

Abstract
Plasmodium falciparum, the major causative agent of human malaria contains three separate genomes. The apicoplast, an intracellular organelle contains a ∼35kb circular DNA genome of unusually high A/T content (>86%) that is replicated by the nuclear encoded replication complex Pfprex. Herein, we have expressed and purified the DNA polymerase domain of Pfprex (KPom1) and measured its fidelity using a LacZ based forward mutation assay. In addition, we analyzed the kinetic parameters for the incorporation of both complementary and non-complementary nucleotides incorporation using Kpom1 lacking the 3′→5′ exonucleolytic activity. KPom1 exhibits a strongly biased mutational spectrum in which the T → C is the most frequent single-base substitution and differs significantly from the closely related E. coli DNA polymerase I (pol I). Using E. coli harboring a temperature sensitive pol I allele, we established that KPom1 can complement the growth defective phenotype at an elevated temperature. We propose that the error bias of KPom1 may be exploited in the complementation assay to identify nucleoside analogs that mimic this base-mispairing and preferentially inhibit apicoplast DNA replication.
Keywords: Replication, Apicoplexan, plDNA, Pom1
Introduction
Plasmodium falciparum, the infectious agent associated with most cases of malaria, is responsible for an estimated 5 million deaths annually throughout the world 1. This infectious parasite contains a nuclear and mitochondrial genome, as well as a third, unique, genome encapsulated in an organelle termed the apicoplast. The apicoplast, thought to be derived from the secondary endocytosis of photosynthetic algae, is involved in a variety of biosynthetic pathways and is required for parasite survival. The apicoplast contains its own plastid derived, ∼35kb closed circular double-stranded DNA genome (plDNA) that is replicated by a bidirectional theta mechanism and segregated into the daughter cells 2; 3. The genome encodes several subunits of rRNA and the accompanying ribosomal proteins, 25 species of tRNA, an RNA polymerase, and several open reading frames coding for chaperones, as well as other proteins of unknown function 4.
The biochemical and cellular processes involved in plDNA replication are poorly understood; however, several proteins that are involved in DNA metabolic processes are encoded by the nuclear genome, synthesized in the cytoplasm and transported to the apicoplast. These include a bacterial-like gyrase, a DNA ligase, and several unclassified open reading frames that are homologous to DNA repair enzymes 5; 6. Of these, the apicoplast gyrase is a known target for inhibition by the drug ciprofloxin, a major therapeutic agent for the treatment of malaria, suggesting that other enzymes involved in plDNA replication and maintenance may be a useful drug target 7; 8; 9; 10.
Apicoplast DNA replication is catalyzed by the nuclear encoded apicoplast-targeted polyprotein, Pfprex, which is a large (2016aa), multifunctional, peptide that contains three distinct domains that exhibit DNA primase, DNA helicase, and DNA polymerase activities, respectively 11 (Fig. 1 A). The helicase and a primase segments are homologous to T7 bacteriophage helicase and primase proteins 11. The third domain contains a DNA polymerase that is evolutionarily related to prokaryotic DNA polymerase I, an A-family polymerase, based on sequence homology. Similar to DNA polymerase γ, which is localized to the mitochondria, Pfprex is believed to be the only DNA synthesizing enzyme in the apicoplast and is thought to be involved in DNA replication, repair, and recombination.
Figure 1.

A, The domain organization of Pfprex. B, the sequence comparison of evolutionarily conserved DNA polymerase motifs of the Plasmodium falciparum Pfprex polymerase domain, E. coli DNA polymerase I, and human Pol ν.
The three genomes present in P. falciparum (i.e. nuclear, mitochondrial, and plastidal) are among the most A/T rich yet sequenced, with the plDNA being the highest (86.9% A/T) 4. Given the highly biased sequence composition of the apicoplast genome, we asked if the plDNA polymerase preferentially incorporated dATP and/or dTTP and thus has a role in the maintenance of the A/T rich genome. To this end, we have expressed and purified the DNA polymerase domain of Pfprex (henceforth referred to as KPom1, where KPom is shorthand for Klenow-like polymerase of malaria) and determined the frequencies of mis-incorporations and the effects of neighboring nucleotides using the M13mp2 forward mutation assay for KPom1 with and without a 3′→5′ exonucleolytic activity. In addition, we also characterized the kinetics of incorporation of complementary and non-complementary nucleotide incorporation. Interestingly, we find that KPom1 exhibits a strongly biased error spectrum with the T→C single-base substitution being the most frequent and thus does not account for the maintenance of the A/T rich plastid genome. Even though the catalytic site motifs are highly conserved between E. coli Pol I and KPom1, the spectrum of mis-incorporation by KPom1is markedly different from that of E.coli Pol I. This finding suggests that residues outside of the active site motifs may influence the fidelity of these two enzymes. Despite these differences, we established that KPom1 is able to substitute for E. coli Pol I, in vivo. We suggest that the error bias of KPom1 may be exploited in the complementation assay to identify nucleoside analogs that mimic base-mispairing and preferentially inhibit plDNA replication.
Methods and Materials
Construction of Recombinant Plasmids
The pHSH576-derivative plasmids pECpolI and pECpol I-3′exo- that carry the E. coli wild-type and 3′→5′ exonuclease-deficient pol I gene, respectively, were constructed as described previously 12. The synthetic codon-optimized KPom1 or exonuclease deficient KPom1D1531A (KPom1exo-) genes (The amino acid numbering system is based on full length Pfprex) (Integrated DNA Technologies, Coralville, IA) (Supplemental Fig. S1) were cloned into the pMAL-c2X expression plasmid using the BamHI and HindIII sites. The pMAL-c2X vector encodes a maltose-binding protein moiety, which was fused to the polymerase gene to expedite purification.
Protein Expression and Purification
E. coli Rosetta2 cells (Novagen, EMD Chemicals) containing a pMAL-c2X-KPom1 plasmid were grown at 37 °C in 3 L of LB medium containing 50 μg/mL carbenicillin and 30 μg/mL chloramphenicol. Protein expression was induced by the addition of 0.2 mM IPTG at OD600 = ∼0.6. Cells were grown for an additional 20 h at 21 °C and harvested by centrifugation. Cell pellets were resuspended in 30 mL Buffer A [20 mM Tris-HCl (pH 7.4), 1 mM DTT, 1 mM EDTA, 5% (w/v) glycerol, 2 mM benzamidine, 500 μg/mL lysozyme and 1 mM PMSF]+200 mM NaCl and incubated on ice for 1 h. The crude cell extract was sonicated and clarified by centrifugation at 15,000 ×g for 20 min (Supplemental Figure 2, Lane I). The supernatant was diluted with Buffer A+200 mM NaCl to 50 mL and loaded onto an amylose column pre-equilibrated in Buffer A. The column was then washed with 100 mL of Buffer A+200 mM NaCl. KPom1 was eluted with 20 mL of Buffer A+200 mM NaCl+20 mM maltose (Supplemental Figure 2, Lane II). The fractions containing KPom1 were pooled and directly diluted with an equal volume of Buffer A without NaCl and loaded onto a heparin column pre-equilibrated in Buffer A+100 mM NaCl. The column was washed with 40 mL of the same buffer. KPom1 was eluted with 30 mL of Buffer A+1M NaCl. Column fractions (2 mL each) containing fusion MBP-KPom1 protein were pooled and dialyzed at 4 °C overnight against the Factor Xa cleavage buffer [20 mM Tris-HCl (pH 8.0), 2 mM CaCl2 and 100 mM NaCl] and then concentrated using an Amicon filter unit (MW cut-off 30,000 Da). The maltose-binding domain was cleaved by addition of Factor Xa per the manufacturer's protocol (New England Biolabs). After cleavage, the sample was diluted with four volumes of Buffer A and loaded onto an amylose column equilibrated with Buffer A. The column was then washed with 15 mL of Buffer A. The column flow-through (Supplemental Figure 2, Lane III) was directly diluted with an equal volume of Buffer A without NaCl and loaded on a heparin column pre-equilibrated in Buffer A+100 mM NaCl and washed with 20 mL of the same buffer. KPom1 was eluted with 15 mL of Buffer A+1M NaCl. Column fractions (1 mL) containing KPom1 proteins were pooled and dialyzed at 4 °C overnight against enzyme storage buffer [20 mM Tris-HCl (pH 7.4), 1 mM DTT, 0.5 mM EDTA, 50 mM NaCl, 2 mM benzamidine, 15% (w/v) glycerol] and concentrated using an Amicon filter unit (MW cut-off 30,000 Da) (Supplemental Figure 2, Lane IV). The mutant KPom1exo- protein was purified with the same protocol as the wild-type KPom1 preparation.
Two-plasmid Based β-lactamase Reversion Assay
The assay was performed as described previously 12. Briefly, the pol Its E. coli strain JS200 (SC-18 recA718 polA12 uvrA155 trpE65 lon-11 sulA1) harboring the reporter plasmid pLA230 was transformed with plasmids encoding the gene for either wild-type pol I, pol Iexo-, KPom1, or KPom1exo-. The recombinant strains were cultured at 30°C for 18 h in LB broth containing 30 mg/ml kanamycin, 12.5 mg/ml tetracycline, and 30 mg/ml chloramphenicol. A 0.01 volume of the pre-cultured broth was inoculated into fresh growth media, then cultured at 37 °C until an A600 of 1.0 was attained. Cells were then plated onto pre-warmed 2xYT agar plates supplemented with 30 μg/ml kanamycin, 12.5 μg/ml tetracycline, and 30 μg/ml chloramphenicol in the presence or absence of 100 μg/ml carbenicillin. After incubation at 37 °C for 24 h, colonies were counted, and reversion frequencies were calculated as the ratio of carbenicillin-resistant to total colonies.
Genetic Complementation Assay
The spiral assay for testing genetic complementation of by exogenously expressed E. coli pol I and KPom1 DNA polymerases was conducted as described previously using the temperature sensitive E. coli strain JS200 13 To quantitatively determine the complementation efficiency of pol Its cells by wild-type and KPom1 DNA polymerases, ∼1,000 cells of JS200 harboring either pHSG576, pEC-polI, pHSG576-KPom1, or pHSG576-KPom1D1798A, a mutant lacking polymerase activity, were plated on 2xYT agar plates containing tetracycline and chloramphenicol at 30 °C or 37 °C. The complementation efficiency of each construct was determined as the ratio of viable colonies at 37 °C on 2xYT agar plates relative to those at 30 °C plates after the 24 h incubation 12. The results shown in all figures represent the average of three experiments that were carried out independently.
Polymerase Activity Assays
The DNA polymerase activity of all purified proteins was quantified using activated calf thymus DNA, as previously described 12. KPom1 and KPom1exo- protein were assayed for 3′-5′ exonuclease activity using a duplex 5′-[32P]-labeled 27/36-mer DNA containing a 3′ terminal G:G mismatch. Reaction mixtures (10 μL) contained 10 nM of 5′-[32P]-labeled 27/36-mer DNA and 0.01 units of wild-type Klenow fragment (New England Biolabs), exonuclease-deficient Klenow fragment (New England Biolabs), 20 nM KPom1, or KPom1exo- protein in the appropriate reaction buffer. Reactions were incubated at 37 °C for 15 min, and then terminated by addition of 10 μl of 2× gel loading buffer (100% formamide, 0.03% bromophenol blue (w/v), 0.03% xylene cyanol (w/v)) and boiled at 95 °C for 5 min. 10 μL of each reaction mixture was analyzed by electrophoresis on an 8M urea, 18% -polyacrylamide gel.
M13 Forward Mutation Assay
The M13mp2 gap-filling forward mutation assay was performed as described previously 21. Briefly, the gap-filling reaction (15 μL) was carried out in polymerase reaction buffer [25 mM Tris·HCl (pH 9.0), 1 mM DTT, 10 mM MgCl2, and 50 mg/mL BSA] containing 4 fmol of purified double-stranded bacteriophage M13mp2 DNA with a 407 nucleotide single stranded gap within the lacZα-complementation target sequence, 5 pmol of purified KPom1 or KPomlexo-, and 100 μM of each dNTP. Reactions were incubated at 37 °C for 10 min and were terminated by the addition of EDTA to a concentration of 10 mM. The completion of gap-filling reactions was confirmed by agarose gel electrophoresis. Aliquots of gap-filling reactions were transformed into MC1061 cells and plated on agar plates containing X-gal (5-bromo-4-chloro-3-indolyl-b-D-galactoside), IPTG (Isopropyl β -D-1-thiogalactopyranoside), and a lawn of CSH50 E. coli host cells. After incubation of plates at 37 °C for 16 h, the number of wild-type (dark blue) and mutant (light blue and white) plaques were scored. Mutant plaques were isolated and individually grown in liquid culture, and the M13mp2 DNA was isolated and sequenced using the sequencing primer 5′-TCGGAACCACCATCAAAC-3′. Error rates were calculated as previously described 14.
Single Nucleotide Insertion Kinetics
The primer extension reaction was performed to determine single nucleotide insertion kinetics as described previously 15; 16. Briefly, the 5′-[32P]-labeled 16-mer primer : 5′-CATGAACTACAAGGAC-3′ was annealed to a 1.5-fold molar excess of the template 36-mer : 5′-GCATTCAGTXGTCCTTGTAGTTCATG-3′, where the bold ‘X’ was either A, C, T, or G. Primer extension reactions (40 μl) were carried out in polymerase reaction buffer [25 mM Tris·HCl (pH 9.0), 1 mM DTT, 10 mM MgCl2, and 50 mg/mL BSA], containing 10 nM 32P-labeled primer/template DNA, 20 nM of purified KPom1exo- and indicated concentrations of dNTPs. Reactions were incubated at 37 °C ranging for 1 to 15 min. The time of each reaction was chosen based on prior experiments that were performed to determine single completed hit conditions with less than 20% of total primer extended 16; 17. Reactions were terminated by adding 40 μL of 2× gel loading buffer [95% formamide, 15 mM EDTA, 0.05% (w/v) bromophenol blue, and 0.05% (w/v) xylene cyanol]. Samples were boiled and loaded onto an 8M urea, 15% -polyacrylamide gel for analysis by electrophoresis.
Results
KPom1 can substitute for DNA polymerase Pol I in E. coli
KPom1 is a member of the A-family of DNA polymerases. There is a high degree of conservation of amino acid sequences at the catalytic sites of DNA polymerases; sequence alignment of KPom1 with E. coli Pol I shows that all the required motifs for polymerase activity in Pol I are present in KPom1 and are highly conserved (Fig. 1B). We have previously shown that both mammalian Pol β 18 and HIV reverse transcriptase 19 can complement E. coli harboring a temperature sensitive mutation in Pol I 18; 19. To determine whether KPom1 can substitute for E. coli Pol I, KPom1 was inserted into an IPTG inducible vector and transformed into E. coli that expresses a temperature-sensitive variant of Pol I (Pol Its). At 37 °C, Pol Its is inactivated and the strain is dependent upon an exogenously supplied polymerase for colony formation. Figure 2A and 2B demonstrate that KPom1 almost fully restores wild-type growth when induced with IPTG. In the absence of IPTG, the KPom1 containing plasmid fails to form colonies at the non-permissive temperature. In addition, essentially no growth is observed in the vector only control or when cells are transformed with a copy of KPom1 harboring an active site mutation, D1798A, that inactivates the polymerase (Fig. 2A,B).
Figure 2. Functional complementation of E. coli DNA polymerase I by Plasmodium falciparum KPom1 in vivo.

(A) E. coli JS200 cells were transformed with an empty pHSG576 vector or vector harboring Pol I, KPom1 or KPom1D1798A gene. (B), The complementation efficiency of Pol Its cells by exogenously expressed Pol I, KPom1 or KPom1D1798A were quantified as described in Camps et al 12. The complementation efficiency of Pol Its cells by KPom1 or KPom1D1798A was normalized to the conditions with Pol I in the absence of IPTG, which was set as 100%. Assays were done in triplicate.
KPom1 is error prone in vivo
We took advantage of the ability of KPom1 to substitute for the endogenous Pol I activity of E. coli in order to determine its in vivo fidelity. We utilized a two-plasmid system detailed by Camps et al that uses a bacterial host that is Pol Its (Figure 3 A)12; 18. In this system, one plasmid encodes the polymerase of interest (Klenow fragment of Pol I or KPom1 and its exonuclease deficient derivatives), while the other contains the β-lactamase gene with an ochre (TAA) mutation. Any mutations that result in a reversion of the ochre codon will lead to active β-lactamase activity and carbenicillin resistance. Thus, the frequency of bacteria rendered carbenicillin resistant reflects the frequency of ochre codon mutagenesis in the target plasmid.
Figure 3. In vivo fidelity of KPom1.
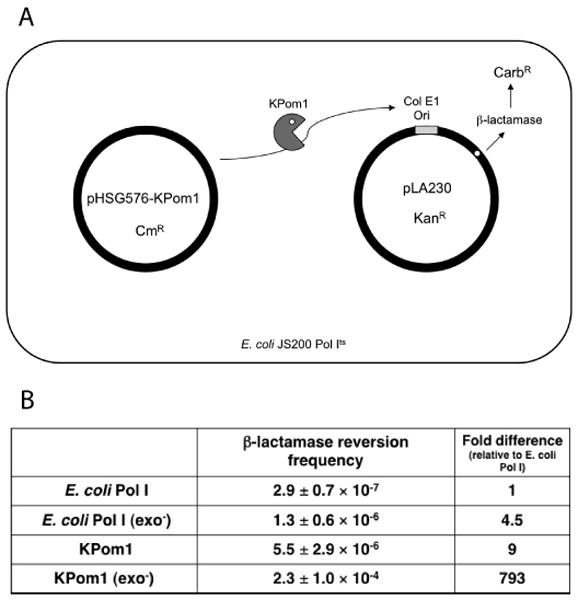
(A) Schematic diagram of the two-plasmid β-lactamase reversion assay. JS200 (Pol Its) cells were transformed with two plasmids. Plasmid pHSG576 is a low copy number plasmid and carries the polA gene under control of the tac promoter. The pLA230 plasmid carries the β-lactamase gene placed in close proximity downstream of a pUC19 (ColEl-type) origin of replication. The polymerase of interest is expressed from pHSG576 and initiates replication of pLA230. If the polymerase makes a misinsertion when copying the ochre codon, it will lead to a reversion a functional β-lactamase enzyme. (B) Reversion frequencies for the β-lactamase reversion assay. The fold-increase in the reversion frequency is relative to E. coli Pol I.
Figure 3B shows the results for the in vivo reversion of mutations in β-lactamase by Klenow, Klenowexo-, KPom1, and KPom1exo-. The reversion frequencies for Klenow and Klenowexo- are consistent with previous reports on β-lactamase reversion with this system 12. Specifically, the reversion frequency with Klenowexo- is 4.5-fold greater than that observed with exonuclease proficient Klenow and is consistent with previous reports 12; 20 In JS200 strains expressing the KPom1 and KPom1exo- enzymes, the reversion frequency is much higher than that of the corresponding controls. The KPom1 enzyme exhibits a 9-fold higher reversion frequency than the Klenow fragment of E. coli Pol I. Surprisingly, KPom1exo- exhibits a 793-fold increase in the in vivo reversion frequency relative to E. coli Pol Iwt and 41-fold higher than KPom1 (Fig. 3B) and suggests that the polymerase domain of KPom1 is very error prone and that the majority of polymerase mistakes are corrected by the exonuclease domain of the enzyme in the context of this system. Interestingly, the extremely high reversion frequency by KPom1exo- occurs in the context of the A/T rich TAA ochre codon and that the plDNA genome that this enzyme replicate is also A/T rich.
Biochemical characterization of KPom1
In order to determine if KPom1 exhibits lower fidelity for nucleotide mis-insertions opposite template dA or dT relative to template dC or dG we used the purified enzyme to measure the in vitro fidelity and incorporation kinetics for all 16 possible base-pairs. We were unable to express the apicoplast DNA polymerase in E. coli or yeast using different high expression vectors. We reasoned that this lack of high expression could be due to the disparity in the codon usage bias between P. falciparum and E. coli. Therefore, we obtained a chemically synthesized gene, optimized for E. coli and coding for the amino acids 1431-2016 of Pfprex (Supplemental Fig. S1), which corresponds to the Pol I-like DNA polymerase domain (KPom1). The expressed N-terminal maltose binding domain fusion protein was purified to near homogeneity using a heparin column followed by amylose resin (Supplemental Fig. S2). After cleavage and removal of the maltose bind peptide, the purified DNA polymerase is highly active, with a specific activity of 77 pmoles of dNMPs incorporated per minute per ng of protein for KPom1WT and 53 pmoles/min·ng for KPom1exo-. KPom1 exhibits maximal activity at ∼10mM Mg+2 (Supplemental Fig. S3A) and pH 9.0 (Supplemental Fig. S3B). Polymerase activity increases 2-fold from 18 to ∼40°C and then rapidly declines; consistent with the idea that enzyme activity has evolved for DNA replication in warm-blooded animals (Supplemental Fig. S3C).
Fidelity of DNA Synthesis by KPom1 DNA Polymerease
The fidelity of DNA synthesis by the wild-type and exonuclease-deficient KPom1 DNA polymerases was determined using the M13mp2 forward mutation assay 21. The substrate is double-stranded M13mp2 with a 407 nucleotide single-stranded gap in the LacZα gene. A purified DNA polymerase is used to fill in the single stranded segment in vitro, which is then transformed into E. coli and plated on a lawn of α-complementation cells in the presence of X-gal and IPTG. Accurate synthesis by the polymerase results in faithful replication of the LacZα gene and the formation of dark blue plaques, while polymerase errors yield light blue or colorless plaques. The fidelity of the polymerase is determined from the ratio of mutant to wild-type plagues; the error spectrum of the polymerase is determined by sequencing the resulting plaques. This assay allows for the monitoring of a broad range of mutations, including all 12 single-nucleotide misinsertion mutations and small insertion-deletion mutations, as well as short duplications, additions and rearrangements 21. With the ‘wild-type’ KPom1, only 14 phenotypic colonies (i.e. light blue or colorless plaques) were observed in the 10,769 observed colonies, resulting in a mutation frequency of 0.13%; less than 2-fold greater than the background of the assay of 0.07% 20; 21. In contrast, with KPom1exo-, 114 mutant plaques were detected in a total of 18,254 plaques, resulting in a mutation frequency of 0.63%. These mutation frequencies for both the wild-type and exonuclease deficient KPom1 are similar to those observed for other high fidelity A-family polymerases, including E. coli Pol I 20, Taq DNA polymerase 22, and human DNA polymerase γ 23. The presence of the exonuclease domain imparts at least a 6-fold increase in the fidelity, thus showing that it is able to correct most misincorporation events that the polymerase active site makes.
The error spectrum of KPom1 was determined by sequencing the observed lacZ mutants and tabulating the types and sequence contexts of the mutations. The results for both KPom1wt and KPom1exo- were used to calculate the error rates for each type of mutation (Fig. 4 and Table 1), and the error spectrum of the KPom1exo- was compared to that of other characterized polymerases (Table 2). For KPom1exo-, 39 of the 115 sequenced verified mutants were single-base insertion or deletions; the error rate was 1.2×l0-5 and 0.2×l0-5, respectively. Of the 34 detected -1 frameshift mutations found, 28 were in nucleotide repeats of two or more identical bases, which is consistent with a mechanism involving a template slippage event 24.
Figure 4. Base substitution error rates of E. coli and Plasmodium falciparum KPom1 DNA polymerase in the LacZ forward mutation assay.
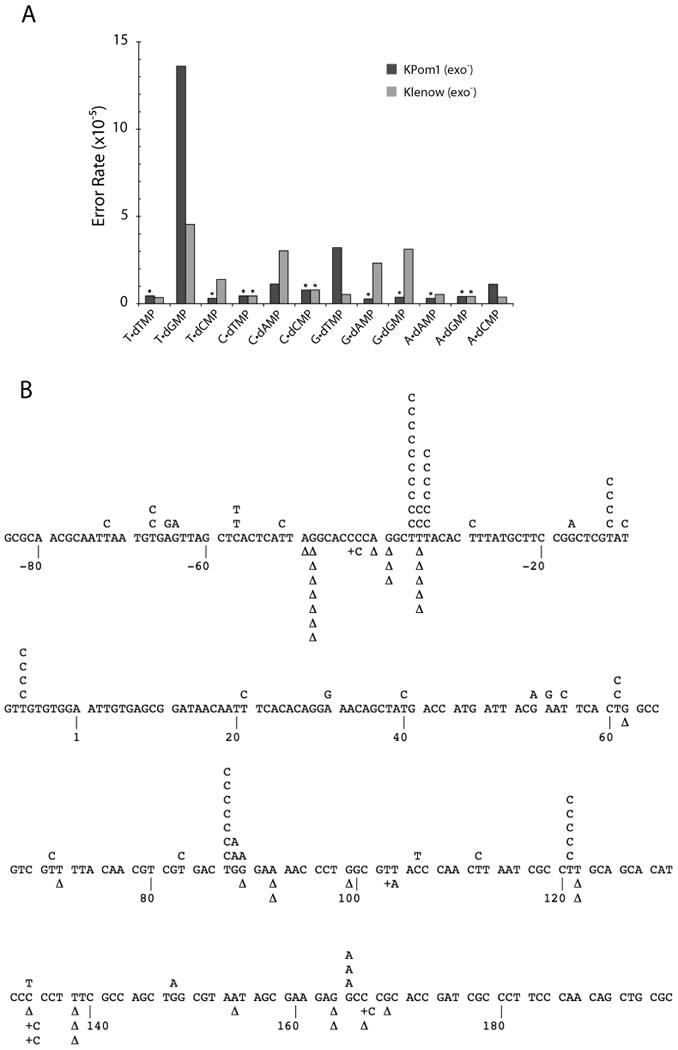
(A) Error rates for all possible base-base mispairs are shown for KPom1exo- (dark bars) and Klenowexo- (light bars). An asterisk (*) indicates less than or equal to values, due to failure to identify any mutations of a particular class. Data for Klenowexo- are from Bebenek and Kunkel 21. (B) Mutation spectrum of Kpom1exo-. The target sequence of the LacZ fragment is shown, with detected single-base substitutions indicated above the target sequence and single nucleotide deletions (Δ) and insertions (+) below.
Table 1. Mutation rates in the LacZ forward mutation assay.
| Mispair | #of Detectable Sites | Wild-Type | Exo- | ||
|---|---|---|---|---|---|
| Number Detected | Error Rate (×10-5) | Number Detected | Error Rate (×10-5) | ||
| T•dTMP | 16 | 0 | ≤0.3 | 0 | ≤0.4 |
| T•dGMP | 27 | 1 | 0.17 | 52 | 14 |
| T•dCMP | 23 | 0 | ≤0.2 | 0 | ≤0.3 |
| C•dTMP | 16 | 0 | ≤0.3 | 0 | ≤0.4 |
| C•dAMP | 25 | 2 | 0.36 | 4 | 1.1 |
| C•dCMP | 9 | 0 | ≤0.5 | 0 | ≤0.8 |
| G•dTMP | 22 | 0 | ≤0.2 | 10 | 3.2 |
| G•dAMP | 25 | 0 | ≤0.2 | 0 | ≤0.3 |
| G•dGMP | 19 | 0 | ≤0.2 | 0 | ≤0.4 |
| A•dAMP | 23 | 0 | ≤0.2 | 0 | ≤0.3 |
| A•dGMP | 17 | 0 | ≤0.3 | 0 | ≤0.4 |
| A•dCMP | 29 | 1 | 0.24 | 3 | 1.1 |
|
| |||||
| Base Substitutions | 125 | 4 | 0.14 | 69 | 3.9 |
|
| |||||
| + Insertions | 199 | 0 | ≤0.02 | 5 | 0.2 |
| -1 Deletions | 199 | 6 | 0.14 | 34 | 1.2 |
| Large Deletiona | Not Defined | 4 | 25 | 7 | 37 |
|
| |||||
| LacZ mutation frequency | 1.3×10-3 | 6.3×10-3 | |||
For large deletions, mutation frequency is reported instead of error rate.
Table 2. Comparison of mutation rates between KPom1exo- and other polymerases.
| DNA polymerase | Single-base deletion error rate (×10-5) | Single-base substitution error rate (×10-5) |
|---|---|---|
| Kpom1 (exo-)a | 1.2 | 3.9 |
| E. coli Klenow (exo-)b | 0.6 | 2.5 |
| Taq pol (exo-)c | 0.6 | 1.7 |
| hPol ν d | 17 | 350 |
| hPol θe | 140 | 240 |
| hPol γ (exo-) (+p140 & p55)f | 0.8 | 4.1 |
| hPol αg | 2.8 | 7.5 |
| hPol δ (exo-)h | 2.0 | 4.4 |
| yPol ε (exo-)i | 5.6 | 24 |
| hPol βj | 14 | 23 |
| hPo λj | 450 | 90 |
| hPol ηk | 240 | 3500 |
| hPol kl | 180 | 580 |
Error rates are from this study.
Single-base deletion are from Minnick et al, 1996 34; Single-base substitutions are from Bebenek et al., 1990 20.
Error rates are from Eckert and Kunkel, 1990 22.
Error rates are from Arana et al., 2007 25.
Error rates are from Arana et al., 2008 29.
Single-base deletions are from Longley, et al., 2001 23; Single-base substitutions are reported in Table 2 of Arana et al., 2007 25.
Error rates are from Schmitt, et al., 2009 15.
Error rates are from Shcherbakova, et al., 2003 35
Error rates are from Bebenek, et al., 2003 36
Error rates are from Matsuda, et al., 2000 37 and Matsuda, et al., 2001 38
Error rates are from Ohashi, et al., 2000 39.
For KPom1exo-, 69 of the 115 phenotypic mutants consisted of single-base substitutions, a base substitution error rate of 3.9×l0-5. Even though the M13mp2 forward mutation assay can detect all of the 12 single-base substitutions, only a subset were observed. Of the 69 sequenced single-base substitutions, the majority (52/69 or 75%) were the result of a T→C transition (i.e. T:dGMP). The frequency of this transtition mutation was 14×l0-5; three-times greater than the frequency generated by the closely related E. coli Pol Iexo- (Fig. 4, Table 1 and 2) 20. The second most common observed base-substitution (10/69 or 14%) was the G→A (i.e G:dTMP) transition, which occurs at a rate of 3.2×l0-5; six-fold higher than what is observed for E. coli Pol Iexo- 20. Both the C→T (C:dAMP) and A→G (A:dCMP) mutations occur at a rate of l.l×l0-5 and, combined, only account for 10% of the observed single base substitutions. No other base substitutions were observed. This error spectrum deviates significantly from E. coli Pol Iexo-, even though these two enzymes share significant sequence identity in their active site motifs (Fig. 1B) and have a similar overall error rate. Specifically, KPom1exo- catalyzes primarily T→C transitions, while E. coli Pol Iexo- misincorporations results predominantly in C→T transitions, and G→T and G→C transversions 20. In addition, a majority of T→C mutations (∼65%) with KPom1exo- DNA polymerase occurred in a sequence context in which the 5′-template base is either a C or a G. The mutation spectrum for KPom1exo- is most similar to that of the A-family lesion bypass polymerase, pol ν (See discussion for details).
Steady-state kinetics of KPom1
Examining the fidelity of KPom1 using steady-state kinetics gives an indication of which step in nucleotide discrimination process is rate limiting for the incorporation of specific deoxynucleoside triposphates. For example, a higher Km would suggest that nucleotide discrimination is due to an increase in substrate dissociation (increase in koff), while a lower Vmax would indicate an unfavorable geometry of the bound nucleotide in the active site 16; 20. We used a steady-state gel-based assay to determine the apparent kinetic parameters (Vmax and Km) for the incorporation of the correct or incorrect nucleotide across for all 16 possible base pairings. These values were then used to calculate the fidelity of KPom1exo- 16.
KPom1exo- efficiently incorporated dAMP, dCMP, dTMP, or dGMP across from their complementary template bases and discriminated against the incorrect nucleotide for all possible nucleotide mismatches (Table 3). The calculated values for the fidelity of nucleotide misinsertion range from 4.6×l0-3 (T:dGMP) to 2.6×l0-5 (A:dCMP) (Table 3). Values for some mismatches could not be calculated due to the lack of observable incorporation. The most prevalent mis-insertions observed in the kinetic assay are the same as the mispairs with the lowest observed fidelity in the M13mp2 forward mutation assay (Table 1). The apparent values for Km ranged from a 325- to 3000-fold increase for incorrect vs correct incorporation, but did not vary substantially for the correct incorporation reactions. The mispairs that were the least frequent in the M13mp2 forward mutation assay also exhibited a relatively high values for Km (>1000μM), whereas base mispairs with the lowest fidelity tended to have much lower values of Km. Specifically, the values for Km were the highest when the identity of the template base was dG or dC or when base-pairing involved an incoming nucleotide with the same identity as the template base. Conversely, when the template base was dA or dT, the values of Km were on the order of 2-3 fold lower than values observed for dG or dC. This observation suggests that KPom1 uses a mechanism whereby nucleotide discrimination for certain base:base mispairs (namely template dG, template dC, and pyr:pyr/pur:pur) involves poor binding (i.e. increased off-rate).
Table 3. Fidelity of single nucleotide insertion by KPom1exo-.
| Base pair | Km(μM) | Vmax(fmol/min) | Vmax/Kma | Fidelityb |
|---|---|---|---|---|
| T•dAMP | 1.6±0.7 | 12.5±0.7 | 7.8 | 1 |
| T•dTMP | N.D.c | N.D.c | - | - |
| T•dGMP | 521±54 | 18.8±0.6 | 0.0361 | 4.61×l0-3 |
| T•dCMP | 648±350 | 0.91±0.13 | 0.0014 | 1.80×l0-4 |
|
| ||||
| C•dGMP | 0.36±0.09 | 8.2±0.4 | 22.8 | 1 |
| C•dTMP | 1300±450 | 0.81±0.09 | .0006 | 2.74×10-5 |
| C•dAMP | N.D.c | N.D.c | - | - |
| C•dCMP | N.D.c | N.D.c | - | - |
|
| ||||
| G•dCMP | 0.72±0.09 | 10.4±0.3 | 14.4 | 1 |
| G•dTMP | 290±120 | 3.2±0.6 | 0.0110 | 7.64×10-4 |
| G•dAMP | 1450±650 | 3.9±0.7 | 0.0027 | 1.86×l0-4 |
| G•dGMP | 2200±620 | 3.0±0.4 | 0.0014 | 9.44×10-5 |
|
| ||||
| A•dTMP | 0.70±0.15 | 11.4±0.6 | 16.30 | 1 |
| A•dAMP | 1500±260 | 1.6±0.2 | 0.0011 | 6.55×l0-5 |
| A•dGMP | 264±117 | 2.9±0.2 | 0.0110 | 6.74×10-4 |
| A•dCMP | 885±228 | 0.92±0.15 | 0.0010 | 6.38×l0-5 |
Units are reported in fmol/μM•min
Fidelity is defined as (Vmax/Km)wrong/(Vmax/Km)right
The reaction was too slow to be determined
In contrast to the values of Km, which exhibit an increase in magnitude for all mismatches relative to the correct base pair, the values for the apparent Vmax ranged from a 13.7-fold decrease to a 1.5-fold increase for incorrect vs correct incorporation. The apparent Vmax for the correct incorporation reactions were very similar to one another, regardless of the template base identity, while the Vmax for most mismatch reactions varied in a range of about 4-fold regardless of the identity of the template base. A notable exception is the T:dGMP mispair, which, surprisingly, exhibits a higher Vmax than the correct base-pair.
Taken together, it appears that, depending on the template base identity, KPom1 uses two different strategies for nucleotide discrimination. When the identity of the template base is dG or dC, the polymerase primarily discriminates between the correct vs incorrect nucleotide based on reduced occupancy of the active site (i.e. the rate of dissociation is much faster than the rate of incorporation). This is most likely due to the inability of these mispairs to form stable hydrogen bonds between the template and the substrate. This is consistent with the idea that fidelity for template dG and dC is mostly governed by substrate dissociation. However, this observation is not the case for the G:dTMP mispair, which exhibits a reduced Km while the value of Vmax for this mismatch is unchanged relative to the other mispairs. This observation suggests that for this base mismatch, the fidelity is increasingly governed by the rate of product formation.
Interestingly, when the template base identity is dA or dT, the values of Km tend to be much lower than that of dG or dC and only vary by less than two-fold, while the Vmax exhibits a greater than 20-fold variation (Table 3). This observation is consistent with the fidelity of mismatches involving template dA or dT being governed mostly by the base-pair geometry in the polymerase active site. As mentioned previously, the T:dGMP base-pair has a higher Vmax than the correct base-pair (Table 3) and indicates that the Vmax for this mismatch does not contribute favorably to the fidelity and that the increased fidelity is primarily due to substrate dissociation.
Discussion
Pfprex is a multi-functional fusion protein targeted to the apicoplast. Even though definitive proof is lacking, it is believed to be responsible for the replication of the apicoplast genome using a DNA Pol I-like domain. In addition, it may also function in apicoplast DNA repair and other DNA synthetic functions. Thus, its presence in the apicoplast is somewhat reminiscent of mammalian DNA polymerase γ, which is also nuclearly encoded and has been shown to carry out a variety of DNA synthetic functions in mitochondria. Because the apicoplast is a unique and required component of the malarial parasite, it could serve as an important target for specifically preventing or treating infections by the malaria parasite. To this end, we have expressed and purified, and present fidelity and kinetic studies of KPom1, the plastid targeted replicative DNA polymerase domain from the malarial parasite P. falciparum. Our results show that the polymerase domain of the apicoplast genome replication enzyme replicates the genome with high fidelity and has an overall fidelity similar to other fidelity A-family polymerases. The high level of fidelity stems from efficient discrimination of mismatch nucleotides at the active site, as well as the presence of a 3′→5′ exonuclease domain involved in proofreading, which is efficient at removing the vast majority of mutations caused by mis-insertion at the active site.
Characterization of the exonuclease deficient form of KPom1 reveals the ‘spectrum’ of errors introduced by the polymerase. The frequencies of insertions and deletions (indels) of only a few nucleotides in length are similar to the evolutionarily related E.coli DNA Pol Iexo-, as well as other high fidelity A-family polymerases 20; 23. The finding that these indels occur at nucleotide repeats suggests that these mutations may be the result of a strand slippage event 24. In addition to indels, single-base substitutions at specific template positions may also be caused by a strand slippage. The M13mp2 forward mutation assay show that a majority of the observed T→C mutations occur after a template dG or a dC. Slippage events could lead to a transient template misalignment with the template dG or dC remaining in the catalytic site followed by incorporation of the ‘correct’ nucleotide (i.e. dCMP and dGMP, respectively). Such a mechanism has been suggested to occur for human pol γ 24
One of the most intriguing aspects of the KPom1 polymerase is the error spectrum for single base substitutions. The most frequent mutations observed with both Klenowexo- and KPom1exo- are T→C transitions; however, the error frequency for T→C transitions is more than three-fold greater during copying with KPom1. The next most frequently scored mutation for KPom1 is the G→A, while with E. coli DNA Pol Iexo-, C→T and G→C mutations are the next most frequent base-substitutions 20. The active site motifs in the polymerase sites for KPom1 and E. coli DNA Pol I are nearly identical, as is the overall accuracy in DNA synthesis; therefore, in order to account for the differences in the error spectrum, it is likely that distant amino acids have a profound effect on the types of mis-incorporations catalyzed at the polymerase site.
Of the DNA polymerases that have been analyzed using the M13mp2 forward mutation assay, the error spectrum of KPom1exo- most closely resembles that of human pol ν, an error-prone lesion bypass polymerase that lacks a proof-reading exonuclease activity 25. While the frequency of misincorporation by human pol ν is greater than KPom1 exo- (Table 2), pol ν makes primarily G→A (G:dTMP) mutations, followed by the T→C (T:dGMP) substitutions 25. In contrast, for KPom1, the primary mutations are T→C mutations followed by G→A substitutions. When considering the flanking sequence immediately 5′ to a mutation, it is interesting to note that G→A mutations by pol ν are primarily preceded by a template dA or dT, while the T→C mutation of KPom1 is primarily preceded by a template dG or dC. These observations suggest that the interaction of a polymerase with the template DNA must also influence the incorporation of specific incorrect nucleotides.
The apicoplast genome base-pair composition is >86% A/T 4 yet, the most frequent mutations produced by KPom1 are T→C transitions, both in the presence or absence of exonuclease activity. This finding would suggest that a genome with a higher G/C content would evolve over time, thus leading to an apicoplast genome with reduced A/T content. One possible explanation as to why a G/C bias is not observed may involve biased nucleoside pools within the parasite. In vivo studies have previously shown that the levels of adenosine and thiamine nucleosides are several times higher in the malarial parasite than are cytosine and guanine 26 and studies have shown that, on a genomic level, there is a distinct trend in the mutational bias towards high A/T content in bacterial obligate endosymbiotes and parasites 27; 28 This enzyme may have evolved to lower the Km value for dGTP and dCTP due to their reduced availability in the pool. This idea is consistent with our observations that mispairs involving dGTP and dCTP tend to have a lower Km. Therefore, the skewed mutational bias exhibited by KPom1 may be the result of a selective advantage that keeps the A/T content of the genome from becoming too skewed and reducing organismal fitness. An alternative possibility as to why a G/C bias is not observed is that the misincorporation bias at the level of the DNA polymerase could be compensated for by a mismatch repair process that preferentially removes dGMP mismatches in the newly replicated DNA strand. However, we are unaware of evidence for a mismatch repair system that functions in apicoplast DNA.
The unique error signature of KPom1 is a product of its specific structural features that govern fidelity. This begs the question as to why KPom1 has an error spectrum similar to human pol ν, even though the amino acid sequence is more closely related to E. coli pol I? A comparison of all six conserved polymerase motifs of KPom1, E.coli Pol I, and human pol ν show that there are no amino acids that are conserved between KPom1 and pol ν that are not also conserved in E.coli Pol I (Fig. 1B). This suggests that these motifs are not the only ones that govern the substrate selection by DNA polymerases and that other structural features or amino acids in the enzyme are also likely to affect substrate selection. Indeed, several multi-amino acid insertions flanking these conserved motifs in human pol ν and pol θ, another A-family bypass polymerase, have been implicated in its reduced fidelity, which suggests that the determinants of fidelity are not strictly confined to the conserved polymerase motifs 29; 30; 31.
Finally, our finding that KPom1 complements the in vivo activity of Pol I in E. coli will allows us to exploit genetic complementary for both structure-function studies and drug screening. Genetic complementation has been shown to facilitate the screening of large libraries of DNA polymerase mutants with random nucleotides at designated positions. An analysis of mutants of KPom1 will be important in identifying amino residues that govern nucleotide selection and that are account for the unique substrate specificity. These mutants can then be compared to other A-family polymerases, as described previously 32; 33.
The ability to substitute KPom1 for Pol I brings about the feasibility of using E. coli as a vehicle for evaluating the potency of DNA polymerase inhibitors in a high throughput manner. Such a system has been used to sensitize E. coli to the antiviral compound AZT by complementing HIV-RT 19. The uniquely biased error spectrum of the KPom1 polymerase suggests that nucleoside analogs could be developed that exploits this bias as a method of terminating apicoplast DNA synthesis. Since the T→C error rate of KPom1 is much higher than that of human pol δ or pol ε, the in replicative DNA polymerases in the nucleus, the design of specific nucleoside analogs that take this fact into account will more specifically target the parasitic polymerase instead of the replicative polymerases, thus minimizing the incorporation into the host genome.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health R01CA115202, R01CA102029, and P01AG033061. We thank Eddie Fox and Sharath Balakrishna for their useful comments and discussion.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Snow RW, Guerra CA, Noor AM, Myint HY, Hay SI. The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature. 2005;434:214–217. doi: 10.1038/nature03342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.He HY, Shaw MK, Pletcher CH, Striepen B, Tilney LG, Roos DS. A plastid segregation defect in the protozoan parasite Toxoplasma gondii. EMBO J. 2001;20:330–339. doi: 10.1093/emboj/20.3.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stanway RR, Witt T, Zobiak B, Aepfelbacher M, Heussler VT. GFP-targeting allows visualization of the apicoplast throughout the life cycle of live malaria parasites. Biol Cell. 2009;101:415–430. doi: 10.1042/BC20080202. [DOI] [PubMed] [Google Scholar]
- 4.Wilson RJM, Denny PW, Preiser PR, Rangachari K, Roberts K, Roy A, Whyte A, Strath M, Moore DJ, Moore PW, Williamson DH. Complete Gene Map of the Plastid-like DNA of the Malaria Parasite Plasmodium falciparum. J Mol Biol. 1996;261:155–172. doi: 10.1006/jmbi.1996.0449. [DOI] [PubMed] [Google Scholar]
- 5.Dahl EL, Rosenthal PJ. Apicoplast translation, transcription and genome replication: targets for antimalarial antibiotics. Trends Parasitol. 2008;24:243–284. doi: 10.1016/j.pt.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 6.Kumar A, Tanveer A, Biswas S, Ram EVSR, Gupta A, Kumar B, Habib S. Nuclear-encoded DnaJ homologue of Plasmodium falciparum interacts with replication ori of the apicoplast genome. Mol Microbiol. 2010;75:942–956. doi: 10.1111/j.1365-2958.2009.07033.x. [DOI] [PubMed] [Google Scholar]
- 7.Ram EVSR, Kumar A, Biswas S, Kumar A, Chaubey S, Siddiqi MI, Habib S. Nuclear gyrB encodes a functional subunit of the Plasmodium falciparum gyrase that is involved in apicoplast DNA replication. Mol Biochem Parasit. 2007;154:30–39. doi: 10.1016/j.molbiopara.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 8.Dahl EL, Rosenthal PJ. Multiple antibiotics exert delayed effects against the Plasmodium falciparum apicoplast. Antimicrob Agents Chemother. 2007;51:3485–3490. doi: 10.1128/AAC.00527-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dahl EL, Shock JL, Shenai BR, Gut J, DeRisi JL, Rosenthal PJ. Tetracyclines specifically target the apicoplast of the malaria parasite Plasmodium falciparum. Antimicrob Agents Chemother. 2006;50:3124–3131. doi: 10.1128/AAC.00394-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goodman CD, Su V, McFadden GI. The effects of anti-bacterials on the malaria parasite Plasmodium falciparum. Mol Biochem Parasit. 2007;152:181–191. doi: 10.1016/j.molbiopara.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 11.Seow F, Sato S, Janssen CS, Riehle MO, Mukhopadhyay A, Phillips RS, Wilson RJM, Barrett MP. The plastidic DNA replication enzyme complex of Plasmodium falciparum. Mol Biochem Parasit. 2005;141:145–153. doi: 10.1016/j.molbiopara.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 12.Camps M, Naukkarinen J, Johnson BP, Loeb LA. Targeted gene evolution in Escherichia coli using a highly error-prone DNA Polymerase I. Proc Natl Acad Sci USA. 2003;100:9727–9732. doi: 10.1073/pnas.1333928100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shinkai A, Loeb LA. In vivo mutagenesis by Escherichia coli DNA polymerase I Ile709 in motif A functions in base selection. J Biol Chem. 2001;276:46759–46764. doi: 10.1074/jbc.M104780200. [DOI] [PubMed] [Google Scholar]
- 14.Glick E, Anderson JP, Loeb LA. In vitro production and screening of DNA polymerase η mutants for catalytic diversity. Biotechniques. 2002;33:1136–1144. doi: 10.2144/02335dd08. [DOI] [PubMed] [Google Scholar]
- 15.Schmitt MW, Matsumoto Y, Loeb LA. High fidelity and lesion bypass capability of human DNA polymerase δ. Biochimie. 2009;91:1163–1172. doi: 10.1016/j.biochi.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boosalis MS, Petruska J, Goodman MF. DNA Polymerase Insertion Fidelity - Gel assay for site-specific kinetics. J Biol Chem. 1987;262:14689–14696. [PubMed] [Google Scholar]
- 17.Creighton S, Bloom LB, Goodman MF. Gel fidelity assay measuring nucleotide misinsertion, exonucleolytic proofreading, and lesion bypass efficiencies. Methods Enzymol. 1995;262:232–256. doi: 10.1016/0076-6879(95)62021-4. [DOI] [PubMed] [Google Scholar]
- 18.Sweasy JB, Loeb LA. Mammalian DNA polymerase β can substitute for DNA polymerase I during DNA replication in Escherichia coli. J Biol Chem. 1992;267:1407–1410. [PubMed] [Google Scholar]
- 19.Kim B, Loeb LA. Human immunodeficiency virus reverse transcriptase substitutes for DNA polymerase I in Escherichia coli. Proc Natl Acad Sci USA. 1995;92:684–688. doi: 10.1073/pnas.92.3.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bebenek K, Joyce CM, Fitzgerald MP, Kunkel TA. The fidelity of DNA synthesis catalyzed by derivatives of Escherichia coli DNA polymerase I. J Biol Chem. 1990;265:13878–13887. [PubMed] [Google Scholar]
- 21.Bebenek K, Kunkel T. Analyzing fidelity of DNA polymerases. Meth Enzymol. 1995;262:217–232. doi: 10.1016/0076-6879(95)62020-6. [DOI] [PubMed] [Google Scholar]
- 22.Eckert KA, Kunkel TA. High fidelity DNA synthesis by the Thermus aquaticus DNA polymerase. Nuc Acids Res. 1990;18:3739–3752. doi: 10.1093/nar/18.13.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Longley MJ, Nguyen D, Kunkel TA, Copeland WC. The fidelity of human DNA polymerase γ with and without exonucleolytic proofreading and the p55 accessory subunit. J Biol Chem. 2001;276:38555–38562. doi: 10.1074/jbc.M105230200. [DOI] [PubMed] [Google Scholar]
- 24.Bebenek K, Kunkel TA. Streisinger revisited: DNA synthesis errors mediated by substrate misalignments. Cold Spring Harbor Symp Quant Biol. 2000;65:81–92. doi: 10.1101/sqb.2000.65.81. [DOI] [PubMed] [Google Scholar]
- 25.Arana ME, Takata Ki, Garcia-Diaz M, Wood RD, Kunkel TA. A unique error signature for human DNA polymerase ν. DNA Repair. 2007;6:213–223. doi: 10.1016/j.dnarep.2006.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dyke Kv, Trush MA, Wilson ME, Stealey PK. Isolation and analysis of nucleotides from erythrocyte-free malarial parasites (Plasmodium berghei) and potential relevance to malaria chemotherapy. B World Health Organ. 1977;55:253–264. [PMC free article] [PubMed] [Google Scholar]
- 27.Hershberg R, Petrov DM. Evidence that mutation is universally biased towards AT in bacteria. PLOS Genet. 2010;6:e1001115. doi: 10.1371/journal.pgen.1001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mann S, Chen YPP. Bacterial genomic G + C composition-eliciting environmental adaptation. Genomics. 2010;95:7–15. doi: 10.1016/j.ygeno.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 29.Arana ME, Seki M, Wood RD, Rogozin IB, Kunkel TA. Low-fidelity DNA synthesis by human DNA polymerase theta. Nuc Acid Res. 2008;36:3847–3856. doi: 10.1093/nar/gkn310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takata Ki, Arana ME, Seki M, Kunkel TA, Wood RD. Evolutionary conservation of residues in vertebrate DNA polymerase N conferring low fidelity and bypass activity. Nuc Acid Res. 2010;38:3233–3244. doi: 10.1093/nar/gkq048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hogg M, Seki M, Wood RD, Doubliè S, Wallace SS. Lesion bypass activity of DNA polymerase θ (POLQ) is an intrinsic property of the pol domain and depends on unique sequence inserts. J Mol Biol. 2011;405:642–652. doi: 10.1016/j.jmb.2010.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loh E, Choe J, Loeb LA. Highly tolerated amino acid substitutions increase the fidelity of Escherichia coli DNA polymerase I. J Biol Chem. 2007;282:1201–12209. doi: 10.1074/jbc.M611294200. [DOI] [PubMed] [Google Scholar]
- 33.Patel PH, Loeb LA. DNA polymerase active site is highly mutable: Evolutionary consequences. Proc Nat Acad Sci USA. 2000;97:5095–5100. doi: 10.1073/pnas.97.10.5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.