similar to the rate-dose response relationship observed with isoproterenol (Fig 1B). The maximum effects of BayK and isoproterenol were equivalent (Fig 1C). These findings showed that BayK and isoproterenol were similarly effective in increasing SANC automaticity. We next asked if the rate response to BayK occurred in larger mammals, by repeating our studies with canine SANCs. We found that BayK increased the spontaneous activity in single SANCs isolated from dogs, similar to our findings in mouse SANCs (Supplementary data Fig S1). The rate increase by BayK in dog SANCs was close to what we found with isoproterenol.12 These data indicate that BayK induces significant rate increases in SANCs isolated from mice and dogs, suggesting that studies of heart rate responses to BayK in mice will also provide information relevant to larger mammals.
Fig 1.
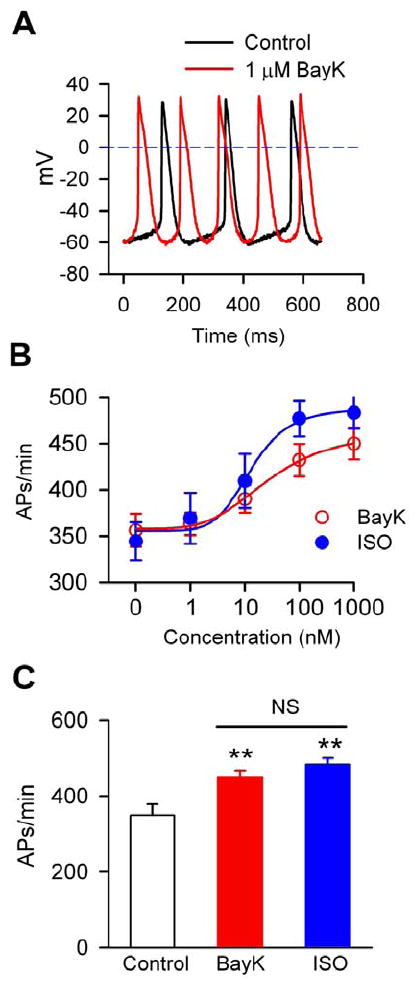
BayK increased the pacemaking activity of SANCs. A. Representative AP traces in a mouse SANC before (black line) and after (red line) 1 μM BayK. B. Dose-response curves of BayK and isoproterenol on the automaticity of mouse SANCs, n=5-22 for each dose. C. Summary data of SANC spontaneous activity under control conditions (n=28), in the presence of 1 μM BayK (n=24) and 1 μM isoproterenol (ISO, n=5) One-way ANOVA **P<0.01 vs control.
CaMKII is required for BayK-triggered increases in SANC rates
We recently reported that CaMKII is required for SAN “fight or flight” physiology during β-AR stimulation.9 In order to test whether acceleration of SAN automaticity by L-type Ca2+ channel activation, independent of β-AR stimulation, requires CaMKII, we measured the effects of BayK on isolated SANCs and Langendorff-perfused hearts from mice with cardiomyocyte-delimited expression of AC3-I. We used AC3-C expressing mice and WT littermates as controls.15 At baseline, the spontaneous activity was similar among AC3-I, AC3-C and WT SANCs, similar to our earlier report (Fig 2A and B).9 However, the rate increase in AC3-I SANCs after BayK was less than half of the BayK-triggered rate increase in controls (Fig 2C). These results showed that BayK-triggered increases in automaticity were significantly (P=0.003) reduced in AC3-I compared to control SANCs. The rate increases by BayK were not eliminated by AC3-I expression, likely because ICa contributes directly to DD and because AC3-I inhibition of CaMKII activity is incomplete.15 Similar to the isolated SANCs, there was no difference in the baseline beating rates in AC3-I, AC3-C and WT Langedorff-perfused hearts. BayK increased the heart rates in all mice, but the increase in AC3-I hearts was significantly (P=0.005) less than that in hearts from WT and AC3-C mice (Supplementary data Fig S2). These results indicated that CaMKII is required for the full range of SANC rate response to BayK.
Fig 2.
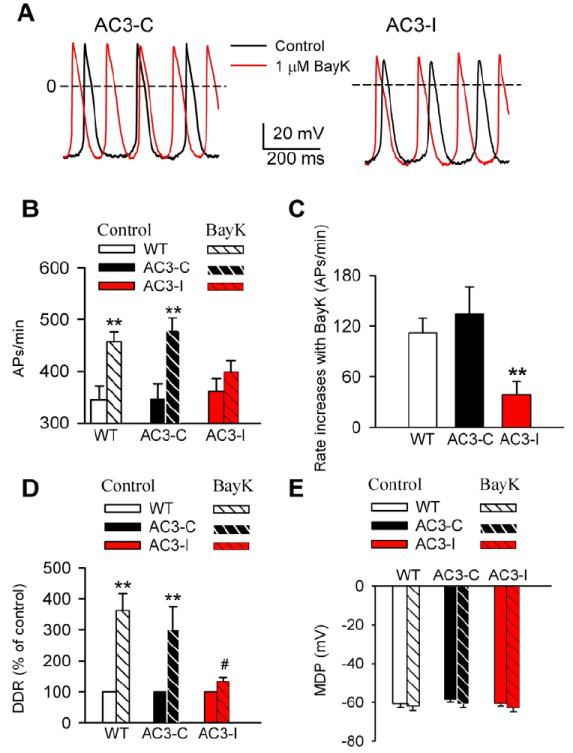
SANC rate increases by BayK are reduced by CaMKII inhibition. A. Representative AP traces recorded before and after BayK in SANCs isolated from AC3-C and AC3-I mice. B. Spontaneous activity of SANCs in the absence and presence of 1 μM BayK. **P<0.01 vs control, n≥16 cells from more than 4 animals for each group. C. SANC rate increases by BayK in WT, AC3-C and AC3-I mice (data from panel B). ** P<0.001 vs WT or AC3-C, n≥16 cells from more than 4 animals for each group. D. BayK (1 μM) significantly increased the DDR in SANCs from WT and AC3-C, but not AC3-I mice. **P<0.01 vs control. # P<0.05 vs WT and AC3-C mice, n≥16 cells from more than 4 animals for each group. E. BayK did not significantly alter the MDP in any groups (data from panel D), n≥16 cells from more than 4 animals for each group.
Reduced DDR response but preserved If and ICa in AC3-I SANCs
Modulating the spontaneous DD is the cell membrane potential mechanism for governing SANC rate.16 At baseline the DD rate (DDR) did not differ among the three genotypes. However, DDR increases by BayK in AC3-I mice were significantly (P=0.01) less than in WT and AC3-C controls (Fig 2D). BayK did not significantly alter MDP (Fig 2E) or APD (data not shown) in any of the groups. Several Ca2+ sensitive adenylyl cylases are expressed in SAN, suggesting the possibility that BayK may enhance the activity of the cAMP/PKA pathway.17-18 We next considered whether If was involved in rate increases by BayK, and measured If from SANCs before and after BayK. BayK (1 μM) did not significantly affect If in any of the groups (Fig 3). These results indicated that If was unlikely to contribute to the reduced efficacy of BayK in AC3-I SANCs, and suggested that increasing SANC Ca2+ by BayK did not activate cAMP19 or PKA20 with access to HCN4. We next measured the PKA phosphorylation site (Ser16) on phospholamban,21-22 to test for evidence of PKA activation by BayK, at a validated PKA target, in SANCs. We did not detect a significant increase in phospholamban Ser16 phosphorylation with BayK treatment (Supplementary data Fig S3) compared to baseline. We interpret our findings to indicate that BayK increases heart rate by a mechanism that is not significantly determined by the cAMP/PKA signaling pathway in SANCs. We next measured ICa to determine if the reduced rate response to BayK in AC3-I SANCs was due to reduced ICa responses in AC3-I compared to control SANCs. Consistent with our previous study,9 there was no difference in the basal ICa among the three genotypes (Fig 4). BayK equivalently increased peak and integrated ICa in each of the genotypes, showing that reduced heart rate responses to BayK in AC3-I mice were independent of BayK actions on ICa. Our data suggested that the reduced BayK response in AC3-I SANCs was determined by a cellular mechanism that was downstream to ICa.
Fig 3.
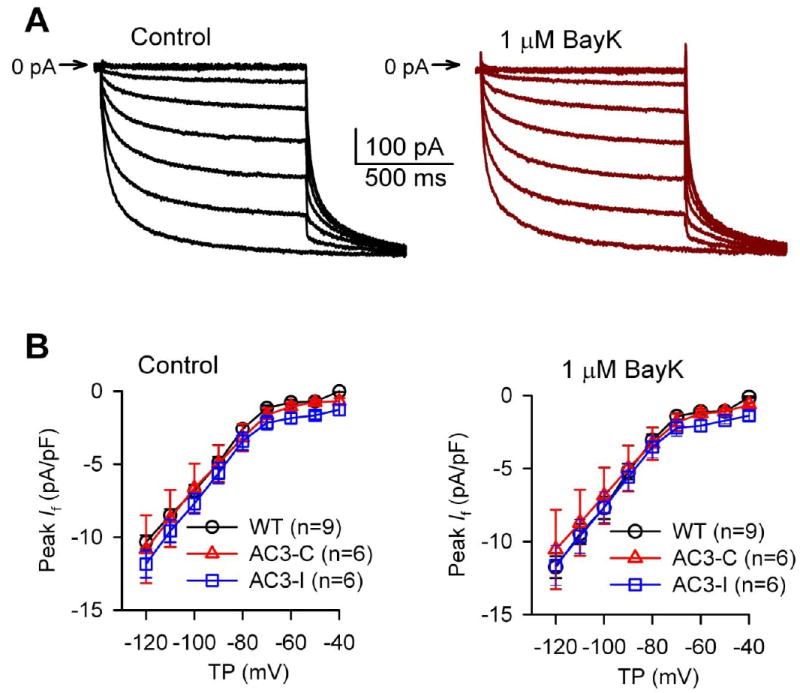
BayK did not affect If. A. Representative If traces recorded in a SANC isolated from a WT mouse before and after 1 μM BayK. B. I-V relationship of If under control conditions and in the presence of 1 μM BayK.
Fig 4.
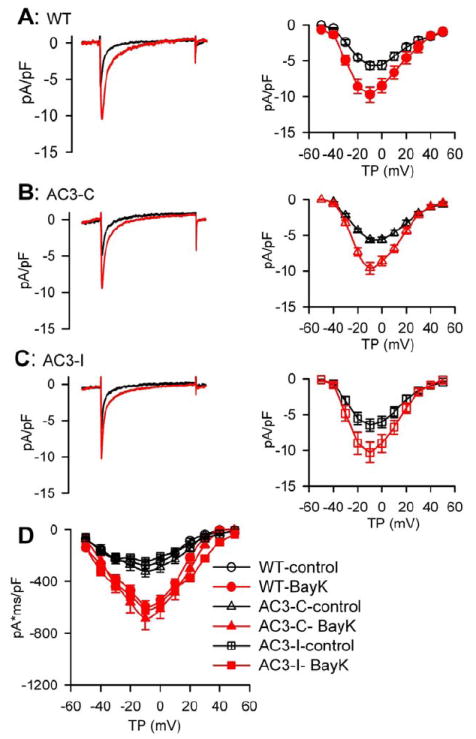
BayK equally increased ICa in AC3-I, AC3-C and WT SANCs. A-C. representative ICa traces (left panels) and I-V relationship (right panels) of peak currents before and after 1 μM BayK from WT (A), AC3-C (B) and AC3-I (C) mice. D. The integrated ICa I-V relationship from WT, AC3-C and AC3-I SANCs, under control conditions and in the presence of BayK, n=8-19 cells from more than 3 animimals for each group.
Ryanodine prevented the SANC response to BayK
Ca2+ influx via ICa is crucial for filling sarcoplasmic reticulum (SR) Ca2+ stores and sustaining the capacity for the local Ca2+ release events from SR that are thought to contribute to SANC DDR and automaticity.4, 9 Because CaMKII promotes SANC SR Ca2+ release in response to isoproterenol,9 we hypothesized that local SR Ca2+ release events may underlie the rate increases by BayK. We used ryanodine, a SR Ca2+ release antagonist, to test if BayK increases would persist in the absence of increases in local SR Ca2+ release events. Ryanodine (2 μM) reduced spontaneous automaticity equally in SANCs from AC3-I, AC3-C and WT mice(Fig 5). The SANC rate increases by BayK were significantly and equivalently blunted in all genotypes, suggesting that SR Ca2+ release was essential for the differential effects of BayK on pacemaking activity in AC3-I compared to AC3-C and WT SANC.
Fig 5.
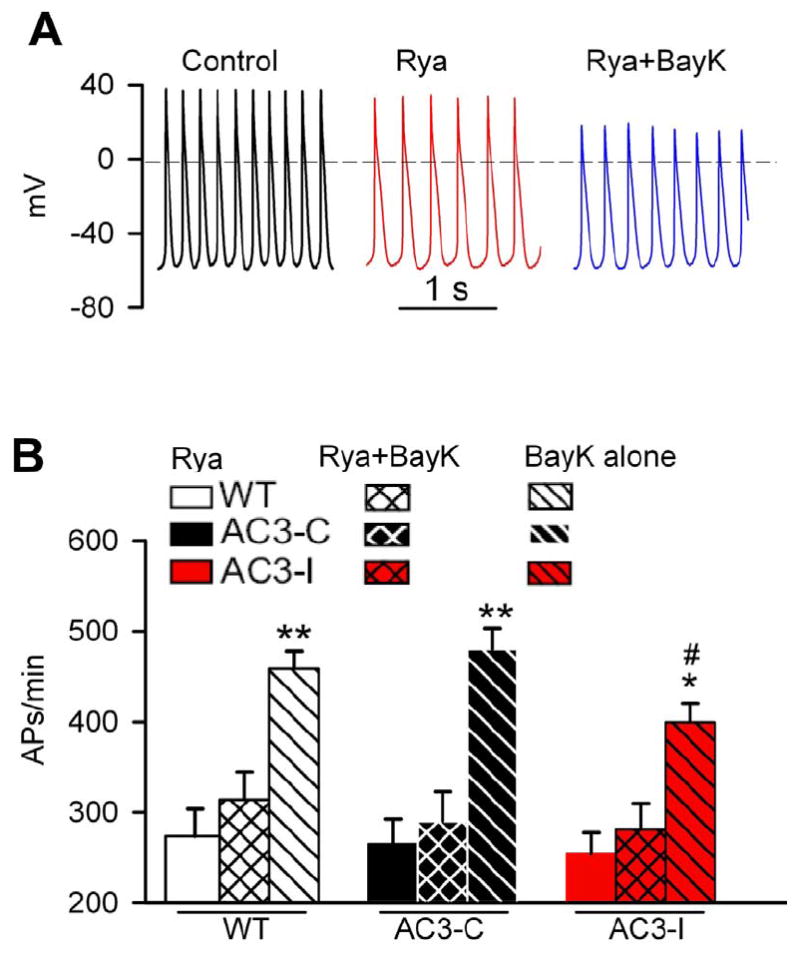
Ryanodine (Rya) pretreatment prevented BayK triggered SANC rate increases. A. Representative APs in control, in the presence of Rya and the combination of Rya plus BayK. B. SANC spontaneous pacemaking activity in the presence of Rya, the combination of Rya plus BayK, and BayK alone. *P<0.05 vs Rya and Rya+BayK, **P<0.001 vs Rya and Rya+BayK, #P<0.05 vs WT and AC3-C in the presence of BayK. n=7-10 cells from 3 animals for each genotypes.
CaMKII inhibition suppressed SR Ca2+ release and BayK triggered increases in INCX
As SR Ca2+ release appeared to contribute to the effects of BayK on SANCpacemaking activity, we acquired confocal Ca2+ images to directly measure SR Ca2+ release events. BayK increased Ca2+ transient amplitudes to a similar level in SANCs from each of the three genotypes (Fig 6A-B). In contrast, BayK significantly increased the frequency of late diastolic local Ca2+ release events in SANCs from WT and AC3-C but not AC3-I mice (Fig 6C). However, Ca2+ spark amplitude, duration and width remained unaltered by BayK in any of the genotypes (data not shown). These results suggested that the reduced response of AC3-I SANCs stimulated with BayK was due to impaired diastolic SR Ca2+ release, similar to the reduction in isoproterenol-stimulated SR Ca2+ release in AC3-I SANCs.9
Fig 6.
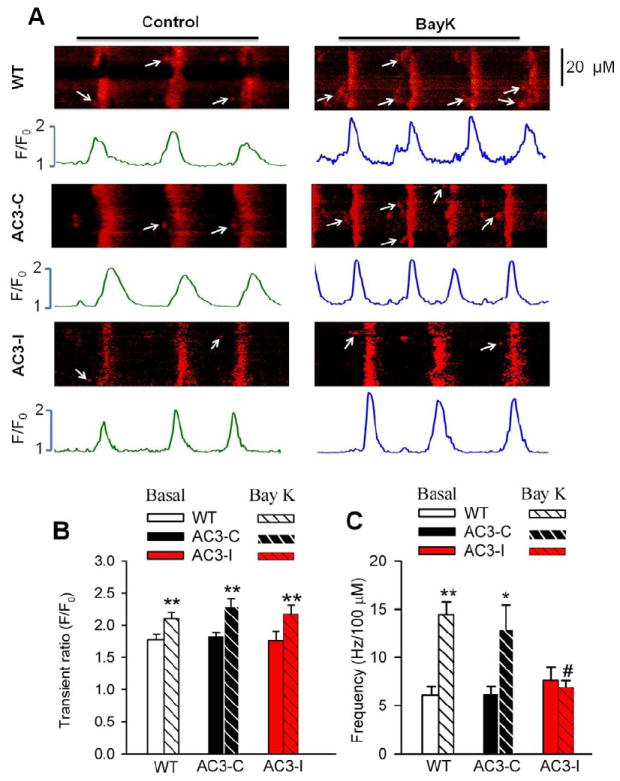
Effect of BayK on the SANC intracellular Ca2+. A. Representative Ca2+ images in control and in the presence of 1 μM BayK. The arrows indicate late diastolic Ca2+ sparks. B. Summary data for Ca2+ transients. n=10-35 cells from more than 5 animals for each group. **P<0.01 vs control. C. Summary data for Ca2+ spark frequency. *P<0.05, vs control, **P<0.01 vs control, #P<0.05 vs WT and AC3-C mice. n≥18 cells from more than 4 animals for each group.
Diastolic SR Ca2+ release enhances INCX in SANCs, and the increase in INCX is a critical for promoting DDR.4, 23-24 We next measured inward currents elicited with a voltage clamp ramp protocol from -60 to -45 mV, mimicking voltage range of spontaneous DD in SANCs, 25 at baseline and after BayK. The inward currents evoked by the voltage command protocol were predominantly INCX, because they were nearly eliminated by replacing Na+ with Li+ in the bath solution (Supplementary data Fig S4). We found BayK increased inward INCX in WT and AC3-C significantly more than in AC3-I mice (Fig 7). These results supported the concept that CaMKII promoted increased diastolic Ca2+ release to enhance INCX and heart rate in response to BayK.25-26
Fig 7.
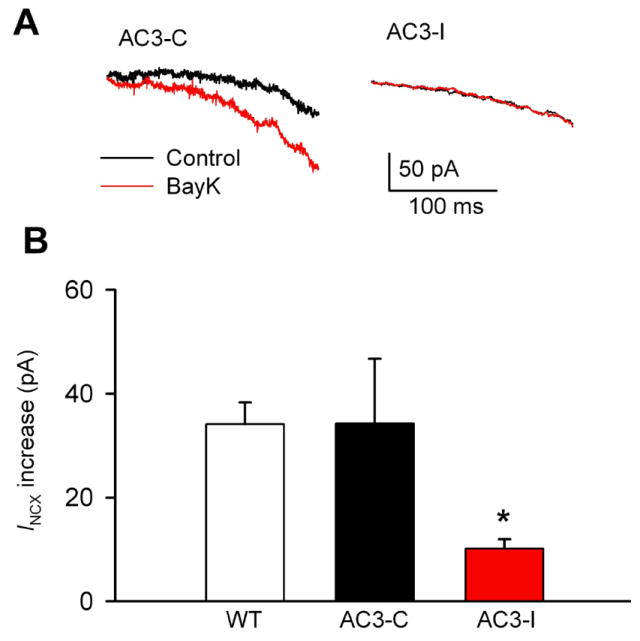
Reduced INCX response to BayK in AC3-I SANCs. A. Representative SANC INCX traces recorded from AC3-C and AC3-I mice before and after 1 μM BayK. B. INCX increases by BayK. **P<0.05 vs WT or AC3-C mice, n=5-10 cells from 3 animals for each group.
BayK enhanced CaMKII activity in SANCs
CaMKII becomes constitutively active in response to prolonged Ca2+ exposure27 by autophosphorylation of a threonine residue (Thr 286 or 287 depending on the isoform) in the CaMKII regulatory domain.28 Our data up to this point strongly supported a concept where CaMKII activation increased DDR and SANC rates by enhancing SR Ca2+ release and INCX. In order to assess the affects of BayK on SANC CaMKII activation, we measured threonine autophosphorylation (p-CaMKII) at baseline and during BayK (Fig 8). p-CaMKII was readily detected after BayK in WT and AC3-C, but not in AC3-I SANCs. We found that p-CaMKII was enriched in a subsarcolemmal distribution after BayK in control SANCs. In contrast, pCaMKII was not evident in SANCs from any of the mice without BayK, under our experimental conditions. We found that BayK significantly increased phosphorylation of a bona fide CaMKII site on phospholamban (pThr17) in WT and AC3-C but not in AC3-I mice (supplementary data Fig S3). Taken together, these results show that BayK activates CaMKII under conditions permissive for increased SR Ca2+ release and SANC rate increases and that AC3-I expression in SANCs is sufficient to inhibit BayK triggered increases in CaMKII activity.
Fig 8.
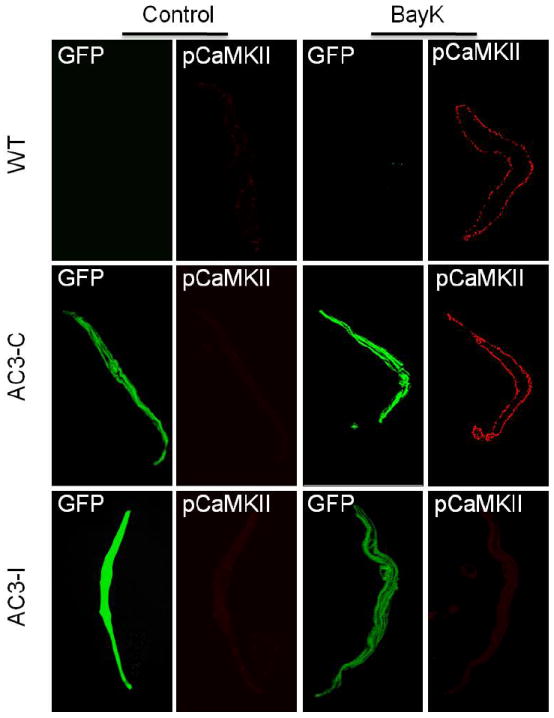
AC3-I expression eliminated CaMKII autophosphorylation response to BayK in mouse SANCs. Representative immunofluorescence images show BayK enhanced CaMKII autophosphorylation (pCaMKII) in SANCs isolated from WT and AC3-C, but not AC3-I mice. Note: eGFP is expressed in AC3-C and AC3-I SANCs. *P<0.05 vs WT and AC3-C
Discussion
The discovery of cAMP-gated channels29 was a major milestone toward developing a molecular understanding of mechanisms for physiological cardiac pacing. The critical importance of If to cardiac pacing in humans was highlighted by findings that patients with defects in If due to mutations in the gene encoding HCN4 required surgical implantation of artificial pacemakers.30 Recent data shows that heart failure patients benefit from new and highly selective If antagonist drugs by reducing heart rate,31 suggesting that hyperactivation of If may contribute to heart disease in humans. We interpret the wealth of cellular and animal studies, in combination with the aforementioned clinical investigations, to provide incontrovertible evidence that If is a fundamental mechanism for cardiac pacing. HCN4 is the predominant cAMP gated channel for If in SANC 32 and new studies show that HCN4 is also activated by PKA phosphorylation.20 Thus, β-AR agonist stimulation increases heart rate in part by enhancing SAN cAMP and PKA activity and increasing If. However, mice with HCN4 knock out show a surprisingly mild phenotype and have normal or near normal heart rate increases in response to isoproterenol.33 These and other findings9, 12, 34 support a view that HCN4-alternative mechanisms also support physiological cardiac pacing.
In this study we activated SANC automaticity by engaging the ‘Ca2+ clock’ mechanism,4 independently of β-AR activation or If by using BayK. We found that BayK and isoproterenol increased SANC rates over a similar dynamic range, suggesting that the Ca2+-based mechanisms for cardiac pacing are capable of supporting a major component of fight or flight heart rate increases. Although the Ca2+ clock is recruited in SANC by β-AR agonist stimulation under physiological conditions, our study showed that the Ca2+ clock can be activated even in the absence of β-AR receptor stimulation. Increased intracellular Ca2+ may increase PKA activity in SANCs due to the relatively enhanced activity of Ca2+-activated adenylyl cyclase isoforms, principally types 1 and 8.7, 18 The potential for Ca2+-activated signaling molecules to affect If has been posited as a mechanism for Ca2+ evoked increases in SAN automaticity.3, 35 Thus, it is possible that BayK-induced increases in SANC automaticity could be a consequence of increased If. However, we found no evidence that BayK evoked increases in cellular Ca2+ entry affected If. We believe our results provide strong evidence that the Ca2+ clock is an independent cellular mechanism that can be clearly distinguished from If.
SANCs show increased basal PKA activity compared to atrial and ventricular myocytes,36 and increased PKA activity is hypothesized to play a critical role in promoting the SR Ca2+ release and inward INCX. 26, 37 Thus, it is possible that hyperactivation of the cAMP/PKA pathway in SANCs, probably due to a relative abundance of Ca2+ activated adenylyl cyclase isoforms, could enhance coupling of the If and Ca2+ clock cellular pacing mechanisms. Several studies now provide evidence showing that β-AR agonist stimulation increases heart rate by coordinate activation of If and a Ca2+ clock mechanism.4-5 Our study contributes new information by showing that the Ca2+ clock can produce significant increases in heart rate and SANC automaticity by a mechanism requiring CaMKII in the absence of β-AR agonist stimulation. We did not identify evidence for Ca2+-activated cAMP or PKA to increase If or to phosphorylate phospholamban. Thus, we interpret our results to show that the Ca2+ clock is highly dependent on CaMKII, while Ca2+ activated increases in cAMP and PKA signaling to If and phospholamban are dispensable, at least under these experimental conditions. The existence of a Ca2+ based mechanism for SANC pacing and the relative importance of If and SR Ca2+ release have become highly polemic issues, with some investigators denying the potential for a Ca2+-dependent, If independent mechanism to drive SANC automaticity.3 We believe that our findings provide new clarity to this controversy, by showing the Ca2+ clock, in the absence of β-AR agonist stimulation, to be a highly effective cellular mechanism for SANC and cardiac pacing. In this study we used BayK as an experimental tool to interrogate the Ca2+ clock. Under physiological conditions, our results suggest that Ca2+ clock activation by enhanced CaMKII with β-AR stimulation may at least partially accout for the heart rate increase in the fight or flight response.
CaMKII now appears to be an essential component of SANC automaticity due to β-AR agonists and BayK. As such, CaMKII represents a highly adaptable connection between If and SR-based SANC pacing mechanisms. CaMKII increases CaV1.2 current and phosphorylation of CaV1.2 channel β subunits, which are presumably shared by CaV1.3, leads to mode 2 gating,38 a high activity state originally identified as a response to BayK.39 By using BayK we were able to circumvent the potentially confounding effects of CaMKII on SANC Ca2+ entry by ICa. Surprisingly, BayK activation of ICa, in the presence of reduced CaMKII activity or impaired SR Ca2+ release, was insufficient to significantly increase SANC automaticity. In contrast, CaMKII activity and SR Ca2+ release coupled to BayK-enhanced ICa effectively increased SANC rates by diastolic SR Ca2+ release favoring DD due to inward INCX. Although BayK failed to increase diastolic SR Ca2+ release and consequent INCX in AC3-I mice, BayK resulted in modest enhancement of SANC automaticity, suggesting that the increases of ICa by BayK also directly contribute to phase 4 DD. SR Ca2+ release is essential for myofilament activation during excitation-contraction coupling in myocardium, whereas increased diastolic SR Ca2+ release is associated with arrhythmias and myopathy in ventricular and atrial myocardium. CaMKII hyperactivation, perhaps partly as a response to increased SR Ca2+, is now recognized to contribute to cardiomyopathy and arrhythmias.40-41 In contrast, diastolic SR Ca2+ leak and CaMKII activation appear to be fundamental aspects of the physiological function of SANC. It will be interesting to learn if hyperactivation of CaMKII in SANCs leads to defects in cardiac pacing, and if CaMKII inhibition could be used therapeutically to prevent excessive SANC firing rates in heart failure, similar to the recent success of If antagonists.
A once sleepy backwater in cardiac biology, the mechanisms of cardiac pacing have become interesting and controversial. Two competing paradigms for cardiac pacing have vied for hegemony: an HCN4 directed pacemaker current (If) and a Ca2+ clock mechanism where the cellular Ca2+ handling apparatus supporting excitation-contraction coupling in atrium and ventricle has been repurposed for sinoatrial nodal cell automaticity. Some research appears to support a view that the Ca2+ clock and If mechanisms are inter-related by Ca2+ activation of cAMP/PKA signaling. In every case, the starting point for studying these mechanisms has been β-adrenergic receptor (β-AR) agonist ligands. Here we employed Bay K8644, a selective agonist of L-type Ca2+ channel, to activate the Ca2+ clock in the absence of catecholamines. We found the Ca2+ clock, in the absence of β-AR agonist stimulation or evidence of cAMP/PKA activation, is a highly effective cellular mechanism for SANC and cardiac pacing and that the Ca2+ clock and β-AR stimulation responses require CaMKII. It will be interesting to learn if hyperactivation of CaMKII in SANCs leads to defects in cardiac pacing, and if CaMKII inhibition could be used therapeutically to prevent excessive SANC firing rates in heart failure, similar to the recent success of If antagonists.
Supplementary Material
Acknowledgments
The authors thank Ms Jinying Yang and Mr William J Kutschke for their expert technical assistance.
Funding Sources: This work was funded by National Institutes of Health (NIH) Grants R01 HL 079031, R01 HL 096652, and R01 HL 070250, the University of Iowa Research Foundation and the Fondation Leducq Transatlantic Alliance for CaMKII Signaling.
Footnotes
Conflict of Interest Disclosurs: None
References
- 1.DiFrancesco D. The role of the funny current in pacemaker activity. Circ Res. 2010;106:434–446. doi: 10.1161/CIRCRESAHA.109.208041. [DOI] [PubMed] [Google Scholar]
- 2.DiFrancesco D, Borer JS. The funny current: cellular basis for the control of heart rate. Drugs. 2007;67:15–24. doi: 10.2165/00003495-200767002-00003. [DOI] [PubMed] [Google Scholar]
- 3.Lakatta EG, DiFrancesco D. What keeps us ticking: a funny current, a calcium clock, or both? J Mol Cell Cardiol. 2009;47:157–170. doi: 10.1016/j.yjmcc.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lakatta EG, Maltsev VA, Vinogradova TM. A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart’s pacemaker. Circ Res. 2010;106:659–673. doi: 10.1161/CIRCRESAHA.109.206078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maltsev VA, Lakatta EG. Dynamic interactions of an intracellular Ca2+ clock and membrane ion channel clock underlie robust initiation and regulation of cardiac pacemaker function. Cardiovasc Res. 2008;77:274–284. doi: 10.1093/cvr/cvm058. [DOI] [PubMed] [Google Scholar]
- 6.Strange PG. Signaling mechanisms of GPCR ligands. Curr Opin Drug Discov Devel. 2008;11:196–202. [PubMed] [Google Scholar]
- 7.Younes A, Lyashkov AE, Graham D, Sheydina A, Volkova MV, Mitsak M, Vinogradova TM, Lukyanenko YO, Li Y, Ruknudin AM, Boheler KR, van Eyk J, Lakatta EG. Ca(2+)-stimulated basal adenylyl cyclase activity localization in membrane lipid microdomains of cardiac sinoatrial nodal pacemaker cells. J Biol Chem. 2008;283:14461–14468. doi: 10.1074/jbc.M707540200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grimm M, Brown JH. Beta-adrenergic receptor signaling in the heart: role of CaMKII. J Mol Cell Cardiol. 2010;48:322–330. doi: 10.1016/j.yjmcc.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu Y, Gao Z, Chen B, Koval OM, Singh MV, Guan X, Hund TJ, Kutschke W, Sarma S, Grumbach IM, Wehrens XH, Mohler PJ, Song LS, Anderson ME. Calmodulin kinase II is required for fight or flight sinoatrial node physiology. Proc Natl Acad Sci U S A. 2009;106:5972–5977. doi: 10.1073/pnas.0806422106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brouha L, Cannon WB, Dill DB. The heart rate of the sympathectomized dog in rest and exercise. J Physiol. 1936;87:345–359. doi: 10.1113/jphysiol.1936.sp003411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cannon WB. Pharmacological Injections and Physiological Inferences. Science. 1929;70:500–501. doi: 10.1126/science.70.1821.500-a. [DOI] [PubMed] [Google Scholar]
- 12.Gao Z, Chen B, Joiner ML, Wu Y, Guan X, Koval OM, Chaudhary AK, Cunha SR, Mohler PJ, Martins JB, Song LS, Anderson ME. I(f) and SR Ca(2+) release both contribute to pacemaker activity in canine sinoatrial node cells. J Mol Cell Cardiol. 2010;49:33–40. doi: 10.1016/j.yjmcc.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen B, Wu Y, Mohler PJ, Anderson ME, Song LS. Local control of Ca2+-induced Ca2+ release in mouse sinoatrial node cells. J Mol Cell Cardiol. 2009;47:706–715. doi: 10.1016/j.yjmcc.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schramm M, Thomas G, Towart R, Franckowiak G. Novel dihydropyridines with positive inotropic action through activation of Ca2+ channels. Nature. 1983;303:535–537. doi: 10.1038/303535a0. [DOI] [PubMed] [Google Scholar]
- 15.Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, Price EE, Jr, Thiel W, Guatimosim S, Song LS, Madu EC, Shah AN, Vishnivetskaya TA, Atkinson JB, Gurevich VV, Salama G, Lederer WJ, Colbran RJ, Anderson ME. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med. 2005;11:409–417. doi: 10.1038/nm1215. [DOI] [PubMed] [Google Scholar]
- 16.Hauswirth O, Noble D, Tsien RW. The mechanism of oscillatory activity at low membrane potentials in cardiac Purkinje fibres. J Physiol. 1969;200:255–265. doi: 10.1113/jphysiol.1969.sp008691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lyashkov AE, Vinogradova TM, Zahanich I, Li Y, Younes A, Nuss HB, Spurgeon HA, Maltsev VA, Lakatta EG. Cholinergic receptor signaling modulates spontaneous firing of sinoatrial nodal cells via integrated effects on PKA-dependent Ca(2+) cycling and I(KACh) Am J Physiol Heart Circ Physiol. 2009;297:H949–959. doi: 10.1152/ajpheart.01340.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mattick P, Parrington J, Odia E, Simpson A, Collins T, Terrar D. Ca2+-stimulated adenylyl cyclase isoform AC1 is preferentially expressed in guinea-pig sino-atrial node cells and modulates the I(f) pacemaker current. J Physiol. 2007;582:1195–1203. doi: 10.1113/jphysiol.2007.133439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DiFrancesco D, Tortora P. Direct activation of cardiac pacemaker channels by intracellular cyclic AMP. Nature. 1991;351:145–147. doi: 10.1038/351145a0. [DOI] [PubMed] [Google Scholar]
- 20.Liao Z, Lockhead D, Larson ED, Proenza C. Phosphorylation and modulation of hyperpolarization-activated HCN4 channels by protein kinase A in the mouse sinoatrial node. J Gen Physiol. 2010;136:247–258. doi: 10.1085/jgp.201010488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chu G, Lester JW, Young KB, Luo W, Zhai J, Kranias EG. A single site (Ser16) phosphorylation in phospholamban is sufficient in mediating its maximal cardiac responses to beta -agonists. J Biol Chem. 2000;275:38938–38943. doi: 10.1074/jbc.M004079200. [DOI] [PubMed] [Google Scholar]
- 22.Simmerman HK, Collins JH, Theibert JL, Wegener AD, Jones LR. Sequence analysis of phospholamban. Identification of phosphorylation sites and two major structural domains. J Biol Chem. 1986;261:13333–13341. [PubMed] [Google Scholar]
- 23.Huser J, Blatter LA, Lipsius SL. Intracellular Ca2+ release contributes to automaticity in cat atrial pacemaker cells. J Physiol. 2000;524:415–422. doi: 10.1111/j.1469-7793.2000.00415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lipsius SL, Huser J, Blatter LA. Intracellular Ca2+ release sparks atrial pacemaker activity. News Physiol Sci. 2001;16:101–106. doi: 10.1152/physiologyonline.2001.16.3.101. [DOI] [PubMed] [Google Scholar]
- 25.Vinogradova TM, Sirenko S, Lyashkov AE, Younes A, Li Y, Zhu W, Yang D, Ruknudin AM, Spurgeon H, Lakatta EG. Constitutive phosphodiesterase activity restricts spontaneous beating rate of cardiac pacemaker cells by suppressing local Ca2+ releases. Circ Res. 2008;102:761–769. doi: 10.1161/CIRCRESAHA.107.161679. [DOI] [PubMed] [Google Scholar]
- 26.Vinogradova TM, Bogdanov KY, Lakatta EG. beta-Adrenergic stimulation modulates ryanodine receptor Ca(2+) release during diastolic depolarization to accelerate pacemaker activity in rabbit sinoatrial nodal cells. Circ Res. 2002;90:73–79. doi: 10.1161/hh0102.102271. [DOI] [PubMed] [Google Scholar]
- 27.De Koninck P, Schulman H. Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations. Science. 1998;279:227–230. doi: 10.1126/science.279.5348.227. [DOI] [PubMed] [Google Scholar]
- 28.Kuret J, Schulman H. Mechanism of autophosphorylation of the multifunctional Ca2+/calmodulin-dependent protein kinase. J Biol Chem. 1985;260:6427–6433. [PubMed] [Google Scholar]
- 29.Ludwig A, Zong X, Jeglitsch M, Hofmann F, Biel M. A family of hyperpolarization-activated mammalian cation channels. Nature. 1998;393:587–591. doi: 10.1038/31255. [DOI] [PubMed] [Google Scholar]
- 30.Milanesi R, Baruscotti M, Gnecchi-Ruscone T, Difrancesco D. Familial sinus bradycardia associated with a mutation in the cardiac pacemaker channel. N Engl J Med. 2006;354:151–157. doi: 10.1056/NEJMoa052475. [DOI] [PubMed] [Google Scholar]
- 31.Swedberg K, Komajda M, Bohm M, Borer JS, Ford I, Dubost-Brama A, Lerebours G, Tavazzi L. Ivabradine and outcomes in chronic heart failure (SHIFT): a randomised placebo-controlled study. Lancet. 2010;376:875–885. doi: 10.1016/S0140-6736(10)61259-7. [DOI] [PubMed] [Google Scholar]
- 32.Shi W, Wymore R, Yu H, Wu J, Wymore RT, Pan Z, Robinson RB, Dixon JE, McKinnon D, Cohen IS. Distribution and prevalence of hyperpolarization-activated cation channel (HCN) mRNA expression in cardiac tissues. Circ Res. 1999;85:e1–6. doi: 10.1161/01.res.85.1.e1. [DOI] [PubMed] [Google Scholar]
- 33.Herrmann S, Stieber J, Stockl G, Hofmann F, Ludwig A. HCN4 provides a ‘depolarization reserve’ and is not required for heart rate acceleration in mice. EMBO J. 2007;26:4423–4432. doi: 10.1038/sj.emboj.7601868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maltsev VA, Lakatta EG. Normal heart rhythm is initiated and regulated by an intracellular calcium clock within pacemaker cells. Heart Lung Circ. 2007;16:335–348. doi: 10.1016/j.hlc.2007.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rigg L, Mattick PA, Heath BM, Terrar DA. Modulation of the hyperpolarization-activated current (I(f)) by calcium and calmodulin in the guinea-pig sino-atrial node. Cardiovasc Res. 2003;57:497–504. doi: 10.1016/s0008-6363(02)00668-5. [DOI] [PubMed] [Google Scholar]
- 36.Vinogradova TM, Lyashkov AE, Zhu W, Ruknudin AM, Sirenko S, Yang D, Deo S, Barlow M, Johnson S, Caffrey JL, Zhou YY, Xiao RP, Cheng H, Stern MD, Maltsev VA, Lakatta EG. High basal protein kinase A-dependent phosphorylation drives rhythmic internal Ca2+ store oscillations and spontaneous beating of cardiac pacemaker cells. Circ Res. 2006;98:505–514. doi: 10.1161/01.RES.0000204575.94040.d1. [DOI] [PubMed] [Google Scholar]
- 37.Vinogradova TM, Brochet DX, Sirenko S, Li Y, Spurgeon H, Lakatta EG. Sarcoplasmic reticulum Ca2+ pumping kinetics regulates timing of local Ca2+ releases and spontaneous beating rate of rabbit sinoatrial node pacemaker cells. Circ Res. 2010;107:767–775. doi: 10.1161/CIRCRESAHA.110.220517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grueter CE, Abiria SA, Dzhura I, Wu Y, Ham AJ, Mohler PJ, Anderson ME, Colbran RJ. L-type Ca2+ channel facilitation mediated by phosphorylation of the beta subunit by CaMKII. Mol Cell. 2006;23:641–650. doi: 10.1016/j.molcel.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 39.Hess P, Lansman JB, Tsien RW. Different modes of Ca channel gating behaviour favoured by dihydropyridine Ca agonists and antagonists. Nature. 1984;311:538–544. doi: 10.1038/311538a0. [DOI] [PubMed] [Google Scholar]
- 40.Bellinger AM, Reiken S, Carlson C, Mongillo M, Liu X, Rothman L, Matecki S, Lacampagne A, Marks AR. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat Med. 2009;15:325–330. doi: 10.1038/nm.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kushnir A, Shan J, Betzenhauser MJ, Reiken S, Marks AR. Role of CaMKIIdelta phosphorylation of the cardiac ryanodine receptor in the force frequency relationship and heart failure. Proc Natl Acad Sci U S A. 2010;107:10274–10279. doi: 10.1073/pnas.1005843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.