

Abstract
Deltamethrin, a pyrethroid insecticide, and BTG 502, an alkylamide insecticide, target voltage-gated sodium channels. Deltamethrin binds to a unique receptor site and causes prolonged opening of sodium channels by inhibiting deactivation and inactivation. Previous 22Na+ influx and receptor binding assays using mouse brain synaptoneurosomes showed that BTG 502 antagonized the binding and action of batrachotoxin (BTX), a site 2 sodium channel neurotoxin. However, the effect of BTG 502 has not been examined directly on sodium channels expressed in Xenopus oocytes. In this study, we examined the effect of BTG 502 on wild-type and mutant cockroach sodium channels expressed in Xenopus oocytes. Toxin competition experiments confirmed that BTG 502 antagonizes the action of BTX and possibly shares a common receptor site with BTX. However, unlike BTX which causes persistent activation of sodium channels, BTG 502 reduces the amplitude of peak sodium current. A previous study showed that BTG 502 was more toxic to pyrethroid-resistant house flies possessing a super-kdr (knockdown resistance) mechanism than to pyrethroid-susceptible house flies. However, we found that the cockroach sodium channels carrying the equivalent super-kdr mutations (M918T and L1014F) were not more sensitive to BTG 502 than the wild-type channel. Instead, a kdr mutation, F1519I, which reduces pyrethroid binding, abolished the action of BTG 502. These results provide evidence the actions of alkylamide and pyrethroid insecticides require a common sodium channel residue.
Keywords: BTG 502, batrachotoxin, pyrethroids, sodium channel, knockdown resistance, site-directed mutagenesis
1. Introduction
Voltage-gated sodium channels are essential for the generation and propagation of action potentials in almost all excitable cells (Catterall, 2000). They are large transmembrane proteins containing four homologous domains (I to IV), each formed by six membrane spanning segments (S1 to S6) connected by intracellular and extracellular loop sequences. The S1-S4 segments serve as the voltage sensing module, whereas the S5 and S6 segments and the loops connecting them function as the pore-forming module. In response to membrane depolarization, sodium channels open (activate) and allow sodium ions to flow into the cell, thereby depolarizing the membrane potential. A few milliseconds after channel activation, the channel pore is occluded through a process known as fast inactivation. Following repolarization (i.e., the membrane potential returns to the initial stage), inactivation is removed and the channel is deactivated (closed), returning back to the resting state.
Sodium channels are targeted by a variety of natural neurotoxins, such as batrachotoxin (BTX) from poison dart frogs and synthetic neurotoxins, such as pyrethroid insecticides and local anesthetics. These neurotoxins bind to distinct receptor sites on the sodium channel and alter sodium channel conductance and gating properties (Zlotkin, 1999; Cestele and Catterall, 2000; Wang and Wang, 2003). For example, BTX binds to site 2, and causes persistent channel activation by inhibiting channel inactivation and shifting the voltage dependence of activation in the hyperpolarizing direction.
Sodium channel-targeting insecticides include DDT, pyrethroids, sodium channel blocker insecticides (SCBIs) and N-alkylamides. DDT and pyrethroids prolong the opening of sodium channels, causing repetitive firing or membrane depolarization in the nervous system (Narahashi, 1988; Soderlund and Bloomquist, 1989; Narahashi, 2000). SCBIs, such as indoxacarb, share a similar mode of action with local anesthetics, which cause state-dependent block of sodium channels, leading to nerve conduction block (Wing et al., 2005; Silver and Soderlund 2007; Silver et al., 2010). Limited information is available on the mode of action of N-alkylamides (Bloomquist, 1996). Effects of an N-alkylamide insecticide, BTG 502, on sodium channels were suggested from studies using mouse brain synaptoneurosomes in 22Na+ influx and [3H]batrachotoxinin A-20-α-benzoate (BTX-B) binding assays (Ottea et al., 1989; 1990). BTG 502 alone did not have any effect on 22Na+ uptake in mouse brain synaptoneurosomes; however, it activated 22Na+ uptake when co-incubated with saturating concentrations of Leiurus quinquestriatus scorpion venom. Furthermore, BTG 502 inhibited BTX-dependent 22Na+ uptake in the absence or presence of Leiurus quinquestriatus scorpion venom. It also inhibited the binding of [3H]BTX-B to sodium channels. These results suggest that BTG 502 and BTX share a common receptor site, site 2, on the sodium channel (Ottea et al., 1989; 1990). However, the effect of BTG 502 has not been directly examined on insect or mammalian sodium channels expressed in Xenopus oocytes.
Among the sodium channel-targeted insecticides, pyrethroids have been extensively used in arthropod pest control. As a result, unfortunately, many pest populations have developed resistance to pyrethroids. One major resistance mechanism, known as knockdown resistance (kdr) or super-kdr (conferring a greater level of pyrethroid resistance than kdr), is caused by mutations in the sodium channel protein. These mutations reduce or abolish the action of pyrethroids (Soderlund, 2005; Davies et al., 2007; Dong, 2007). In many cases, once a pest population develops resistance to one pyrethroid, the population often becomes cross-resistant to the entire class of pyrethroids. Pyrethroid resistant insects have not been reported to be cross-resistant to SCBIs, such as indoxacarb. However, a previous study, based on laboratory insecticide bioassay, shows that super-kdr house flies are hypersusceptible to BTG 502 (Elliott et al., 1986). This negative cross-resistance of super-kdr flies to BTG 502 is interesting because although N-alkylamides are not currently used in pest control due to chemical instability and/or unfavorable mammalian toxicity, they may represent an excellent probe to investigate potential interactions of different receptor sites on the sodium channel at the molecular level. Therefore, in this study, we examined the effect of BTG 502 on cockroach sodium channels expressed in Xenopus oocytes and compare the responses of pyrethroid-sensitive and resistant sodium channels to BTG 502 to gain insights into possible molecular interactions between N-alkylamides and pyrethroids on insect sodium channels.
2. Materials and Methods
2.1 Site-Directed Mutagenesis
cDNA from the pyrethroid-sensitive cockroach sodium channel, BgNav1-1a (Song et al, 2004), was used to generate mutant constructs. Site-directed mutagenesis was performed by polymerase chain reaction (PCR) using mutant primers and Pfu Turbo DNA polymerase (Stratagene, La Jolla, CA). All mutagenesis results were verified by DNA sequencing.
2.2 Expression of BgNav Sodium Channels in Xenopus Oocytes
The procedures for oocyte preparation and cRNA injection were identical to those described previously (Tan et al., 2002). For robust expression of BgNav sodium channels, cRNA was co-injected into oocytes with cRNA encoding the Drosophila melanogaster TipE auxiliary subunit (1:1 ratio), which enhances the expression of insect sodium channels in oocytes.
2.3 Electrophysiological Recording and Analysis
Methods for electrophysiological recording and data analysis were identical to those described previously (Tan et al., 2002; 2005). To measure the effect of BTG 502 on the sodium channel, sodium currents were elicited by a 20-ms test pulse to -10 mV from a holding potential of -120 mV after 100 repetitive depolarizing pulses to -10 mV at 10 Hz. Data are presented as mean ± S.D. A one-way ANOVA with Scheffe’s post hoc analysis was used to evaluate the significance of changes in mean values. P values <0.05 were considered statistically significant.
2.4 Chemicals
BTG 502 and deltamethrin were from Rothamsted Research, Harpenden, UK. BTX was a generous gift from John Daly (National Institutes of Health, Bethesda, MD). Stock solutions of BTX (1 mM), BTG 502 (50 mM) and deltamethrin (100 mM) were dissolved in dimethyl sulfoxide (DMSO). The working concentration was prepared in ND96 recording solution immediately prior to experiments. The concentration of DMSO in the final solution was < 0.5%, which had no effect on the function of sodium channels. The method for application of chemicals in the recording system was identical to that described by Tan et al. (2002). Effects of deltamethrin, BTX, and BTG 502 were measured 10 min after toxin application.
3. Results and discussion
3.1 BTG 502 inhibits sodium currents
We first examined the effect of BTG 502 on a pyrethroid-sensitive cockroach sodium channel variant, BgNav1-1a, expressed in Xenopus oocytes using two-electrode voltage clamp. BTG 502 reduced the peak sodium current when a train of repetitive depolarizing prepulses was delivered at a frequency of 10 Hz (Fig. 1B). No peak current inhibition was observed in the absence of the depolarizing prepulses (Fig. 1C). BTG 502, therefore, appears to reduce sodium current by binding to the open-state sodium channels. The maximal peak current inhibition was 50% by 10 μM of BTG 502 at 100 repetitive prepulses (Fig. 1D). BTG 502 had no effect on the voltage dependence of activation or fast inactivation of BgNav1-1a channels (Fig. 1E and F). The BTG 502 effect is strikingly different from the effects of BTX, which inhibits channel inactivation and causes persistent activation by shifting the voltage dependence of activation in the hyperpolarizing direction (Tan et al., 2005).
Fig. 1.
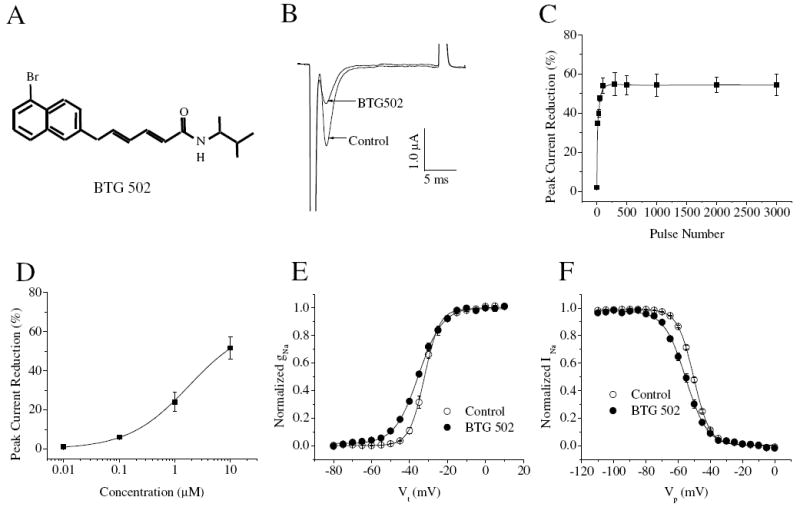
Effects of BTG 502 on the BgNav1-1a channel. (A) The chemical structure of BTG 502. (B) BTG 502 reduced peak sodium current. (C) Use-dependent inhibition of peak current by BTG 502 (10 μM). (D) Dose-response curve of BTG 502 action. (E and F) The voltage dependence of activation (E) and inactivation (F) before and after the application of 10 μM of BTG 502. After 100 repetitive depolarizing pulses at 10 Hz, sodium currents were elicited by a 20-ms test pulse to -10 mV from the holding potential of -120 mV before and after the application of 10 μM of BTG 502. The activation and inactivation curves were fitted with two-state Boltzmann equations.
3.2 BTX and BTG 502 competition experiments
To explore possible competition between BTX and BTG 502 for the same binding site on the sodium channel, we conducted toxin competition experiments by measuring the effects of BTG 502 on BgNav1-1a channels after BTX pre-treatment (Fig. 2A-C), and vice versa (Fig. 2D-F). For both types of experiments, we chose a recording protocol that included a 20-ms test pulse to -10 mV from a holding potential of -120 mV after 3,000 repetitive depolarizing pulses to -10 mV at a frequency of 10 Hz. This protocol has been previously used to describe the effects of BTX on cockroach sodium channels (Tan et al., 2005) and is suitable for observing the effects of BTG 502.
Fig. 2.
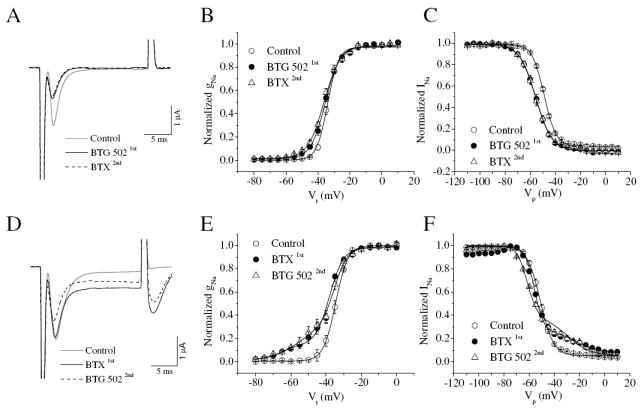
Toxin competition experiments. (A-C) BTX treatment followed by BTG 502 application. (D-F) BTG 502 treatment followed by BTX application. A and D: sodium current traces; B and E: voltage-dependence of activation; C and F: voltage-dependence of inactivation. 10 μM BTG 502 and 500 nM BTX were applied in these experiments. Sodium current was elicited by a 20-ms test pulse to -10 mV from a holding potential of -120 mV after 3,000 repetitive depolarizing pulses to -10 mV at a frequency of 10 Hz.
As expected, pretreatment of oocytes with BTG 502 (10 μM) reduced peak sodium current of BgNav1-1a channels (Fig. 2A). However, subsequent application of BTX (500 nM) did not affect the action of BTG 502, indicating that BTX cannot alter the action of BTG 502 when added after BTG 502 (Fig. 2A, B and C). It is possible that the pretreatment of BTG 502 could have modified 100% of channels, which prevents the binding and/or action of BTX.
Pretreatment of BgNav1-1a channels with BTX (500 nM) induced a large tail current as well as a non-inactivating component of inactivation, which are characteristic of BTX effects on the sodium channel (Fig. 2D). Following subsequent treatment with BTG 502 (10 μM), both the BTX-induced tail current and non-inactivating current were reduced and inhibition of peak sodium current by BTG 502 was observed, suggesting that BTG 502 could partially antagonize the BTX action even when added after BTX. Alternatively, BTX dissociates rapidly enough so that subsequent BTG 502 can modify the channels. However, the negative shift in the voltage dependence of activation by BTX was not reduced by application of BTG 502 (Fig. 2E and F).
The partial BTG 502 antagonism of the action of BTX on insect sodium channels expressed in oocytes is consistent with the findings by Soderlund and colleagues who showed that BTG 502 inhibits BTX binding in mouse brain synaptoneurosomes (Ottea et al., 1989; 1990). These results suggest that BTX and BTG 502 may share overlapping binding sites on sodium channels. However, the two toxins must have somewhat distinct binding and/or action properties that translate into distinct electrophysiological effects.
3.3 Sensitivity of the BgNav1-1a channel to BTG 502 was not altered by super-kdr mutations, but by another kdr mutation, F1519I in IIIS6
Although super-kdr house flies were reported to be hyper-susceptible to BTG 502 than pyrethroid-susceptible house flies (Elliott et al., 1986), an earlier electrophysiological study using isolated axonal preparations showed that the nerve sensitivity of super-kdr house flies to BTG 502 was not different from that of pyrethroid-susceptible house flies (Gibson et al., 1990). To determine whether the super-kdr mutations enhanced the action of BTG 502 on cockroach sodium channels expressed in Xenopus oocytes, we compared the effects of BTG 502 on a pyrethroid-sensitive BgNav1-1a channel and a mutant channel carrying both the M897T and L993F mutations, which are equivalent to the house fly super-kdr mutations M918T and L1014F (Fig. 3A). The double mutations almost completely abolished the action of deltamethrin, a type II pyrethroid, on cockroach sodium channels (Fig. 3B), similar to previous reports in house fly and Drosophila sodium channels (Lee et al., 1990; Vais et al., 2000). The double mutations did not enhance the sensitivity of cockroach sodium channels to BTG 502 (Fig. 3C), which is consistent with the earlier electrophysiological report (Gibson et al., 1990). Our results suggest that the enhanced susceptibility of super-kdr house flies to BTG 502 is not caused by the super-kdr mutations. Alternatively, the effect of super-kdr mutations on the action of BTG 502 depends on insect species.
Fig. 3.
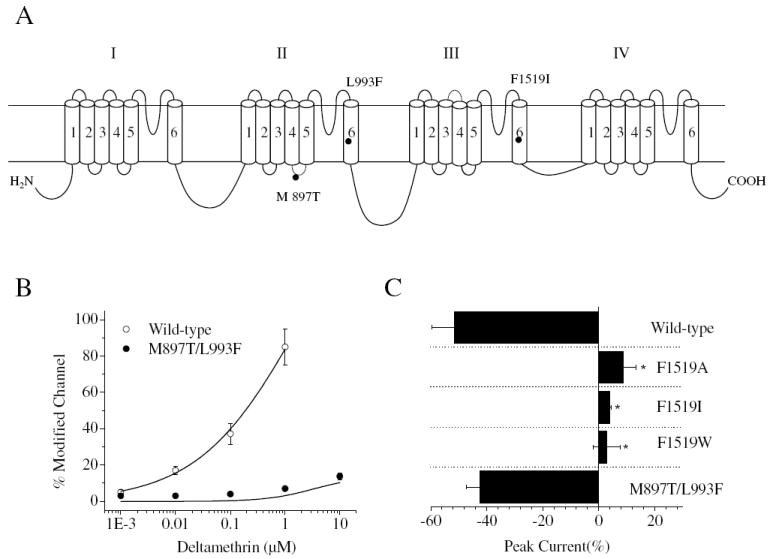
Effects of naturally occurring sodium channel mutations on the action of deltamethrin and BTG 502. (A) Positions of pyrethroid-resistant mutations in this study are indicated in the topology of the cockroach sodium channel protein. Sodium channels have four homologous domains (I-IV) each with six transmembrane segments (S1-S6). M897T and L993F correspond to the super-kdr mutations M918T and L1014F in the house fly sodium channel Vssc1. F1519I corresponds to the F to I mutation detected in pyrethroid resistant southern cattle tick (He et al., 1999). (B) Double mutations, M897T/L993F, abolished the action of deltamethrin on BgNav1-1a channels. (C) Effects of pyrethroid-resistant mutations, M897T/L993F, F1519I, F1519A and F1519W, on peak current reduction by BTG 502 (10 μM).
* Statistically significant different compared with BgNav1-1a.
We examined the effect of another kdr mutation, F1519I in IIIS6 (He et al., 1999), on the action of BTG 502. The F1519I mutation abolished the action of pyrethroids on cockroach sodium channels expressed in Xenopus oocytes (Tan et al., 2005). Surprisingly, the F1519I substitution also abolished the inhibitory action of BTG 502 (Fig. 3C). We then examined the effect of two additional substitutions, F1519A and F1519W. The F1519A channel, like the F1519I channel, was completely insensitive to deltamethrin, whereas the F1519W channel exhibited 10-fold reduced sensitivity to deltamethrin (Tan et al., 2005). F1519A/W substitutions also completely abolished the inhibitory action of BTG 502, indicating that F1519 is critical for the action of BTG 502. BTG 502 (10 μM) slightly enhanced the amplitude of peak current of all three mutant channels (Fig. 3C).
The requirement of F1519 for the activities of both deltamethrin and BTG 502 provides a molecular link between the actions of pyrethroids and BTG 502 on insect sodium channels. Computer modeling of the pyrethroid-binding site using the X-ray structure of the Kv1.2 potassium channel as a template predicts that the pyrethroid receptor site is located in a hydrophobic pocket formed by the IIS4-S5 linker and IIS5 and IIIS6 helices (O’Reilly et al., 2006). Based on this model, in the open-state sodium channel, several kdr mutation sites, including the super-kdr mutation M918T (i.e., M897T in BgNav) in the IIS4-S5 linker, L925I and T929I in IIS5 and F1538I (i.e., F1519I in BgNav) in IIIS6, are located in or near the hydrophobic pocket. Most of these sites are likely in contact with pyrethroids, although the details of contact may vary depending on the chemical structures of pyrethroids. Furthermore, for the F1519I mutation, Schild analysis showed that this mutation reduced the binding of an inactive 1S cis permethrin isomer, which competes for the same receptor site with an active 1R cis permethrin isomer, suggesting that F1519 is part of the pyrethroid receptor site (Tan et al., 2005). Our results in this study show that F1519, but not M897, is involved in BTG 502 action, suggesting distinct but possibly overlapping receptor sites of BTG 502 and pyrethroids.
The receptor sites for BTX and pyrethroids are distinct and are localized at the opposite sides of the IIIS6 helix (Tan et al, 2005; Du et al., 2009; Du et al., 2011). The F1519I mutation in IIIS6 did not alter the action of BTX (Tan et al., 2005), indicating that F1519 is not part of the BTX receptor site. Further experiments using systematic site-directed mutagenesis of residues in IIIS6 and computer modeling will provide valuable insight into the molecular basis of BTG 502 binding and action on sodium channels.
Acknowledgments
We thank Dr. Kris Silver and anonymous reviewers for critical review of this manuscript; and Dr. Zhiqi Liu and Ms. Yoshiko Nomura for making the mutant constructs used in this study. This study was supported by a grant to KD from National Institutes of Health (GM057440).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Catterall WA. From ionic currents to molecular mechanisms: The structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- Cestele S, Catterall WA. Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie. 2000;82:883–892. doi: 10.1016/s0300-9084(00)01174-3. [DOI] [PubMed] [Google Scholar]
- Davies TGE, Field LM, Usherwood PNR, Williamson MS. DDT, pyrethrins, pyrethroids and insect sodium channels. IUBMB Life. 2007;59:151–162. doi: 10.1080/15216540701352042. [DOI] [PubMed] [Google Scholar]
- Dong K. Insect sodium channels and insecticide resistance. Invert Neurosci. 2007;7:17–30. doi: 10.1007/s10158-006-0036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Lee JE, Nomura Y, Zhang T, Zhorov BS, Dong K. Identification of a cluster of residues in transmembrane segment 6 of domain III of the cockroach sodium channel essential for the action of pyrethroid insecticides. Biochem J. 2009;419:377–385. doi: 10.1042/BJ20082082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Garden D, Wang L, Zhorov BS, Dong K. identification of new batrachotoxin-sensing residues in segment IIIS6 of sodium channel. J Biol Chem. 2011 doi: 10.1074/jbc.M110.208496. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott M, Farnham AW, Janes NF, Johnson DM, Pulman DA. Insecticidal amides with selective potency against a resistant (Super-kdr) strain of houseflies (Musca domestica L.) Agric Biol Chem. 1986;50:1347–1349. [Google Scholar]
- Gibson AJ, Osborne MP, Ross HF, Sawicki RM. An electrophysiological study of susceptible (Cooper) and resistant (kdr; super-kdr) strains of the adult housefly (Musca domesticaL.) using an isolated mesothoracic leg preparation. Pestic Sci. 1990;30:379–396. [Google Scholar]
- He H, Chen AC, Davey RB, Ivie GW, George JE. Identification of a point mutation in the para-type sodium channel gene from a pyrethroid-resistant cattle tick. Biochem Biophys Res Comm. 1999;261:558–561. doi: 10.1006/bbrc.1999.1076. [DOI] [PubMed] [Google Scholar]
- Lee SH, Smith TJ, Knipple DC, Soderlund DM. Mutations in the housefly Vssc1 sodium channel gene associated with super-kdr resistance abolish the pyrethroid sensitivity of Vssc1/tipE sodium channels expressed in Xenopus oocytes. Insect Biochem Mol Biol. 1999;29:185–194. doi: 10.1016/s0965-1748(98)00122-2. [DOI] [PubMed] [Google Scholar]
- Linford NJ, Cantrell AR, Qu Y, Scheuer T, Catterall WA. Interaction of batrachotoxin with the local anesthetic receptor site in transmembrane segement IVS6 of the voltage-gated sodium channel. Proc Natl Acad Sci. 1998;95:13947–13952. doi: 10.1073/pnas.95.23.13947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narahashi T. Molecular and cellular approaches to neurotoxicology: past, present and future. In: Lunt GG, editor. Neurotox ’88: Molecular Basis of Drug & Pesticide Action. Elsevier; New York: 1988. pp. 563–582. [Google Scholar]
- Narahashi T. Neuroreceptors and ion channels as the basis for drug action: past, present, and future. J Pharmacol Exp Ther. 2000;294:1–26. [PubMed] [Google Scholar]
- O’Reilly AQ, Khambay BPS, Williamson MS, Field LM, Wallace BA, Davies TGE. Modeling insecticide binding sites at the voltage-gated sodium channel. Biochem J. 2006;396:255–263. doi: 10.1042/BJ20051925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottea JA, Payne GT, Soderlund DM. Action of insecticidal N-alkylamides at site 2 of the voltage-sensitive sodium channel. J Agric Food Chem. 1990;38:1724–1728. [Google Scholar]
- Ottea JA, Payne GT, Bloomquist TR, Soderlund DM. Activation of sodium channels and inhibition of [3H]-batrachotoxinin A-20-α-benzoate binding by an N-alkylamide neurotoxin. Mol Pharmacol. 1989;36:280–284. [PubMed] [Google Scholar]
- Silver K, Soderlund DM. Point mutations at the local anesthetic receptor site modulate the state-dependent block of rat Nav1.4 sodium channels by pyrazoline-type insecticides. Neurotoxicology. 2007;28:655–663. doi: 10.1016/j.neuro.2007.02.001. [DOI] [PubMed] [Google Scholar]
- Silver KS, Song W, Nomura Y, Salgado VL, Dong K. Mechanism of action of sodium channel blocker insecticides on insect sodium channels. Pestic Biochem Physiol. 2010;97:87–92. doi: 10.1016/j.pestbp.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderlund DM. Sodium channels. In: Gilbert LI, Iatrou K, Gill SS, editors. Comprehensive Molecular Insect Science. Vol. 6. Elsevier; New York: 2005. pp. 1–24. [Google Scholar]
- Soderlund DM, Bloomquist JR. Neurotoxic actions of pyrethroid insecticides. Annu Rev Entomol. 1989;34:77–96. doi: 10.1146/annurev.en.34.010189.000453. [DOI] [PubMed] [Google Scholar]
- Song W, Liu Z, Dong K. Molecular basis of differential sensitivity of insect sodium channels to DCJW, a bioactive metabolite of the oxadiazine insecticide indoxacarb. Neurotoxicology. 2006;27:237–244. doi: 10.1016/j.neuro.2005.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Liu Z, Nomura Y, Goldin AL, Dong K. Alternative splicing of an insect sodium channel gene generates pharmacologically distinct sodium channels. J Neurosci. 2002;22:5300–5309. doi: 10.1523/JNEUROSCI.22-13-05300.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Liu Z, Wang R, Huang ZY, Chen AC, Gurevitz M, Dong K. Identification of amino acid residues in the insect sodium channel critical for pyrethroid binding. Mol Pharmacol. 2005;67:513–522. doi: 10.1124/mol.104.006205. [DOI] [PubMed] [Google Scholar]
- Vais H, Williamson MS, Goodson SJ, Devonshire AL, Warmke JW, Usherwood PNR, Cohen C. Activation of Drosophila sodium channels promotes modification by deltamethrin: reductions in affinity caused by knock-down resistance mutations. J Gen Physiol. 2000;115:305–318. doi: 10.1085/jgp.115.3.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SY, Wang GK. Voltage-gated sodium channels as primary targets of diverse lipid-soluble neurotoxins. Cellular Signaling. 2003;15:151–159. doi: 10.1016/s0898-6568(02)00085-2. [DOI] [PubMed] [Google Scholar]
- Wing KD, Andaloro JT, McCann SF, Salgado VL. Indoxacarb and the sodium channel blocker insecticides: chemistry, physiology, and biology in insects. In: Gilbert LI, Iatrou K, Gill SS, editors. Comprehensive Molecular Insect Science. Vol. 6. Elsevier; New York: 2005. pp. 31–53. [Google Scholar]
- Zlotkin E. The insect voltage-gated sodium channel as target of insecticides. Annu Rev Entomol. 1999;44:429–455. doi: 10.1146/annurev.ento.44.1.429. [DOI] [PubMed] [Google Scholar]