

Abstract
A regiospecific silver-mediated fluorination of aryl silanes is reported. The reaction is operationally simple, and employs Ag2O as readily available, inexpensive silver source, which can be recovered.
Keywords: Silver oxide, Fluorine, Aryl silane, Selectfluor
1. Introduction
Fluorinated molecules, especially aryl fluorides, are used as pharmaceuticals, agrochemicals, materials, and tracers for positron emission tomography (PET).1 Although significant research has focused on the synthesis of functionalized aryl fluorides, they are still more challenging to prepare than the heavier halogen homologs.2 Conventional arene fluorination reactions, such as nucleophilic aromatic substitution, electrophilic fluorination using fluorine gas, and the pyrolysis of diazonium tetrafluoroborates can afford simple aryl fluorides.3 The electrophilic fluorination of aryl Grignard reagents has been significantly improved over the years but is still limited by the lower functional group tolerance of Grignard reagents compared to other less basic organometallics such as aryl boron and aryl silane reagents.4 Transition-metal-mediated and -catalyzed arene fluorination reactions have been developed over the past decade.5 Most of these fluorination reactions required the presence of an ortho directing group or the use of stoichiometric amounts of transition metal complexes. In 2009, Buchwald developed an elegant palladium-catalyzed aromatic fluorination reaction of aryl triflates with AgF or CsF.6 Although mixtures of constitutional iosmers were formed in some cases, this reaction was a major breakthrough in transition-metal-catalyzed nucleophilic aromatic fluorination.
Based on the hypothesis that transition metal complexes can afford aryl fluorides by reductive elimination from high-valent aryltransition metal fluorides, we have previously identified a silver-catalyzed fluorination reaction that can afford aryl fluorides in up to 92% yield from the corresponding aryl stannanes.7 In our opinion, this silver-catalyzed aromatic fluorination reaction exhibits the broadest demonstrated functional group tolerance of all fluorination reactions reported to date. However, aryl stannanes are toxic, and in some cases, the formation of hydrodestannylated byproducts was observed in up to 10% yield. We also developed a Ag-mediated fluorination reaction of arylboronic acid derivatives, which are less toxic than stannanes and do not afford sideproducts resulting from hydrodeborylation.8 Boronic acid fluorination is functional-group-tolerant and benefits from the ready availability of boronic acid derivatives. However, the incompatibility of the fluorinating reagent F-TEDA-BF4 (1) with the reaction conditions used for transmetalation from arylboronic acids required a one-pot-two-step process. After evaporation of the solvent methanol used for transmetalation, acetone was used as solvent for fluorination. The two step process lowered the overall practicality of the reaction. To improve the practicality of arene fluorination, we sought to identify aryl nucleophiles that efficiently transmetalate to Ag(I), and do not participate in unproductive background reactions with electrophilic fluorination reagents such as F-TEDA-BF4 (1). In this manuscript we describe a practical one-step process, in which aryltrialkoxysilanes and F-TEDA-BF4 (1) react in the presence of Ag2O to give functionalized aryl fluorides.
Aryl silane derivatives have been used in catalysis,9 for example in the Hiyama reaction––the Pd- or Ni-catalyzed cross-coupling reaction of organosilanes with organohalides or -triflates.10 Silver(I) oxide (Ag2O) has been used by Hiyama and others as an activator for aryl silanols that undergo Pd-catalyzed biaryl formation.11 Transmetalation from aryl silanes to palladium is believed to be accelerated by silicophilic additives such as fluoride, which can generate nucleophilic pentacoordinated silicates.10a,12 Aryl silanes possess low toxicity, are inexpensive, and are stable to a variety of reaction conditions; most of the silanes shown in this study are stable toward chromatography on silica gel. Several methods are available to make arylsilanes: Transmetalation from aryl Grignard reagents to tetraalkoxysilanes provides aryl silanes.13 For molecules that contain functional groups that are not compatible with Grignard reagents, Pd(0)- or Rh(I)-catalyzed silylation of aryl iodides and bromides with tetraethoxysilanes can afford aryl silanes.14
2. Results and discussion
The direct electrophilic fluorination reaction of arylsilanes with electrophilic fluorinating reagents such as 1 is not general, synthetically useful reaction; for example 4-(biphenyl)triethoxysilane (2) afforded 4-biphenyl fluoride (3) in less than 4% yield upon treatment with 1 in various solvents in the presence and absence of fluoride and hydroxide, respectively (see Supporting Information). However, upon the addition of Ag(I) salts, regioselective fluorination was observed (Table 1). Ag(I) oxide was identified as the silver salt that resulted in the highest yield of aryl fluoride. Ag2O is an attractive reagent because it is easy to handle, straightforward to remove by filtration, and less expensive ($1/g) than other silver salts and all platinum group metal salts.
Table 1.
Evaluation of silver(I) source
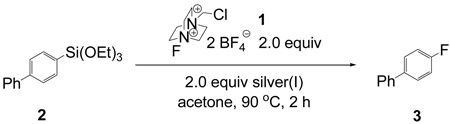 | |||
|---|---|---|---|
| Silver salt | Yield [%]a | Silver salt | Yield [%]a |
| Ag2O | 69 | AgOCN | 0 |
| AgOAc | 12 | AgSCN | 0 |
| AgBF4 | 11 | AgOTf | 6 |
| AgClO4 | 5 | AgPF6 | 10 |
| AgNO3 | 0 | AgSbF6 | 0 |
| Ag2CO3 | 5 | AgF | 21 |
| AgCN | 0 | None | 0 |
Yield was determined by intergration of the 19F NMR using 1-fluoro-3-nitrobenzene as an internal standard.
Evaluation of different silanes showed that aryl trihydroxy-and aryl trialkoxysilanes afforded the highest yield of fluorination (79%–81%, Table 2). Aryl dimethylsilanols and aryl dimethylsilanolates yielded 31% and 22% of aryl fluoride, respectively, while aryl trimethylsilanes gave no fluorination product. F-TEDA-BF4 (1) uniquely afforded fluorination; no fluorination was observed with other commercially available fluorination reagents. Acetone as solvent afforded the highest yield, while acetonitrile, DMF, THF, dioxane, benzene, and CH2Cl2 did not afford any fluorination product. Although the reaction yield is highly dependent on solvent, addition of one equivalent of water did not result in lower yields. The reaction can be carried out under ambient atmosphere. Fluorination in acetone requires heating; a yield of 65% was observed when the acetone suspension was heated at reflux. The yield could be increased to 81% when the temperature was raised to 90 °C in a sealed vessel.
Table 2.
Evaluation of organosilicon reagents
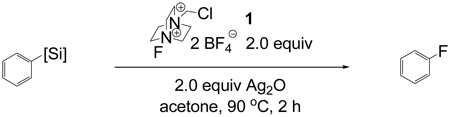 | |
|---|---|
| Organosilicon reagents [Si] | Yield[%]a |
| SiMe3 | 0 |
| SiMe2OH | 31 |
| SiMe2ONa | 22 |
| Si(OEt)3 | 81 |
| Si(OMe)3 | 80 |
| Si(OH)3 | 79 |
Yield was determined by intergration of the 19F NMR using 1-fluoro-3-nitrobenzene as an internal standard.
During our initial optimization of reaction conditions (Table 1), we observed 10% of hydrodesilylated starting material (biphenyl). Hydrodesilylation does not only reduce the yield of the desired product, but also complicates product purification because fluoroarenes and their corresponding hydrocarbons typically exhibit similar physical properties, such as boiling point and Rf value. During the development of the silver-mediated fluorination of aryltrialkyltin compounds we learned that trialkyltin triflate was involved in hydrodestannalylation.6 Although we did not observe potentially Lewis acidic triethoxysilyl derivatives during or after the reactions shown in Table 2, we hypothesized that the silicon product after transmetalation to silver was responsible for byproduct formation. We evaluated different bases that could potentially sequester silicon-based Lewis acids formed during the reaction to reduce byproduct formation and increase the yield of fluorinated product. Addition of one equivalent of BaO, MgO, and 2,6-lutidine increased the yield by up to 10 % (Table 3). Importantly, biphenyl formation was decreased to less than 5% when BaO was employed. Additional BaO beyond one equivalent reduced the yield, presumably due to unproductive reaction of BaO with the fluorinating reagent. In a control experiment, 50% F-TEDA-BF4 had reacted when treated with one equivalent of BaO after two hours at 90 °C in acetone.
Table 3.
Effect of basic additives on fluorination yield
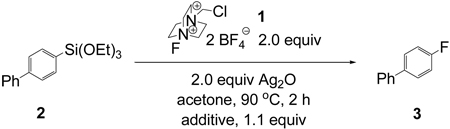 | |
|---|---|
| Additive | Yield [%]a |
| None | 75 |
| BaO | 85 |
| MgO | 82 |
| 2,6-lutidine | 81 |
| Ba(OH)2 | 35 |
| NaOH | 30 |
| K3PO4 | 34 |
| TBAF | 79 |
Yield was determined by intergration of the 19F NMR using 1-fluoro-3-nitrobenzene as an internal standard.
After reaction optimization, we investigated the substrate scope of aryl triethoxysilane fluorination with F-TEDA-BF4, mediated by Ag2O (Table 4). Fluorination proceeded for electron-rich, electron-poor, and ortho, ortho-disubstituted arenes and heteroarenes. Electron-rich arenes afforded about 10% lower yield than electron-deficient and –neutral arenes. For example, p-fluoroanisole (15) was obtained in 76% yield, while p-trifluoromethylfluorobenzene (4) and fluorobenzene (7) were obtained in 90% yield. For some electron-rich arenes we identified secondary fluorination products. For example, 5% of 2,4-difluoroanisole was observed during fluorination to form 15. Fortunately, the formation of secondary fluorination products could be circumvented by addition of one equivalent of 2,6-lutidine to the reaction mixture. The beneficial effect of BaO to reduce the formation of hydrodesilylated material was general for electron-neutral and electron-poor arenes, but less pronounced for electron-rich arenes (see Supporting Information).
Table 4.
Electrophilic fluorination of aryl silanes
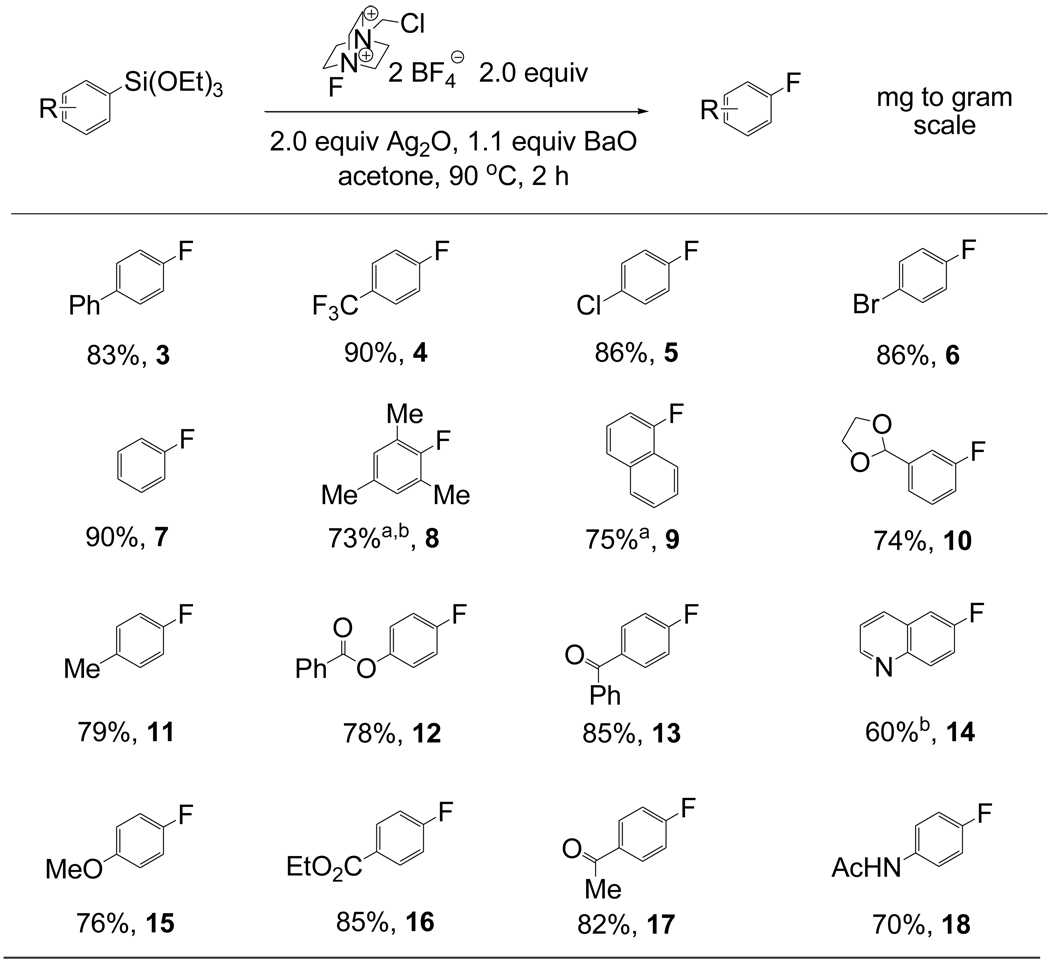 |
2,6-lutidine was used instead of BaO
3.0 equiv Ag2O was used.
Highest yields for fluorination were observed when two equivalents of Ag2O were employed. While Ag2O is one of the least expensive silver sources and less than half the price of F-TEDA-BF4, we want to point out that our procedure requires four equivalents of silver with respect to silane for optimal results. Excess Ag2O and all silver-containing products were conveniently removed by filtration. Ag2O could be regenerated by dissolving the solid silver salts in dilute nitric acid and subsequent addition of NaOH solution to precipitate Ag2O in 80% yield. The precipitated Ag2O was recycled and afforded fluorination in comparable yield (80% vs 83%, Equation 1).
 |
(1) |
The silver-mediated fluorination reaction presented herein has a larger substrate scope than traditional fluorination reactions such as nucleophilic aromatic substitution and nucleophilic fluorination using fluorine gas.3 Compared to other functional-group-tolerant fluorination reactions, the advantage of the presented fluorination is the straightforward one-step procedure. Aryl silane, Ag2O, and F-TEDA-BF4 can be combined together in reagent grade acetone under ambient conditions and afford product upon heating. Silver salts at the end of the reaction can be filtered off and recycled.
The mechanism of fluorination is currently unknown. We have previously speculated7 that redox chemistry at a multinuclear silver complex with metal-metal redox interaction15 plays an important role in efficient C–F bond formation by reductive elimination.
3. Conclusion
We have developed a functional group tolerant, one-step fluorination of aryltriethoxysilanes with F-TEDA-BF4 mediated by Ag2O. Although the silver salt is used in superstoichoimetric amounts for optimal results, it can be recovered after the reaction. A silver-catalyzed fluorination of aryl silanes would be a useful advance but has so far not been developed.
4. Experimental section
4.1. General
Reactions were carried out under ambient atmosphere unless otherwise specified. Solvents were dried by passage through alumina.16 Except as indicated otherwise, reactions were monitored by thin layer chromatography (TLC) using EMD TLC plates pre-coated with 250 µm thickness silica gel 60 F254 plates and visualized by fluorescence quenching under UV light. Flash chromatography was performed on Whatman Silica Gel 60 µm particle size using a forced flow of eluant at 0.3–0.5 bar pressure.17 NMR spectra were recorded on either a Varian Unity/Inova 500 spectrometer operating at 500 MHz and 125 MHz for 1H and 13C acquisitions, respectively, or a Varian Mercury 400 spectrometer operating at 400 HMz and 375 MHz for 1H and 19F acquisitions, respectively. Chemical shifts are reported in ppm with the solvent resonance as the internal standard. Data is reported as follows: s = singlet, d = doublet, t = triplet, q = quartet, h = heptet, m = multiplet, br = broad; coupling constants in Hz; integration. High-resolution mass spectra were obtained on Jeol AX-505 or SX-102 spectrometers at the Harvard University Mass Spectrometry Facilities. Triethylamine was distilled over calcium hydride. Silver oxide was purchased from Strem. Acetone (CHROMASOLV® Plus, for HPLC, ≥ 99.9%), 1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate), tetraethyl orthosilicate, triethoxysilane, and bis(acetonitrile)(1,5-cyclooctadiene)rhodium(I) tetrafluoroborate were purchased from Aldrich and used as received. Commercially available aryl silanes (phenyltriethoxysilane, p-tolyltriethoxysilane, 4-chlorophenyltriethoxysilane, p-methoxyphenyltriethoxysilane, 4-trifluomethylphenyltriethoxysilane, ethyl 4-triethoxysilylbenzoate, 2-(3-triethoxylsilylphenyl)-1,3-dioxolane, and 1-naphthyltriethoxylsilane) were purified by distillation prior to use. NMR spectroscopic data of known compounds correspond to the data given in the appropriate references. NMR spectra of new compounds are provided in the Supporting Information.18
4.2. Synthesis of aryl silanes
4.2.1. (4-Biphenyl)triethoxysilane(2)19
To tetraethyl orthosilicate (6.70 mL, 30.0 mmol, 3.00 equiv) in 20 mL of THF at −30 °C was added biphenylmagnesium bromide solution (0.50 M in THF, 20 mL, 10 mmol, 1.0 equiv) dropwise over 10 min. After stirring at −30 °C for 1 h, the reaction mixture was warmed to 23 °C and was stirred for 12 h. The reaction mixture was poured onto 100 mL of pentane, the phases were separated and the organic phase was washed three times with water (3 × 20 mL), and dried over Na2SO4. After filtration, the solvent was removed under reduced pressure. Bulb-to-bulb distillation (125 °C, 0.5 Torr) afforded 2.52 g of the title compound as a colorless oil (80% yield). Rf = 0.50 (hexanes). NMR Spectroscopy: 1H NMR (500 MHz, CDCl3, 23 °C, δ): 7.75 (d, J = 8.0 Hz, 2H), 7.62–7.60 (m, 4H), 7.45 (d, J = 7.5 Hz, 2H), 7.36 (t, J = 7.5 Hz, 1H), 3.90 (q, J = 7.0 Hz, 6H), 1.27 (t, J = 7.0 Hz, 9H). 13C NMR (125 MHz, CDCl3, 23 °C, δ): 142.96, 140.95, 135.28, 129.59, 128.76, 127.50, 127.17, 126.56, 58.76, 18.23.
4.2.2. (4-Bromophenyl)triethoxysilane20
[Rh(cod)(MeCN)2]BF4 (22.0 mg, 0.0600 mmol, 0.0300 equiv) and 1-bromo-4-iodobenzene (563 mg, 2.00 mmol, 1.00 equiv) were charged in a 20 mL vial capped with a rubber septum. The vial was evacuated and backfilled with nitrogen. To this vial, DMF (8 mL), triethylamine (0.830 mL, 6.00 mmol, 3.00 equiv) and triethoxysilane (0.730 mL, 4.00 mmol, 2.00 equiv) were added. The reaction mixture was stirred at 80 °C for 2 h, then cooled to 23 °C. The mixture was diluted with ether (100 mL) and washed three times with water (3 × 40 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by Kugelrohr distillation to give 508 mg of the title compound as a colorless oil (80% yield). Rf = 0.63 (hexanes). NMR Spectroscopy: 1H NMR (500 MHz, CDCl3, 23 °C, δ): 7.53–7.52 (m, 4H), 3.85 (q, J = 7.0 Hz, 6H), 1.24 (t, J = 7.0 Hz, 9H). 13C NMR (125 MHz, CDCl3, 23 °C, δ):136.34, 131.04, 129.86, 125.33, 58.77, 18.16.
4.2.3. (2, 4, 6-Trimethylphenyl)triethoxysilane21
To tetraethyl orthosilicate (3.30 mL, 15.0 mmol, 3.00 equiv) in 10 mL of THF at −30 °C was added 2,4,6-trimethylphenylmagnesium bromide solution (1.0 M in THF, 5.0 mL, 5.0 mmol, 1.0 equiv) dropwise over 10 min. After stirring at −30 °C for 1 h, the reaction mixture was warmed to 23 °C and was further stirred for 12 h. The reaction mixture was poured onto 100 mL of pentane, and was washed three times with water (3 × 20 mL) and dried over Na2SO4. After filtration, the solvent was removed under reduced pressure. Bulb-to-bulb distillation (125 °C, 0.5 Torr) afforded 0.87 g of the title compound as a colorless oil (62% yield). Rf = 0.14 (hexanes). NMR Spectroscopy: 1H NMR (500 MHz, CDCl3, 23 °C, δ): 6.80 (s, 2H), 3.83 (q, J = 7.0 Hz, 6H), 2.51 (s, 6H), 2.26 (s, 3H), 1.24 (t, J = 7.0 Hz, 9 H). 13C NMR (125 MHz, CDCl3, 23 °C, δ): 145.92, 139.79, 128.81, 124.99, 58.10, 23.73, 21.08, 18.15.
4.2.4. 4-(Triethoxysilyl)phenylbenzoate
[Rh(cod)Cl]2 (15.0 mg, 0.0300 mmol, 0.0300 equiv) and 4-iodophenyl benzoate (323 mg, 1.00 mmol, 1.00 equiv) were charged in 10 mL vial capped with a rubber septum. The vial was evacuated and backfilled with nitrogen. To this vial, DMF (4 mL), triethylamine (0.420 mL, 3.00 mmol, 3.00 equiv) and triethoxysilane (0.360 mL, 2.00 mmol, 2.00 equiv) were added. The reaction mixture was stirred at 80 °C for 2 h, then cooled to 23 °C. The mixture was diluted with ether (50 mL) and washed three times with water (3 × 20 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by Kugelrohr distillation to give 252 mg of the title compound as a colorless oil (70% yield). Rf = 0.30 (hexanes/EtOAc 1:1 (v/v)). NMR Spectroscopy: 1H NMR (500 MHz, CDCl3, 23 °C, δ): 8.24–8.21 (m, 2H), 7.78–7.75 (m, 2H), 7.68–7.64 (m, 1H), 7.56–7.52 (m, 2H), 7.28–7.25 (m, 2H), 3.90 (q, J = 7.0 Hz, 6H), 1.27 (t, J = 7.0 Hz, 9H). 13C NMR (125 MHz, CDCl3, 23 °C, δ): 164.93, 152.82, 136.25, 133.63, 130.19, 129.52, 128.63, 128.58, 121.22, 58.78, 18.22. Mass Spectrometry: HRMS-FIA (m/z): Calcd for [M + Na]+, 378.17313. Found, 378.17314.
4.2.5. 6-(Quinolinyl)triethoxysilane
[Rh(cod)Cl]2 (15.0 mg, 0.0300 mmol, 0.0300 equiv), 6-(quinolinyl)trifluoromethanesulfonate (307 mg, 1.00 mmol, 1.00 equiv) and tetra-n-butylammonium iodide (369 mg, 1.00 mmol, 1.00 equiv) were charged in 10 mL vial capped with a rubber septum. The vial was evacuated and backfilled with nitrogen. To this vial, DMF (4 mL), triethylamine (0.420 mL, 3.00 mmol, 3.00 equiv) and triethoxysilane (0.360 mL, 2.00 mmol, 2.00 equiv) were added. The reaction mixture was stirred at 80 °C for 2 h, then cooled to 23 °C. The mixture was diluted with ether (50 mL) and washed three times with water (3 × 20 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by Kugelrohr distillation to give 218 mg of the title compound as a colorless oil (75% yield). Rf = 0.50 (hexanes/EtOAc 3:1 (v/v)). NMR Spectroscopy: 1H NMR (500 MHz, CDCl3, 23 °C, δ): 8.94 (dd, J = 4.0, J = 1.5 Hz, 1H), 8.19–8.18 (m, 2H), 8.10 (d, J = 8.5 Hz, 1H), 7.95 (d, J = 8.5 Hz, 1H), 7.41 (dd, J = 8.5, 4.5 Hz, 1 H), 3.92 (q, J = 7.0 Hz, 6H), 1.27 (t, J = 7.0 Hz, 9H). 13C NMR (125 MHz, CDCl3, 23 °C, δ): 151.26, 149.18, 136.46, 136.18, 134.11, 129.87, 128.67, 127.69, 121.21, 58.91, 18.23.
4.2.6. 4-(Triethoxysilyl)acetophenone22
[Rh(cod)(MeCN)2]BF4 (11.0 mg, 0.0300 mmol, 0.0300 equiv) and 4-iodoacetonphenone (246 mg, 1.00 mmol, 1.00 equiv) were charged in 10 mL vial capped with a rubber septum. The vial was evacuated and backfilled with nitrogen. To this vial, DMF (4 mL), triethylamine (0.420 mL, 3.00 mmol, 3.00 equiv) and triethoxysilane (0.360 mL, 2.00 mmol, 2.00 equiv) were added. The reaction mixture was stirred at 80 °C for 2 h, then cooled to 23 °C. The mixture was diluted with ether (50 mL) and washed three times with water (3 × 20 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by Kugelrohr distillation to give 197 mg of the title compound as a colorless oil (70% yield). Rf = 0.56 (hexanes). NMR Spectroscopy: 1H NMR (500 MHz, CDCl3, 23 °C, δ): 7.93 (dd, J = 6.5, J = 1.5 Hz, 2H), 7.78 (dd, J = 6.5, J = 1.5 Hz, 2H), 3.88 (q, J = 7.0 Hz, 6H), 2.61 (s, 3H), 1.25 (t, J = 7.0 Hz, 9H). 13C NMR (125 MHz, CDCl3, 23 °C, δ): 198.37, 138.33, 137.32, 135.02, 127.28, 58.88, 26.68, 18.19.
4.2.7. 4-(Triethoxysilyl)acetanilide23
[Rh(cod)(MeCN)2]BF4 (11.0 mg, 0.0300 mmol, 0.0300 equiv) and 4-iodoacetanilide (260 mg, 1.00 mmol, 1.00 equiv) were charged in 10 mL vial capped with a rubber septum. The vial was evacuated and backfilled with nitrogen. To this vial, DMF (4 mL), triethylamine (0.420 mL, 3.00 mmol, 3.00 equiv) and triethoxysilane (0.360 mL, 2.00 mmol, 2.00 equiv) were added. The reaction mixture was stirred at 80 °C for 2 h, then cooled to 23 °C. The mixture was diluted with ether (50 mL) and washed three times with water (3 × 20 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by Kugelrohr distillation to give 238 mg of the title compound as a colorless oil (80% yield). Rf = 0.25 (hexanes/EtOAc 1:1 (v/v)). NMR Spectroscopy: 1H NMR (500 MHz, CDCl3, 23 °C, δ): 7.69 (d, J = 8.0 Hz, 2H), 7.59 (d, J = 8.5 Hz, 2H), 7.42 (br s, 1H), 3.85 (q, J = 7.0 Hz, 6H), 2.17 (s, 3H), 1.23 (t, J = 7.0 Hz, 9H). 13C NMR (125 MHz, CDCl3, 23 °C, δ): 168.39, 139.79, 135.77, 126.28, 118.89, 58.69, 24.66, 18.18.
4.3. Synthesis of aryl fluorides
4.3.1. General Procedure A (for volatile compounds)
To aryl silane (0.100 mmol, 1.00 equiv) in acetone (2.0 mL) at 23 °C was added silver oxide (46.4 mg, 0.200 mmol, 2.00 equiv), barium oxide (15.6 mg, 0.100 mmol, 1.00 equiv) and 1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate) (1) (70.8 mg, 0.200 mmol, 2.00 equiv). The reaction mixture was stirred for 2 h at 90 °C in a sealed vial, then cooled to 23 °C. To the reaction mixture was added 3-nitrofluorobenzene (10.0 µL, 0.0939 mmol). The yields were determined by comparing the integration of the 19F NMR (375 MHz, acetone-d6, 23 °C) resonance of an aryl fluoride and that of 3-nitrofluorobenzene (–112.0 ppm).
4.3.2. General Procedure B (for non-volatile compounds)
To aryl silane (0.100 mmol, 1.00 equiv) in acetone (2.0 mL) at 23 °C was added silver oxide (46.4 mg, 0.200 mmol, 2.00 equiv), barium oxide (17.2 mg, 0.110 mmol, 1.10 equiv) or 2,6-lutidine (12.8 µL, 0.110 mmol, 1.10 equiv) and 1-chloromethyl-4-fluoro-1,4-diazoniabicyclo [2.2.2]octane bis(tetrafluoroborate) (1) (70.8 mg, 0.200 mmol, 2.00 equiv). The reaction mixture was stirred for 2 h at 90 °C in a sealed vial. The reaction mixture was cooled to 23 °C and concentrated under reduced pressure. To the residue was added CH2Cl2 and the mixture was filtered through a pad of Celite eluting with CH2Cl2. The filtrate was concentrated under reduced pressure and the residue was purified by chromatography on silica gel or preparative TLC.
4.3.3. General Procedure C (for heterocyclic compounds)
To aryl silane (0.100 mmol, 1.00 equiv) in acetone (2.0 mL) at 23 °C was added silver oxide (69.6 mg, 0.300 mmol, 3.00 equiv), barium oxide (17.2 mg, 0.110 mmol, 1.10 equiv) and 1-chloromethyl-4-fluoro-1,4-diazoniabicyclo-[2.2.2]octane bis(tetrafluoroborate) (1) (70.8 mg, 0.200 mmol, 2.00 equiv). The reaction mixture was stirred for 2 h at 90 °C in a sealed vial. The reaction mixture was cooled to 23 °C, passed through a pad of Celite and concentrated under reduced pressure. To the residue was added CH2Cl2 (20 mL) and a saturated aqueous solution of NaHCO3 (20 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified on preparative TLC.
4.3.4. 4-Fluorobiphenyl (3)
Yield: 14.3 mg (83%). Rf = 0.60 (hexanes/EtOAc 19:1 (v/v)). NMR Spectroscopy: 1H NMR (500 MHz, CDCl3, 23 °C, δ): 7.60–7.54 (m, 4H), 7.47 (dd, J = 7.5 Hz, 7.0 Hz, 2H), 7.36 (t, J = 7.5 Hz, 1H), 7.14 (dd, J = 8.0 Hz, 7.5 Hz, 2H). 13C NMR (125 MHz, CDCl3, 23 °C, δ): 162.44 (d, J = 244 Hz), 140.25, 137.30, 128.80, 128.75 (d, J = 8.5 Hz), 127.24, 127.00, 115.59 (d, J = 21 Hz). 19F NMR (375 MHz, CDCl3, 23 °C, δ): –117.2. These spectroscopic data correspond to previously reported data.5c
4.3.5. 1-Fluoronaphthalene (9)
Yield: 10.9 mg (75%). Rf = 0.40 (hexane/EtOAc 3:1 (v/v)). NMR Spectroscopy: 1H NMR (500 MHz, CDCl3, 23 °C, δ): 8.13–8.11 (m, 1H), 7.88–7.86 (m, 1H), 7.63 (d, J = 8.5 Hz, 1H), 7.56–7.53 (m, 1H), 7.43–7.38 (m, 1H), 7.17–7.13 (m, 1H). 13C NMR (125 MHz, CDCl3, 23 °C, δ): 158.78 (d, J = 250 Hz), 134.87 (d, J = 4.5 Hz), 127.50 (d, J = 3.6 Hz), 126.80, 126.15 (d, J = 1.9 Hz), 125.58 (d, J = 9.1 Hz), 123.76, 123.62 (d, J = 3.6 Hz), 120.53 (d, J = 5.5 Hz), 109.39 (d, J = 20 Hz). 19F NMR (375 MHz, CDCl3, 23 °C, δ): –125.6. These spectroscopic data correspond to previously reported data.24
4.3.6. 4-Fluorophenyl benzoate (12)
Yield: 16.9 mg (78%). Rf = 0.20 (hexane/EtOAc 3:1 (v/v)). NMR Spectroscopy: 1H NMR (500 MHz, CDCl3, 23 °C, δ): 8.21–8.18 (m, 2H), 7.66–7.63 (m, 1H), 7.54–7.51 (m, 2H), 7.20–7.17 (m, 2H), 7.13–7.09 (m, 2H). 13C NMR (125 MHz, CDCl3, 23 °C, δ): 165.19, 160.30 (d, J = 242 Hz), 146.75 (d, J = 2.8 Hz), 133.71, 130.17, 129.29, 128.61, 123.10 (d, J = 9.0 Hz), 116.14 (d, J = 24 Hz). 19F NMR (375 MHz, CDCl3, 23 °C, δ): –119.2. These spectroscopic data correspond to previously reported data.25
4.3.7. 4-Fluorobenzophenone (13)
Yield: 17.0 mg (85%). Rf = 0.50 (hexane/EtOAc 3:1 (v/v)). NMR Spectroscopy: 1H NMR (500 MHz, CDCl3, 23 °C, δ): 7.86–7.84 (m, 2H), 7.78–7.76 (m, 2H), 7.61–7.58 (m, 1H), 7.51–7.48 (m, 2H), 7.18–7.15 (m, 2H). 13C NMR (125 MHz, CDCl3, 23 °C, δ): 195.27, 165.39 (d, J = 252 Hz), 137.51, 133.79, 132.66 (d, J = 9.1 Hz), 132.45, 129.87, 128.35, 115.45 (d, J = 22 Hz). 19F NMR (375 MHz, CDCl3, 23 °C, δ):–108.7. These spectroscopic data correspond to previously reported data.26
4.3.8. 6-Fluoroquinoline (14)
Yield: 8.8 mg (60%). Rf = 0.47 (EtOAc). NMR Spectroscopy: 1H NMR (500 MHz, CDCl3, 23 °C, δ): 8.91 (dd, J = 4.5 Hz, 1.5 Hz, 1H), 8.18 (d, J = 8.0 Hz, 1H), 8.15 (dd, J = 9.0 Hz, J = 5.5 Hz, 1H), 7.53 (ddd, J = 9.0 Hz, 8.5 Hz, 2.0 Hz, 1H), 7.50–7.45 (m, 2H). 13C NMR (125 MHz, CDCl3, 23 °C, δ): 160.43 (d, J = 247 Hz), 149.56, 145.11, 135.70 (d, J = 5.3 Hz), 131.80 (d, J = 9.1 Hz), 128.86, 121.79, 119.94 (d, J = 26 Hz), 110.74 (d, J = 21 Hz). 19F NMR (375 MHz, CDCl3, 23 °C, δ):–113.0. These spectroscopic data correspond to previously reported data.27
4.3.9. Ethyl 4-fluorobenzoate (16)
Yield: 14.3 mg (85%). Rf = 0.30 (hexane/EtOAc 3:1 (v/v)). NMR Spectroscopy: 1H NMR (500 MHz, CDCl3, 23 °C, δ): 8.06 (dd, J = 9.0 Hz, J = 5.5 Hz, 2H), 7.10 (dd, J = 9.0 Hz, J = 8.5 Hz, 2H), 4.37 (q, J = 7.0 Hz, 2H), 1.39 (t, J = 9.0 Hz, 3H). 13C NMR (125 MHz, CDCl3, 23 °C, δ): 165.68 (d, J = 252 Hz), 165.65, 132.04 (d, J = 10 Hz), 126.72, 115.42 (d, J = 22 Hz), 61.07, 14.30. 19F NMR (375 MHz, CDCl3, 23 °C, δ):–108.4. These spectroscopic data correspond to previously reported data.28
4.3.10. 4-Fluoroacetophenone (17)
Yield: 11.3 mg (82%). Rf = 0.30 (hexane/EtOAc 3:1 (v/v)). NMR Spectroscopy: 1H NMR (500 MHz, CDCl3, 23 °C, δ): 7.99–7.96 (m, 2H), 7.14–7.11 (m, 2H), 2.58 (s, 3H). 13C NMR (125 MHz, CDCl3, 23 °C, δ): 196.68, 165.99 (d, J = 253 Hz), 133.84, 131.16 (d, J = 9.1 Hz), 115.88 (d, J = 22 Hz), 26.75. 19F NMR (375 MHz, CDCl3, 23 °C, δ):–108.4. These spectroscopic data correspond to previously reported data.29
4.3.11. 4-Fluoroacetanilide (18)
Yield: 10.7 mg (70%). Rf = 0.30 (hexane/EtOAc 3:1 (v/v)). NMR Spectroscopy: 1H NMR (500 MHz, CDCl3, 23 °C, δ): 7.99–7.96 (m, 2H), 7.14–7.11 (m, 2H), 2.58 (s, 3H). 13C NMR (125 MHz, CDCl3, 23 °C, δ): 168.43, 159.35 (d, J = 242 Hz), 133.83, 121.81 (d, J = 7.3 Hz), 115.56 (d, J = 23 Hz), 24.32. 19F NMR (375 MHz, CDCl3, 23 °C, δ):–121.4. These spectroscopic data correspond to previously reported data.8
4.4. 5-mmol-Scale fluorination of 4-(biphenyl)triethoxysilane
To 4-(biphenyl)triethoxylsilane (2) (1.58 g, 5.00 mmol, 1.00 equiv) in acetone (100 mL) at 23 °C was added silver oxide (2.32 g, 10.0 mmol, 2.00 equiv), barium oxide (0.780 g, 5.00 mmol, 1.10 equiv) and 1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]-octane bis(trifluoroborate) (1) (3.54 g, 10.0 mmol, 2.00 equiv). The reaction mixture was stirred at 90 °C for 2 h in a 350 mL sealed vessel. The reaction mixture was cooled to 23 °C and concentrated under reduced pressure. To the residue was added CH2Cl2 and the mixture was filtered through a pad of Celite eluting with CH2Cl2. The filtrate is concentrated under reduced pressure and the residue is purified by chromatography on silica gel eluting with hexane, to afford 714 mg of the title compound as a white solid (83% yield).
4.5. 1-mmol-Scale fluorination of 4-(triethoxysilyl)benzophenone
To 4-(triethoxysilyl)benzophenone (344 mg, 1.00 mmol, 1.00 equiv) in acetone (20 mL) at 23 °C was added silver oxide (464 mg, 2.00 mmol, 2.00 equiv), barium oxide (168 mg, 1.10 mmol, 1.10 equiv) and 1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]-octane bis(trifluoroborate) (1) (706 mg, 2.00 mmol, 2.00 equiv). The reaction mixture was stirred at 90 °C for 2 h in a sealed vessel. The reaction mixture was cooled to 23 °C and concentrated under reduced pressure. To the residue was added CH2Cl2 and the mixture was filtered through a pad of Celite eluting with CH2Cl2. The filtrate is concentrated under reduced pressure and the residue is purified by chromatography on silica gel eluting with hexanes/EtOAc 10:1 (v/v), to afford 160 mg of the title compound as a white solid (80% yield).
4.6. Regeneration of Ag2O
To 4-(biphenyl)triethoxylsilane (2) (1.58 g, 5.00 mmol, 1.00 equiv) in acetone (100 mL) at 23 °C was added silver oxide (2.32 g, 10.0 mmol, 2.00 equiv), barium oxide (0.780 g, 5.00 mmol, 1.10 equiv) and 1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]-octane bis(trifluoroborate) (1) (3.54 g, 10.0 mmol, 2.00 equiv). The reaction mixture was stirred at 90 °C for 2 h in a 350 mL sealed vessel. The reaction mixture was cooled to 23 °C and concentrated under reduced pressure. The residue was washed with CH2Cl2 (3 × 20 mL) and the solid was dissolved in 250 mL HNO3 (10%, v/v in H2O). After stirring for 30 min at 23 °C, the reaction mixture was filtered. To the filtrate was added NaOH (10%, v/v in H2O, 250 mL). The suspention was filtered and the solid residue washed with water (3 × 20 mL) to afford 1.86 g Ag2O (80%) as a brown powder.
Supplementary Material
Acknowledgments
We thank the NSF (CHE-0952753) and the NIH-NIGMS (GM088237) for financial support. We thank Air Products and Chemicals, Inc. for a generous donation of F-TEDA-BF4.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
Supplementary data includes detailed experimental procedures and spectroscopic data for all new compounds. Supplementary data related to this article can be found online at doi: xxxx.
References and notes
- 1.(a) Jeschke P. ChemBioChem. 2004;5:570–589. doi: 10.1002/cbic.200300833. [DOI] [PubMed] [Google Scholar]; (b) Müller K, Faeh C, Diederich F. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]; (c) Ametamey SM, Honer M, Schubiger PA. Chem. Rev. 2008;108:1501–1516. doi: 10.1021/cr0782426. [DOI] [PubMed] [Google Scholar]; (d) Shimizu M, Hiyama T. Angew. Chem. Int. Ed. 2005;44:214–231. doi: 10.1002/anie.200460441. [DOI] [PubMed] [Google Scholar]
- 2.(a) Chambers RD. Fluorine in organic chemistry. New York: Oxford; 2004. [Google Scholar]; (b) Furuya T, Kuttruff CA, Ritter T. Curr. Opin. Drug. Disc. Dev. 2008;11:803–819. [PubMed] [Google Scholar]; (c) Furuya T, Klein JEMN, Ritter T. Synthesis. 2010;11:1804–1821. doi: 10.1055/s-0029-1218742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Balz G, Schiemann G. Ber. Deutsch. Chem. Ges. 1927;60:1186–1190. [Google Scholar]; (b) Adams DJ, Clark JH. Chem. Soc. Rev. 1999;28:225–231. [Google Scholar]; (c) Sun H, Dimagno SG. Angew. Chem. Int. Ed. 2006;45:2720–2725. doi: 10.1002/anie.200504555. [DOI] [PubMed] [Google Scholar]; (d) Sandford G. J. Fluorine Chem. 2007;128:90–104. [Google Scholar]
- 4.(a) Barnette WE. J. Am. Chem. Soc. 1984;106:452–454. [Google Scholar]; (b) Differding E, Wehrli M. Tetrahedron Lett. 1991;32:3819–3822. [Google Scholar]; (c) Lal GS. J. Org. Chem. 1993;58:2791–2796. [Google Scholar]; (d) Davis FA, Han W, Murphy CK. J. Org, Chem. 1995;60:4730–4737. [Google Scholar]; (e) Silverman GS, Rakita PE. Handbook of Grignard Reagents. New York: Marcel Dekker; 1996. [Google Scholar]; (f) Yamada S, Gavryushin A, Knochel P. Angew. Chem. Int. Ed. 2010;49:2215–2218. doi: 10.1002/anie.200905052. [DOI] [PubMed] [Google Scholar]; (g) Anbarasan P, Neumann H, Beller M. Angew. Chem. Int. Ed. 2010;49:2219–2222. doi: 10.1002/anie.200905855. [DOI] [PubMed] [Google Scholar]
- 5.(a) Tredwell M, Gouverneur V. Org. Biomol. Chem. 2006;4:26–32. doi: 10.1039/b513399h. [DOI] [PubMed] [Google Scholar]; (b) Hull KL, Anani WQ, Sanford MS. J. Am. Chem. Soc. 2006;128:7134–7135. doi: 10.1021/ja061943k. [DOI] [PubMed] [Google Scholar]; (c) Furuya T, Kaiser HM, Ritter T. Angew. Chem. Int. Ed. 2008;47:5993–5996. doi: 10.1002/anie.200802164. [DOI] [PubMed] [Google Scholar]; (d) Furuya T, Ritter T. J. Am. Chem. Soc. 2008;130:10060–10061. doi: 10.1021/ja803187x. [DOI] [PubMed] [Google Scholar]; (e) Kaspi AW, Yahav-Levi A, Goldberg I, Vigalok A. Inorg. Chem. 2008;47:5–7. doi: 10.1021/ic701722f. [DOI] [PubMed] [Google Scholar]; (f) Furuya T, Strom AE, Ritter T. J. Am. Chem. Soc. 2009;131:1662–1663. doi: 10.1021/ja8086664. [DOI] [PubMed] [Google Scholar]; (g) Ball ND, Sanford MS. J. Am. Chem. Soc. 2009;131:3796–3797. doi: 10.1021/ja8054595. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Wang X, Mei T-S, Yu JQ. J. Am. Chem. Soc. 2009;131:7520–7521. doi: 10.1021/ja901352k. [DOI] [PubMed] [Google Scholar]; (i) Furuya T, Benitez D, Tkatchouk E, Strom AE, Tang P, Goddard WA, III, Ritter T. J. Am. Chem. Soc. 2010;132:3793–3807. doi: 10.1021/ja909371t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watson DA, Su M, Teverovskiy G, Zhang Y, García-Fortanet J, Kinzel T, Buchwald SL. Science. 2009;325:1661–1664. doi: 10.1126/science.1178239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang P, Furuya T, Ritter T. J. Am. Chem. Soc. 2010;132:12150–12154. doi: 10.1021/ja105834t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Furuya T, Ritter T. Org. Lett. 2009;11:2860–2863. doi: 10.1021/ol901113t. [DOI] [PubMed] [Google Scholar]
- 9.(a) Hiyama T. In: Metal-Catalyzed Cross-Coupling Reactions. Diederich F, Stang PJ, editors. Chapter 4. Weinheim: Wiley-Vch; 1998. [Google Scholar]; (b) Denmark SE, Sweis RF. In: Metal-Catalyzed Cross-Coupling Reactions. de Meijere A, Diederich F, editors. Chapter 4. Weinheim: Wiley-Vch; 2004. [Google Scholar]; (c) Brook MA. Silicon in Organic, Organometallic, and Polymer Chemistry. New York: John Wiley & Sons; 2000. [Google Scholar]; (d) Denmark SE, Sweis RF. Acc. Chem. Res. 2002;35:835–846. doi: 10.1021/ar020001r. [DOI] [PubMed] [Google Scholar]; (e) Denmark SE, Butler CR. Chem. Commun. 2009:20–33. doi: 10.1039/b809676g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Hiyama T, Hatanaka Y. Pure & Apple. Chem. 1994;66:1471–1478. [Google Scholar]; (b) Hiyama T. J. Organomet. Chem. 2002;653:58–61. [Google Scholar]
- 11.(a) Hirabayashi E, Mori A, Kawashima J, Suguro M, Nishihara Y, Hiyama T. J. Org. Chem. 2000;65:5342–5349. doi: 10.1021/jo000679p. [DOI] [PubMed] [Google Scholar]; (b) Malm J, Björk P, Gronowitz S, Hörnfeldt AB. Tetrahedron Letter. 1992;33:2199–2202. [Google Scholar]; (c) Uenishi J, Beau JM, Armstrong RW, Kishi YJ. J. Am. Chem. Soc. 1987;109:4756–4758. [Google Scholar]; (d) Gillmann T, Weeber T. Synlett. 1994:649–650. [Google Scholar]; (e) Mateo C, Fernández-Rivas C, Echavarren AM, Cárdenas DJ. Organometallics. 1997;16:1997–1999. [Google Scholar]
- 12.For efficient cross-coupling reactions of silanolates with aromatic halides under mild conditions, see: Denmark SE, Smith RC, Chang WT, Muhuhi JM. J. Am. Chem. Soc. 2009;131:3104–3118. doi: 10.1021/ja8091449.
- 13.(a) Lee ASY, Chang YT, Chu SF, Tsao KW. Tetrahedron Letter. 2006;47:7085–7087. [Google Scholar]; (b) Manoso AS, Ahn C, Soheili A, Handy CJ, Correia R, Seganish WM, DeShong P. J. Org. Chem. 2004;69:8305–8314. doi: 10.1021/jo048667h. [DOI] [PubMed] [Google Scholar]
- 14.(a) Murata M, Suzuki K, Watanabe S, Masuda Y. J. Org. Chem. 1997;62:8569–8571. doi: 10.1021/jo971143f. [DOI] [PubMed] [Google Scholar]; (b) Murata M, Ishikura M, Nagata M, Watanabe S, Masuda Y. Org. Lett. 2002;4:1843–1845. doi: 10.1021/ol025770p. [DOI] [PubMed] [Google Scholar]; (c) Manoso AS, Deshong P. J. Org. Chem. 2001;66:7449–7455. doi: 10.1021/jo010621q. [DOI] [PubMed] [Google Scholar]; (d) Murata M, Yamasaki H, Ueta T, Nagata M, Ishikura M, Watanabe S, Masuda Y. Tetrahedron. 2007;63:4087–4094. [Google Scholar]
- 15.(a) Powers DC, Ritter T. Nat. Chem. 2009;1:302–309. doi: 10.1038/nchem.246. [DOI] [PubMed] [Google Scholar]; (b) Powers DC, Geibel MAL, Klein JEMN, Ritter T. J. Am. Chem.Soc. 2009;131:17050–17051. doi: 10.1021/ja906935c. [DOI] [PubMed] [Google Scholar]; (c) Powers DC, Benitez D, Tkatchouk E, Goddard WA, III, Ritter T. J. Am. Chem. Soc. 2010;132:14092–14103. doi: 10.1021/ja1036644. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Powers DC, Xiao DY, Geibel MAL, Ritter T. J. Am. Chem. Soc. 2010;132:14530–14536. doi: 10.1021/ja1054274. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Chuang GJ, Wang W, Lee E, Ritter T. J. Am. Chem. Soc. 2011;133:1760–1762. doi: 10.1021/ja108396k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518–1520. [Google Scholar]
- 17.Still WC, Kahn M, Mitra A. J. Org. Chem. 1978;43:2925–2927. [Google Scholar]
- 18.Fulmer GR, Miller AJM, Sherden NH, Gottlieb HE, Nudelman A, Stoltz BM, Bercaw JE, Goldberg KI. Organometallics. 2010;29:2176–2179. [Google Scholar]
- 19.Hargrave JD, Herbert J, Bish G, Frost CG. Org. Biomol. Chem. 2006;4:3235–3241. doi: 10.1039/b606977k. [DOI] [PubMed] [Google Scholar]
- 20.Maegawa Y, Nagano T, Yabuno T, Nakagawa H, Shimada T. Tetrahedron. 2007;63:11467–11474. [Google Scholar]
- 21.Gilman H, Smart GNR. J. Org. Chem. 1950;15:720–740. [Google Scholar]
- 22.Murata M, Yamasaki H, Ueta T, Nagata M, Ishikura M, Watanabe S, Masuda Y. Tetrahedron. 2007;63:4087–4094. [Google Scholar]
- 23.Murata M, Suzuki K, Watanabe S, Masuda Y. J. Org. Chem. 1997;62:8569–8571. doi: 10.1021/jo971143f. [DOI] [PubMed] [Google Scholar]
- 24.Yokoka M, Fujita D, Ichikawa J. Org. Lett. 2007;22:4639–4642. doi: 10.1021/ol702279w. [DOI] [PubMed] [Google Scholar]
- 25.Conte L, Napoli M, Gambaretto GP, Guerrato A. J. Fluor. Chem. 1994;67:41–45. [Google Scholar]
- 26.Xing D, Guan B, Cai G, Fang Z, Yang L, Shi Z. Org. Lett. 2006;8:693–696. doi: 10.1021/ol052830t. [DOI] [PubMed] [Google Scholar]
- 27.Sveinbjornsson A, Bradlow HL, Oae S, Vanderwerf CA. J. Org. Chem. 1951;16:1450–1457. [Google Scholar]
- 28.Cai C, Rivera NR, Balsells J, Sidler RR, Mcwilliams JC, Shultz CS, Sun Y. Org. Lett. 2006;8:5161–5164. doi: 10.1021/ol062208g. [DOI] [PubMed] [Google Scholar]
- 29.Liu S, Berry N, Thomson N, Pettman A, Hyder Z, Mo J, Xiao J. J. Org. Chem. 2006;71:7467–7470. doi: 10.1021/jo0609632. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.