

Abstract
Excitatory amino acid transporters (EAATs) control the glutamate concentration in the synaptic cleft by glial and neuronal glutamate uptake. Uphill glutamate transport is achieved by the co-/countertransport of Na+ and other ions down their concentration gradients. Glutamate transporters also display an anion conductance that is activated by the binding of Na+ and glutamate but is not thermodynamically coupled to the transport process. Of the five known glutamate transporter subtypes, the retina-specific subtype EAAT5 has the largest conductance relative to glutamate uptake activity. Our results suggest that EAAT5 behaves as a slow-gated anion channel with little glutamate transport activity. At steady state, EAAT5 was activated by glutamate, with a Km= 61 ± 11 μM. Binding of Na+ to the empty transporter is associated with a Km = 229 ± 37 mM, and binding to the glutamate-bound form is associated with a Km = 76 ± 40 mM. Using laser-pulse photolysis of caged glutamate, we determined the pre-steady-state kinetics of the glutamate-induced anion current of EAAT5. This was characterized by two exponential components with time constants of 30 ± 1 ms and 200 ± 15 ms, which is an order of magnitude slower than those observed in other glutamate transporters. A voltage-jump analysis of the anion currents indicates that the slow activation behavior is caused by two slow, rate-limiting steps in the transport cycle, Na+ binding to the empty transporter, and translocation of the fully loaded transporter. We propose a kinetic transport scheme that includes these two slow steps and can account for the experimentally observed data. Overall, our results suggest that EAAT5 may not act as a classical high-capacity glutamate transporter in the retina; rather, it may function as a slow-gated glutamate receptor and/or glutamate buffering system.
Introduction
Excitatory amino acid transporters (EAATs) 1–5 are secondary-active transporters that contribute to the regulation of the glutamate concentration in the synapse. Glutamate transporters use the free energy stored in the transmembrane concentration gradients of the cotransported Na+ and K+ ions to maintain an up to 106-fold glutamate concentration gradient across the cell membrane. Glutamate transport is coupled to the movement of three Na+ ions and one H+, and, in an independent step, the countertransport of one K+ ion. Therefore, two positive charges move into the cell for each transported glutamate molecule, generating a steady-state transport current. In addition to this transport current, glutamate transporters also catalyze a thermodynamically uncoupled anion conductance (1). This conductance may not absolutely require the presence of extracellular Na+ (2), but it is of much higher magnitude in the presence of both Na+ and glutamate. For EAAT3, it has been shown that the anion conductance reports on transitions in the transport cycle (3,4). Therefore, investigators proposed that the anion conductance is kinetically linked to glutamate transport (3,5,6). Out of the five known glutamate transporter subtypes, EAAT4 and EAAT5 are thought to have the most prominent anion conductance relative to their transport capacity, representing >95% of the observed steady-state currents (7,8). In the absence of permeable anions, glutamate uptake can still take place, but the anion conductance cannot be observed (9,10).
The subtypes EAAT1 and EAAT2 are mostly expressed in glia cells, whereas EAATs 3–5 are neuronal. EAAT5 was first cloned by Arriza and co-workers (11) and has been identified in bipolar cone and rod photoreceptor terminals, as well as in axon terminals of rod bipolar cells (12,13). It was also found in salamander Müller cells and neurons, and, more recently, in the brain of zebrafish (14). It has been hypothesized that the large EAAT5 glutamate-gated anion conductance can serve to regulate neuronal excitability by clamping the membrane potential at an optimal level. According to this hypothesis, the transporter acts as a glutamate receptor that mediates a fast inhibitory feedback regulation of the release of glutamate via activation of this conductance, resulting in hyperpolarization of synaptic membranes (15–18). However, although the transporters EAAT1–4 have been functionally characterized in significant detail, with EAAT4 showing different properties from the EAATs 1–3 (4,19,20), the functional properties of the retinal subtype EAAT5 are not known.
By studying the mechanism of glutamate transport/anion conductance activation by EAAT5, we may be able to obtain a better understanding of the function of this transporter and its importance for neuronal excitability in the retina. For this purpose, we expressed EAAT5 in HEK293 cells and performed whole-cell current recordings coupled with laser photolysis of 4-methoxy-7-nitroindolinyl (MNI)-glutamate. In addition, we conducted voltage-jump experiments. The results show that EAAT5 and the other glutamate transporter subtypes share a similar mechanism, but EAAT5 has very different transport kinetics and a lower affinity for glutamate compared with the other subtypes. Furthermore, pre-steady-state kinetics with glutamate concentration jumps showed that the turnover rate of this transporter is at least an order of magnitude lower than that of the other subtypes. The voltage-jump experiments showed that the voltage dependence of the anion current does not display the same behavior as EAAC1 (EAAT3) or EAAT4. Our results suggest that EAAT5 behaves as a slow-gated, glutamate-dependent anion channel with little glutamate uptake activity. It also has the potential to control glutamate concentration by buffering at the visual synapse.
Materials and Methods
Cell culture and molecular biology
HEK293 cells (No. CRL 1573; American Type Culture Collection) were cultured as described previously (21). The cell cultures were transiently transfected with EAAT5 cDNA (Origene, Rockville, MD) inserted into a pBK-CMV-expression plasmid (Agilent Technologies, Santa Clara, CA) with the use of a Fugene HD transfection reagent according to the protocol supplied by the manufacturer (Roche, Indianapolis, IN), and 24–48 h after transfection the cells were used for electrophysiological measurements. Immunocytochemistry is described in the Supporting Material.
Whole-cell current recording
Glutamate-induced currents were measured in the whole-cell current-recording configuration as described in the Supporting Material.
Laser-pulse photolysis and rapid solution exchange
Laser-pulse photolysis experiments were performed as previously described (4). In brief, glutamate or 4-methoxy-7-nitroindolinyl (MNI)-caged glutamate (TOCRIS) was applied to the bath solution surrounding the cells. Photolysis of caged glutamate was initiated with a light flash (355 nm Nd:YAG laser, Minilite series; Continuum, Santa Clara, CA) that was delivered to the cell with an optical fiber (350 μm diameter). Laser energies were varied in the range of 50–570 mJ/cm2 with neutral density filters. To estimate the concentration of photolytically released glutamate, we calibrated the yield from the photolysis of 1 mM MNI-glutamate to ∼200 μM liberated glutamate (20%) for the highest laser energy used (550 μJ) at the end of the fiber that photolyzes the caged glutamate applied to the cell, as determined by comparing the laser-induced signal with that obtained by applying a standard concentration of glutamate, and the known dose response curve. With the application of neutral density filters, the laser energy was attenuated, resulting in lower percentage of release. This procedure is described in detail elsewhere (21).
The data were low-pass-filtered at 1–20 kHz, digitized with a sampling rate of 5–50 kHz, and recorded with the use of pClamp8 software (Molecular Devices, Sunnyvale, CA).
Data analysis and modeling
We analyzed the data using Origin software (OriginLab, Northampton, MA) as described in detail elsewhere (19).
To further interpret our data, we performed simulations using Berkeley Madonna software (www.berkeleymadonna.com) and numerically integrating the differential equations pertaining to the simplified mechanisms in Scheme 1. The details of the modeling can be found in the Supporting Material.
Scheme 1.
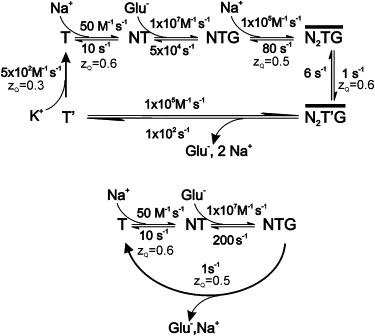
Simplified kinetic schemes used in the modeling of EAAT5 kinetics, including the respective kinetic parameters and valences. T, transporter, N, sodium, G, glutamate.
Results
Transport activity and glutamate-dependent anion conductance
Functional characterization of the EAAT5 glutamate transporter subtype was performed after transient expression in HEK293 cells. None of the polymerase chain reaction, immunocytochemical, immunoblot, or electrophysiological analyses indicated the expression of endogenous EAATs 1–5 in HEK293 cells. A colocalization analysis (Fig. 1 A) of AcGFP-coupled-EAAT5 and the cell surface label, Alexa Fluor 546 WGA, confirmed 60% plasma membrane localization of EAAT5 in HEK293 cells. An immunoblot analysis (Fig. 1 B) demonstrated heterologous expression of EAAT5 with a similar electrophoretic mobility as determined for the native EAAT5 (13). Membrane vesicles prepared from HEK293 cells expressing EAAT5 mediated Na+-dependent glutamate uptake (Fig. 1 C). Uptake was linear within the first minute, with a rate of 7.8 ± 0.4 pmol/(mg/min). It is difficult to compare these uptake rates with previous results from EAATs 1–3 in the HEK293 expression system (19); however, they are roughly two orders of magnitude lower.
Figure 1.
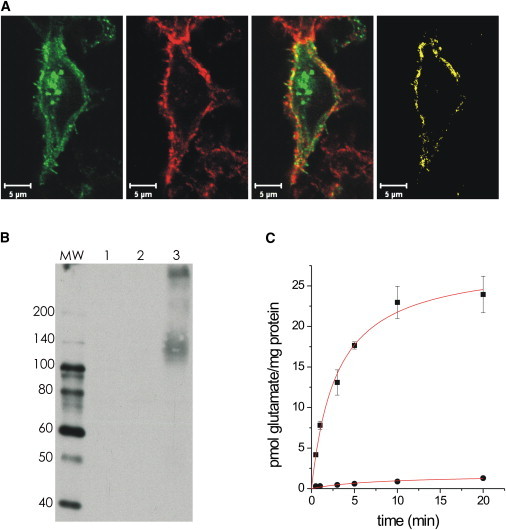
Heterologous protein expression and functional characterization of EAAT5. (A) AcGFP-EAAT5 expressing cells (green), counterstained with TRITC wheat germ agglutinin (red) exhibit 63% ± 6% membrane localization of EAAT5 (yellow, n = 5). (B) Immunoblot analysis reveals the typical broad electrophoretic mobility of EAATs (lane 1: nontransfected; lane 2: EAAC1-transfected; and lane 3: EAAT5-transfected cells). Molecular mass markers (lane MW) are indicated in kDa. (C) EAAT5 expression (solid squares) significantly increases L-[3H]-glutamate uptake in HEK293 cells (nontransfected HEK293, solid circles), which is in the same range as that measured for EAAT4 (19).
We performed whole-cell current-recording experiments to characterize the steady-state properties of EAAT5 transiently expressed in HEK293 cells. When methanesulfonate (Mes–) was used as the sole anion, no currents were observed in response to glutamate up to 1 mM (0 ± 3 pA). This suggests that EAAT5 turnover is too slow and/or expression levels in the membrane are too low to generate a measurable transport current. Therefore, we used the glutamate-induced anion conductance in the forward transport mode to obtain information about the glutamate transport process. The observations that SCN– is a highly permeable ion in EAAT5, and EAAT5 currents are mainly carried by anions are in agreement with previous work (11,17), and we used this anion conductance as a tool to characterize the functional properties of this transporter.
In the presence of potassium thiocyanate (KSCN) inside the pipette, we observed an inward current (−80 ± 25 pA, n = 5; Fig. 2 A) due to the outward movement of SCN– ions (see “Voltage dependence of the anion conductance” below), and this current was dependent on the concentration of glutamate. The apparent Km for glutamate in the presence of 140 mM Na+ is 61 ± 11 μM, with a Hill coefficient of n = 0.6 ± 0.1 (Fig. 2 A). This result is in agreement with values previously measured in oocytes (11,22). These data suggest that EAAT5 has a significantly lower affinity for glutamate than the other glutamate transporter subtypes (3,5,19,21).
Figure 2.
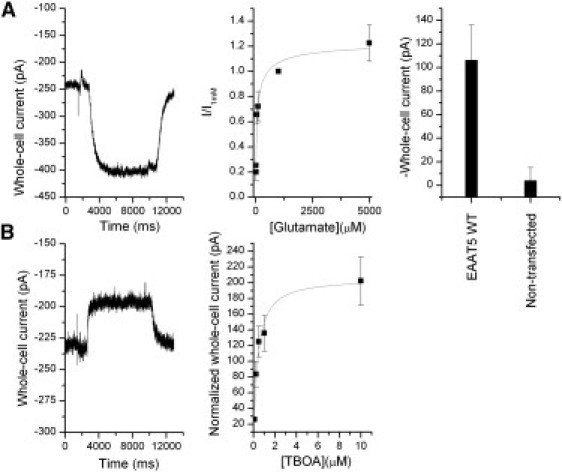
(A) Left: Typical anion current induced by the application of 60 μM glutamate. Right: Comparison of average responses to 100 μM glutamate application of nontransfected control cells and cells transfected with EAAT5 WT. Middle: Glutamate concentration dependence of EAAT5 anion whole-cell currents. The currents were normalized to the current at 1 mM glutamate. The data were fitted to the Hill equation with an apparent Km= 61 ± 11 μM, n = 0.6 ± 0.1. (B) TBOA inhibition of the leak anion current of EAAT5, yielding Ki = 0.4 ± 0.1 μM. The transmembrane potential in A and B was 0 mV. Anion currents were recorded in the presence of intracellular KSCN.
No significant currents were observed under more-physiological conditions, i.e., in the presence of 140 mM extracellular chloride and 30 mM KCl/110 mM KMes in the intracellular solution (ICl− = 11 ± 9 pA, n = 5, at 0 mV). This result is expected if the permeability ratio PSCN-/PCl− ≥ 70 (e.g., as in EAAT1 (3)). With an average SCN–-carried current of −80 pA, the chloride current would be expected to be negligible.
Apparent Na+ affinity
In all other glutamate transporter subtypes of the solute carrier 1 (SLC1) family (EAATs 3–5), a leak anion conductance was observed (reviewed in Grewer and Rauen (23)). Likewise, for EAAT5, in addition to the glutamate-dependent anion current, we observed a leak anion current in the absence of glutamate (11). This leak anion conductance is inhibited by TBOA (22), resulting in an outward current in the presence of intracellular anions. For EAAT5, this inhibition is dependent on the [TBOA], with an apparent Ki of 0.4 ± 0.1 μM and n = 1.1 ± 0.3 (Fig. 2 B) in the same range as that of the other glutamate transporter subtypes (22,24).
The activation of the leak anion conductance requires the presence of extracellular Na+ (Fig. S1 A). To determine the apparent affinity of Na+ binding to the empty transporter, we measured the [Na+] dependence of the leak anion current in the absence of glutamate (Fig. S1 A). A small current observed in nontransfected cells due to endogenous conductances (−8 pA for [Na+] = 20 mM to −28pA, for [Na+] = 300 mM) was subtracted from the total current to yield the EAAT5-specific leak current. The data were fitted to the Hill equation with an apparent Km = 229 ± 37 mM and a Hill coefficient of n = 1.6 ± 0.2. This Km-value is slightly higher than (but on the same order of magnitude as) that observed in the other transporter subtypes (19,25).
We studied binding of Na+ to the glutamate-loaded transporter in isolation by using a saturating glutamate concentration (10 mM) as described previously (4). The experiments were performed in the forward transport mode with 140 mM KSCN in the pipette solution, yielding a Km = 76 ± 38 mM (Fig. S1), which is in the same range as the values obtained for the other transporter subtypes of this family (19,25).
Time dependence of glutamate-induced anion currents
The time course of EAAT5 anion currents was determined in the forward transport mode (KSCN inside the pipette) after a glutamate concentration jump induced by laser-pulse photolysis of MNI-glutamate. In contrast to other transporter subtypes (4,19), the resulting current did not show an overshoot component; instead, it exhibited a slow rise to reach a steady-state level after ∼2 s (Fig. 3 A). The rise of the current exhibited two phases and was best fit with a two-exponential function, except at low concentrations of glutamate, at which only one exponential component was observed (Fig. 3, A–D). The fast phase of the current rise was dependent on the glutamate concentration, with an increasing rate at higher glutamate concentrations. The rate constant associated with the slow phase, τ1 = 200 ± 15 ms, was found to be independent of glutamate concentration. The time constants associated with these currents are one order of magnitude slower than those observed for the other glutamate transporter subtypes. These results indicate that a comparatively slow step in the transport cycle limits the overall turnover rate as well as the speed of anion current activation. The time course of the current decay after glutamate removal is shown in Fig. S4.
Figure 3.
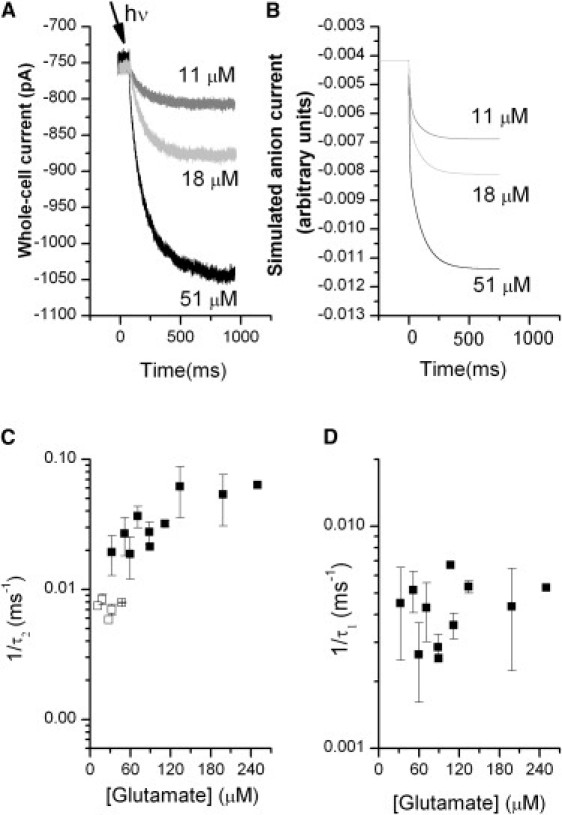
Photolysis of MNI-glutamate rapidly activates currents in EAAT5. Photolysis of 1 mM MNI glutamate results in activation of an inwardly directed anion current (KSCN is present in the pipette solution). The concentration of glutamate photolyzed can be controlled by the application of neutral density filters. (A) Current resulting from the release of 51 μM glutamate (black), 18 μM glutamate (gray), and release of 11 μM glutamate (light gray). The experiments were done at V = 0 mV. For glutamate concentrations > 60 μM, for which the traces show two exponential components, the ratio of the components was A1(τ1)/A2(τ2) = 1.91 ± 0.17. (B) Simulated currents for the same concentrations, according to the model and parameters in Scheme 1. (C and D) Dependence of EAAT5 pre-steady-state anion currents on the glutamate concentration. (C) Dependence of the rate constant 1/τ2 on the glutamate concentration. The solid and open squares correspond to the rate constants for the high concentrations (where the fit had two phases) and low concentrations (only one phase) of glutamate, respectively. (D) Time constant, τ1, for the high glutamate concentration current releases.
Voltage-jump-induced pre-steady-state currents
As an alternative to obtaining pre-steady-state kinetic data, we perturbed the steady state by applying step changes in the membrane potential, and then measured the current relaxations to the new steady state (Fig. 4 A and Fig. S2 A). The experiments were performed either with SCN− in the pipette solution or SCN− in the externally applied solutions. With SCN– inside, the voltage protocol consisted of a prepulse to +60 mV followed by a voltage step to the final voltage, ranging from −90 mV to +60 mV. The prepulse was used to obtain a larger change in anion channel open probability due to the larger ΔV. With SCN– outside, the protocol was reversed, with a prepulse voltage of −90 mV instead of +60 mV.
Figure 4.
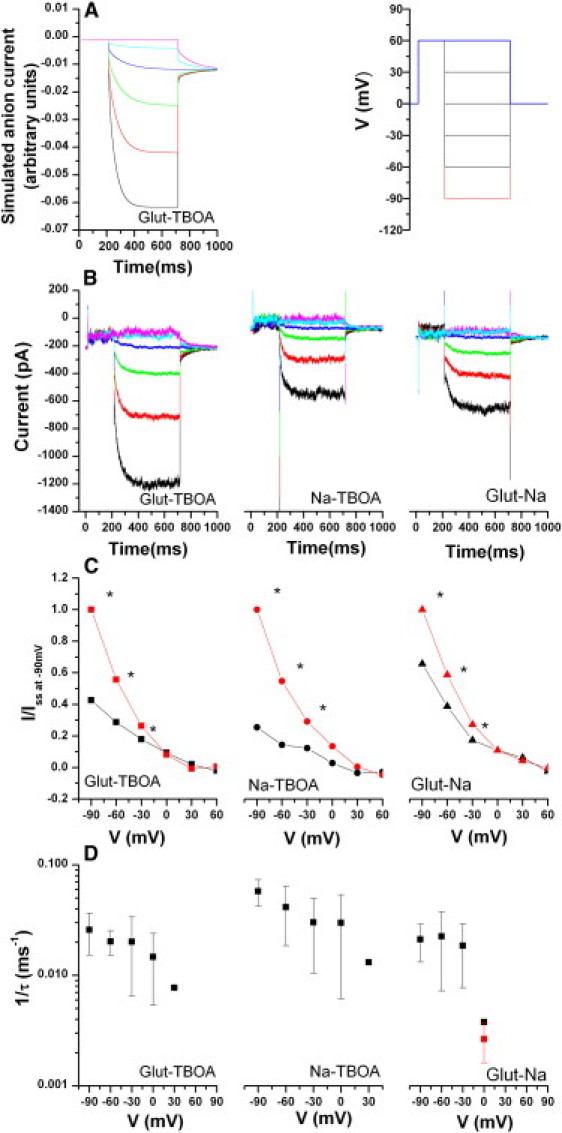
(A) Left trace: Simulation of the voltage dependence of anion current traces in the forward transport mode. The simulation was done by numerical integration of the pertinent differential equations, according to the mechanism in Scheme 1. The right panel illustrates the voltage jump protocol used to measure the current traces shown in B. (B and C) Voltage-jump-induced current relaxations in EAAT5, in forward mode, with SCN−-inside (KSCN) in the pipette solution. The protocol starts at 0 mV, has a jump to +60 mV, and then jumps to −90 mV and finally to 0 mV, as depicted in A. The purpose of the initial jump is to increase the amplitude of the observed currents. (B) Typical signals obtained through subtraction of current traces were recorded as indicated at the bottom of each picture: (left) subtraction of the traces with glutamate from the traces recorded in the presence of TBOA, (center) subtraction of the traces in the absence of glutamate (extracellular 140 mM sodium chloride solution; see Materials and Methods) from the traces with TBOA, and (right) subtraction of the traces in the presence of glutamate (sodium solution as in center) from the traces in the absence of glutamate. The Glut-Na traces were included to enable comparison with the large amount of data available for glutamate transporters in this mode. (C) Voltage dependence of I (initial, black) and ISS (steady-state, red) currents for each of the plots. The currents are normalized to the observed steady-state current at −90 mV, for each of the conditions. The stars denote significance. The glutamate concentration was 60 μM (close to the Km-value) and the TBOA concentration was 100 μM (saturating). (D) Voltage dependence of the additional relaxation component of the anion current, due to a slow relaxation to a new steady state with different open probability, τ-values for the voltage induced increase in conductance (relaxation) to a new steady state, with SCN–-inside in the pipette solution (as in B and C). For comparison, the 1/τ-value obtained using a glutamate concentration jump in the laser photolysis experiments, under the same experimental conditions, is plotted as the red data point.
Representative results of the voltage-jump experiments in the presence of extracellular glutamate are shown in Fig. 4 B (left panel). When SCN− was on the inside, jumps to a more negative membrane potential resulted in an instantaneous increase in the inward current due to the increase of the electrical driving force for anion outflow. The time constant of this process was ∼200 ± 100 μs, in the same range as that observed for charging the membrane (26). The voltage dependence of the instantaneous phase of the current relaxation is consistent with this interpretation (Fig. 4 C). After this instantaneous phase, we observed a slow rising phase of the inward current, most likely due to a slow increase of the open probability of the anion channel at more negative membrane potentials. At 0 mV, the time constant of this phase was 265 ms, which is comparable to the time constant observed in the concentration jump experiments (the voltage dependence is shown in Fig. 4 C).
When the membrane potential was stepped from −90 mV back to 0 mV, we observed a fast phase of the current deactivation, indicative of the reduced driving force for SCN− outflow, followed by a slow decay to a new steady-state level. This decay was fitted best with two exponentials: a slow component with a time constant of 31 ± 29 ms, and a fast component with a time constant of 0.9 ± 0.8 ms. This slow decay is caused by a slow, time-dependent reduction of the anion channel open probability back to its steady-state level at 0 mV.
As shown in Fig. 2 B, EAAT5 also catalyzes an anion leak current that is activated by Na+ in the absence of glutamate and is inhibitable by TBOA. The leak current is associated with a biphasic time dependence in response to voltage jumps (Fig. 4 B, middle panel), with a fast, instantaneous phase (change in the driving force for SCN− outflow) and a slow relaxation to a new steady state (τ = 18 ± 5 ms; – 90 mV and 72 ± 40 ms; 0 mV). This relaxation is indicative of an increase in the open probability of the anion channel with increasing negative transmembrane potential.
Binding of Na+ ions to the glutamate-free form of other glutamate transporter subtypes was shown to be voltage-dependent, with negative membrane potentials favoring the Na+-bound state (19,27).This raises the possibility that the slow, voltage-dependent current relaxation is caused by Na+ binding/unbinding from the EAAT5 transporter. In other subtypes, these binding/unbinding events were associated with a transient charge movement induced by voltage jumps. When EAAT5-expressing cells were subjected to voltage jumps in the absence of permeant anions, no transient currents were observed (I = 0 ± 26 pA). Therefore, either Na+ binding is not electrogenic in EAAT5 or, more likely, it is too slow to be observed as a measurable current. Because the time constant of the EAAT5 Na+ binding event is expected to be in the 40 ms range (at 0 mV), which is 100 times larger than the time constant of this event in EAAC1 (4,21), the EAAT5 peak transient current amplitude per transporter is expected to be 100 times smaller than that observed in EAAC1. Therefore, the resulting current is expected to be at or below the detection limit of our patch-clamp apparatus.
The presence of permeant anions can potentially affect the transport properties, as previously suggested for EAAT4 (19). To test this possibility for EAAT5, we performed a voltage-jump analysis after reversing the SCN– gradient (SCN− only present in the extracellular solution (Fig. S2 B), with Cl− inside). As expected for SCN− inflow, anion currents were outwardly directed at all membrane potentials (Fig. S2 C) and increased with increasing membrane potential. The leak anion currents (absence of glutamate) showed the time dependence expected from the SCN−-inside experiments, with a slow increase in anion channel open probability with increasingly negative transmembrane potential (Fig. 4 B, middle panel). However, for the glutamate-induced anion current, the slow phase of the anion current present in SCN–-inside conditions (Fig. 4 B) was not observed in SCN–-outside conditions (Fig. S2). This result indicates that the kinetics of anion current activation by glutamate depend on the direction of the [SCN−] gradient across the membrane, suggesting that the anion has other roles than just passively reporting on transitions in the transport cycle. The time constants associated with these voltage-jump-induced relaxation currents are voltage-dependent (Fig. 4 D and Fig. S2 D), with the relaxation being accelerated by negative transmembrane potentials. For SCN− inside, a single-exponential fit of the traces yielded time constants between τ = 17 (for V = −90 mV) and 330 ms (for V = 0 mV). These time constants are on the order of magnitude of the time constants measured after a rapid glutamate concentration jump (performed at 0 mV; red square in Fig. 4 D).
To test whether the slow phase of the anion current activation is only caused by slow Na+ binding, which precedes glutamate binding, we performed experiments in the homoexchange mode (Fig. S3). The aim of these experiments was to restrict the transporter to transitions associated with translocation only, although the low affinity for the extracellular Na+ binding step after glutamate binding prevented us from fully saturating this Na+ binding site. Under these conditions, the slow rising phase of the anion current was reduced in amplitude but still present (Fig. S3), suggesting that processes associated with translocation and/or Na+ binding to the glutamate-bound form of EAAT5 are also slow (τ = 88 ± 61 ms, V= −60 mV).
Discussion
The glutamate transporter family consists of five members (EAATs 1–5), and three of these (EAATs 1–3) were previously found to have similar functional properties (4,6,27). In contrast, it was proposed that EAAT4 and EAAT5 behave differently because they have a much higher anion conductance, relative to their transport activity, than the other subtypes (3,11,16,19,27). It is also known that EAAT4 has a much slower transport rate than EAAT3 (19). The main new (to our knowledge) finding of this study is that these functional and kinetic properties are even more different for EAAT5. Currents carried by this transporter are carried predominantly by the anion conductance, and the activation of this current is more than an order of magnitude slower than it is for all the other transporters of this family (3–6,19,21). Furthermore, the apparent Km-value for glutamate interaction with EAAT5 is higher than for the other EAATs, but the apparent Na+ affinity is similar. Finally, the voltage dependence of the anion conductance is different, with a time-dependent increase of the channel open probability and/or conductance at increasingly negative membrane potentials. Below, we discuss some possible reasons for its slow activation kinetics by using modeling to aid interpretation of the experimental results.
EAAT5 has a lower affinity for glutamate
The apparent affinity of this transporter for glutamate, Km = 61 μM, is lower than that observed for all of the other subtypes. The apparent affinity of EAAT5 for glutamate is eightfold lower than that of EAAT3 (Km = 8 μM (21)) and 100-fold lower than that of EAAT4 (Km = 0.6 μM (19)). It is also lower than that of EAAT1 (Km = 7 μM) and EAAT2 (Km = 12 μM). Our simulations suggest that the main cause of the reduced apparent affinity is an increase in the unbinding rate of glutamate. Overall, the glutamate affinity data suggest that EAAT5 is not optimized for tight binding of glutamate, and may have a different binding microenvironment for the substrate than the other subtypes. However, the apparent affinity of EAAT5 would make it well suited as a buffering system for glutamate at the rod bipolar cell synapse, in which the glutamate concentration is in the 20–200 μM range (28).
The apparent affinity of EAAT5 for Na+ is not significantly different from that of the other glutamate transporters of this family. The binding of Na+ to the empty transporter, in the absence of glutamate, is of low apparent affinity (Km = 230 mM). This suggests that only ∼38% of the transporters are immediately available for glutamate binding and translocation at physiological conditions. For 62% of the transporters, glutamate binding must await additional Na+ binding, which we propose to be slow (see below).
Pre-steady-state kinetic properties of EAAT5
Current responses to rapid glutamate concentration jumps reflect early transitions in the transport cycle. Fig. 3 indicates that the activation of the majority of the anion current in EAAT5 in the forward transport mode is slow. It is at least an order of magnitude slower than is observed for the other glutamate transporter subtypes (4,19). We fitted the current rise with two exponentials (see Results and Fig. 3). The slow process (τ1) is independent of the concentration of glutamate. The fast process (τ2) is dependent on the glutamate concentration, with the relaxation rate constant 1/τ2 leveling off at a high concentration of glutamate. If 1/τ2 were associated with glutamate binding, one would expect the 1/τ2 versus [glutamate] relationship to be linear, which is not consistent with the experimental data. Therefore, glutamate binding is a fast process and is not rate-limiting for anion current activation, at least at high glutamate concentrations. Our simulations of the experimental data are consistent with a bimolecular rate constant of glutamate association with EAAT5 of 1 × 107 M−1 s−1, in the same range as that reported for other glutamate transporter subtypes.
The fact that 1/τ2 saturates at high [glutamate] suggests that rapid glutamate binding is followed by a slower step, possibly a conformational change, that becomes independent of [glutamate] once all binding sites are saturated. One possibility is that the fast phase of the anion current rise is caused by Na+ binding to the glutamate-bound form of EAAT5, in analogy to what has been proposed for EAAC1 (4). A second possibility is that it is associated with the translocation step. However, this second scenario is unlikely, for the following reason: The experiments were conducted under forward transport conditions, in which glutamate and Na+ are expected to dissociate rapidly and irreversibly after translocation. Because the glutamate transporter anion conductance is associated with the translocation but not the relocation branch of the transport cycle, the translocation rate constant should be associated with a decaying phase of the anion current, but not a rising phase. Since no overshoot, decaying component of the anion current was observed, glutamate translocation cannot account for the fast phase of the current rise.
We propose that the rate constant of the [glutamate]-independent, slow step of the anion current rise is dominated by a slow step that precedes glutamate binding to its transporter binding site. This analysis is consistent with the simulation results and can be quantified by Eq. S1. A step that precedes glutamate binding is Na+ binding to the empty transporter, and/or the conformation change(s) associated with it. This assignment is in agreement with the results from the voltage-jump analysis, which display a slow, voltage-dependent current change with a time constant similar to that observed in the glutamate-concentration-jump experiment. Furthermore, no Na+-dependent transient currents in response to voltage jumps were observed in EAAT5, suggesting that the Na+-dependent steps of the empty transporter are too slow to generate a detectable current.
For EAAT3, it has been demonstrated that the binding of Na+ induces a conformational change (26,29), although this conformational change of EAAT3 is much faster than that of EAAT5 and is associated with transient transport currents. This different behavior of the two transporters may be due to different transport domain interactions in EAAT5 (compared with the other subtypes) that make this conformational change slow. At present, we cannot determine which residue(s) are involved. A chimera approach might be helpful to address this issue.
Voltage dependence of the anion conductance
For the glutamate transporter EAAT3, the probability of occupying the anion conducting state(s) is virtually voltage-independent (4), i.e., after voltage jumps, the magnitude of the current is affected only by changes in the driving force for the anion, and not by changes in the channel open probability and/or single-channel conductance. In contrast, the anion conductance of EAAT4 was found to be strongly dependent on the transmembrane potential, and inhibited by negative membrane potentials (19). For EAAT5, we also find a strong voltage dependence of the probability of occupying the anion conducting state(s), with a behavior opposite to that of EAAT4. The EAAT5 anion conductance is not inhibited, but is activated by negative membrane potentials.
This voltage-dependent behavior of EAAT5 can be interpreted as follows: As the membrane potential is changed to a more negative voltage, the probability of populating anion-conducting states increases, because it encounters a more favorable conformation for populating an anion-conducting state; alternatively, the unitary conductance for the anion current may be increased at more negative membrane potentials. These two possibilities can potentially be distinguished by a noise analysis approach, as previously described for glutamate transporters in native cells (30). Results from a previous noise analysis of glutamate transporter chloride currents in photoreceptors (cones and rods, presumably expressing EAAT5) suggest that glutamate activates a channel with a single-channel conductance of 0.5–0.7 pS (30). In EAAT1 patches, the noise induced by glutamate was much smaller (3). The results were interpreted as indicating that the conductance of EAAT5 is much larger than that of EAAT1, or the open times for EAAT1 are smaller (3). On the basis of the results presented, however, we cannot be certain that the transporters observed in the rods and cones are indeed EAAT5, since GLT1 is also expressed in these tissues (13,17,31–33). More recently, Torres-Salazar and Fahlke (34), also using noise analysis, determined the unitary anion conductance and absolute open probabilities for EAAT3 and EAAT4, and demonstrated that they are similar despite the much higher macroscopic anion conductance present in EAAT4. In our hands, noise levels in EAAT5-expressing cells were similar in the absence and presence of glutamate, which prevented us from performing a detailed noise analysis. However, this finding is not surprising given that the presumably slow gating of the EAAT5 anion conductance generates low-frequency changes of current levels that cannot be recorded as specific noise without interference from low-frequency, unspecific noise components resulting from whole-cell instability.
Together, these findings suggest that the EAAT5 anion conductance is significantly voltage-dependent, indicating the existence of a voltage sensor controlling channel activity. The voltage-sensing process is most likely related to electrogenic steps in the transport cycle, in which a net positive charge is transferred into the cell. Although the coupling stoichiometry of EAAT5 has not been determined, there is no reason to believe that it is different from that of the other glutamate transporter subtypes. Our experiments directly demonstrate the coupling of glutamate gating of the anion channel to at least two Na+ ions. Binding of Na+ to the empty (glutamate-free) transporter, and/or conformational changes of that transporter, could be responsible for voltage sensing. Consistent with this hypothesis, the rate of anion conductance activation in the absence of glutamate increases with more negative transmembrane potential (Fig. 4 D and Fig. S2 D). This behavior is expected if Na+ binding is electrogenic, as for the other glutamate transporter subtypes (3,4,19,27), such that a negative transmembrane potential will favor the Na+-bound state.
Anion conductance and the transport rate
The transport rates and the magnitude of the anion conductance seem to be inversely correlated for the glutamate transporter family (19,34). Using noise analysis, Torres-Salazar and Fahlke (34) demonstrated that unitary anion channel amplitudes were comparable for the EAATs included in their study (EAAT3 and EAAT4). Therefore, although the anion-conducting properties of these transporters are conserved within the family, the differences in anion currents seem to be directly and inversely related to the actual rate of glutamate transport. In other words, the electrophysiological behavior of transporters with low transport rates, such as EAAT4 and EAAT5, appears to be dominated by their anion conductance, whereas for transporters with higher transport rates (EAATs 1–3), the transport current becomes more important.
For EAAT5, the current caused by the anion conductance was the only current we were able to observe experimentally. We were not able to measure transport currents for this transporter, probably because it has a low turnover rate. According to our simulations, for EAAT5 at steady state, a transport current of ∼−1 pA at 0 mV and not more than −15 pA at −90 mV would be expected. This is at about the limit of detectability of currents in the whole-cell recording configuration. This result is in agreement with a previous study by Arriza and colleagues (11), who showed that EAAT5 currents in Xenopus oocytes were abolished after dialysis with nonpermeant anions. Therefore, the lack of EAAT5-coupled transport current is not restricted to the HEK293 expression system.
Our analysis of anion current kinetics under conditions of asymmetric anion concentration gradients across the membrane revealed a distinct kinetic behavior depending on whether the anion was present on the intra- or extracellular side of the membrane. This result suggests not only that the anion conductance passively reports on transitions in the transport cycle, but also that anion binding may actively alter the rate constants of these transitions. This finding is in contrast to results reported for EAAT3, in which the rates for individual reaction steps of the cycle were the same in the absence and presence of permeable anions (4,23). Therefore, the EAAT5 anion conductance, in contrast to EAAT3, displays gating-permeation coupling, in analogy to other Cl− channels, such as the CLCs (35,36).
Mechanism of transport by EAAT5
Simplified mechanisms of glutamate transport by EAAT5 are shown in Scheme 1 and discussed in detail in the Supporting Material. The kinetic parameters used for modeling are also included in Scheme 1. The models account very well for the experimental data, as shown in Figs. 3 B and 4 A. For the purposes of modeling, we only include one Na+ ion binding before and one after the glutamate binds, but the stoichiometry of this transporter may include three Na+ ions. The most recent evidence (2) indicates that two Na+ ions bind before glutamate, creating a high-affinity binding site for glutamate on the transporter, and substrate binding enables the last Na+ ion to bind. The fully loaded transporter can then proceed to a full cycle. For the sake of simplicity, we exclude the proton-binding step(s) from our model. In the absence of any other information, we assume that EAAT5 has the same stoichiometry (11) and general mechanism but different kinetics compared with the other glutamate transporter subtypes. We propose that two reaction steps are responsible for the very slow anion current activation kinetics of this transporter: 1), the slow first Na+-binding step or a slow conformational change related to this step; and 2), the glutamate/Na+ translocation step. The fact that the slow component of the current relaxation is reduced in amplitude but is still present when the experiments are performed in exchange mode, when the Na+-binding step(s) to the empty transporter and the glutamate-binding site are saturated (see Fig. S3), further supports the notion that a slow first Na+ binding step is partially responsible for the slower kinetics of this transporter.
Physiological significance
In mammals, EAAT5 is retina-specific. In addition to EAAT5, GLAST1/EAAT1, different splice variants of GLT1/EAAT2 (GLT1a, GLT1c, and GLT1v), and EAAC1/EAAT3 are expressed in the retina (33,37–39). There is no expression of EAAT4 in the retina. Although GLAST1/EAAT1 is localized in glial Müller cells, EAAT5, the GLT1/EAAT2 isoforms, and EAAC1/EAAT3 are neuronal. Presynaptic localization has been demonstrated for EAAT5 and GLT1c in both cone and rod photoreceptor terminals (13,17,33). In cone and rod bipolar cells, EAAT5 is again presynaptically localized (13,17). GLT1c is not expressed in bipolar cells; however, GLT1v is localized in these cells primarily postsynaptically, and GLT1a appears to be presynaptically expressed in them (37).
Hasegawa et al. (28) indicated that presynaptic EAATs in rod photoreceptors play a major role in removing tonically released glutamate from the synaptic cleft, whereas EAATs in Müller cells and postsynaptic neurons make a negligible contribution, suggesting a predominant role for EAAT5 in glutamate reuptake at the rod photoreceptor-to-rod bipolar cell synapse. However, this would require EAAT5 to have high-capacity glutamate uptake properties. Further on in the vertical signaling pathway in presynaptic rod bipolar terminals, Veruki et al. (40) and Wersinger et al. (17) demonstrated a presynaptic transporter-associated anion current (EAAT5) that suppresses further transmitter release by hyperpolarization or shunting inhibition of presynaptic axon terminals. However, this proposed inhibitory receptor function requires a high-capacity charge movement by the EAAT5-mediated anion current.
Here, we report that heterologously expressed EAAT5 has a low uptake capacity, with an estimated turnover rate of <1 s−1. From this turnover rate and our current recording data, we can estimate an upper limit of the density of EAAT5 in the cell membrane as 3000 μm−2, assuming an average cell surface area of 1000 μm2 (41). This density is at least a factor 3 lower than that estimated by Hasegawa et al. (28). Although this reduced expression density in 293 cells compared with the visual synapse can account for our inability to measure transport current, it cannot explain the rapid transporter kinetics observed by Veruki et al. (40), which may point to a presynaptic rod bipolar transporter other than EAAT5. However, because GLT1 isoforms are also expressed in bipolar cells, they may account for the rapidly gated anion current signals seen by those authors.
Our EAAT5 results suggest that a maximum transport rate/surface area of 2,400 μm−2 s−1 can be supported, in contrast to the simulations of Hasegawa et al. (28) (370,000 μm−2 s−1). Even if heterologously expressed EAAT5 could be inserted at a threefold higher density, approaching that of other glutamate transporter subtypes, this transport rate cannot account for the high-capacity uptake suggested by Hasegawa et al. (28). To account for these discrepancies, we cannot exclude the possibility that differential cellular environments modulate individual EAAT5 functions, such as the high glutamate uptake capacity in rod photoreceptors and the high-capacity charge movement in bipolar cells versus the low-capacity uptake and insignificant chloride current in heterologously expressed EAAT5. The unambiguous assignment of the molecular identity of the glutamate transporters under observation in previous studies (17,28,40) is therefore of critical importance to resolve these issues. Overall, therefore, our data suggest that EAAT5 does not function like a classical high-capacity glutamate transporter, but instead serves as a slow-gated glutamate receptor or a buffer system for tonically released glutamate at the rod bipolar cell synapse.
Acknowledgments
This study was supported by the National Institutes of Health (grant 2R01NS049335-06A1 to C.G.), the Binational Science Foundation (grant 2007051 to C.G. and Baruch I. Kanner), and the Deutsche Forschungsgemeinschaft (grant RA 753/1-3 to T.R.).
Footnotes
The title of this work was adapted from Nadolny, S., The Discovery of Slowness, 1997, Penguin Group USA, New York.
Supporting Material
References
- 1.Zerangue N., Kavanaugh M.P. Flux coupling in a neuronal glutamate transporter. Nature. 1996;383:634–637. doi: 10.1038/383634a0. [DOI] [PubMed] [Google Scholar]
- 2.Tao Z., Rosental N., Grewer C. Mechanism of cation binding to the glutamate transporter EAAC1 probed with mutation of the conserved amino acid residue Thr101. J. Biol. Chem. 2010;285:17725–17733. doi: 10.1074/jbc.M110.121798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wadiche J.I., Kavanaugh M.P. Macroscopic and microscopic properties of a cloned glutamate transporter/chloride channel. J. Neurosci. 1998;18:7650–7661. doi: 10.1523/JNEUROSCI.18-19-07650.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Watzke N., Bamberg E., Grewer C. Early intermediates in the transport cycle of the neuronal excitatory amino acid carrier EAAC1. J. Gen. Physiol. 2001;117:547–562. doi: 10.1085/jgp.117.6.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bergles D.E., Tzingounis A.V., Jahr C.E. Comparison of coupled and uncoupled currents during glutamate uptake by GLT-1 transporters. J. Neurosci. 2002;22:10153–10162. doi: 10.1523/JNEUROSCI.22-23-10153.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Otis T.S., Kavanaugh M.P. Isolation of current components and partial reaction cycles in the glial glutamate transporter EAAT2. J. Neurosci. 2000;20:2749–2757. doi: 10.1523/JNEUROSCI.20-08-02749.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seal R.P., Shigeri Y., Amara S.G. Sulfhydryl modification of V449C in the glutamate transporter EAAT1 abolishes substrate transport but not the substrate-gated anion conductance. Proc. Natl. Acad. Sci. USA. 2001;98:15324–15329. doi: 10.1073/pnas.011400198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sonders M.S., Amara S.G. Channels in transporters. Curr. Opin. Neurobiol. 1996;6:294–302. doi: 10.1016/s0959-4388(96)80111-5. [DOI] [PubMed] [Google Scholar]
- 9.Grewer C., Gameiro A., Rauen T. Glutamate forward and reverse transport: from molecular mechanism to transporter-mediated release after ischemia. IUBMB Life. 2008;60:609–619. doi: 10.1002/iub.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kanner B.I., Zomot E. Sodium-coupled neurotransmitter transporters. Chem. Rev. 2008;108:1654–1668. doi: 10.1021/cr078246a. [DOI] [PubMed] [Google Scholar]
- 11.Arriza J.L., Eliasof S., Amara S.G. Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance. Proc. Natl. Acad. Sci. USA. 1997;94:4155–4160. doi: 10.1073/pnas.94.8.4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bringmann A., Pannicke T., Reichenbach A. Role of retinal glial cells in neurotransmitter uptake and metabolism. Neurochem. Int. 2009;54:143–160. doi: 10.1016/j.neuint.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 13.Pow D.V., Barnett N.L. Developmental expression of excitatory amino acid transporter 5: a photoreceptor and bipolar cell glutamate transporter in rat retina. Neurosci. Lett. 2000;280:21–24. doi: 10.1016/s0304-3940(99)00988-x. [DOI] [PubMed] [Google Scholar]
- 14.Rico E.P., de Oliveira D.L., Bogo M.R. Expression and functional analysis of Na(+)-dependent glutamate transporters from zebrafish brain. Brain Res. Bull. 2010;81:517–523. doi: 10.1016/j.brainresbull.2009.11.011. [DOI] [PubMed] [Google Scholar]
- 15.Billups B., Rossi D., Attwell D. Anion conductance behavior of the glutamate uptake carrier in salamander retinal glial cells. J. Neurosci. 1996;16:6722–6731. doi: 10.1523/JNEUROSCI.16-21-06722.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fairman W.A., Vandenberg R.J., Amara S.G. An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature. 1995;375:599–603. doi: 10.1038/375599a0. [DOI] [PubMed] [Google Scholar]
- 17.Wersinger E., Schwab Y., Roux M.J. The glutamate transporter EAAT5 works as a presynaptic receptor in mouse rod bipolar cells. J. Physiol. 2006;577:221–234. doi: 10.1113/jphysiol.2006.118281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Picaud S.A., Larsson H.P., Werblin F.S. Glutamate-gated chloride channel with glutamate-transporter-like properties in cone photoreceptors of the tiger salamander. J. Neurophysiol. 1995;74:1760–1771. doi: 10.1152/jn.1995.74.4.1760. [DOI] [PubMed] [Google Scholar]
- 19.Mim C., Balani P., Grewer C. The glutamate transporter subtypes EAAT4 and EAATs 1-3 transport glutamate with dramatically different kinetics and voltage dependence but share a common uptake mechanism. J. Gen. Physiol. 2005;126:571–589. doi: 10.1085/jgp.200509365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watzke N., Grewer C. The anion conductance of the glutamate transporter EAAC1 depends on the direction of glutamate transport. FEBS Lett. 2001;503:121–125. doi: 10.1016/s0014-5793(01)02715-6. [DOI] [PubMed] [Google Scholar]
- 21.Grewer C., Watzke N., Rauen T. Glutamate translocation of the neuronal glutamate transporter EAAC1 occurs within milliseconds. Proc. Natl. Acad. Sci. USA. 2000;97:9706–9711. doi: 10.1073/pnas.160170397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shigeri Y., Shimamoto K., Amara S.G. Effects of threo-β-hydroxyaspartate derivatives on excitatory amino acid transporters (EAAT4 and EAAT5) J. Neurochem. 2001;79:297–302. doi: 10.1046/j.1471-4159.2001.00588.x. [DOI] [PubMed] [Google Scholar]
- 23.Grewer C., Rauen T. Electrogenic glutamate transporters in the CNS: molecular mechanism, pre-steady-state kinetics, and their impact on synaptic signaling. J. Membr. Biol. 2005;203:1–20. doi: 10.1007/s00232-004-0731-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shimamoto K., Shigeri Y., Nakajima T. Syntheses of optically pure β-hydroxyaspartate derivatives as glutamate transporter blockers. Bioorg. Med. Chem. Lett. 2000;10:2407–2410. doi: 10.1016/s0960-894x(00)00487-x. [DOI] [PubMed] [Google Scholar]
- 25.Tao Z., Zhang Z., Grewer C. Neutralization of the aspartic acid residue Asp-367, but not Asp-454, inhibits binding of Na+ to the glutamate-free form and cycling of the glutamate transporter EAAC1. J. Biol. Chem. 2006;281:10263–10272. doi: 10.1074/jbc.M510739200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mim C., Tao Z., Grewer C. Two conformational changes are associated with glutamate translocation by the glutamate transporter EAAC1. Biochemistry. 2007;46:9007–9018. doi: 10.1021/bi7005465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wadiche J.I., Amara S.G., Kavanaugh M.P. Ion fluxes associated with excitatory amino acid transport. Neuron. 1995;15:721–728. doi: 10.1016/0896-6273(95)90159-0. [DOI] [PubMed] [Google Scholar]
- 28.Hasegawa J., Obara T., Tachibana M. High-density presynaptic transporters are required for glutamate removal from the first visual synapse. Neuron. 2006;50:63–74. doi: 10.1016/j.neuron.2006.02.022. [DOI] [PubMed] [Google Scholar]
- 29.Larsson H.P., Tzingounis A.V., Kavanaugh M.P. Fluorometric measurements of conformational changes in glutamate transporters. Proc. Natl. Acad. Sci. USA. 2004;101:3951–3956. doi: 10.1073/pnas.0306737101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Larsson H.P., Picaud S.A., Lecar H. Noise analysis of the glutamate-activated current in photoreceptors. Biophys. J. 1996;70:733–742. doi: 10.1016/S0006-3495(96)79613-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kugler P., Beyer A. Expression of glutamate transporters in human and rat retina and rat optic nerve. Histochem. Cell Biol. 2003;120:199–212. doi: 10.1007/s00418-003-0555-y. [DOI] [PubMed] [Google Scholar]
- 32.Wiessner M., Fletcher E.L., Rauen T. Localization and possible function of the glutamate transporter, EAAC1, in the rat retina. Cell Tissue Res. 2002;310:31–40. doi: 10.1007/s00441-002-0612-1. [DOI] [PubMed] [Google Scholar]
- 33.Sullivan R., Rauen T., Pow D.V. Cloning, transport properties, and differential localization of two splice variants of GLT-1 in the rat CNS: implications for CNS glutamate homeostasis. Glia. 2004;45:155–169. doi: 10.1002/glia.10317. [DOI] [PubMed] [Google Scholar]
- 34.Torres-Salazar D., Fahlke C. Neuronal glutamate transporters vary in substrate transport rate but not in unitary anion channel conductance. J. Biol. Chem. 2007;282:34719–34726. doi: 10.1074/jbc.M704118200. [DOI] [PubMed] [Google Scholar]
- 35.Chen T.Y. Coupling gating with ion permeation in ClC channels. Sci. STKE. 2003;2003:pe23. doi: 10.1126/stke.2003.188.pe23. [DOI] [PubMed] [Google Scholar]
- 36.Chen T.Y. Structure and function of clc channels. Annu. Rev. Physiol. 2005;67:809–839. doi: 10.1146/annurev.physiol.67.032003.153012. [DOI] [PubMed] [Google Scholar]
- 37.Rauen T., Rothstein J.D., Wässle H. Differential expression of three glutamate transporter subtypes in the rat retina. Cell Tissue Res. 1996;286:325–336. doi: 10.1007/s004410050702. [DOI] [PubMed] [Google Scholar]
- 38.Rauen T., Taylor W.R., Wiessner M. High-affinity glutamate transporters in the rat retina: a major role of the glial glutamate transporter GLAST-1 in transmitter clearance. Cell Tissue Res. 1998;291:19–31. doi: 10.1007/s004410050976. [DOI] [PubMed] [Google Scholar]
- 39.Rauen T., Wiessner M., Pow D.V. A new GLT1 splice variant: cloning and immunolocalization of GLT1c in the mammalian retina and brain. Neurochem. Int. 2004;45:1095–1106. doi: 10.1016/j.neuint.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 40.Veruki M.L., Mørkve S.H., Hartveit E. Activation of a presynaptic glutamate transporter regulates synaptic transmission through electrical signaling. Nat. Neurosci. 2006;9:1388–1396. doi: 10.1038/nn1793. [DOI] [PubMed] [Google Scholar]
- 41.Thomas P., Smart T.G. HEK293 cell line: a vehicle for the expression of recombinant proteins. J. Pharmacol. Toxicol. Methods. 2005;51:187–200. doi: 10.1016/j.vascn.2004.08.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.