

Abstract
Recent studies have implicated an N-terminal caspase-6 cleavage product of mutant huntingtin (htt) as an important mediator of toxicity in Huntington's disease (HD). To directly assess the consequences of such fragments on neurologic function, we produced transgenic mice that express a caspase-6 length N-terminal fragment of mutant htt (N586) with both normal (23Q) and disease (82Q) length glutamine repeats. In contrast to mice expressing N586-23Q, mice expressing N586-82Q accumulate large cytoplasmic inclusion bodies that can be visualized with antibodies to epitopes throughout the N586 protein. However, biochemical analyses of aggregated mutant huntingtin in these mice demonstrated that the inclusion bodies are composed largely of a much smaller htt fragment (terminating before residue 115), with lesser amounts of full-length N586-82Q fragments. Mice expressing the N586-82Q fragment show symptoms typical of previously generated mice expressing mutant huntingtin fragments, including failure to maintain weight, small brain weight and reductions in specific mRNAs in the striatum. Uniquely, these N586-82Q mice develop a progressive movement disorder that includes dramatic deficits in motor performance on the rotarod and ataxia. Our findings suggest that caspase-6-derived fragments of mutant htt are capable of inducing novel HD-related phenotypes, but these fragments are not terminal cleavage products as they are subject to further proteolysis. In this scenario, mutant htt fragments derived from caspase 6, or possibly other proteases, could mediate HD pathogenesis via a ‘hit and run' type of mechanism in which caspase-6, or other larger N-terminal fragments, mediate a neurotoxic process before being cleaved to a smaller fragment that accumulates pathologically.
INTRODUCTION
Huntington's disease (HD) is a genetic, neurodegenerative and fatal disorder resulting from an expanded glutamine repeat within the first exon of the huntingtin (htt) gene (1). Glutamine (Q) repeats ≥40 will result in adult onset HD and repeats ≥60 typically result in juvenile onset; the repeat length is indirectly proportional to the age of onset (2). The htt gene encodes a 3144 amino acid protein, and it is the expression of this polyQ domain within the htt protein that causes HD. Symptoms include cognitive deficits with psychiatric abnormalities, and movement disabilities characterized by chorea progressing to rigidity (reviewed in 3). Cell loss is most prevalent in the cortex and striatum (4,5), whereas subcellular inclusion pathology can be found throughout the central nervous system (CNS) (6–9). The subcellular inclusions are generally reactive to htt antibodies with epitopes in the N-terminal, polyQ-containing region of htt, but not to more C-terminal antibodies (6,8,10–12). Thus, there is little doubt that the pathophysiology of HD involves proteolysis of mutant htt, but the role of specific proteolytic fragments in neurotoxic processes in HD remains unresolved.
Huntingtin is subject to proteolytic processing by a number of proteases to produce a series of N-terminal fragments. The fragment that produces the inclusion bodies found in CNS neurons of mouse models and human HD are thought to terminate N-terminal to amino acids 115–125 (residue numbering from HTT GenBank Accession #NM_002111). This fragment has been referred to as cleavage product-A (Cp-A or Cp-1) (11,13). Another htt fragment results from cleavage at residue 167 and is referred to as Cp-B or Cp-2 (11,13). The protease responsible for generating Cp-B/2 is unknown, whereas recent studies suggest that Cp-A/1 may be generated by γ-secretase activity (14). Multiple additional cleavage products, generated by caspase, calpain and metalloproteinase activities, have been identified to produce fragments that terminate between amino acids 400 and 600 (15–18), and it is largely unclear what role these longer cleavage products may play in disease pathogenesis. In studies of caspase-generated fragments, mutational analyses of a full-length human htt yeast artificial chromosome (YAC) with 128Q has suggested that a fragment terminating at residue 586 that is generated by caspase-6 is critical in mediating the phenotypes observed in mice harboring htt YAC128Q (19–21). Mice transgenic for htt YAC128Q encoding mutations that block caspase-6 cleavage at residue 586 (C6R YAC128Q) lacked the motor and neurologic abnormalities seen in YAC128Q mice (19). This outcome prompted us to directly assess the neurologic consequences of the production of mutant htt N586 fragments in transgenic mice.
In this study, we generated transgenic mice co-expressing enhanced green fluorescent protein (eGFP) in skin and the N-terminal 586 amino acids of htt with 23 or 82 glutamines in the brain. The approach we describe here has been used to produce mice that express htt N118-82Q and htt N171-82Q (22), yielding animals (HD118-eGFP and HD171-eGFP) that develop phenotypes similar to the R6/2 and N171-82Q mouse models of HD, including premature death, failure to gain weight and profound hypoactivity (23–25). In contrast to these models and mice expressing htt N586-23Q, the most obvious phenotype in mice expressing htt N586-82Q was profound dyskinesia with ataxia-like movements. These mice did, however, also show phenotypes more typical of the R6/2 and HD-N171-82Q models, including failure to gain weight, poor performance on the rotarod task and premature death. The ataxia-like abnormalities in N586-82Q mice were accompanied by profound atrophy of the cerebellum with obvious loss of cerebellar granule cells. Obvious degeneration of the hippocampus was also observed. Additionally, throughout the CNS, N586-82Q mice accumulated large, thioflavin S-positive, cytoplasmic inclusions that were immunoreactive to antibodies that recognize epitopes through amino acids 400–500, suggesting incorporation of N586-82Q fragments in these inclusions. However, biochemical analyses of insoluble htt in these mice indicated that N586-82Q is subject to cleavage to produce a much smaller N-terminal htt fragment (terminating N-terminal to residues 115–125), with this smaller fragment representing the most abundant form of mutant htt in the insoluble fraction. This finding indicates that caspase-6-derived fragments of mutant htt are not terminal cleavage products, but rather are substrates for further proteolysis. Although we do not yet understand the basis for the profound cerebellar phenotypes in the N586-82Q mice, our data indicate that mutant htt N-terminal fragments generated by caspase-6 are capable of mediating novel phenotypes.
RESULTS
To facilitate identification of transgenic mice, we co-injected MoPrP.Xho vectors (26) encoding htt N586-23Q or N586-82Q with a vector to express eGFP in skin [Keratin-14 (K14)-eGFP] (22). We have recently used this strategy to produce transgenic mice that express two N-terminal fragments of mutant htt (N171-82Q or N118-82Q via the Mo.PrP vector), demonstrating that the strategy results in co-integration of the two transgenes to provide efficient and correct identification of transgenic mice harboring htt transgenes (22).
Using this co-injection strategy, we obtained four founder mice expressing eGFP in skin, and used northern blot to screen for expression of htt transgene mRNA in the brain. The two highest expressing lines, designated lines C62 and C63, both expressed lower levels of transgene htt mRNA than the previously described HD-N171-82Q line 81 mice (25) (Supplementary Material, Fig. S1). N586-82Q mice from line C63 appeared to express the htt transgene mRNA at higher levels than line C62 mice (Supplementary Material, Fig. S1), and mice from line C63 developed the phenotypes described below earlier than mice from line C62. Because of the earlier phenotypic onset, most of the studies detailed below focused on mice from line C63 (designated, N586-82Q-C63). We also generated mice that express N586-23Q htt fragments (normal glutamine repeat length), using the same co-injection strategy with the same vectors (MoPrP.N586-23Q+K14-eGFP). We were able to establish one line of mice expressing N586-23Q at levels that were equivalent to our highest expressing line of N586-82Q mice (line designated A2, N586-23Q-A2) (Fig. 1). Immunoblots of soluble proteins from forebrain of young (<3 months old) mice demonstrated relatively high and equivalent levels of these recombinant proteins in brains of the N586-23Q-A2 and N586-82Q-C63 mice (Fig. 1; see Supplementary Material, Fig S2 for a replicate gel of lower exposure).
Figure 1.

Immunoblot analysis of N586-82Q and N586-23Q expression. Mouse forebrain homogenate (60 μg) was analyzed by SDS–PAGE and immunoblot using the anti-Htt antibody 2B4. Htt N586 protein levels of line A2 and line C63 are comparable. A Cp-A/1-sized cleavage product from the N586-82Q protein was detected at low levels (arrowhead).
In the brains of mice expressing the N586-82Q protein, we detected a smaller N-terminal fragment that appeared to be produced by proteolytic cleavage of the N586-82Q protein (Fig. 1, arrow). We were not able to detect this smaller fragment in brains from the N586-23Q mice.
Dyskinesia, failure to gain weight and premature death in N586-82Q mice
The most obvious phenotype we observed in our N586-82Q-C63 mice was a profound dyskinesia with ataxia-like movements (Supplementary Material, Video S1). The first signs of abnormal gait appeared at 4 months of age in N586-82Q-C63 mice, with the severity of dyskinesia progressing to end-stage by 8 months of age, at which time the movement disorder became so severe that the animals could not adequately acquire food and water. We thus developed criteria for a humane endpoint that included severity of movement abnormalities and dehydration. N586-82Q-C63 reached these endpoints at an average of 247 ± 10 days of age (Fig. 2). N586-23Q-A2 mice did not develop ataxia, at least out to an age of 10 months (Supplementary Material, Video S2). At the time of writing, we have not aged the N586-23Q-A2 mice beyond 10 months of age, but at 10 months of age the transgenic N586-23Q-A2 mice cannot be distinguished from non-transgenic littermates.
Figure 2.

N586-82Q-C63 mice reach humane endpoints prematurely. A Kaplan–Meier plot shows the ages at which mice reach humane endpoints. Line C63 mice reach endpoints between 200 and 300 days with an average time to endpoint of 247.3 ± 10.4 days, SEM (n = 13).
As one might expect, N586-82Q-C63 mice, exhibiting the dyskinesia phenotypes, performed very poorly on the rotarod task, which assesses coordination and balance (Fig. 3). Compared with their NTg littermates, and similarly aged N586-23Q-A2 mice, 8-month-old N586-82Q-C63 mice exhibited a 10–50-fold deficit in latency to fall from the rotarod (Fig. 3). Younger N586-82Q-C63 mice (3 months of age) showed moderate rotarod deficits that progressed to severe deficits by 6 months of age (Fig. 3). End-stage line C63 mice exhibited body weights that were approximately one-third of their NTg littermates with brain weights that were approximately half of their NTg littermates (Fig. 4A and B). Brain size differences were profound and could be appreciated by eye immediately after extraction (Fig. 4C). Collectively, these findings demonstrate that N586-82Q fragments of htt produce phenotypes similar to the N171-82Q and R6/2 models (failure to gain weight, rotarod deficits and brain atrophy) (23–25,27), with the added novel phenotype of profound dyskinesia and ataxia.
Figure 3.

Progressive motor dysfunction on the rotarod task. NTg (black square) and N586-82Q-C63 (C63, grey diamond) mice were assessed by rotarod at 3 (3 m/o), 6 (6m/o) and 8 (8 m/o) months of age. For comparison, 8 m/o mice transgenic for N586-23Q (line A2) were tested (light grey circles). At each age tested, N586-82Q-C63 mice were statistically different from non-transgenic littermates (P < 0.05, n = 6–8 for each group). In contrast, the performance of N586-23Q-A2 mice was not different from NTg mice.
Figure 4.

N586-82Q-C63 mice fail to thrive. (A) At end-stage, the body weight of N586-82Q-C63 mice (dashed bars, 15.85 ± 0.56 g, n = 14) was less than half of their NTg littermates (white bars, 41.48 ± 3.86 g, n = 6; unpaired t-test P< 0.0001). (B) Brain weight immediately after extraction is also significantly less in end-stage C63 mice (0.2733 ± 0.0058 g, n = 9) than NTg littermates (0.4917 ± 0.0054 g, n = 6; unpaired t-test P < 0.0001). (C) A digital picture demonstrates the difference in brain size between NTg (left) and line C63 (right) mice.
N586-83Q mice from line C62 also developed the dyskinesia and ataxia phenotypes, but at significantly later ages. Obvious dyskinesia and ataxia were not evident until after 12 months of age with a slower rate of progression. First-generation N586-82Q-C62 mice developed obvious dyskinesia by 15 months of age (Supplementary Material, Video S3), but mice from subsequent generations show even later onset of gait abnormalities and milder ataxia. Although we are certain that the dyskinesia and ataxia phenotypes were reproduced in both lines of N586-82Q mice, at present only N586-82Q-C63 mice show robust early onset dyskinesia.
Large cytoplasmic inclusions and cerebellar degeneration in N586-82Q mice
To determine the neuropathologic correlates of the progressive movement phenotype in the N586-82Q-C63 mice, we performed extensive pathologic examinations. In standard hematoxylin and eosin stains of cerebellum from ataxic N586-82Q-C63 mice, we observed an obvious loss of cerebellar granule cells (Fig. 5A). In contrast, age-matched mice expressing N586-23Q showed granule cell densities similar to non-transgenic mice (Fig. 5B and C). Note that Purkinje cells (Fig. 5A, arrows) were relatively spared when compared with granule cells. This finding is not unexpected because previous studies have established that the Mo.PrP vector does not express in Purkinje cells (25,26). Profound degeneration of the hippocampus was also noted in the N586-82Q-C63 mice when compared with N586-23Q-A2 and non-transgenic mice (Supplementary Material, Fig. S3).
Figure 5.
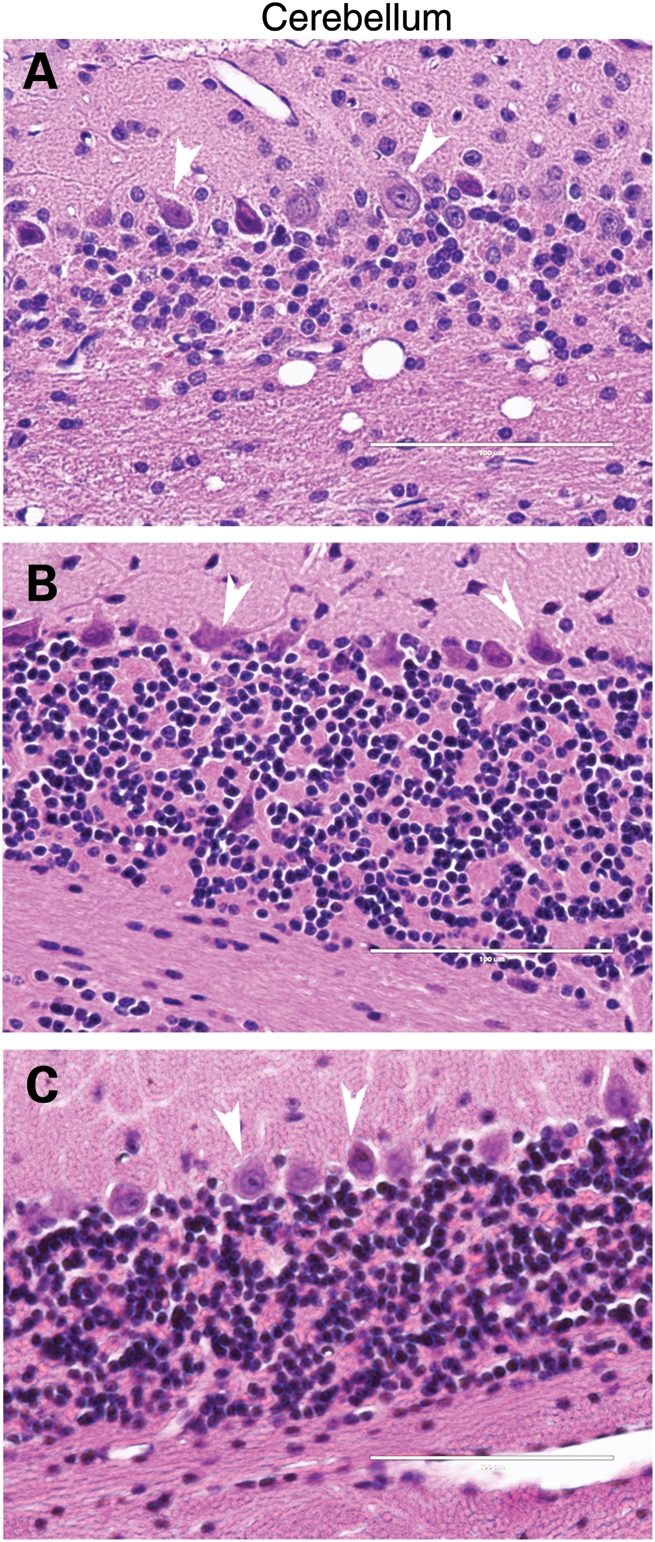
Cerebellar degeneration in N586-82Q-C63 mice. Hematoxylin and eosin staining show that the granule cell layer of N586-82Q-C63 mice (A) is less densely packed with cells and less organized than cerebellum of age-matched N586-23Q-A2 mice (B) or NTg littermates (C). These images are representative of what was visualized in three different symptomatic animals on at least three tissue sections from each animal.
Immunohistochemical staining of brains from N586-82Q-C63 mice revealed abundant htt-immunoreactive inclusions in the cerebellar granule cell layer (Fig. 6A). Large inclusions were also seen in the striatum (Fig. 6B). In contrast, we found diffusely distributed immunoreactivity in the cerebellar granule cells of mice expressing N586-23Q-A2 with much fainter staining in the striatum (Fig. 6D). No immunoreactivity was detected in non-transgenic mice that were stained in parallel for equivalent times (Fig. 6E and F). In both lines of the N586-82Q mice, we found large htt immunoreactive inclusions in virtually every brain structure (Supplementary Material, Figs S4–S6).
Figure 6.

Inclusion pathology in the cerebellum and striatum of N586-82Q-C63 mice. End-stage line N586-82Q-C63 mice (8 months old) exhibit punctate immunostaining in the cerebellum and striatum (A and B). In contrast, age-matched N586-23Q-A2 mice exhibit diffuse, non-aggregated htt staining (C and D). Age-matched NTg littermates of line C63 mice exhibit very low levels of htt immunoreactivity when stained and processed in parallel (E and F). These images are representative of what was visualized in three different symptomatic animals on at least three tissue sections from each animal immunostained with the 2B4 antibody (see Materials and Methods).
The appearance of inclusions and the accompanying degeneration of granule cells in the cerebellum were progressive. In 2-month-old N586-82Q-C63 mice, immunoreactivity for the N586-82Q protein was largely diffuse and the density of the cerebellar granule cell layer appeared normal (Supplementary Material, Fig. S7). However, at 8 months of age, inclusion pathology was abundant and an obvious loss in granules cells was also evident (Supplementary Material, Fig. S7).
In the cerebellum, it was difficult to determine whether the htt inclusions were nuclear or cytoplasmic (Fig. 6). However, in the striatum, it appeared that most of the inclusions were cytoplasmic. To assess the location of inclusions, we inspected higher power images of the cortex, which we noted to have a high density of inclusion pathology (see Supplementary Material, Fig. S5). In htt immunostains of the cortex, we saw a variety of structures. There were numerous small punctate immunoreactive structures that appeared to be located in neuropil (Fig. 7A). We also saw a number of very large inclusions, some of which were located very near nuclei. However, most nuclei lacked an obvious immunoreactive inclusion (Fig. 7A) and moreover, these nuclei lacked the diffuse accumulation of huntingtin immunoreactivity that has previously been described in HD-N171-82Q mice (25). In the cortex of N586-23Q-A2 mice, we observed a diffuse distribution of immunoreactivity throughout the cell body of a subset of neurons. Reactivity in dendritic projections was also visible (Fig. 7B). Non-transgenic mice stained, in parallel, lacked obvious immunoreactivity to the htt antibody. Overall, these data show that pathology in N586-82Q-C63 mice includes very large inclusions that are predominantly localized to cytoplasm.
Figure 7.
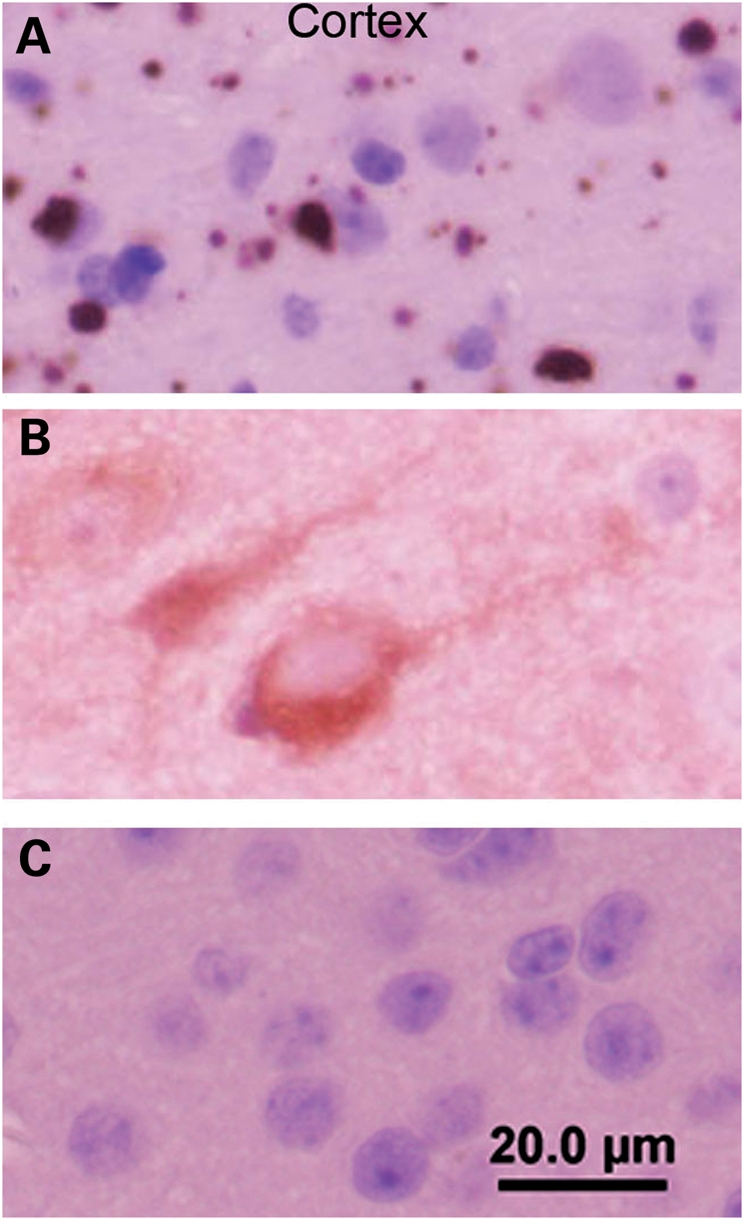
Cortical inclusions are predominantly cytoplasmic in N586-82Q-C63 mice. End-stage line N586-82Q-C63 mice (8 months old) exhibit punctate immunostaining in the neuropil of the cortex with larger perinuclear aggregates also visible (A). In contrast, age-matched N586-23Q-A2 mice exhibit diffuse staining of the cell bodies of a subset of cortical neurons (B). Age-matched NTg littermates of line C63 mice exhibit very low levels of htt immunoreactivity when stained and processed in parallel (C). These images are representative of what was visualized in three different symptomatic animals on at least three tissue sections from each animal immunostained with the 2B4 antibody (see Materials and Methods).
The aggregation of htt N-terminal fragments in vitro has been reported to produce amyloid-like structures rich in beta-sheet conformation (28). These mutant htt structures bind Thioflavin compounds in vitro (29) and in tissues (30). Consistent with these studies, inclusions in symptomatic N586-82Q-C63 mice were detectable with Thioflavin-S staining (Supplementary Material, Fig. S8 for examples of inclusions in the cerebellum and striatum).
An additional pathological feature of human HD is astrogliosis (31), which has also been reported in knock-in mouse models of HD (32,33). To assess astrogliosis, we examined the level of glial fibrillary acid protein (GFAP) immunoreactivity in the cerebellum, striatum and cortex of N586-82Q-C63, N586-23Q-A2 and non-transgenic mice (Fig. 8). When compared with non-transgenic and N586-23Q-A2 mice, robust astrogliosis was detected in all three structures in N586-82Q-C63 mice (Fig. 8; see Supplementary Material, Fig. S9 for low power images).
Figure 8.

Astrocytic reaction in N586-82Q mice. Cerebellum, striatum and cortex, stained with anti-GFAP antibody and visualized with DAB, show robust astrogliosis in N586-82Q-C63 mice (A, D, G, respectively). In contrast, line A2 mice (B, E, H) show patterns of GFAP immunoreactivity that are similar to control NTg mice (C, F, I). These images are representative of what was visualized in three different symptomatic animals on at least three tissue sections from each animal.
To determine whether the cytoplasmic, or neuropil, inclusions where localized to astroglia, we used double immunofluorescent labeling with the htt antibody (2B4) and GFAP antibodies. Inclusions were generally not found in a perinuclear location in the cell bodies of astroglia where it is easiest to definitively demonstrate astroglial localization (Supplementary Material, Fig. S10). Most inclusions were localized to neuropil where they could be in processes derived from either neurons or glia.
Another established phenotype of the HD-N171-82Q and R6/2 mouse models of HD is that the levels of mRNA for dopamine and c-AMP-regulated phosphoprotein-32kD (Darpp-32), phosphodiesterase 1b (Pde1b), phosphodiesterase 10a (Pde10a), preproenkephalin (Penk), dopamine receptor D2 (Drd2) and early growth response 1 (Egr1) are reduced in the striatum (34,35). To assess expression of these mRNAs in the striatum of N586-82Q-C63 mice, we performed in situ hybridization on coronal sections of the brain. In comparison to non-transgenic littermates, the levels of all of these transcripts except Darpp-32 were significantly lower in the symptomatic line C63 mice (Fig. 9).
Figure 9.

Reduced levels of specific mRNA in the striatum of N586-82Q-C63 mice. In situ hybridization shows that relative mRNA intensities of Pde1b, Pde10a, Penk, Drd2 and Egr1 mRNAs are decreased in the striatum, compared with NTg mice. Darpp-32 was not significantly decreased (n.s.), nor was the control gene actin. n = 3 for each NTg and line C63 mice.
Collectively, these data show that the N586-82Q-C63 mice we developed by co-injection with K14-eGFP vectors develop cytoplasmic/neuropil inclusion pathology, obvious degeneration of cerebellar granule cells, obvious hippocampal degeneration, widespread reactive gliosis and reductions in the levels of specific mRNAs in the striatum.
Proteolytic processing of htt N586-82Q
To examine the composition of the inclusion pathology in the N586-82Q-C63 mice, we used a combination of immunohistochemistry and biochemical analysis. For immunohistochemical analyses, we used antibodies that recognize three regions of the N586-82Q protein (Fig. 10A). In sections of cerebellum stained with the 2B4 antibody (amino acids 59–65), we observed inclusions of an array of sizes that were predominately in neuropil (Fig. 10B). Similar structures were recognized by the 1H6 (amino acids 115–129) (Fig. 10C) and the 2166 (amino acids 414–503) antibodies (Fig. 10D). These findings suggest that the entire N586 fragment is at least a partial component of these inclusions.
Figure 10.

The N586-82Q protein is found in cytoplasmic inclusions. (A) The diagram shows where the known epitopes of htt antibodies 2B4, 1H6 and 2166 are located within the N586 protein (the black box represents the polyQ domain). (B–D) Immunocytochemistry using 2B4, 1H6 and 2166, respectively, shows htt inclusions (red) contain the full N586-82Q protein. Nuclear DNA is stained blue with DAPI. These images are representative of what was visualized in three different symptomatic animals on at least three tissue sections from each animal.
To better quantify the contribution of the entire N586-82Q fragment to inclusion pathology, we performed detergent extraction and sedimentation to isolate insoluble protein components of inclusions. Tissues were extracted in buffers containing 2% sodium dodecyl sulfate (SDS) yielding an SDS soluble fraction. The insoluble pellet was then solubilized in formic acid (FA) and both fractions were analyzed by SDS–polyacrylamide gel electrophoresis (PAGE) analysis and immunoblotting with 2B4, 1H6 and 2166 antibodies. For comparison, we analyzed brain tissue from the HD-N171-82Q (line 81) model in parallel to N586-82Q-C63 mice. With equal protein loading, we were largely unable to detect htt N171-82Q or smaller Cp-A/1 length fragments in the brains of the HD-N171-82Q mice. It is our impression that the htt N586-82Q protein is a relatively stable protein because of the ease with which we detect it on immunoblots when compared with smaller N171-82Q fragments despite the higher level of transgene mRNA in the N171-82Q line 81 mice (see Supplementary Material, Fig. S1). The greater stability of the N586-82Q protein may contribute to the large size of the inclusion pathology described above.
In the FA solubilized fraction from the N586-82Q-C63 mice, we detected an htt fragment that was reactive with the 2B4 antibody (amino acids 59–65) (Fig. 11A, arrow), but not with the 1H6 or 2166 antibodies (Fig. 11B and C). This immunoreactive profile is characteristic of a Cp-A/1-sized fragment (11,12). In the SDS-insoluble fractions that had been denatured in FA, the 1H6 and 2166 antibodies detected the full-length N586-82Q protein (Fig. 11B and C, arrowhead); a finding that confirms the immunohistochemistry which detected these epitopes in inclusions. In the SDS-soluble fractions, the 2B4 antibody primarily recognized a smear of apparently aggregated htt protein (Fig. 11A, bracket). This smear was less prominent in immunoblots with the 1H6 or 2166 antibodies (Fig. 11B and C). One interpretation of this outcome is that the predominant htt fragment in the aggregated material is a Cp-A/1-sized fragment. However, we can best estimate the relative amounts of the Cp-A/1 and N586-82Q fragments in inclusions by analysis of the FA denatured preparations of SDS-insoluble proteins that were analyzed by immunoblot with the 2B4 antibody, which recognizes both Cp-A/1 and full-length N586. In this case, the Cp-A/1-sized fragment was much more abundant than the full-length N586-82Q fragment. Thus, we conclude that Cp-A/1-sized fragments are likely to be the major components of the inclusion pathology observed in the HD-586-82Q-eGFP mice.
Figure 11.

Immunoblots of detergent-insoluble htt in N586-82Q-C63 mice. (A) NTg, HD-N171-82Q line 81 and N586-82Q-C63 brain lysates were fractionated into SDS-soluble and -insoluble fractions with the insoluble fraction subsequently denatured and solubilized in FA (see Materials and Methods) and analyzed by western blot with the 2B4 (A), 1H6 (B) and 2166 (C) antibodies. Brackets represent high molecular weight, insoluble species. Arrowheads identify the N586 fragment and the arrow in (A) identifies Cp-A/1, which is not reactive to 1H6 or 2166 antibodies. A representative image is shown for an experiment that was repeated twice.
To determine whether there might be inclusions in which the shorter fragments are the only component, we performed double immunolabeling with an antibody to amino acids 64–82 (htt64–82), which recognizes epitopes in the Cp-A/1-sized fragment and antibodies 1H6 and 2166, which recognize epitopes in full-length N586-82Q protein (Supplementary Material, Fig. S11). All inclusions appeared to co-label with both the N-terminal htt64–82 antibody and either of the two more C-terminal antibodies (1H6 or 2166). Thus, we conclude that inclusions are composed of a mixture of Cp-A/1-sized fragments and full-length N586-82Q protein.
DISCUSSION
An N-terminal cleavage fragment of mutant htt, generated by caspase-6 cleavage at residue 586, has been suggested to play a pivotal role in HD pathogenesis based on the phenotypes that appear in mice harboring a modified htt YAC with 128Q (19). To directly assess the neurologic consequences of this N-terminal 586 amino acid fragment, we generated transgenic mice expressing an engineered htt N586-82Q protein. The highest expressing line of these mice recapitulates many features that have previously been identified in the R6/2 and HD-N171-82Q transgenic mouse models of HD (23–25,27), including failure to gain weight, general brain atrophy, htt inclusion body pathology and premature death. However, these mice also exhibited several phenotypes novel to HD mouse models, including profound dyskinesia and ataxia, large inclusions that are exclusively cytoplasmic and profound degeneration of the cerebellum and hippocampus. Thus, our findings provide evidence to support the notion that mutant N586 htt fragments could mediate specific types of neurologic abnormalities.
One important feature of this model is that we demonstrate that the N586-82Q fragment is not a terminal cleavage product, but rather is subject to further proteolysis. Although studies of htt proteolysis using the YAC128Q transgenic mice implicated caspase-6 cleavage of mutant htt as potentially essential in HD pathogenesis (19), they did not exclude the possibility that caspase-6-derived htt fragments are subject to further proteolysis. In neuropathologic studies of these models, the antibodies that were used to detect htt inclusion pathology had epitopes within the Cp-A/1 fragment (19,36,37). Thus, the original studies of the YAC128Q did not provide evidence that caspase-6-derived htt fragments accumulate to any appreciable level. In our model, in which the N586-82Q fragment is directly over-expressed, we find this fragment accumulating in cytoplasmic/neuropil inclusion bodies along with a more abundant Cp-A/1-sized fragment, which is the fragment that appears to be the primary component of inclusion pathology of human HD (6,8,10–12). In a normal setting in human patients, in which production of mutant N586 fragments would be expected to be much lower than achieved in our mouse model, it is likely that any N586 fragments generated would be completely proteolyzed to smaller fragments. Thus, it becomes very possible that caspase-6-derived N-terminal fragments of mutant htt exist only transiently in human HD.
The most unique feature of N586-82Q mice is the progressive dyskinesia and ataxia with coincident cerebellar atrophy. Early in the course of these studies, we provided our Mo.PrP.N586-82Q vectors to Dr Christopher Ross (Johns Hopkins University School of Medicine) as a joint venture within the Huntington's Disease Society of America Coalition for the Cure, and these vectors were used to produce transgenic mice without the co-injection of the K14-eGFP vectors. The primary difference in phenotypes between mice generated with and without the K14-eGFP co-injection was that the mice produced without the co-injection did not develop dyskinesia and ataxia and did not show obvious degeneration of cerebellar granule cells (E. Waldron et al., submitted for publication). Otherwise, the two models develop similar types of cytoplasmic inclusion pathology in similar locations. Thus, the K14-eGFP transgene functioned as not only a marker for transgenesis but as an inadvertent disease modifier. We do not yet understand the mechanism for disease modification. Importantly, mice that express htt N171-82Q or htt N118-82Q made by co-injection of Mo.PrP vectors and K14-eGFP vectors do not develop dyskinesia and ataxia, or show degeneration of cerebellar granule cells or the hippocampus (22). Thus, the K14-eGFP modifier effect is specific to the co-expression of htt N586-82Q.
There are several possible explanations for the unique ataxia of the N586-82Q + K14-eGFP mice we describe here. We do not think it is likely that the modifier effect is due to an alteration of an endogenous gene by transgene integration because the initial offspring of the N586-82Q-C62 mice exhibited an ataxia similar to N586-82Q-C63 mice (Supplementary Material, Video S3). The occurrence of the same phenotype in a second independent line is very unlikely to be a consequence of random integration. However, it is bothersome that subsequent offspring from N586-82Q-C62 mice have shown later onset of the ataxia with diminished severity. A second consideration is whether some type of gene re-arrangement produced a novel eGFP fusion protein or that the expression of eGFP in the brain (from the K14-vector; 22) contributes to the phenotype. The eGFP present in the brains of symptomatic N586-82Q-C63 mice migrates as expected on immunoblots, is entirely soluble and does not localize to inclusions (Supplementary Material, Fig. S12). Thus, we cannot find evidence to implicate the presence of eGFP in the brains as playing a role in the novel phenotypes. We think the most likely explanation is that enhancer elements in the K14 promoter affect the temporal or spatial pattern of htt N586-82Q expression in some manner that modifies disease phenotype. A careful study of the expression of transgenes in our N586-82Q+K14-eGFP mice and the N586-82Q mice made without the co-injection may reveal an explanation. However, other explanations are possible and it may take considerable effort to determine the basis for the different phenotypes.
Although the modifier effect we observe in our N586-82Q + K14-eGFP mice appears to be artificial, the appearance of the novel ataxic behavior in the highest expressing mice provides evidence that mutant htt N586-82Q production has the potential to induce unique neurologic consequences.
In addition to the motor deficit, the other profound feature of N586-82Q-C63 mice is the robust cytoplasmic inclusion pathology that develops throughout the brain, particularly the cerebellum. Cytoplasmic inclusions, also referred to as extranuclear inclusions or neuropil inclusions, are a prominent pathology of human HD (6,8). In the N586-82Q-C63 mice, we find that the cytoplasmic inclusions contain the full-length N586-82Q protein as well as htt fragments that are Cp-A/1 sized, which have previously been described as principal components of intranuclear inclusions (11,12). One scenario to explain the scarcity of intranuclear inclusions in this model is that the htt N586-82Q fragment aggregates in the cytosol first and is then subsequently cleaved to the smaller Cp-A/1-sized fragments. In this scenario, the protein would be trapped in cytosol and unable to diffuse at any appreciable level to the nucleus. We also note that the N586-23Q protein showed a predominant cytosolic location, which is consistent with this larger protein being unable to diffuse into the nucleus. Thus, it is possible that the cytosolic location of the N586-82Q protein leads to cytoplasmic inclusions because any Cp-A/1-sized cleavage product produced in the cytosol aggregates before it can diffuse to the nucleus. In either case, our findings establish the potential for a caspase-6-derived fragment of mutant htt to be a precursor to cytoplasmic inclusions containing a shorter Cp-A/1-sized fragment. It is notable that mice harboring htt YAC128Q genes with mutations that eliminate the caspase-6 cleavage site at residue 586 (C6R YAC128Q mice) still develop intranuclear inclusion pathology (19). Whether cytoplasmic inclusion pathology was altered in these C6R YAC128Q mice was not directly addressed (19), but side by side comparisons of nuclear and cytoplasmic accumulations of htt immunoreactivity in multiple HD mouse models including the C6R YAC128Q mice has been reported (37). In reviewing this latter work, we note that images of htt inclusion pathology in C6R YAC128Q mice appear to show much less obvious cytoplasmic inclusion pathology. Clearly, further study is required to clarify the origins of intranuclear and cytoplasmic (also referred to as neuropil) inclusion pathology.
In summary, we have generated a transgenic mouse model that expresses an N-terminal fragment of mutant htt corresponding to fragments generated by caspase-6 (19). Mice expressing the N586-82Q fragments exhibit several novel phenotypes including widespread astrogliosis, dyskinesia/ataxia, cerebellar and hippocampal atrophy, and inclusion pathology dominated by large cytoplasmic aggregates. We also demonstrate that caspase-6-derived fragments of mutant htt are not terminal cleavage products but instead are substrates for further proteolytic processing. The novel phenotypes of this model suggest that different cleavage products of htt have the capacity, under certain conditions, to elicit specific neurologic abnormalities. Overall, these findings provide a framework in which to consider the possibility that caspase-6, and possibly other proteases, produce N-terminal fragments mutant htt that exist only transiently and yet have the capacity to mediate specific neurologic consequences.
MATERIALS AND METHODS
All experiments described herein have been approved by the University of Florida Institutional Animal Care and Use Committee. Mice were kept on a 14 h:10 h light-to-dark ratio and had ad libitum access to food and water.
Generation of plasmids
The Mo.PrP N171-18Q and -82Q vectors, which have been described (25), were the basis for the generation of expression vectors for htt N586 cDNA genes. A plasmid was generated by GenScript (Piscataway, NJ, USA) that contained cDNA encoding htt amino acids 171–586. This 171–586 cDNA was excised using XhoI (at 171) and SalI (which was engineered after a ‘Stop’ codon following amino acid 586) restriction enzymes and ligated into Mo.PrP N171 vectors linearized with XhoI at the 171 site (XhoI and SalI produce compatible overhanging sequence for ligation). A similar strategy was utilized to modify pcDNA3.1(+) vectors encoding htt N171-18Q and -82Q (12), to encode htt N586-23Q and N586-82Q (note that in cloning the 18Q repeat of htt N171-18Q expanded to 23Q). All vectors were sequenced at the University of Florida Interdisciplinary Center for Biotechnology Research to confirm sequence and number of glutamines. The K14-eGFP expression vector has previously been described (22).
Genotyping
The co-injection of Mo.PrP.N586 vectors with K14-eGFP vectors allowed us to use visual detection of eGFP to genotype most of the animals (22). To confirm the co-integration of K14-eGFP with the htt vectors, genomic DNA was extracted from tail biopsies and used to confirm integration of the Mo.PrP-N586-82Q transgene. Approximately 200 ng of genomic DNA was amplified with the following primers in one reaction: PrP-SJ, 5′-GGGACTATGTGGACTGATGTC GG-3′; PrP-ASJ, 5′-CCAAGCCTAGACCACGAGAATGC-3′; Htt-78–79 SmaI-S, 5′-CCACCCGGGCCGGCTGTGGCT-3′. The primer pair PrP-SJ and PrP-ASJ amplifies the endogenous PrP allele (∼750 bp) and the primer pair Htt-78-79 SmaI-S and PrP-ASJ amplifies the htt transgene (∼1500 bp). After initial confirmations of co-integration and co-segregation of Mo.PrP.N586 and K14-eGFP transgenes, visual genotyping was used to identify transgenic animals.
Immunoblotting
Mice were anesthetized then perfused with cold phosphate buffered saline (PBS). Extracted mouse brains were divided into forebrain and cerebellum, and then homogenized in 10-fold volumes of 10 mm Tris, pH 7.5; 1 mm ethylene-diamine-tetra-acetic acid, pH 8.0; 100 mm NaCl (TEN) + protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA), followed by centrifugation at 800g for 5 min. Resulting supernatants were mixed with 1/10th volume of 10% NP-40, sonicated and centrifuged at 100 000g for 30 min. The resulting supernatant was kept as S1 and defined as soluble proteins.
Mouse brain S1 lysates were mixed with Laemmli buffer (38), boiled and electrophoresed through Tris-glycine 4–20% polyacrylamide gels (Invitrogen) until the dye front reached the bottom of the gel. Proteins were then transferred to nitrocellulose membranes, blocked in 5% milk/Tris–buffered saline (TBS) +0.05% Tween-20 and incubated with htt antibody 2B4 (1:1000, Millipore, Billerica, MA, USA). Goat anti-rabbit secondary antibodies conjugated to horseradish-peroxidase (HRP) followed by chemiluminescence was used to detect proteins on a Fuji imaging system (FUJIFILM Life Science, Stamford, CT, USA).
Aggregate solubilization
Mouse brains were homogenized in 10-fold volumes of TEN + protease inhibitor cocktail and then diluted with SDS to a final concentration of 2%. This mixture was sonicated until it appeared clear and no longer viscous, followed by centrifugation at 100 000g for 5 min. The resulting supernatant was saved as SDS-S, and the pellet was resuspended by sonication in 98% FA. This resuspended protein mixture was incubated at 37°C for 2 h and then centrifuged in a speed-vac (DyNA Vap, Labnet International, Woodbridge NJ, USA) to near complete dryness. A small volume (∼30 μl) of water was added followed by another centrifuging to dry the sample and then the sample was resuspended in 4× Laemmli buffer followed by heating at 95°C for 5 min. Laemmli buffer was also added to the samples from SDS-soluble fractions and mixed well, but not boiled. SDS–PAGE and protein detection were performed as previously stated. Primary antibodies used were 2B4 (1:1000, Millipore), 1H6 (1:2000, Millipore) and 2166 (1:1000, Millipore).
Rotarod analysis
Mice were placed on a rotarod (IITC Life Sciences Inc., Series 8, Woodland Hills, CA, USA) that was set to accelerate from 4 to 30 rpm over 5min. This test was repeated four times a day for four consecutive days with a 10min rest interval between each trial (39). Times for each animal were averaged and then the average per animal was averaged again by genotype for each day and plotted as ±SEM with unpaired Student's t-test statistical analysis.
Immunohistochemistry
Mice were anesthetized followed by perfusion with PBS, pH 7.4. Brains were removed and fixed by incubation in 4% paraformaldehyde in PBS for 48 h. For immunofluorescence studies on free-floating frozen tissue sections, brains were incubated in 30% sucrose in PBS for >48 h at 4°C. Brains were cryosectioned at 30 µm intervals and stored in anti-freeze solution (100 mm sodium acetate pH 6.5, 1% polyvinylpyrrolidone, 40% ethylene glycol in dH2O). Sections were then washed in PBS, heated to 90°C for 10 min in 10 mm sodium citrate and 0.01% Tween-20, washed twice in PBS and blocked in 5% normal goat serum (NGS) in PBS + 0.01% Tween-20. Sections were then incubated in primary antibodies diluted in blocking buffer (5% NGS + Tween-20), washed 3× for 5 min in PBS, incubated in secondary antibodies with 4′,6-diamidino-2-phenylindole (DAPI) (1:2000, Invitrogen) diluted in blocking buffer, washed and then mounted on to glass microscope slides. Sections were covered with Aqua Poly/Mount (Polysciences, Inc., Warrington, PA, USA) and glass cover slips. Images were taken on an Olympus DSU-IX81 Spinning Disc Confocal Microscope or Olympus BX60 light microscope. Primary antibodies used were 2B4 (1:1000), 1H6 (1:1000), 2166 (1:1000), htt64-82 (1:1000, Sigma) and anti-GFAP (1:1000, Dako). Secondary antibodies used were goat anti-mouse Alexa Fluor 488 and Alexa Fluor 568, and goat anti-rabbit Alexa Fluor 488 and Alexa Fluor 568 (1:2000, Molecular Probes, Invitrogen, Carlsbad, CA, USA).
Some tissues were embedded in paraffin for sectioning and staining in the Molecular Pathology Core (University of Florida College of Medicine). Tissues were embedded following standard procedures and then cut in 6μm sections according to a standard protocol. Standard procedures for hematoxylin/eosin staining were used. For immunostaining, slides were deparaffinized by oven heating at 60°C followed by immersion in xylene and then alcohol. Endogenous peroxidase activity was quenched by incubation with 3% hydrogen peroxide in methanol for 10 min. Sections were then rehydrated through sequential incubations in 100% ethanol, 95% ethanol, 70% ethanol and water. The slides were incubated in Antigen Retrieval Citra Solution (BioGenex Laboratories Inc., Fremont, CA, USA) in TBS; 50 mm Tris–HCl, pH 7.6, 150 mm NaCl) for 30 min. Non-specific epitopes were then blocked with Mouse Background Sniper (Biocare Medical, Concord, CA, USA) in TBS. Primary antibodies anti-Huntingtin (2B4, monoclonal, Millipore, Temecula, CA, USA) or GFAP (GA5, monoclonal, Cell Signal Technology, Inc., Danvers, MA, USA) were diluted 1:1000 in TBS and incubated overnight at 4°C in a humid chamber. After being washed of excess primary antibody with TBS, slides were incubated with MACH 2TM HRP Polymer (Biocare Medical) for 30 min. Antibody binding was visualized with diaminobenzidene (DAB; Vector Laboratories, Burlingame, CA, USA) according to the manufacturer's directions. Finally, the slides were counterstained with hematoxylin and permanently mounted.
In situ hybridization
Fresh-frozen mouse brain tissue was sectioned at 14 µm on a cryostat. Approximately 25 slides with four sections per slide for each animal were generated. In situ hybridization was performed on representative coronal mouse brain sections (bregma +1.70 to −0.50) by using radio-labeled (33P) gene-specific anti-sense oligonucleotide probes. The probes targeted dopamine and cyclic AMP-regulated phosphoprotein with molecular weight 32 kDa (Darpp-32), Penk, Pde10a, Pde1b, Drd2, Egr1 (also known as neuronal growth factor I-A) and β-actin. The methods employed for in situ hybridization have been described previously in detail (40–43). Briefly, slides were allowed to thaw to room temperature, fixed with 4% PFA for 5min and rinsed with RNase-free PBS for 5 min twice. A subsequent rinse was done with 2× RNase-free sodium saline citrate (SSC) for 20 min. A solution containing 10 pmol/μl of the oligonucleotide probe, 30 U/μl of TdT (Promega, Madison, WI, USA) and 33P dATP at a concentration of 3000 curies/mmol heated to 37°C for 90 min and then mixed with hybridization buffer and applied to the slides. The slides were incubated overnight at 37°C. Slides were washed 4× for 30 min in 1×, 0.5× and .25× SSC, allowed to dry overnight, then placed with BioMax MR X- film from Kodak in cassettes. After 3–4-week development, the films were scanned and quantification of signal intensity was done using the Quantity One analysis software from Bio-Rad (Hercules, CA, USA). Background intensity was subtracted from the striatal readings to get the raw data.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
ACKNOWLEDGEMENTS
We thank Rick Morimoto, Ron Kopito, Erich Wanker and Gill Bates for discussions relating to the design of these analyses. We thank Elaine Waldron and Christopher Ross for sharing their preliminary findings in analyses of N586-82Q mice that were made without co-injection of K14-eGFP vectors.
Conflict of Interest statement. None declared.
FUNDING
This work was supported by the Huntington's Disease Society of America and the CHDI Coalition for a Cure (A.T.N.T. and D.R.B.) and the Intramural Research Program of the NIH, Center for Cancer Research, National Cancer Institute (for D.S. and L.T.).
REFERENCES
- 1.Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 2.Brinkman R.R., Mezei M.M., Theilmann J., Almqvist E., Hayden M.R. The likelihood of being affected with Huntington disease by a particular age, for a specific CAG size. Am. J. Hum. Genet. 1997;60:1202–1210. [PMC free article] [PubMed] [Google Scholar]
- 3.Walker F.O. Huntington's disease. Lancet. 2007;369:218–228. doi: 10.1016/S0140-6736(07)60111-1. [DOI] [PubMed] [Google Scholar]
- 4.de la Monte S.M., Vonsattel J.-P., Richardson E.P. Morphometric demonstration of atrophic changes in the cerebral cortex, white matter, and neostriatum in Huntington's disease. J. Neuropathol. Exp. Neurol. 1988;47:516–525. doi: 10.1097/00005072-198809000-00003. [DOI] [PubMed] [Google Scholar]
- 5.Myers R.H., Vonsattel J.P., Stevens T.J., Cupples L.A., Richardson E.P., Martin J.B., Bird E.D. Clinical and neuropathologic assessment of severity in Huntington's disease. Neurology. 1988;38:341–347. doi: 10.1212/wnl.38.3.341. [DOI] [PubMed] [Google Scholar]
- 6.DiFiglia M., Sapp E., Chase K.O., Davies S.W., Bates G.P., Vonsattel J.P., Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 7.Becher M.W., Kotzuk J.A., Sharp A.H., Davies S.W., Bates G.P., Price D.L., Ross C.A. Intranuclear neuronal inclusions in Huntington's disease and dentatorubral and pallidoluysian atrophy: correlation between the density of inclusions and IT15 CAG triplet repeat length. Neurobiol. Dis. 1998;4:387–397. doi: 10.1006/nbdi.1998.0168. [DOI] [PubMed] [Google Scholar]
- 8.Gutekunst C.A., Li S.H., Yi H., Mulroy J.S., Kuemmerle S., Jones R., Rye D., Ferrante R.J., Hersch S.M., Li X.J. Nuclear and neuropil aggregates in Huntington's disease: relationship to neuropathology. J. Neurosci. 1999;19:2522–2534. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herndon E.S., Hladik C.L., Shang P., Burns D.K., Raisanen J., White C.L., III Neuroanatomic profile of polyglutamine immunoreactivity in Huntington disease brains. J. Neuropathol. Exp. Neurol. 2009;68:250–261. doi: 10.1097/NEN.0b013e318198d320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sieradzan K.A., Mechan A.O., Jones L., Wanker E.E., Nukina N., Mann D.M. Huntington's disease intranuclear inclusions contain truncated, ubiquitinated huntingtin protein. Exp. Neurol. 1999;156:92–99. doi: 10.1006/exnr.1998.7005. [DOI] [PubMed] [Google Scholar]
- 11.Lunkes A., Lindenberg K.S., Ben Haiem L., Weber C., Devys D., Landwehrmeyer G.B., Mandel J.L., Trottier Y. Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol. Cell. 2002;10:259–269. doi: 10.1016/s1097-2765(02)00602-0. [DOI] [PubMed] [Google Scholar]
- 12.Schilling G., Klevytska A., Tebbenkamp A.T., Juenemann K., Cooper J., Gonzales V., Slunt H., Poirer M., Ross C.A., Borchelt D.R. Characterization of huntingtin pathologic fragments in human Huntington disease, transgenic mice, and cell models. J. Neuropathol. Exp. Neurol. 2007;66:313–320. doi: 10.1097/nen.0b013e318040b2c8. [DOI] [PubMed] [Google Scholar]
- 13.Ratovitski T., Gucek M., Jiang H., Chighladze E., Waldron E., D'Ambola J., Hou Z., Liang Y., Poirer M.A., Hirschhorn R.R., et al. Mutant Huntingtin N-terminal fragments of specific size mediate aggregation and toxicity in neuronal cells. J. Biol. Chem. 2009;284:10855–10867. doi: 10.1074/jbc.M804813200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kegel K.B., Sapp E., Alexander J., Reeves P., Bleckmann D., Sobin L., Masso N., Valencia A., Jeong H., Krainc D., et al. Huntingtin cleavage product A forms in neurons and is reduced by gamma-secretase inhibitors. Mol. Neurodegener. 2010;5:58. doi: 10.1186/1750-1326-5-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wellington C.L., Singaraja R., Ellerby L., Savill J., Roy S., Leavitt B., Cattaneo E., Hackam A., Sharp A., Thornberry N., et al. Inhibiting caspase cleavage of huntingtin reduces toxicity and aggregate formation in neuronal and nonneuronal cells. J. Biol. Chem. 2000;275:19831–19838. doi: 10.1074/jbc.M001475200. [DOI] [PubMed] [Google Scholar]
- 16.Wellington C.L., Ellerby L.M., Gutekunst C.A., Rogers D., Warby S., Graham R.K., Loubser O., van Raamsdonk J., Singaraja R., Yang Y.Z., et al. Caspase cleavage of mutant huntingtin precedes neurodegeneration in Huntington's disease. J. Neurosci. 2002;22:7862–7872. doi: 10.1523/JNEUROSCI.22-18-07862.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gafni J., Hermel E., Young J.E., Wellington C.L., Hayden M.R., Ellerby L.M. Inhibition of calpain cleavage of huntingtin reduces toxicity: accumulation of calpain/caspase fragments in the nucleus. J. Biol. Chem. 2004;279:20211–20220. doi: 10.1074/jbc.M401267200. [DOI] [PubMed] [Google Scholar]
- 18.Miller J.P., Holcomb J., Al Ramahi I., de Haro M., Gafni J., Zhang N., Kim E., Sanhueza M., Torcassi C., Kwak S., et al. Matrix metalloproteinases are modifiers of huntingtin proteolysis and toxicity in Huntington's disease. Neuron. 2010;67:199–212. doi: 10.1016/j.neuron.2010.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graham R.K., Deng Y., Slow E.J., Haigh B., Bissada N., Lu G., Pearson J., Shehadeh J., Bertram L., Murphy Z., et al. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006;125:1179–1191. doi: 10.1016/j.cell.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 20.Warby S.C., Doty C.N., Graham R.K., Carroll J.B., Yang Y.Z., Singaraja R.R., Overall C.M., Hayden M.R. Activated caspase-6 and caspase-6-cleaved fragments of huntingtin specifically colocalize in the nucleus. Hum. Mol. Genet. 2008;17:2390–2404. doi: 10.1093/hmg/ddn139. [DOI] [PubMed] [Google Scholar]
- 21.Graham R.K., Deng Y., Carroll J., Vaid K., Cowan C., Pouladi M.A., Metzler M., Bissada N., Wang L., Faull R.L., et al. Cleavage at the 586 amino acid caspase-6 site in mutant huntingtin influences caspase-6 activation in vivo. J. Neurosci. 2010;30:15019–15029. doi: 10.1523/JNEUROSCI.2071-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tebbenkamp A.T., Swing D., Tessarollo L., Borchelt D.R. Premature death and neurologic abnormalities in transgenic mice expressing a mutant huntingtin exon-2 fragment. Hum. Mol. Genet. 2011 doi: 10.1093/hmg/ddr040. epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mangiarini L., Sathasivam K., Seller M., Cozens B., Harper A., Hetherington C., Lawton M., Trottier Y., Lehrach H., Davies S.W., et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 24.Carter R.J., Lione L.A., Humby T., Mangiarini L., Mahal A., Bates G.P., Dunnett S.B., Morton A.J. Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation. J. Neurosci. 1999;19:3248–3257. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schilling G., Becher M.W., Sharp A.H., Jinnah H.A., Duan K., Kotzuk J.A., Slunt H.H., Ratovitski T., Cooper J.K., Jenkins N.A., et al. Intranuclear inclusions and neuritic pathology in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum. Mol. Genet. 1999;8:397–407. doi: 10.1093/hmg/8.3.397. [DOI] [PubMed] [Google Scholar]
- 26.Borchelt D.R., Davis J., Fischer M., Lee M.K., Slunt H.H., Ratovitsky T., Regard J., Copeland N.G., Jenkins N.A., Sisodia S.S., et al. A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet. Anal. (Biomed. Eng.) 1996;13:159–163. doi: 10.1016/s1050-3862(96)00167-2. [DOI] [PubMed] [Google Scholar]
- 27.Schilling G., Jinnah H.A., Gonzales V., Coonfield M.L., Kim Y., Wood J.D., Price D.L., Li X.J., Jenkins N., Copeland N., et al. Distinct behavioral and neuropathological abnormalities in transgenic mouse models of HD and DRPLA. Neurobiol. Dis. 2001;8:405–418. doi: 10.1006/nbdi.2001.0385. [DOI] [PubMed] [Google Scholar]
- 28.Scherzinger E., Sittler A., Schweiger K., Heiser V., Lurz R., Hasenbank R., Lehrach H., Wanker E.E. Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: implications for Huntington's disease pathology. Proc. Natl Acad. Sci. USA. 1999;96:4604–4609. doi: 10.1073/pnas.96.8.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen S., Berthelier V., Yang W., Wetzel R. Polyglutamine aggregation behavior in vitro supports a recruitment mechanism of cytotoxicity. J. Mol. Biol. 2001;311:173–182. doi: 10.1006/jmbi.2001.4850. [DOI] [PubMed] [Google Scholar]
- 30.Diaz-Hernandez M., Torres-Peraza J., Salvatori-Abarca A., Moran M.A., Gomez-Ramos P., Alberch J., Lucas J.J. Full motor recovery despite striatal neuron loss and formation of irreversible amyloid-like inclusions in a conditional mouse model of Huntington's disease. J. Neurosci. 2005;25:9773–9781. doi: 10.1523/JNEUROSCI.3183-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vonsattel J.P.G., DiFiglia M. Huntington disease. J. Neuropath. Exp. Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- 32.Lin C.H., Tallaksen-Greene S., Chien W.M., Cearley J.A., Jackson W.S., Crouse A.B., Ren S., Li X.J., Albin R.L., Detloff P.J. Neurological abnormalities in a knock-in mouse model of Huntington's disease. Hum. Mol. Genet. 2001;10:137–144. doi: 10.1093/hmg/10.2.137. [DOI] [PubMed] [Google Scholar]
- 33.Heng M.Y., Duong D.K., Albin R.L., Tallaksen-Greene S.J., Hunter J.M., Lesort M.J., Osmand A., Paulson H.L., Detloff P.J. Early autophagic response in a novel knock-in model of Huntington disease. Hum. Mol. Genet. 2010;19:3702–3720. doi: 10.1093/hmg/ddq285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luthi-Carter R., Strand A.D., Hanson S.A., Kooperberg C., Schilling G., La Spada A.R., Merry D.E., Young A.B., Ross C.A., Borchelt D.R., et al. Polyglutamine and transcription: gene expression changes shared by DRPLA and Huntington's disease mouse models reveal context-independent effects. Hum. Mol. Genet. 2002;11:1927–1937. doi: 10.1093/hmg/11.17.1927. [DOI] [PubMed] [Google Scholar]
- 35.Hodges A., Strand A.D., Aragaki A.K., Kuhn A., Sengstag T., Hughes G., Elliston L.A., Hartog C., Goldstein D.R., Thu D., et al. Regional and cellular gene expression changes in human Huntington's disease brain. Hum. Mol. Genet. 2006;15:965–977. doi: 10.1093/hmg/ddl013. [DOI] [PubMed] [Google Scholar]
- 36.Slow E.J., van Raamsdonk J., Rogers D., Coleman S.H., Graham R.K., Deng Y., Oh R., Bissada N., Hossain S.M., Yang Y.Z., et al. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum. Mol. Genet. 2003;12:1555–1567. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- 37.Wang C.E., Tydlacka S., Orr A.L., Yang S.H., Graham R.K., Hayden M.R., Li S., Chan A.W., Li X.J. Accumulation of N-terminal mutant huntingtin in mouse and monkey models implicated as a pathogenic mechanism in Huntington's disease. Hum. Mol. Genet. 2008;17:2738–2751. doi: 10.1093/hmg/ddn175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 39.Schilling G., Coonfield M.L., Ross C.A., Borchelt D.R. Coenzyme Q10 and remacemide hydrochloride amerliorate motor deficits in a Huntington's disease transgenic mouse model. Neurosci. Lett. 2001;315:149–153. doi: 10.1016/s0304-3940(01)02326-6. [DOI] [PubMed] [Google Scholar]
- 40.Luthi-Carter R., Strand A., Peters N.L., Solano S.M., Hollingsworth Z.R., Menon A.S., Frey A.S., Spektor B.S., Penney E.B., Schilling G., et al. Decreased expression of striatal signaling genes in a mouse model of Huntington's disease. Hum. Mol. Genet. 2000;9:1259–1271. doi: 10.1093/hmg/9.9.1259. [DOI] [PubMed] [Google Scholar]
- 41.Rodriguez-Lebron E., Denovan-Wright E.M., Nash K., Lewin A.S., Mandel R.J. Intrastriatal rAAV-mediated delivery of anti-huntingtin shRNAs induces partial reversal of disease progression in R6/1 Huntington's disease transgenic mice. Mol. Ther. 2005;12:618–633. doi: 10.1016/j.ymthe.2005.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hebb A.L., Robertson H.A., Denovan-Wright E.M. Striatal phosphodiesterase mRNA and protein levels are reduced in Huntington's disease transgenic mice prior to the onset of motor symptoms. Neuroscience. 2004;123:967–981. doi: 10.1016/j.neuroscience.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 43.Hu H., McCaw E.A., Hebb A.L., Gomez G.T., Denovan-Wright E.M. Mutant huntingtin affects the rate of transcription of striatum-specific isoforms of phosphodiesterase 10A. Eur. J. Neurosci. 2004;20:3351–3363. doi: 10.1111/j.1460-9568.2004.03796.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.