

Abstract
Motivation: Condition-specific networks capture system-wide behavior under varying conditions such as environmental stresses, cell types or tissues. These networks frequently comprise parts that are unique to each condition, and parts that are shared among related conditions. Existing approaches for learning condition-specific networks typically identify either only differences or only similarities across conditions. Most of these approaches first learn networks per condition independently, and then identify similarities and differences in a post-learning step. Such approaches do not exploit the shared information across conditions during network learning.
Results: We describe an approach for learning condition-specific networks that identifies the shared and unique subgraphs during network learning simultaneously, rather than as a post-processing step. Our approach learns networks across condition sets, shares data from different conditions and produces high-quality networks that capture biologically meaningful information. On simulated data, our approach outperformed an existing approach that learns networks independently for each condition, especially for small training datasets. On microarray data of hundreds of deletion mutants in two, yeast stationary-phase cell populations, the inferred network structure identified several common and population-specific effects of these deletion mutants and several high-confidence cases of double-deletion pairs, which can be experimentally tested. Our results are consistent with and extend the existing knowledge base of differentiated cell populations in yeast stationary phase.
Availability and Implementation: C++ code can be accessed from http://www.broadinstitute.org/~sroy/condspec/
Contact: sroy@broadinstitute.org
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
All cells on earth have the potential to sense and respond to different extracellular conditions defined by different environmental stresses, growth factors or cellular differentiation signals. Response to different conditions or condition-specific response is orchestrated by changes in the concentration of cellular components (mRNAs, proteins and metabolites) as well as the interactions among these components. Condition-specific networks capture the interactions among cellular components under different conditions, providing a system-wide view of condition-specific behavior. Understanding these networks can provide important insight into how organisms function, adapt and evolve.
Although technological advances are allowing us to capture the concentrations of the system components, our ability to quantify the condition-specific interactions is still limited. Fortunately, network inference algorithms based on probabilistic graphical models that have been used successfully to infer a functional network from one gene expression (mRNA concentration) dataset (Friedman et al., 2000; Pe'er et al., 2006; Segal et al., 2003, 2005; Werhli et al., 2006; Yu et al., 2004), provide a starting point for inferring condition-specific networks.
In principle, a probabilistic network for each condition could be inferred separately from each condition. In practice, however, such an approach is limited because it fails to recognize an important aspect of condition-specific network learning: this is a multiple-network learning problem, where the networks for each condition are related (with both shared and unique subnetworks). To extend existing network inference methods to infer condition-specific networks, it is important to explicitly capture and exploit the shared information during network learning.
In this article, we develop a novel approach, Network Inference with Pooling Data (NIPD), based on probabilistic graphical models that explicitly incorporates the shared information across conditions by simultaneously learning multiple, high-quality networks across a small number of conditions. NIPD treats the condition as an additional random variable, different values of which induce different network structures (Fig. 1). Edges in NIPD are statistical dependencies that abstract the true condition-specific, physical network (protein–protein, protein–DNA interactions) and are inferred from condition-specific mRNA concentrations. Modeling the condition variable within the learning framework allows the condition information to directly influence which edges occur in the final inferred networks, and also the condition-specific roles of the edges.
Fig. 1.
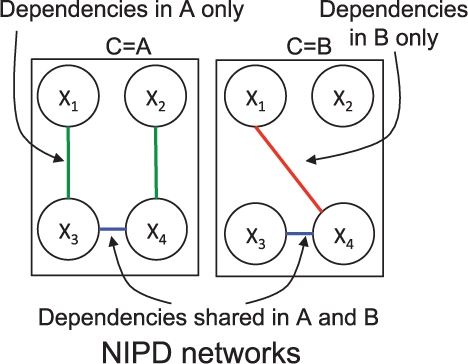
NIPD framework for two conditions, A and B. C represents conditions in NIPD. NIPD learns dependencies that are shared between A and B, as well as those unique to A or to B.
Existing network-based approaches for condition-specific responses can be grouped into module-centric and gene-centric approaches. Module-centric approaches identify transcription modules (set of transcription factors regulating a set of target genes) that are coexpressed in a condition-specific manner (Kim et al., 2006; Segal et al., 2005; Tuck et al., 2006). However, these approaches assume identical behavior for all genes within a module, and do not provide fine-grained interaction structure that explains the condition-specific behavior of individual genes. Further, condition-specific patterns are identified via a post-learning gene set enrichment analysis.
Gene-centric approaches identify the set of condition-specific edges on a per-gene basis. Such approaches have been developed for capturing condition-specific behavior in diseases (Chuang et al., 2007), in yeast stress responses (Rokhlenko et al., 2007; Roy et al., 2009) and also in different species (Bergmann et al., 2004; Stuart et al., 2003). However, these approaches are not probabilistic in nature (Chuang et al., 2007; Rokhlenko et al., 2007), often rely on the network being known (Chuang et al., 2007) and are restricted to pairwise coexpression relationships rather than general statistical dependencies (Bergmann et al., 2004; Stuart et al., 2003). Most of these approaches infer a network for each condition separately, and then compare the networks from different conditions to identify the edges unique or shared across conditions. The approach in Myers et al. does integrate context more directly within a Bayesian network, but relies on an initial training set to learn model parameters (Myers and Troyanskaya, 2007). Other approaches such as differential dependency networks (Zhang et al., 2008) and mixture of subgraphs (Sanguinetti et al., 2008) construct probabilistic models, but focus on differences rather than both differences and similarities.
The NIPD approach is a gene-centric approach with the following benefits: (i) infers general statistical dependencies including pairwise and higher order dependencies (dependencies among more than two genes); (ii) does not rely on the network being known; (iii) is probabilistic in nature, providing a system-wide description of the condition-specific behavior as a probabilistic network; (iv) simultaneously learns networks across multiple conditions allowing the learning procedure to be informed by the shared information across conditions; and (v) does not bias the networks toward only differences or similarities, thus identifying both conserved and unique aspects of condition-specific responses.
Results on simulated data from known ground truth networks suggested that networks inferred by NIPD were of significantly higher quality than networks learned separately for each condition. Results on microarray compendia from two yeast cell populations, quiescent and non-quiescent, both under glucose stress (Aragon et al., 2008), demonstrated that NIPD identified dependencies capturing shared processes (respiration) that agree with the global starvation stress experienced by these cells, as well as population-specific processes (chromatin modeling in quiescent), that are consistent with the physiological state of these cells. Finally, NIPD networks were used to extract candidates of double deletion experiments, that can lead to insight into this important stage in the life cycle of yeast, and for processes such as cancer and aging (Gray et al., 2004).
2 METHODS
2.1 Probabilistic graphical models for functional networks
NIPD is based on the framework of probabilistic graphical models (Lauritzen, 1996). These models have been widely used for modeling biological networks (Friedman et al., 2000; Pe'er et al., 2006; Segal et al., 2005; Yu et al., 2004), because they can represent complex interaction patterns and also model noisy data, both typical of biological systems. A probabilistic graphical model has two components: a graph G and a set of potentials functions Ψ={ψ1,…,ψ|Ψ|} (Lauritzen, 1996). The nodes of G represent random variables, X={X1,…,Xn}, encoding the mRNA expression level of genes. The edges of G represent pairwise and higher order (among >2 genes) statistical dependencies among random variables. Each ψi specifies the mathematical form of the dependency between Xi and its neighbors. The graph can have directed edges as in Bayesian networks (Heckerman, 1999) or undirected edges as in Markov random fields (Lauritzen, 1996). We focus on undirected models, because they can represent cyclic dependencies, which arise in biological networks and are difficult to represent in directed models.
Learning in these models typically optimizes a likelihood-based score by greedily searching the space of candidate graphs (Heckerman, 1999). In undirected models, because likelihood cannot be exactly computed, we use pseudo-likelihood to learn the structure (Besag, 1977). Briefly, pseudo-likelihood decomposes as a product over conditional distributions per variable, P(Xi|Mi), where Mi denotes the immediate neighborhood or Markov Blanket of Xi. To learn G, we need to find the best Mi for each Xi.
2.2 Learning multiple condition-specific networks
Assume we have a set of k conditions, 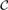 . Let |Dc|={xc1,…,xc|
. Let |Dc|={xc1,…,xc| c|} denote the dataset for the c-th condition, 1≤c≤k. xcd, 1≤d≤|c|, denotes the mRNA expression values in the d-th microarray in condition c. The problem of condition-specific network learning is defined as: given k datasets, {1,…,k}, learn k graphs, {G1,…,Gk}, one for each condition.
c|} denote the dataset for the c-th condition, 1≤c≤k. xcd, 1≤d≤|c|, denotes the mRNA expression values in the d-th microarray in condition c. The problem of condition-specific network learning is defined as: given k datasets, {1,…,k}, learn k graphs, {G1,…,Gk}, one for each condition.
A simple approach for learning multiple networks is to learn each graph Gc, independent of all Gc′,c′≠c), using dataset c only. We refer to this approach as the independent learner (INDEP). INDEP is a generalization of several existing approaches (Bergmann et al., 2004; Rokhlenko et al., 2007; Stuart et al., 2003), which can all be considered as instances of INDEP with a specific network learning algorithm. We use the Markov blanket search algorithm as the learning algorithm for INDEP, which finds the best Markov blanket of every node using a greedy search of edge additions (Roy et al., 2009).
2.2.1 Exploiting shared information for learning multiple networks (NIPD):
To motivate NIPD, let us first consider the ‘condition’ semantics of an edge for the two condition case ={A,B}. An edge between two variables, Xi and Xj, can occur only in condition A but not in B, only in condition B but not in A, or in both A and B. Thus, edges in condition A's network are the union of edges that occur in the singleton set {A}, and the edges that occur in the set {A, B}. Our goal is to explicitly capture these condition subset semantics of each edge. Such information can be useful to characterize transcription factors that regulate overlapping target sets in different conditions (Harbison et al., 2004). An edge that occurs in a non-singleton condition subset is a shared edge, and its parameters can be learned by pooling the data from all the conditions in the subset. Of course, both the edges and their condition-specific semantics are unknowns and must be inferred automatically from the data.
NIPD uses a novel formulation of the conditional distribution, P(Xi|Mci), where Mci is the Markov blanket (MB) of Xi in condition c, such that shared edges impose a cross-condition dependence in the MBs and their parameters. NIPD incorporates this formulation within the pseudo-likelihood score and evaluates candidate networks with respect to data from any subset of conditions. This enables NIPD to identify the condition-subset semantics of MBs and also exploit shared information across conditions.
Let θci denote the parameters of P(Xi|Mci). Mci in turn is determined by all neighbors of Xi in any subset of that includes c. Let E⊆ be a subset of condition. Let M*Ei) be the set of variables that are connected to Xi only in condition set E and θEi*) denote the parameters associated with M*Ei. For example, if ={A, B}, then M*{A}i denotes the set of variables that are connected to Xi in A only and not B, M*{A,B}i) denotes the set of variables that are connected to (Xi) in both A and B and so on. The overall MB of Xi in condition A is MAi=M*Ai ∪ M*{A,B}i). Similarly, overall MB of Xi in condition B is MBi=M*{B}i ∪ M*{A,B}i. θAi and θBi are therefore concatenations of θ{A}i* and θ{A,B}i*). Here, the shared component θ{A,B}i* associated with M*{A,B}i enforces cross-condition dependence among the θAi and θBi.
More generally, for any  , where M*Ei) denotes the neighbors of Xi only in condition set E. We assume P(Xi|M*i to be conditional Gaussians. We now need to define P(Xi|Mci) such that it takes into account all subsets E, c∈E. We use a product formulation: P(Xi|Mci)∝∏E∈powerset():c∈EP(Xi|M*Ei). The proportionality sign can be eliminated by dividing by a normalization constant. However, we work with the unnormalized product because it decomposes over condition subsets (Supplementary Material). Other formulations of P(Xi|Mci), such as a weighted sum of mixtures (Hastie et al., 2001), are not decomposable over condition subsets and did not have significant advantages in our preliminary experiments.
, where M*Ei) denotes the neighbors of Xi only in condition set E. We assume P(Xi|M*i to be conditional Gaussians. We now need to define P(Xi|Mci) such that it takes into account all subsets E, c∈E. We use a product formulation: P(Xi|Mci)∝∏E∈powerset():c∈EP(Xi|M*Ei). The proportionality sign can be eliminated by dividing by a normalization constant. However, we work with the unnormalized product because it decomposes over condition subsets (Supplementary Material). Other formulations of P(Xi|Mci), such as a weighted sum of mixtures (Hastie et al., 2001), are not decomposable over condition subsets and did not have significant advantages in our preliminary experiments.
θEi is estimated by pooling the data for all non-singleton E, which in turn makes more data available for estimating these parameters. This enables NIPD to robustly estimate parameters, especially those of higher order dependencies, which are harder to estimate relative to pairwise dependencies.
2.2.2 Structure learning algorithm of NIPD in detail:
Our structure learning algorithm maintains a conditional distribution for every variable, Xi for every set E∈powerset() and computes score improvement of adding an edge {Xi,Xk} in every set E. This addition will affect the conditionals of Xi and Xj in all conditions e∈E. The net score improvement of adding an edge {Xi,Xj} to a condition set E is given by:
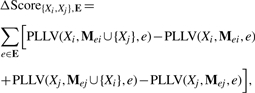 |
(1) |
where PLLV(Xi,Mei,e) is the conditional log likelihood of Xi given its neighbors, and is defined as 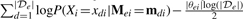 , where the second term is MDL penalty. xdi and mdi are assignments to Xi and Mei, respectively, from the d-th data point xd in dataset e. Because of our product formulation of P(Xi|Mei), the pseudo-likelihood contribution of each e to PLLV(Xi,Mei,e) decomposes as ∑Fs.t:e∈FPLLV(Xi,M*Fi,e). Because the edge {Xi,Xj} is being added to ME*, all terms not involving E remain unchanged producing the score improvement:
, where the second term is MDL penalty. xdi and mdi are assignments to Xi and Mei, respectively, from the d-th data point xd in dataset e. Because of our product formulation of P(Xi|Mei), the pseudo-likelihood contribution of each e to PLLV(Xi,Mei,e) decomposes as ∑Fs.t:e∈FPLLV(Xi,M*Fi,e). Because the edge {Xi,Xj} is being added to ME*, all terms not involving E remain unchanged producing the score improvement:
This score allows us to score an edge in condition sets in a decomposable manner.
Our structure learning algorithm begins with k empty graphs and proposes edge additions for all variables, for all subsets of the condition set (Algorithm 1). The outermost for loop (Steps 4–14 ) iterates over variables Xi to identify new candidate MB variables, Xj, in a condition set E. We iterate over all candidate MBs Xj (Steps 5–12) and condition sets E (Steps 6–11) and compute the score improvement for each pair {Xj,E} (Step 10). If the current condition set under consideration has more than one condition, data from these conditions are pooled and parameters for the new distribution P(Xi|M*Ei) are estimated using the pooled dataset (Steps 7–9). A candidate move for a variable Xi is composed of a pair {Xj′,E′} with the maximal score improvement over all variables and conditions (Step 13). After all candidate moves have been identified, moves are attempted in the order of decreasing score improvement (Step 15), to enable moves with the highest score improvements to be attempted first. A move connecting two variables, Xi and Xj, can fail if a previous move updated the neighborhoods of either Xi or Xj. The algorithm converges when no edge addition improves the score of the k graphs. Although we implement NIPD with undirected, probabilistic graphical models, the NIPD framework is applicable to directed graphs as well.

2.3 Dataset description
2.3.1 Microarray data from yeast stationary phase:
Each microarray measures the mRNA expression of all yeast (Saccharomyces cerevisiae) genes in response to ≈100 genetic deletions from quiescent and non-quiescent populations, isolated from glucose-starved stationary phase cultures (Aragon et al., 2008), We first filtered the microarray data to exclude genes with > 80% missing values. We then considered only those genes whose expression changed significantly (|z|-score ≥ 4) compared with wild type, in either quiescent or non-quiescent populations. This resulted in a final dataset of 2639 genes exhibiting downstream knockout effects of 1 of the 88 deletions. Although we selected genes that were significantly affected by at least by one deletion, neither NIPD nor INDEP knew which genes were affected by which deletion.
2.3.2 Simulated data:
We simulated data from six networks, one network per condition. One of these was a subnetwork of 99 nodes from the Escherichia coli regulatory network (Salgado et al., 2006). The other five were generated by flipping 10, 30, 50, 70 and 100% of the edges of the first network. A simulated dataset was generated per network, using a differential equation-based simulator, by perturbing all transcription factor nodes and measuring the steady state of all genes (Mendes et al., 2003). This was repeated 1000 times per network.
2.4 Validation of network structure
2.4.1 Inferred from simulated data:
We used three methods to infer networks on simulated data: NIPD, INDEP with a Markov blanket search algorithm (Roy et al., 2009) and GeneNet (Schäfer and Strimmer, 2005). GeneNet also infers a network per condition independently by learning a shrinkage-based partial correlations graph, and is well-suited for sparse data situations. Note the GeneNet algorithm outputs a weight for every edge corresponding to the statistical significance of an edge. To obtain a network, one needs to specify an input number of edges. We used the number of edges from NIPD because this was when GeneNet had the highest performance. We compared the structure of the networks inferred by these algorithms to the true network structure using standard precision and recall of edge match, and also the neighborhood match of every node (Supplementary Material). For edge match, we report the F-score, which is the harmonic mean of precision and recall. We evaluate neighborhood structure quality by obtaining the number of nodes on which one approach was significantly better than another approach (t-test P<0.05) in capturing node neighborhood as a function of training data size (See Supplementary Material). This comparison captures a more localized picture of the types of random variables that may be contributing most to errors.
We partitioned each dataset into q equal partitions, where q∈{3, 4, 5, 6, 7, 8, 9, 10}. The training data size for each partition is  and decreases with increasing q. For each q, we learned a network for each partition and report the average F-score.
and decreases with increasing q. For each q, we learned a network for each partition and report the average F-score.
2.4.2 Inferred from microarray data:
Network inference from microarray data is known to be a notoriously difficult problem because of the relatively few samples (microarrays) compared with the number of genes. A concern that arises in this situation is distinguishing true from spurious dependencies. We took several measures to ensure the dependencies captured by our networks represented meaningful dependencies: (i) we use an MDL-based score that penalizes complex structures, (ii) genes were connected to only the 88 genes for which we had single deletion strains (this was done uniformly for both NIPD and INDEP), (iii) genes without deletions were not allowed to have more than eight neighbors (the 88 genes with deletion mutants were unconstrained), (iv) edges inferred by INDEP or NIPD were assigned a confidence score based on a well-known bootstrap analysis (Friedman et al., 2000; Pe'er et al., 2001), where we subsampled the data 20 times. We considered only those edges that had a confidence ≥τ, which in turn was determined based on the probability of observing an edge with this confidence by random (Supplementary Fig. S1). We selected τ=0.3 because this represented the point of most dramatic change in the probability for random edges. All GO Process enrichments were performed in Genatomy (http://www.c2b2.columbia.edu/danapeerlab/html/genatomy.html).
3 RESULTS
The goals of our experiments were as follows: (i) to compare our approach against independent network learning approaches, on simulated data from known ground-truth networks and (ii) to assess the value of our approach on real microarray data. For (i) we used two independent learners, INDEP with a Markov blanket search algorithm and GeneNet (Schäfer and Strimmer, 2005). Experiments on simulated data allowed us to systematically compare the quality of the inferred networks and, therefore, assess the benefit of pooling data and sharing information across conditions versus learning networks independently. To address (ii) we applied NIPD and INDEP on microarray data from two yeast cell populations isolated from glucose-starved stationary phase cultures (Aragon et al., 2008). These data are good case studies for condition-specific network learning (hundreds of samples per condition), and the inferred quiescent and non-quiescent networks can provide potentially new insight into stationary phase, a process known to be intricately related to cancer and aging.
3.1 NIPD had superior performance on networks with known ground truth
We first evaluated overall network structure using two sets of networks, HIGHSIM and LOWSIM. Networks in HIGHSIM shared a large portion (75%) of the edges, and networks in LOWSIM shared a small (20%) portion of the edges. On HIGHSIM, NIPD performed significantly better than INDEP and GeneNet for all training data sizes (Fig. 2). On LOWSIM, NIPD was significantly better than INDEP, and comparable to GeneNet performing better on one network and at par on the second network.
Fig. 2.

Performance comparison using F-score of NIPD, INDEP and GeneNet on simulated data of two networks. Shown are mean and SDs of F-scores estimated from partitions of different sizes,  , where 3≤q≤10. Top is for HIGHSIM (75% edges shared) and bottom is for LOWSIM (20% edges shared). The top and bottom graphs are for networks from the individual conditions. Higher is better.
, where 3≤q≤10. Top is for HIGHSIM (75% edges shared) and bottom is for LOWSIM (20% edges shared). The top and bottom graphs are for networks from the individual conditions. Higher is better.
In addition to standard F-score, we also compared the algorithms based on how well neighborhoods of individual variables were identified (Supplementary Figs S5-S8). These ‘per-variable’ scores provide a more granular understanding of algorithm performance. In particular, nodes with high degree may be contributing to the majority of the errors, but standard precision–recall values will not be able to capture this. NIPD consistently outperformed other algorithms, especially when there is some sharing of structure between conditions.
To demonstrate that NIPD is not specific to two conditions, we varied the number of conditions from two to six. We found that NIPD and GeneNet were generally better than INDEP, which does not handle sparse data conditions, and the performance margin between NIPD and GeneNet decreased with increasing number of networks (Fig. 3, Supplementary Figs S2-S4). NIPD seemed to suffer most on networks with the fewest number of shared edges, whereas GeneNet suffered most when there were more shared edges. However, GeneNet did not significantly outperform NIPD in any case. Overall, our results show that when the individual condition-specific networks are similar, NIPD has a significantly better performance than other algorithms. When the underlying networks are different, NIPD performs at par with methods suited for small training data sizes.
Fig. 3.

Performance comparison INDEP, NIPD and GeneNet as a function of decreasing training data for six networks. Shown are mean and SD of F-score of inferred network structure from each partition size. Higher is better.
3.2 Application to yeast quiescence
We applied NIPD to microarray datasets from yeast quiescent and non-quiescent cell populations separated from stationary phase cultures (Aragon et al., 2008). The cell populations have experienced the same glucose starvation stress, but have differentiated physiologically (Davidson et al., 2011), suggesting that each population responds to starvation stress differently. Each microarray within each dataset measures the expression profile of 1 of 88 deletion mutants, selected based on existing knowledge of yeast stationary phase.
We applied NIPD and INDEP approaches to learn quiescent and non-quiescent-specific networks, treating each cell population as a condition. One of the goals of our study was to further characterize these deletion mutants, many of which have no known phenotype. Hence, we constrained the networks such that only genes with deletion mutants are connected to the remaining genes1. The neighborhood of each deletion mutant determined by the network topology was analyzed for Gene Ontology (GO) process enrichment (FDR <0.05), and the network quality was considered proportional to the number of unique GO processes enriched within a neighborhood.
3.2.1 NIPD captures both common response as well as population-specific starvation responses:
To determine if one method was superior than another, we examined the GO process categories enriched in the neighborhood of every deletion mutant in the inferred networks. On quiescent, 21 of the deletion mutants were enriched in 68 different biological processes using the NIPD-inferred network, whereas using INDEP only 2 mutants were associated with a biological process, all of which were identified by the NIPD algorithm already (Supplementary Material 1). Similar behavior was observed on non-quiescent.
To identify similarities and differences between the two populations, we asked which deletion mutants affect the same processes in both populations, and which mutants affect different processes (Supplementary Fig. S10). We found several processes that were affected by the same deletion in quiescent and non-quiescent populations, suggesting a conserved, starvation stress response (response to stimulus, cellular respiration). Both these processes were previously identified to be associated with quiescent cells (Aragon et al., 2008). NIPD also identified several processes that were associated exclusively with quiescent cells (purine metabolic process, cellular aging, chromatin modification). Although respiration was present in both populations, quiescent was associated with more processes related to respiration, consistent with our recent finding that quiescent cells exhibit higher rates of respiration (Davidson et al., 2011). The only process that we did not recover was signal transduction. This may be because of our per-mutant analysis, whereas previous work considered upregulated genes in the entire compendium of quiescent cells. In contrast, INDEP did not identify any shared process, and identified a small subset of these population-specific processes.
3.2.2 NIPD identified several deletion combinations:
We next analyzed neighborhood genes of individual deletion mutants to identify pairs of deletion mutants that had more overlapping neighbors (Hypergeometric P<1E−3). Such combinations represent candidates for double deletion analysis, that are generally harder to experimentally test, but are necessary to identify genetic interactions and cross-talk between pathways. We found 18 and 21 deletion pairs in quiescent and non-quiescent populations, respectively. These numbers are significantly higher than in random networks with identical degree distributions (z-test, P<1E−4, Supplementary Table ST1). In quiescent cells, we found several deletion pairs likely to affect TCA cycle and aerobic respiration. In non-quiescent cells, several knock-out combinations involved fatty acid metabolism and aerobic respiration. Our predictions included genes either with known phenotypes in stationary phase or related to genes with such phenotypes. For example, QCR7, QCR8 and QCR10 are subunits of ubiqunol-cytochrome c oxidoreductase complex; QCR7 is essential for viability in stationary phase (Aragon et al., 2008; Martinez et al., 2004); , and both QCR8 and QCR10 were in our predictions. Our predictions also included ALD4 and ADH2, both dehydrogenases, which may affect population heterogeneity of non-quiescent cells and reproductive capacity of both cell types (A. Dodson, personal communication). Overall, 17 of the 39 genes in our predicted deletion combinations are required for viability and survival in stationary phase, which suggests these combinations are likely to have significant phenotypes.
4 DISCUSSION
We have introduced a novel, probabilistic graphical modeling approach, NIPD, for learning fine-grained interaction patterns of condition-specific responses. The crux of our approach is to recognize that condition-specific network learning poses a multiple network learning problem where the networks are related and, therefore, share information among them. NIPD solves this problem by simultaneously learning networks across all conditions using a novel score that allows the learning process to be aware of the shared information across conditions.
Small training datasets, which are common for biological data, present significant challenges for any network learning approach. In particular, standard approaches that infer networks for each condition independently (Roy et al., 2009) suffer from low sensitivity because they miss shared dependencies that are too weak within individual datasets. In contrast, NIPD is able to recover these shared dependencies because by pooling data across conditions during network learning, NIPD effectively has more data for estimating parameters for the shared parts of the network. This allows NIPD to have more discovery power than INDEP, and our results confirmed that the additional dependencies inferred by NIPD represent shared, biologically meaningful dependencies.
One of the strengths of NIPD was its ability to identify networks with the most biologically meaningful structure. This allowed us to characterize deletion mutants based on their affects on quiescent and non-quiescent cells, and also to predict combinations of such individual gene deletions. Several of these predictions are supported by literature, including genes that are known to have a phenotypic effect on stationary phase cultures (Aragon et al., 2008; Martinez et al., 2004). Importantly, these predictions are a drastic reduction of the possible double deletion combinations of 88 single gene deletions and provide directions for future experiments.
The probabilistic framework of NIPD can be easily extended to automatically infer the condition variable to handle situations with uncertainty about conditions. The run time of NIPD scales exponentially with the number of conditions (Supplementary Fig. S9) which makes the algorithm prohibitively slow for higher number of conditions. Another direction of future research is to incorporate condition hierarchies or temporally related conditions, to focus on a few, instead of all, condition subsets. NIPD can also be extended to integrate data from other levels of cellular organization such as the proteome, metabolome (Bradley et al., 2009) and the epigenome (Kaplan et al., 2008).
An important feature of NIPD is that it is gene centric, allowing us to derive condition specificity of individual pairwise and higher order dependencies, which is feasible for conditions with enough samples for such a fine-grained analysis. While module-centric approaches have had much success in dissecting the regulatory program across diverse conditions, such approaches are amenable to situations where a fine-grained analysis is not possible. Thus, both approaches have their merits and the suitability of each approach is dependent upon the data at hand. Next-generation technologies are making large-scale transcript data per cell type and condition a near possibility. We expect our approach to be beneficial for interrogating these datasets and deciphering mechanisms of cell type and tissue specificity.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Manolis Kellis, Daniel Marbach and Ana Paula Leite for helpful discussions.
Funding: This work is supported by NIMH (1R01MH076282-03) and NSF (IIS-0705681) to T.L.; NIH (GM-060201, GM-67593) and NSF (MCB0734918) to M.W.W. S.R. was supported by NSF (0937060) to the Computing Research Association for the CIFellows project.
Conflict of Interest: none declared.
Footnotes
1This is not a bi-partite graph because the genes with deletion mutants are allowed to connect to each other.
REFERENCES
- Aragon A.D., et al. Characterization of differentiated quiescent and non-quiescent cells in yeast stationary-phase cultures. Mol. Biol. Cell. 2008;19:1271–1280. doi: 10.1091/mbc.E07-07-0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann S., et al. Similarities and differences in genome-wide expression data of six organisms. PLoS Biol. 2004;2:E9. doi: 10.1371/journal.pbio.0020009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besag J. Efficiency of pseudolikelihood estimation for simple gaussian fields. Biometrika. 1977;64:616–618. [Google Scholar]
- Bradley P.H., et al. Coordinated concentration changes of transcripts and metabolites in Saccharomyces cerevisiae. PLoS Comput. Biol. 2009;5:e1000270. doi: 10.1371/journal.pcbi.1000270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang H.-Y., et al. Network-based classification of breast cancer metastasis. Mol. Syst. Biol. 2007;3 doi: 10.1038/msb4100180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson G.S., et al. The proteomics of quiescent and non-quiescent cell differentiation in yeast stationary-phase cultures. Mol. Biol. Cell. 2011;22:988–998. doi: 10.1091/mbc.E10-06-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman N., et al. Using bayesian networks to analyze expression data. J. Comput. Biol. 2000;7:601–620. doi: 10.1089/106652700750050961. [DOI] [PubMed] [Google Scholar]
- Gray J.V., et al. ‘sleeping beauty’: Quiescence in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 2004;68:187–206. doi: 10.1128/MMBR.68.2.187-206.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbison C.T., et al. Transcriptional regulatory code of a eukaryotic genome. Nature. 2004;431:99–104. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastie T., et al. The Elements of Statistical Learning. New York: Springer; 2001. [Google Scholar]
- Heckerman D. A Tutorial on Learning with Bayesian Networks. In: Jordan M., editor. Learning in Graphical Models. Cambridge, MA: MIT Press; 1999. [Google Scholar]
- Kaplan N., et al. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature. 2008;458:362–366. doi: 10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H., et al. Unraveling condition specific gene transcriptional regulatory networks in saccharomyces cerevisiae. BMC Bioinformatics. 2006 doi: 10.1186/1471-2105-7-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauritzen S.L. Oxford Statistical Science Series. New York, USA: Oxford University Press; 1996. Graphical Models. [Google Scholar]
- Martinez M.J., et al. Genomic analysis of stationary-phase and exit in Saccharomyces cerevisiae: gene expression and identification of novel essential genes. Mol. Biol. Cell. 2004;15:5295–5305. doi: 10.1091/mbc.E03-11-0856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes P., et al. Artificial gene networks for objective comparison of analysis algorithms. Bioinformatics. 2003;19:122–129. doi: 10.1093/bioinformatics/btg1069. [DOI] [PubMed] [Google Scholar]
- Myers C.L., Troyanskaya O.G. Context-sensitive data integration and prediction of biological networks. Bioinformatics. 2007;23:2322–2330. doi: 10.1093/bioinformatics/btm332. [DOI] [PubMed] [Google Scholar]
- Pe'er D., et al. Inferring subnetworks from perturbed expression profiles. Bioinformatics. 2001;17(Suppl. 1):S215–S224. doi: 10.1093/bioinformatics/17.suppl_1.s215. [DOI] [PubMed] [Google Scholar]
- Pe'er D., et al. Minreg: a scalable algorithm for learning parsimonious regulatory networks in yeast and mammals. J. Mach. Learn. Res. 2006;7:167–189. [Google Scholar]
- Rhein R.O., Strimmer K. From correlation to causation networks: a simple approximate learning algorithm and its application to high-dimensional plant gene expression data. BMC Syst. Biol. 2007;1:37. doi: 10.1186/1752-0509-1-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokhlenko O., et al. Similarities and differences of gene expression in yeast stress conditions. Bioinformatics. 2007;23:e184–e190. doi: 10.1093/bioinformatics/btl308. [DOI] [PubMed] [Google Scholar]
- Roy S., et al. Proceedings of Pacific Symposium on Biocomputing. 2009. Inference of functional networks of condition-specific response–a case study of quiescence in yeast; pp. 51–62. Correct citation of Roy et al. 2009 is: Roy et al. (2009) [PubMed] [Google Scholar]
- Salgado H., et al. Regulondb (version 5.0): Escherichia coli k-12 transcriptional regulatory network, operon organization, and growth conditions. Nucleic Acids Res. 2006;34:D394. doi: 10.1093/nar/gkj156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti G., et al. Mmg: a probabilistic tool to identify submodules of metabolic pathways. Bioinformatics. 2008;24:1078–1084. doi: 10.1093/bioinformatics/btn066. [DOI] [PubMed] [Google Scholar]
- Schäfer J., Strimmer K. An empirical bayes approach to inferring large-scale gene association networks. Bioinformatics. 2005;21:754–764. doi: 10.1093/bioinformatics/bti062. [DOI] [PubMed] [Google Scholar]
- Segal E., et al. Module networks: identifying regulatory modules and their condition-specific regulators from gene expression data. Nat. Genet. 2003;34:166–176. doi: 10.1038/ng1165. [DOI] [PubMed] [Google Scholar]
- Segal E., et al. Learning module networks. J. Mach. Learn. Res. 2005;6:557–588. [Google Scholar]
- Stuart J.M., et al. A gene-coexpression network for global discovery of conserved genetic modules. Science. 2003;302:249–255. doi: 10.1126/science.1087447. [DOI] [PubMed] [Google Scholar]
- Tuck D.P., et al. Characterizing disease states from topological properties of transcriptional regulatory networks. BMC Bioinformatics. 2006;7 doi: 10.1186/1471-2105-7-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werhli A.V., et al. Comparative evaluation of reverse engineering gene regulatory networks with relevance networks, graphical gaussian models and Bayesian networks. Bioinformatics. 2006;22:2523–2531. doi: 10.1093/bioinformatics/btl391. [DOI] [PubMed] [Google Scholar]
- Yu J., et al. Advances to Bayesian network inference for generating causal networks from observational biological data. Bioinformatics. 2004;20:3594–3603. doi: 10.1093/bioinformatics/bth448. [DOI] [PubMed] [Google Scholar]
- Zhang B., et al. Differential dependency network analysis to identify condition-specific topological changes in biological networks. Bioinformatics. 2008 doi: 10.1093/bioinformatics/btn660. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.