

Abstract
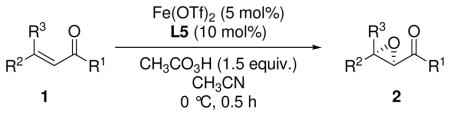
The combination of Fe(OTf)2 and novel phenanthroline ligands enables the catalytic asymmetric epoxidation of acyclic β,β-disubstituted enones which have been heretofore inaccessible substrate classes. The reactions provide highly enantioenriched α,β-epoxyketones (up to 92% ee) which are further converted to functionalized β-ketoaldehydes with an all-carbon quaternary center.
The catalytic asymmetric epoxidation of olefins presents a powerful strategy for the synthesis of chiral molecules. Thus, numerous efforts have been dedicated to achieving more efficiency with less expensive and environmentally benign catalysts and oxidants as well as the applicability to a variety of substrate classes.1 Among various useful methods, the development of an iron-catalyzed asymmetric epoxidation should provide us with many advantages, as iron is the most abundant transition metal on the earth and relatively non-toxic.2, 3, 4 In addition, understanding the mechanism of iron-catalyzed oxidations, which play important roles in biological metabolism, may lead to new insights in biocatalysis and a resulting drug design.5 Actually, the biomimetic asymmetric epoxidation of styrene derivatives with iron porphyrin complexes was first reported in 1999,6,7 although it has drawbacks such as the difficult synthesis of the required chiral porphyrin ligands. After studies to pursue non-heme iron catalysts which could be easily prepared and modified, Beller and co-workers reported that the best results have been from iron-catalyzed asymmetric epoxidation of stilbene derivatives with excellent enantioselectivity.4c, d However, the high selectivity was obtained only for one specific substrate with 10 mol% catalyst loading. Therefore, it is apparent that iron has yet to be fully introduced in asymmetric epoxidations.
Obviously, the extension of the accessible substrate classes for catalytic asymmetric epoxidation has been desirable. To the best of our knowledge, a general method for the catalytic asymmetric epoxidation of acyclic β,β-disubstituted enones is still lacking, probably due to the stereocongestion at the β-carbon in the Weitz-Scheffer type epoxidation, which is commonly employed to access α,β-epoxy carbonyl compounds (Scheme 1, eq 1).1d, 8 In the case of acyclic enones, a β-substituent (R3 group) increases the steric repulsion not only between the β-carbon and nucleophile but also between the R3 group and the acyl group, which causes the substrate to break conjugation to avoid repulsion. As a result, electrophilicity of the double bond in acyclic β,β-disubstituted enones is thought to be lower than that in cyclic or β-non-substituted enones.9 In contrast, the deconjugation described above should increase reactivity toward electrophilic epoxidation (eq. 2).9c, i Herein is a solution to the catalytic asymmetric epoxidation of acyclic β,β-disubstituted enones with a newly designed iron complex.
Scheme 1.

The epoxidation of acyclic β,β-disubstituted carbonyl compounds.
We initially investigated the epoxidation of the readily available (E)-dypnone (1a) as a model substrate with a variety of complexes consisting of iron metals and phenanthroline ligands attached to binaphthyl moieties (Table 1). After preliminary screening of reaction conditions, peracetic acid as a terminal oxidant was recognized as being crucial to afford epoxides.10 When 1a was reacted with 5 mol % of FeCl2 and monophenanthroline ligand (L1) in the presence of peracetic acid solution in acetonitrile, the epoxidation resulted in only low conversion of starting material and low selectivity (entry 1). Replacing FeCl2 with Fe(OTf)2 led to a significant improvement in terms of reactivity and enantioselectivity (entry 2). Thus, we turned our attention to the effects of the several monophenanthroline ligands. Introduction of a methyl group on the 2′-position in binaphthyl group dramatically diminished reactivity as well as selectivity (entry 3). To our delight, we found that the introduction of a phenyl group on the 3,8-positions in the phenanthroline moiety, which is expected to restrict the rotation of the bond between binaphthyl group and phenanthroline moiety, resulted in increased enantioselectivity significantly (entry 4). After testing various of ligands bearing aromatic groups on the phenanthroline rings (entry 4–7), we identified L5 as the ligand providing the excellent yield and enantioselectivity (entry 6). Study of the ligand-metal ratio implied that an iron complex coordinated by two phenanthroline ligands induces high enantioselectivity (entries 6, 8 and 9). Furthermore, catalyst loading was successfully lowered to 2.5 mol % with only a slight decrease of yield and selectivity (entry 10). It should be noted that only one diastereomer of epoxide was observed during the course of this study, while Weitz-Scheffer type epoxidation of trisubstituted α,β-carbonyl compounds often suffered from low diastereocontrol.9e, g, 11 In addition, this diastereospecific epoxide formation in our catalytic system implies a concerted pathway unlike the stepwise mechanism proposed previously.12n
Table 1.
Screening of reaction conditions
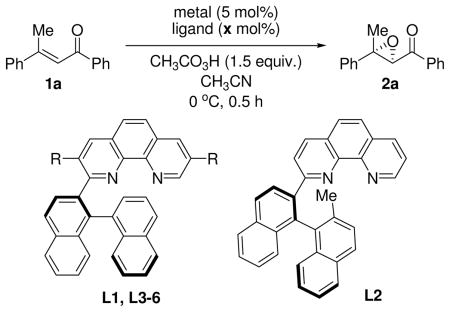 | |||||
|---|---|---|---|---|---|
| entry | metal | ligand L (R) | x | 2a | |
| % yielda | % eeb | ||||
| 1 | FeCl2 | L1 (R=H) | 10 | 17 | 3 |
| 2 | Fe(OTf)2 | L1 (R=H) | 10 | 95 | 57 |
| 3 | Fe(OTf)2 | L2 | 10 | <5 | 17 |
| 4 | Fe(OTf)2 | L3 (R=Ph) | 10 | 93 | 83 |
| 5 | Fe(OTf)2 | L4 (R=o-Tol) | 10 | 72 | 75 |
| 6 | Fe(OTf)2 | L5 (R=m-xylyl) | 10 | 88 (80) | 91 |
| 7 | Fe(OTf)2 | L6 (R=m-Et2C6H3) | 10 | 64 | 86 |
| 8 | Fe(OTf)2 | L5 (R=m-xylyl) | 5 | 73 | 78 |
| 9 | Fe(OTf)2 | L5 (R=m-xylyl) | 15 | 25 | 53 |
| 10c | Fe(OTf)2 | L5 (R=m-xylyl) | 5 | 81 | 86 |
Yields were determined using 1H-NMR analysis with 1,1,2,2-tetrachloroethane as an internal standard. The value in parentheses represents isolated yield
Determined by chiral HPLC analysis.
2.5 mol% of Fe(OTf)2 was used.
Next, X-ray crystallographic analysis was carried out on single-crystal grown in an acetonitrile solution of Fe(OTf)2 and two equivalent of rac-L3. As illustrated in Figure 1, an octahedral, mononuclear [Fe(L3)2(CH3CN)(OTf)](OTf) was identified in which two homo-chiral phenanthroline ligands coordinate the iron center in a cis topology to construct a pseudo C2-symmetric iron complex. Although the bidentate phenanthroline ligands can, in principle, adopt many possible diastereomers on the iron center, including the hetero- or homo-combination of rac-L3, only the diastereomer shown above was selectively crystallized.13 This selective complexation can be explained by the π–π interactions between the naphthyl groups and diphenyl phenanthroline. However the relationship between this selective formation of iron-ligand complex and enantioselectivities, as well as the actual structure of the catalyst in the reaction medium, is still unclear.12
Figure 1.

X-ray structure of [Fe(L3)2(CH3CN)(OTf)](OTf) is shown as CPK model. Thermal ellipsoids correspond to 50 % probability. Hydrogen atoms and non-coordinating molecules are omitted for clarity. Triflate group and CH3CN are replaced by a green atom and a yellow atoms for clarity, respectively. See supporting information for details. C: gray, N: blue, Fe: red.
With optimized conditions in hand, we next examined the scope of substrates using Fe(OTf)2-L5 complex (Table 2). The epoxidations of β,β-disubstituted enones having different electronic characters on the phenacyl groups proceeded smoothly with good yield and with excellent enantioselectivities (entries 1–5). Sterically different types of aromatic substituents were also tolerated (entries 6–8). While an electron-deficient aromatic group on the β-position had no influence on the epoxidation (entry 9), an electron-rich group such as naphthyl group on the β-position showed the deleterious effect on the yield of the product, probably due to the instability of the epoxide obtained under the acidic condition, although the high enantioselectivity was still maintained (entry 10). On the other hand, the substrate bearing an alkyl substituent on the β-position turned out to have inferior reactivity and selectivity compared to aromatic substrates (entry 11). The substrate with an ethyl group as the R3 substituent kept the high enantiomeric excess (entry 12). Notably, a single diastereomer was obtained even in employing (Z)-dypnone with poor enantioselectivity. This fact could further support the reaction proceeds via a concerted pathway (entry 13).
Table 2.
Substrate scope of epoxidations
 | |||||
|---|---|---|---|---|---|
| entry | R1 | R2 | R3 | % yielda | % eeb |
| 1 | 1a Ph | Ph | Me | 2a (80) | 91 |
| 2 | 1b p-MeOC6H4 | Ph | Me | 2b (78) | 90 |
| 3 | 1c p-MeC6H4 | Ph | Me | 2c (77) | 92 |
| 4 | 1d p-FC6H4 | Ph | Me | 2d (78) | 92 |
| 5 | 1e p-CF3C6H4 | Ph | Me | 2e (70) | 89 |
| 6 | 1f m-MeC6H4 | Ph | Me | 2f (67) | 90 |
| 7 | 1g o-MeC6H4 | Ph | Me | 2g (61) | 92 |
| 8 | 1h 2-Naphthyl | Ph | Me | 2h (88) | 90 |
| 9 | 1i Ph | p-ClC6H4 | Me | 2i (88) | 92 |
| 10 | 1j Ph | 2-Naphthyl | Me | 2j (45) | 92 |
| 11 | 1k Ph | n-C3H7 | Me | 2k (20) | 50 |
| 12 | 1l Ph | Ph | Et | 2l (72) | 92 |
| 13 | 1m Ph | Me | Ph | 2m (33) | 6 |
Isolated yields.
Determined by chiral HPLC analysis.
Gratifyingly, we realized this chiral iron-phenanthroline system can also be applied not only to α,β-enones but also to a non-activated olefin such as trans-α-methyl stilbene with good enantioselectivity (Scheme 2, eq. 1).14 With this result in hand, we conducted an intermolecular competition reaction to prove the nature of the active oxidant (eq. 2).15 A competitive reaction of electron-deficient alkene 1a and electron-rich one 1n shows a 2.4:1 preference for the latter with comparable enantioselectivities, confirming the electrophilic nature of the active oxidant.
Scheme 2.

The asymmetric epoxidation with a non-activated olefin and a competitive experiment using electron-rich and electron-deficient olefins.
The utility of chiral α,β-epoxyketones was demonstrated as shown in Scheme 3. The obtained α,β-epoxyketones (2a and 2l) were converted into β-ketoaldehydes (3a and 3l) bearing all-carbon quaternary centers without significant loss of enantiomeric excess by utilizing the Lewis acid-mediated rearrangement (Scheme 3, eq. 1).16 Unlike the substrate having a phenacyl group, the iron-catalyzed epoxidation of alkyl substituted substrate 1o gave the rearranged product 3o as a major compound concomitant with the corresponding epoxide (eq. 2). Eventually, the pure rearranged product 3o could be successfully obtained in 35% yield over two steps by treatment of the mixture with BF3·OEt2 at room temperature. Furthermore, the chiral α,β-epoxyketones can be transformed to 2-isoxazolidines,17 which are important intermediates in the preparation of a variety of compounds with 1,3-difunctional groups such as β-hydroxy ketones18 and γ-amino alcohols.19 The treatment of 2a with hydroxylamine hydrochloride in the presence of pyridine furnished a highly substituted 2-isoxazoline 4a in optically pure form (eq.3).20
Scheme 3.

The transformations of optically active α,β-epoxyketones.
In summary, we have developed the first iron-catalyzed asymmetric epoxidation of acyclic β,β-disubstituted enones. Essential for success was the use of the iron complex consisting of Fe(OTf)2 and two equivalents of carefully designed phenanthroline ligand. X-ray crystallography revealed a pseudo C-2 symmetric iron/ligand complex. Moreover, the construction of the chiral all-carbon quaternary center was also realized by the Lewis acid mediated rearrangement of the obtained chiral epoxides. Furthermore, this work provides a new strategy for designing pseudo C2-symmetric orthophenanthroline ligand-based catalysts, which should have a vast potential for transition metal catalyzed organic synthesis in general.
Supplementary Material
Acknowledgments
This work was partly supported by the Uehara Memorial Foundation, National Science Foundation (CHE-0717618) and National Institutes of Health (5R01GM068433). We thank Dr. Ian M. Steele for X-ray structure determination and Antoni Jurkiewicz for his NMR expertise.
Footnotes
Supporting Information Available: Experimental procedures, characterization of compounds L1-6, 1a-m, 1o, 2a-n, 3a, 3l, 3o, 4a and including these 1H NMR and 13C NMR spectra and HPLC analysis. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For recent reviews on the asymmetric epoxidation, see: Adolfsson H, Shi Y. In: Modern Oxidation Methods. Backvall JE, editor. Wiley-VCH; Weinheim: 2004. p. 21.Matsumoto K, Katsuki T. In: Catalytic Asymmetric Synthesis. 3. Ojima I, editor. Wiley-VCH; New York: 2010. p. 839.Lane BS, Burgess K. Chem Rev. 2003;103:2457. doi: 10.1021/cr020471z.Díez D, Núñez MG, Antón AB, García P, Moro RF, Garrido NM, Marcos IS, Basabe P, Urones JG. Curr Org Synth. 2008;5:186.Matsumoto K, Sawada Y, Katsuki T. Pure Appl Chem. 2008;80:1071.Wong OA, Shi Y. Chem Rev. 2008;108:3958. doi: 10.1021/cr068367v.
- 2.For reviews on iron catalysis, see: Enthaler S, Junge K, Beller M. Angew Chem Int Ed. 2008;47:3317. doi: 10.1002/anie.200800012.Bolm C, Legros J, Le Paih J, Zani L. Chem Rev. 2004;104:6217. doi: 10.1021/cr040664h.
- 3.For the seminal work on non-heme iron-catalyzed epoxidation: White MC, Doyle AG, Jacobsen EN. J Am Chem Soc. 2001;123:7194. doi: 10.1021/ja015884g.
- 4.For examples of non-heme iron-catalyzed asymmetric epoxidation, see: Francis MB, Jacobsen EN. Angew Chem Int Ed. 1999;38:937. doi: 10.1002/(SICI)1521-3773(19990401)38:7<937::AID-ANIE937>3.0.CO;2-O.Marchi-Delapierre C, Jorge-Robin A, Thibon A, Ménage S. Chem Commun. 2007:1166. doi: 10.1039/b616172c.Gelalcha F, Bitterlich B, Anilkumar G, Tse M, Beller M. Angew Chem Int Ed. 2007;46:7293. doi: 10.1002/anie.200701235.Gelalcha F, Anilkumar G, Tse M, Brückner A, Beller M. Chem Eur J. 2008;16:7687. doi: 10.1002/chem.200800595.Yeung HL, Sham KC, Tsang CS, Lau TC, Kwong HL. Chem Commun. 2008:3801. doi: 10.1039/b804281k.
- 5.Costas M, Mehn MP, Jensen MP, Que L. Chem Rev. 2004;104:939. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 6.(a) Collman JP, Wang Z, Straumanis A, Quelquejeu M, Rose E. J Am Chem Soc. 1999;121:460. [Google Scholar]; (b) Rose E, Ren QZ, Andrioletti B. Chem Eur J. 2004;10:224. doi: 10.1002/chem.200305222. [DOI] [PubMed] [Google Scholar]
- 7.For the chiral porphyrin catalysts, see: Rose E, Andrioletti B, Zrig S, Quelquejeu-Ethève M. Chem Soc Rev. 2005;34:573. doi: 10.1039/b405679p.and references therein.
- 8.The asymmetric epoxidation of (E)-dypnone with a stoichiometric amount of chiral peroxide was reported (40 %, 72 %ee): Adam W, Rao PB, Degen HG, Saha-Möller CR. Eur J Org Chem. 2002:630. doi: 10.1021/jo0162078.
- 9.For examples of catalytic asymmetric epoxidations of trisubstituted α,β-carbonyl compounds, see: Bentleya PA, Bickleya JF, Roberts SM, Steinera A. Tetrahedron Lett. 2001;42:3741.Adam W, Rao PB, Degen HG, Levai A, Patonay T, Saha-Möller CR. J Org Chem. 2002;67:259. doi: 10.1021/jo0162078.Wu XY, She X, Shi Y. J Am Chem Soc. 2002;124:8792. doi: 10.1021/ja020478y.Ooi T, Ohara D, Tamura M, Maruoka K. J Am Chem Soc. 2004;126:6844. doi: 10.1021/ja048600b.Marigo M, Franzén J, Poulsen TB, Zhuang W, Jørgensen KA. J Am Chem Soc. 2005;127:6964. doi: 10.1021/ja051808s.Chen Z, Morimoto H, Matsunaga S, Shibasaki M. Synlett. 2006:3529.Wang X, List B. Angew Chem Int Ed. 2008;47:1119. doi: 10.1002/anie.200704185.Wang X, Reisinger CM, List B. J Am Chem Soc. 2008;130:6070. doi: 10.1021/ja801181u.Wang B, Wu XY, Wong OA, Nettles B, Zhao MX, Chen D, Shi Y. J Org Chem. 2009;74:3986. doi: 10.1021/jo900330n.
- 10.mCPBA also afforded a epoxide in a comparable yield, while other oxidants such as H2O2, sodium percarbonate, tert-butyl hydroperoxide, and cumene hydroperoxide were ineffective.: Dubois G, Murphy A, Stack TDP. Org Lett. 2003;5:2469. doi: 10.1021/ol0347085.
- 11.Moiseev AG, Neckers DC. Synthesis. 2005:2901. [Google Scholar]
- 12.For selected mechanistic studies, see Chen K, Costas M, Kim J, Tipton AK, Que L. J Am Chem Soc. 2002;124:3026. doi: 10.1021/ja0120025.Quiñonero D, Musaev DG, Morokuma K. Inorg Chem. 2003;42:8449. doi: 10.1021/ic0348392.Fujita M, Que L. Adv Synth Catal. 2004;346:190.Mairata i Payeras A, Ho RYN, Fujita M, Que L. Chem Eur J. 2004;10:4944. doi: 10.1002/chem.200400480.Bassan A, Blomberg MRA, Siegbahn PEM, Que L. Angew Chem Int Ed. 2005;44:2939. doi: 10.1002/anie.200463072.Quiñonero D, Morokuma K, Musaev DG, Mas-Ballesté R, Que L. J Am Chem Soc. 2005;127:6548. doi: 10.1021/ja051062y.Bukowski MR, Comba P, Lienke A, Limberg C, Lopez de Laorden C, Mas-Ballesté R, Merz M, Que L. Angew Chem Int Ed. 2006;45:3446. doi: 10.1002/anie.200504357.Mas-Ballesté R, Costas M, van den Berg T, Que L. Chem Eur J. 2006;12:7489. doi: 10.1002/chem.200600453.Duban EA, Bryliakov KP, Talsi EP. Eur J Inorg Chem. 2007;852Bautz J, Comba P, Lopez de Laorden C, Menzel M, Rajaraman G. Angew Chem Int Ed. 2007;46:8067. doi: 10.1002/anie.200701681.Mas-Ballesté R, Que L. J Am Chem Soc. 2007;129:15964. doi: 10.1021/ja075115i.Comba P, Rajaraman G. Inorg Chem. 2008;47:78. doi: 10.1021/ic701161r.Company A, Feng Y, Güell M, Ribas X, Luis J, Que L, Costas M. Chem Eur J. 2009;15:3359. doi: 10.1002/chem.200802597.Benet-Buchholz J, Comba P, Llobet A, Roeser S, Vadivelu P, Wadepohl H, Wiesner S. Dalton Trans. 2009;5910 doi: 10.1039/b902037c.Lyakin OY, Bryliakov KP, Britovsek GJP, Talsi EP. J Am Chem Soc. 2009;131:10798. doi: 10.1021/ja902659c.Li F, England J, Que L. J Am Chem Soc. 2010;132:2134. doi: 10.1021/ja9101908.
- 13.(a) Knof U, Zelewsky Av. Angew Chem Int Ed. 1999;38:302. doi: 10.1002/(SICI)1521-3773(19990201)38:3<302::AID-ANIE302>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]; (b) Knight PD, Scott P. Coord Chem Rev. 2003;242:125. [Google Scholar]
- 14.See supporting information for another example.
- 15.It has been reported that non-heme iron complexes may generate either nucleophilic intermediates or electrophilic ones when treated with peroxide based oxidants.: Fujita M, Costas M, Que L. J Am Chem Soc. 2003;125:9912. doi: 10.1021/ja029863d.
- 16.(a) Domagala JM, Bach RD. J Am Chem Soc. 1978;100:1605. [Google Scholar]; (b) Domagala JM, Bach RD. J Org Chem. 1984;49:4181. [Google Scholar]; (c) Sankaararaman S, Nesakumar JE. J Chem Soc, Perkin Trans I. 1999:3173. [Google Scholar]
- 17.3-arylisoxazolidine derivatives showed a variety of biological activities, see, for examples: Olson RE, Sielecki TM, Wityak J, Pinto DJ, Balt DG, Frietze WE, Liu J, Tobin AE, Orwat MJ, Di Meo SV, Houghton GC, Lalka GK, Mousa SA, Racanelli AL, Hausner EA, Kapil RP, Rabel SR, Thoolen MJ, Reilly TM, Anderson PS, Wexler RR. J Med Chem. 1999;42:1178. doi: 10.1021/jm980348t.Pruitt JR, Pinto DJ, Estrella MJ, Bostrom LL, Knabb RM, Wong PC, Wright MR, Wexler RR. Bioorg Med Chem. 2000;10:685. doi: 10.1016/s0960-894x(00)00097-4.Burgey CS, Nguyen DN, Paone DV, Potteiger CM, Vacca JP. 2011/0003822 A1. U S patent. 2011 Jan 6;Mihara J, Murata T, Yamazaki D, Yoneta Y, Shibuya K, Shimojo E, Goergens U. 2008/122375. Patent WO. 2008 Oct 16;:A2.
- 18.(a) Curran DP. J Am Chem Soc. 1983;105:5826. [Google Scholar]; (b) Kozikowski AP, Adamczyk M. Tetrahedron Lett. 1982;23:3123. [Google Scholar]
- 19.(a) Jäger V, Schwab W, Buss V. Angew Chem Int Ed. 1981;20:601. [Google Scholar]; (b) Schwab W, Jäger V. Angew Chem Int Ed. 1981;20:603. [Google Scholar]
- 20.Ito S, Sato M. Bull Chem Soc Jpn. 1990;63:2739. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.