

Abstract
Apolipoprotein (apo) A-I-containing lipoproteins in the form of high density lipoproteins (HDL) are inversely correlated with atherosclerosis. Because HDL is a soft form of condensed matter easily deformable by thermal fluctuations, the molecular mechanisms for HDL remodeling are not well understood. A promising approach to understanding HDL structure and dynamics is molecular dynamics (MD). In the present study, two computational strategies, MD simulated annealing (MDSA) and MD temperature-jump, were combined with experimental particle reconstitution to explore molecular mechanisms for phospholipid (PL)-rich HDL particle remodeling. The N-terminal domains of full length apoA-I were shown to be “sticky”, acting as a molecular latch largely driven by salt bridges, until, at a critical threshold of particle size, the associated domains released to expose extensive hydrocarbon regions of the PL to solvent. The “sticky” N-termini also associate with other apoA-I domains, perhaps being involved in N-terminal loops suggested by other laboratories. Alternatively, the overlapping helix 10 C-terminal domains of apoA-I were observed to be extremely mobile or “promiscuous”, transiently exposing limited hydrocarbon regions of PL. Based upon these models and reconstitution studies, we propose that separation of the N-terminal domains, as particles exceed a critical size, trigger fusion between particles or between particles and membranes, while the C-terminal domains of apoA-I drive the exchange of polar lipids down concentration gradients between particles. This hypothesis has significant biological relevance since lipid exchange and particle remodeling are critically important processes during metabolism of HDL particles at every step in the anti-atherogenic process of reverse cholesterol transport.
High density lipoproteins (HDL) are inversely correlated with the risk of coronary artery disease (CAD) and atherosclerosis (1) and so HDL represents an important target for pharmacological therapy of CAD. This lipoprotein represents a heterogeneous population of particles with apolipoprotein (apo) A-I as the major protein. Whether HDL plays a direct role in CAD prevention (e.g., removal of cholesterol from clogged arteries (1)) or an indirect one (e.g., acts as a platform for the clustering of protective molecules (2), such as anti-inflammatory or antioxidant proteins), knowledge of HDL structure and dynamics is desirable.
Of the possible mechanisms that have been suggested to explain the protective role of apoA-I, a process called reverse cholesterol transport is most completely understood at the molecular level. Reverse cholesterol transport consists of three relatively well-understood steps: First, the transmembrane phospholipid (PL) pump protein, ATP-binding cassette transporter A1 (ABCA1) (3), is required for the assembly of newly synthesized (lipid-poor) apoA-I into discoidal, PL-rich HDL, resulting, as a consequence, in unesterified cholesterol (UC) efflux from cells. Second, the activation of the enzyme lecithin:cholesterol acyl transferase (LCAT) (4) by apoA-I is necessary for esterification of the cholesterol molecules of HDL to cholesteryl ester (CE) and leads to the conversion of PL-rich to spheroidal, CE-rich (circulating) HDL. Third, interaction of CE-rich HDL with receptors, such as the scavenger receptor B, type I (SR-BI) (5), leads to CE uptake by the liver and excretion into the bile. This interaction results in remodeling of the CE-rich HDL particle, a process that regenerates lipid-poor HDL (6).
A fuller understanding of reverse cholesterol transport requires knowledge of the detailed structure and dynamics of the various HDL particles and the intermediates in their assembly, knowledge that should help illuminate the structural mechanisms involved in HDL particle remodeling. However, since HDL, a supramolecular assembly of lipid and protein, is a nanoscale soft form of condensed matter easily deformable by thermal fluctuations (7), direct experimental methods for studying HDL structure-function have had only limited success. Adding to the problem of determining HDL structure, and partly the result of the soft matter nature of HDL, the conformation of apoA-I is highly plastic. Since it appears to have a broad conformational space, a more complete understanding of HDL structure and dynamics necessitates liberal use of computer simulations cross-checked by experimental testing through approaches such as site-directed mutagenesis and physical chemical methods for establishing distance constraints.
In 1999, our lab developed a detailed molecular double belt model for discoidal HDL (8, 9). The general features of this model, the so-called LL5/5 registration, have been confirmed by several laboratories using a variety of physical chemical methods (10-24). Recognizing the profound constraints imposed on the conformations of lipid-associated apolipoproteins by lipid (25), we turned to molecular dynamics (MD) simulations of the detailed molecular double belt model to aid in solving HDL structure/dynamics (25-29). While MD simulation has been used by several other research groups to examine HDL structure (30-39), a feature of our approach that distinguishes it from all but three of these studies is that we considered double belt registration in creating our initial models. Only an all-atom MD simulation of apoA-I:Milano in discoidal HDL (32), a registration that differs considerably from the “native” LL5/5 registry, an all-atom MD simulation of a non-discoidal structure, the double superhelix model (37), and a coarse grained MD simulation of spherical HDL (38) have considered double belt registration in model creation. Give our emphasis on interactions between terminal domains of the double belt model, formation of particles with “native” double belt registration is critical.
One approach for overcoming kinetic trapping is MD simulated annealing (MDSA) (40-43). This procedure, when used to refine x-ray crystallography and NMR structures, after placing constraints on the initial protein structure in implicit solvent, applies temperature jumps (to 500-1000 K or so), followed by slow cooling to physiologic temperatures. Since we wanted to overcome energy barriers during the simulations, we developed optimal conditions for unconstrained (29), as opposed to constrained (40-43) MDSA. We reasoned that the lipid in the HDL assemblies would place its own considerable restraints on apoA-I (44).
In a recent publication (25), we used temperature-jump (T-jump) MD simulations to 500 K and found that, upon bilayer expansion at the elevated temperature, the conformations of the overlapping N- and C-terminal domains in the particles were unusually mobile, exposing hydrocarbon regions of the PL to solvent. On the basis of these results, we proposed that the terminal domains of apoA-I function as polar lipid remodeling switches for HDL particles (25).
In the current study we use a combination of MDSA and T-jump MD to test our remodeling hypothesis in PL-rich HDL particles that differ from those of our previous T-jump study (25) in that the particles are formed from full length (FL)-apoA-I and contain, in addition to palmitoyloleoylphosphatidylcholine (POPC), UC. Through a combination of computation and experimentation, we provide evidence that: i) The overlapping N-terminal proline-rich domains of FL-apoA-I are “sticky”; they are latched together predominantly by salt bridges until, at a critical threshold of particle size, sufficient hydrocarbon regions of the PL are exposed to solvent to drive particle-particle and/or particle-membrane fusion. ii) The C-terminal (helix 10) domains of apoA-I are quite mobile (‘promiscuous’) and may drive an exchange of polar lipid between particles through creation of metastable exposure of lipid acyl chains to solvent.
Experimental Procedures
Computational
Description of PL-rich apoA-I/HDL particles
Fig. 1A is a schematic diagram that illustrates the size and composition of the six known reconstituted PL-rich apoA-I/HDL particles containing two apoA-I per particle (R2-0 to R2-5) and their relationship to PL type and degree of truncation of apoA-I (9, 44). Briefly, DMPC is unable to form the smallest particle (R2-0), POPC cannot form the larger two particles (R2-4 and R2-5) (44), all but the smallest particle (R2-0) can be reconstituted from FL-apoA-I and DMPC (9), only the two smallest particles (R2-0 and R2-1) can be reconstituted with truncated Δ43 apoA-I and POPC, while three particles (R2-0, R2-1, R2-2 and R2-3) can be reconstituted from truncated Δ10 apoA-I and DMPC) (44).
FIGURE 1. Schematics of the creation, characteristics, and modeling of apoA-I/HDL R2 particles.


A. Schematics of the composition and size of apoA-I/HDL R2 particles. B. Flow diagram of the creation of the different R2 particles and ensembles by MD, MDSA (30 ns total simulation time), and T-jump (20 ns total simulation time).
MD simulations
All-atom MD, MDSA and T-jump simulations were performed using NAMD (45) as described (25, 27). Each system was ionized and charge neutralized with NaCl to 0.15 M with the Autoionize plug-in of Visual Molecular Dynamics (VMD) (46). The TIP3P water model was used (47). The CHARMM 22 (48, 49) and 27 (50, 51) force fields were used for protein and lipid molecules, respectively. For simulations at a fixed temperature, velocity reassignments occurred every 1 ns to prevent the “flying ice cube” effect (52). MDSA and T-jump simulations were replicated to give ensembles of trajectories and final structures for each experiment (see Fig. 1B).
Creation of seven ensembles of PL-rich apoA-I/HDL from an initial R2-20 particle
Seven ensembles were created from an initial particle (R2-20) containing 160 POPC, 24 UC, and 2 apoA-I molecules (160:24:2); R2-20 itself was created as described elsewhere (44). As shown in Fig. 1B, four ensembles, R2-24, R2-216, R2-14 and R2-04, were created by MDSA and three ensembles, X2-24, X2-14 and X2-04, were created by T-jump. For creation of the smaller particles, R2-10 and R2-00, POPC and UC were removed centrally (27) and MD equilibration was applied as described in detail elsewhere (44).
Creation of a larger particle, 240:36:2
Starting with a particle containing 252 POPC and 2 apoA-I molecules, 12 POPC were added to each side of the disc (totaling 276) and were then reabsorbed by 5 ns MD simulation at 310K. Then 36 of the POPC were mutated to UC and the particle simulated for an additional 5 ns at 310K. The resulting particle was subjected to two replications of 10 ns 500K T-jump.
Average Cα coordinate structures
Structures created by averaging the Cα coordinates over all final structures were calculated for the MDSA and T-jump ensembles by using the “measure avpos” command in VMD after aligning over the Cα atoms of the solvent inaccessible salt bridges (residues 77-188).
Generation of salt bridge and molecular contact maps
To obtain the salt bridge analyses over the last 20% of the sixteen 160:24:2 MDSA simulations (R2-216), the VMD “saltbr” command was performed to itemize all intrahelical and interhelical salt bridges found within that portion of the simulation as well as calculate the average distances. These salt bridge distances were collected for each pair of residues forming salt bridges for all sixteen replications of the ensemble.
To obtain the molecular contact maps over the last 10 ns (33%) of the R2-216 ensemble trajectories, the number of times that interhelical Cα atoms were within 25 Å of each other were counted and then plotted relative to their frequency using GNUPLOT (www.gnuplot.info).
Calculation of surface areas
In our previous T-jump paper (25), we used a surface triangulation algorithm to derive a T-jump MD simulation lipid expansion factor of 40%. More recently, we have used calculations of the convex hull of the lipid surfaces to calculate a lipid expansion factor of 50% for our T-jump simulations (Fig. S1 in Supporting Information). In the current paper, we use a range of 40-50% for the expansion factor.
Experimental
Purification of human plasma apoA-I and recombinant (Δ43)apoA-I
Human plasma apoA-I was isolated from the HDL fraction of fresh human plasma and purified according to the procedure described previously (53). To produce the N-terminus truncated (Δ43)apoA-I, human (Δ43)apoA-I cDNA in plasmid pGEMEX was expressed in E. coli BL21/DE3 cells and the recombinant protein was purified as described by Rogers et al (54). Briefly, the expression of apoA-I was monitored by Western blotting analysis of the bacterial lysate with anti-human apoA-I antibody. The bacterial lysate containing mutant apoA-I was loaded onto a preparative reversed-phase HPLC (C4) column and proteins were eluted and separated by a gradient of acetonitrile with 0.1% trifluoroacetic acid. Preparative HPLC fractions containing apoA-I (identified by immuno-dot-blots) were then subjected to purity and identity analyses. The purity of expressed proteins was examined by analytical HPLC (C18) and SDS-PAGE. The identity of the proteins was confirmed by mass spectrometry and N-terminal amino acid sequencing. Purified proteins were lyophilized and stored at −20°C or -80°C.
Preparation of wild type and mutant apoA-I solutions
Lyophilized protein was solubilized in 6 M guanidine hydrochloride. The solubilized protein solution was loaded on a desalting column (Econo-Pac 10DG Disposable Chromatography Columns, BioRad, Hercules, CA) and the protein was eluted by PBS buffer (0.02 M phosphate, 0.15 M NaCl, pH 7.4). An extinction coefficient of 1.13 ml/(mg-cm) and 0.991 ml/(mg-cm) at 280 nm was used for determining the concentration of full-length apoA-I and (Δ43)apoA-I, respectively, in 6 M guanidine hydrochloride (54).
Preparation and analysis of reconstituted HDL (apoA-I/HDL) assemblies
POPC was purchased from Avanti Polar Lipids, Inc. (Alabaster, AL) and used without further purification. Reconstituted HDL assemblies were prepared by the sodium cholate dialysis procedure as described previously (9, 55). The incubation temperature was set above the transition temperature for POPC at 4°C. The particles formed were analyzed initially by non-denaturing gradient gel electrophoresis (NDGGE) on a 4-20% gradient Tris-glycine acrylamide gel (Invitrogen, Carlsbad, CA). High molecular weight marker (Amersham Cat # 17-0445-01) with known Stokes diameters (Sd) were used as standards: thyroglobulin (170 Å), ferritin (122 Å), catalase (104 Å), lactate dehydrogenase (82 Å), and albumin (71 Å). The gels were stained with the colloidal blue staining kit (Invitrogen, Carlsbad, CA). With reference to protein molecular markers, the Stokes diameters of different sized particles in the complexes were calculated based on density scans of the gels by LabWorks image acquisition and analysis software (UVP Inc., Upland, CA).
Results
Experimental Studies
Evidence for mechanisms involved in remodeling of PL:apoA-I particles
In a recent publication (25) we proposed that increased mobility of the terminal overlap domains was an important factor leading to remodeling of PL-rich apoA-I/HDL particles. A limitation of this hypothesis was the use of truncated rather than FL-apoA-I.
Fig. 2 explores the ability of reconstituted POPC-rich HDL particles containing FL-apoA-I to be remodeled. In Fig. 2A we show that addition of apoA-I to pre-formed POPC:FL-apoA-I particles drives the formation of smaller particles. In Fig. 2B we demonstrate that addition of bulk POPC as multilamellar vesicles (MLV) has no observable effect on size and distribution of the particles, even after incubation overnight at 37 °C. Finally, in Fig. 1C we show that co-incubation of smaller- and larger-sized POPC:apoA-I particles (R2-0 and R2-2) results in particle remodeling through the formation of an intermediate-sized particle (R2-1) at higher incubation temperatures.
FIGURE 2. Remodeling of pre-formed POPC:apoA-I particles by NDGGE analysis.

The gels (4-20% polyacrylamide) were run for 48 hrs and stained with colloidal blue. A. Preformed POPC:apoA-I complexes at the 160:2 molar ratio were subjected to further incubation for 16 hrs at 4, 25, or 37°C without (-) (lane 1, 3, & 5) or with (+) (lane 2, 4, & 6) additional apoA-I to reach a final POPC:apoA-I ratio of 80:2. The results showed that the addition of apoA-I drove the formation of smaller particles with an increase of incubation temperature. B. Preformed POPC:apoA-I complexes at the 200:2 molar ratio were subjected to further incubation for 16 hrs at 4, 25, or 37°C without (-) (lane 1, 3, & 5) or with (+) (lane 2, 4, & 6) additional POPC in the form of MLV to reach a final POPC:apoA-I ratio of 400:2. The results showed that further incubations at increased temperatures sharpened the distribution of POPC:apoA-I assemblies in 16 hrs. but addition of POPC to preformed assemblies (predominantly R2-2) did not change the size distribution of these particles regardless of incubation temperatures C. Preformed R2-0 POPC:Δ43apoA-I particles at a 50:2 ratio and R2-2 POPC:apoA-I particles at a 160:2 ratio were incubated separately (lane 4-6 and lane 7-9) or together (lane 1-3) for an additional 16 hrs at 4, 25, or 37°C, respectively. The results showed that a new intermediate-sized particle (R2-1) (solid arrowhead) was formed at the highest incubation temperature.
We have shown that lipid transfer between bulk dimyristoylphosphatidylcholine (DMPC) and pre-formed DMPC:apoA-I apoA-I/HDL assemblies occurs readily (9). On the basis of the results of Fig. 2B, lipid transfer occurs slowly, if at all, between bulk POPC and pre-formed POPC:apoA-I apoA-I/HDL particles. These results are important since they suggest that bulk POPC membrane bilayers have a high energy barrier for interaction with preformed POPC:FL-apoA-I apoA-I/HDL particles, while preformed POPC:apoA-I particles have a lower energy barrier (high chemical potential) for remodeling through apoA-I/HDL particle-particle interactions probably following particle collisions.
Experimental selection of controls for computational testing of the polar lipid remodeling-switch hypothesis
To address the major limitation of our previous T-jump study, the use of truncated apoA-I, we made a series of PL-rich apoA-I/HDL particles from FL-apoA-I containing a range of POPC concentrations. The goal of this study was to identify particle stoichiometries to serve as positive or negative controls for MD simulations; we defined positive or negative controls as particle stoichiometries at which 40-50% expansion results in unstable particles and fusion or stable particles without fusion, respectively.
Analysis by NDGGE indicated that a 40-50% increase in POPC for 160:2 apoA-I/HDL particles, creating a stoichiometry of 220-240:2 (Fig. 3A, lower arrow), results in large R4 and R3 fusion products. Increase in POPC in the 180:2 apoA-I/HDL particles by 40-50% results in a further increase in concentration of the fusion products compared to the 160:2 apoA-I/HDL particles (Fig. 3B, lower arrow). Particles with these stoichiometries were therefore selected as positive (fusion-ready) controls for MD simulations.
FIGURE 3. Effects of POPC concentration on particle size distribution of POPC:FL-apoA-I recombinants.
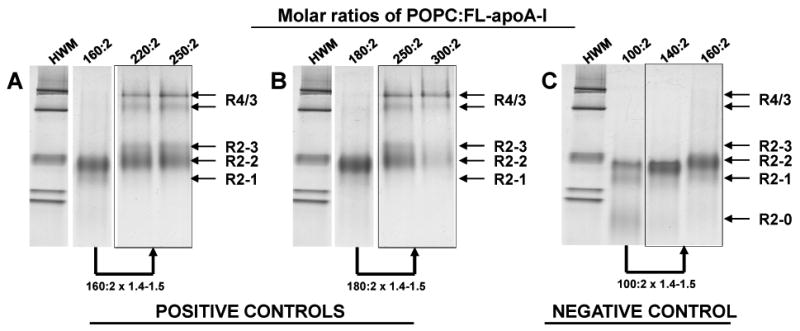
The gels (4-20% polyacrylamide) were run for 48 hrs and stained with colloidal blue. NDGGE of reconstituted particles having POPC:FL-apoA-I molar ratios of: (positive controls) A. 160:2, 220:2 and 250:2; B. 180:2, 250:2 and 300:2; and (negative control): C. 100:2, 140:2 and 160:2. Fusion products are represented by the R4/3 bands.
The particle R2-1 was selected on the basis of Fig. 3C as a negative (fusion-resistant) control. Analysis by NDGGE of a PL-rich apoA-I/HDL particle containing FL-apoA-I with a POPC:apoA-I molar ratio of 100:2 (R2-1) indicated that a 40-50% increase in POPC for this apoA-I/HDL particle, creating a stoichiometry of 140-160:2, results in larger R2 apoA-I/HDL particles but no fusion products such as R4 or R3 (Fig. 3C, lower arrow).
The rationale for these choices for positive and negative controls was that, since T-jump produces a 40-50% increase in particle surface area, the simulations would be equivalent to the experimental addition of 40-50% more POPC. Our ultimate goal for these studies was to develop an understanding of the structural mechanisms regulating particle fusion and polar lipid exchange.
Computational Studies
MDSA (R2-24) and T-jump (X2-24) simulations of positive (fusion-ready) starting control particle, R2-20
Fig. 4A shows cross-eyed stereo images of one example of the final structures of the four MDSA simulations of the starting FL-apoA-I-containing 160:24:2 HDL particle, R2-20. In the structure illustrated, the N- and C-terminal segments of the terminal domain (lower panel, residues 1-43, blue and helix 10, red, respectively) are more mobile than the central domain (helix 5, green) and helix 10 pairs are the more spatially separated of the two termini. Motion of each helix 10 is associated with exposure of a small patch of acyl chain surface area to solvent (middle panel, white arrowhead). Most importantly, the N-terminal proline-rich domains (Fig. 4A, lower panel, yellow arrowhead), residues 1-10, while mobile compared to the helix 5 pair, are in contact in three of the structures and near one another in the fourth (Fig. S2A in Supporting Information).
FIGURE 4. Structural features of one example each of the four MDSA and four 500 K MD T-jump simulations of the 160:24:2 (R2-2) particle.

A. Final structure of one example from the ensemble of four MDSA simulations (R2-24) of the R2-2 particle. B. Final structure of one example from the ensemble of four 500 K MD T-jump simulations (X2-24) of the R2-2 particle. (Upper panel) Cross-eyed stereo image of spacefilling models, including hydrogens, viewed from the face of the particles with the central domain (helix 5) at the top. (Middle panel) Spacefilling models, excluding hydrogens, viewed from the terminal overlap domain side of the particles. Each particle has been rotated 90° from the upper row view as indicated by the arrows. The white arrowheads indicate areas of exposure of acyl chains to solvent. Protein: Skyblue except proline (yellow). POPC: Phosphorous atoms (gold), phosphate oxygen atoms (red), choline nitrogens and methyls (blue), acyl chains (black), hydrogens (white). UC: (cpk). (Lower panels) Same view as middle panel showing protein only. Protein: Silver blue except proline (yellow), N-terminal G* domain, residues 1-43 (blue), C-terminal helix 10, residues 220-243 (red), helix 5, residues 121-142 (green. The yellow arrowheads indicate clustering of the N-terminal proline-rich pairs. C. Cross-eyed stereo images of all the final structures from the ensembles of all four MDSA (R2-24, upper panel) and 500 K MD T-jump (X2-24, lower panel) simulations aligned from residues 77 to 188. POPC and UC are represented in line format (cyan). The protein is represented as a Cα backbone tube model except prolines that are spacefilling. The structures are viewed from the terminal overlap domain side of the ensemble particle. N-terminal G* domains, residues 1-43 (blue); C-terminal helix 10 domains, residues 220-243, (red); and remainder of protein (gray). D. Cross-eyed stereo images of average final structures of FL-apoA-I double belt for the four MDSA (R2-24, upper panel) and 500 K MD T-jump (X2-24, lower panel) simulations of the 160:24:2 particle created from calculated average coordinates. The apoA-I double belts are viewed from the helix 5 domain. The protein is represented as a Cα backbone tube model except Cα proline (spacefilling yellow). Helix 5 (green), C-terminal helix 10 domains, residues 220-243 (red), N-terminal G* domains, residues 1-43 (blue), and remainder of protein (gray). The N-terminal (proline-rich) domains are indicated by red arrowheads.
Cross-eyed stereo images of one example of the final structures of the four MD T-jump simulations of the FL-apoA-I-containing 160:24:2 HDL particles (X2-24) are shown in Fig. 4B. As the result of increased particle size, there have been significant changes in the dynamics of the N-terminal domains; they no longer are locked together but are spatially separated (yellow arrowheads, lower panel). Spatial separation and increased mobility has resulted in exposure of large patches of acyl chain surface area to solvent (white arrowhead, middle panel) in all four simulations (Fig. S2B in Supporting Information).
Fig. 4C represents alignment of the final structures of the R2-24 and X2-24 ensembles. This figure dramatically illustrates the hyper-mobility of both the helix 10 C-terminal and N-terminal proline-rich domains (in red and blue, respectively) in both types of MD simulations. The figure also illustrates coverage of the space between the C-terminal domains by the N-terminal domains (in blue) during MDSA simulations and exposure of that space during T-jump simulations.
Finally, Fig. 4D shows average Cα structures for the final structures of the R2-24 and X2-24 ensembles (shown in Fig. S2 in Supporting Information). This figure illustrates very clearly the spatial separation of helix 10 domains (in red) in both simulations and the self-association of the N-terminal proline-rich domains (in blue) during MDSA and their wide spatial separation following T-jump-induced particle expansion (proline-rich N-terminal domains are indicated with red arrowheads).
The structures of two examples of a larger R2-2 HDL particle after T-jump are shown in Fig. S3 in Supporting Information. Not only have the N-terminal domains separated (red arrowheads, lower row) but the particles have disintegrated with POPC molecules spilling into the solvent.
MDSA (R2-14, R2-04) and T-jump (X2-14and X2-04) simulations of negative (fusion-resistant) starting control particles, R2-10 and R2-00
Fig. 5A-B shows cross-eyed stereo images of one example of the final structures of the four MDSA and four T-jump simulations of the 100:15:2 PL-rich apoA-I/HDL starting particle R2-10; see Fig. S4 in Supporting Information for images of all replicas. It is worthy of note, even in these more compact structures, that helix 10 C-terminal domains (in red) are spatially separated in all structures, while the N-terminal proline-rich domains (red arrowheads) remain in close contact both before and after T-jump-induced particle expansion.
FIGURE 5. Cross-eyed stereo images of structural features of the four MDSA and four 500 K MD T-jump simulations of the 100:15:2 (R2-1) and 50:8:2 (R2-0) particles.

A. Final structure of one example from the ensemble of four MDSA simulations (R2-14) of the R2-1 particle. B. Final structure of one example from the ensemble of four 500 K MD T-jump simulations (X2-14) of the R2-1 particle. (Top panels) Spacefilling models, excluding hydrogens, viewed from the terminal overlap domain side of the particles. Protein: Skyblue except proline (yellow). POPC: Phosphorous atoms (gold), phosphate oxygen atoms (red), choline nitrogens and methyls (blue), acyl chains (black). UC: (cpk). The white arrowheads indicate areas of exposure of acyl chains to solvent. (Bottom rows) Same view as top row with protein in ribbon representation, except for prolines (yellow spacefilling) and POPC and UC in line representation. Protein: Gray except proline (yellow), N-terminal G* domain, residues 1-43 (blue), C-terminal helix 10, residues 220-243 (red), helix 5, residues 121-142 (green) visible behind the POPC bilayer. The red arrowheads indicate clustering of the N-terminal proline-rich pairs. C. All the final structures from the ensemble of four R2-0 MDSA simulations (R2-04) aligned at residues 77 to 188. D. All the final structures from the ensemble of four R2-0 500 K MD T-jump simulations (X2-04) aligned at residues 77 to 188. POPC and UC are represented in line format (cyan). The protein is represented as a Cα backbone tube model except prolines that are spacefilling. The structures are viewed from the terminal overlap domain side of the particle. N-terminal G* domains, residues 1-43 (blue); C-terminal helix 10 domains, residues 220-243, (red); and remainder of protein (gray).
Fig. 5C-D shows cross-eyed stereo images of alignment of the final structures of the four MDSA and four MD T-jump simulations for the 50:8:2 PL-rich HDL starting particle R2-00. This figure graphically illustrates the hyper-mobility of both the helix 10 C-terminal and N-terminal proline-rich domains (in red and blue, respectively). In spite of this hyper-mobility, the lipid surfaces of the particles are well covered by the terminal domains. This suggests that PL-rich HDL particle surface area expansion by 40-50% during T-jump is absorbed in these smaller particles, not by increased separation of the overlap region as occurs for R2-2 (Fig. 4D), but by changes in flexibility along the apoA-I chain (see Figs. S5A-C in Supporting Information).
Mobility of terminal domains in MDSA and T-jump ensembles R2-24, R2-14 R2-04, and X2-24, X2-14, X2-04, respectively
RMSF of individual residues during the trajectories of the R2-24, R2-14 and R2-04 MDSA ensembles were analyzed as measures of local mobility (Figs. S5A-C in Supporting Information). Average RMSF analyses of the last 20% of MDSA simulations for the three sizes of PL-rich HDL particles confirm that the N- and C-terminal domains of each type of particle even at 310 K are more mobile than their central domains (Fig. S5A-C in Supporting Information, open circles). This increased mobility is confined to the N-terminal proline-rich repeat (residues 1-10) and the C-terminal half of helix 10. In each particle, the C-terminal domain was slightly more mobile than the N-terminal domain (see also Fig. S5D in Supporting Information).
RMSF analyses over the trajectories of the X2-24, X2-14 and X2-04 T-jump ensembles for the three sizes of PL-rich apoA-I/HDL particles also show increased mobility of the terminal domains compared with the rest of apoA-I (Fig. S5A-C in Supporting Information, closed circles). For the ensembles of the two smaller particles, X2-14 and X2-04, the C-terminal domains are somewhat more mobile than the N-terminal domains. However, for the ensemble of the largest PL-rich HDL particle, X2-24, during T-jump particle expansion both N- and C-terminal domains become equally mobile (Fig. S5A in Supporting Information, closed circles), suggesting that disruption of the previous interaction between the N-terminal proline-rich domain allows increased mobility.
Distance between ends of terminal domain pairs in MDSA and T-jump ensembles R2-24, R2-14, R2-04 and X2-24, X2-14, X2-04, respectively
The distances between the ends of the N- or C-terminal domain pairs were measured as a function of time over the last 10 ns of the trajectories of each of the four MDSA and T-jump simulations. Fig. S6 in Supporting Information is a plot of the average spatial separation calculated for each terminal domain for each ensemble. The plots for ensembles R2-14 and R2-04, of the smaller PL-rich HDL particles, show that, while spatial separation between ends of C-terminal pairs tends to be greater than between N-terminal pairs, there is little change in separation of the N-terminal or C-terminal domain pairs following T-jump.
For the largest PL-rich HDL particle, R2-2, particle expansion following T-jump causes an increase in separation between ends of both terminal domain pairs (Fig. S6 in Supporting Information). While separation between ends of the helix 10 C-terminal pair is greater than between ends of the N-terminal pair, the percent increase in separation following T-jump is much greater for the ends of the N-terminal pair than for the ends of the C-terminal pair, 139% versus 27%, respectively. One can calculate that a 40-50% increase in particle surface area is equivalent to an 18-22% increase in circumference, close to the 27% increase in separation that occurs in the C-terminal pair following T-jump.
Enlarged MDSA ensemble of sixteen R2-2 particles, R2-216
To expand our exploration of conformational space and increase the robustness of our MDSA simulations, we created an enlarged ensemble of R2-2 particles in the LL5/5 registry with the molar ratio of 160-POPC:24-UC:2-apoA-I by performing sixteen replications of MDSA (R2-216). Fig. 6 shows detailed stereo images of the double belt structure of FL-apoA-I for the final structures of the R2-216 ensemble after aligning the protein chains within the domain containing the interhelical solvent-inaccessible salt bridges, residues 77-188. Fig. 6A that represents the aligned structures containing both protein and lipid viewed from three different directions illustrates the soft matter nature of the particles. Fig. 6B, showing the aligned protein only viewed from two different directions illustrates the relative immobility of the central domain and much greater mobility of the terminal domains. Finally, Fig. 6C is a molecular graphic representation of the average Cα backbone structure for the ensemble showing that, in spite of the dramatic increase in motion of the termini, the head to head association between the pair of proline-rich N-terminal domains seen in Fig. 4D is maintained, on average, in the larger ensemble, as is the wider separation of the C-terminal helix 10 pair.
FIGURE 6. Detailed stereo images of the double belt structure of FL-apoA-I in the R2-216 ensemble after MDSA.

The protein chains of the final structures of the sixteen member ensemble were aligned between residues 77-188, the domain containing the interhelical solvent-inaccessible salt bridges. A. Cross-eyed stereo images of the aligned structures viewed from three different directions. Upper panel, view from helix 5 side; middle panel, view from terminal domain side; lower panel, view from disc top. C-terminal helix 10 domains, residues 220-243 (red licorice), N-terminal domains, residues 1-43 (blue licorice), helix 5 domain (green licorice), remainder of protein (gray licorice), and lipid (cyan line). B. Cross-eyed stereo image of the aligned structures showing the relative immobility of the central domain (green licorice) and mobility of the terminal domains (gray licorice). C. Cross-eyed stereo image of an average final structure created from calculated average coordinates. The apoA-I double belt is viewed from the helix 5 domain. The protein is represented as a Cα backbone licorice model except Cα proline (spacefilling yellow). Helix 5 (green), C-terminal helix 10 domains, residues 220-243 (red), N-terminal domains, residues 1-43 (blue), and remainder of protein (gray). The N-terminal proline-rich domains are indicated by red arrowheads.
Average RMSF of individual residues within the R2-216 ensemble trajectories (Fig. S5D in Supporting Information) shows even more clearly than the RMSF data for the smaller R2-24 ensemble (Fig. S5A in Supporting Information, open circles) that the C-terminal domains are slightly, but significantly more mobile than the N-terminal domains (note standard error bars in Fig. S5D in Supporting Information). Further, as noted also in the protein alignments of the R2-216 ensemble (Fig. 6) the relatively immobile central region of the apoA-I double belt correlates with the location of the solvent inaccessible salt bridges (Fig. S5D in Supporting Information, open circles).
Mean per residue helicity calculations of individual residues during the R2-216 ensemble trajectories were analyzed as another measure of the robustness of the MDSA (Fig. S5E in Supporting Information). Reflecting the robustness of an ensemble of sixteen replicas, these results show that the pattern of helicity is reproducible (note standard error bars in Fig. S5E in Supporting Information) and, as noted for the R2-24 ensemble in Gu, et al (44), the most helical region is correlated with the location of the solvent inaccessible salt bridges (Fig. S5E in Supporting Information, open circles).
Intermolecular interactions between the terminal domains
To further understand the nature of the forces acting on the terminal domains, we then analyzed intermolecular interactions between the terminal N-N, C-C and N-C domains of the R2-216 ensemble during MD simulation. The R2-216 ensemble trajectories were analyzed for the presence of both inter- and intrahelical salt bridges over the last 20% of the MDSA protocol and for molecular contacts. The resulting sequence matrix plots in Fig. 7 illustrate the dynamic nature of salt bridges and molecular contacts between terminal domains during MD simulations of the R2-216 ensemble.
FIGURE 7. Molecular contact maps between N-terminal pairs, C-termini pairs and N-terminal to C-terminal pairs plotted for the R2-216 ensemble of sixteen MDSA simulations of the 160:24:2 (R2-2) particle.
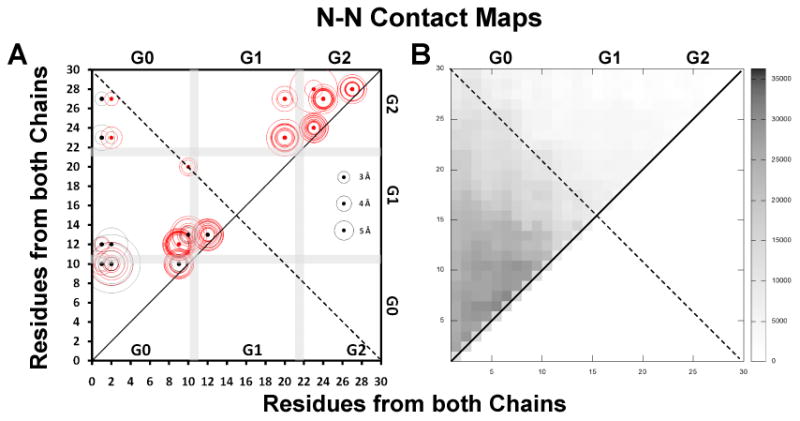
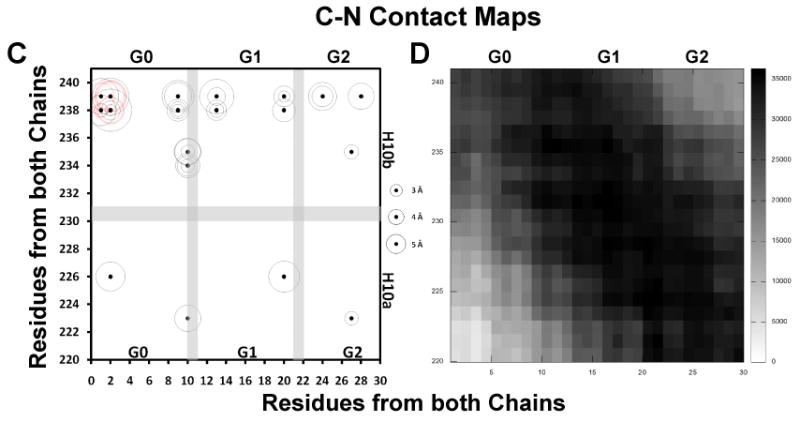
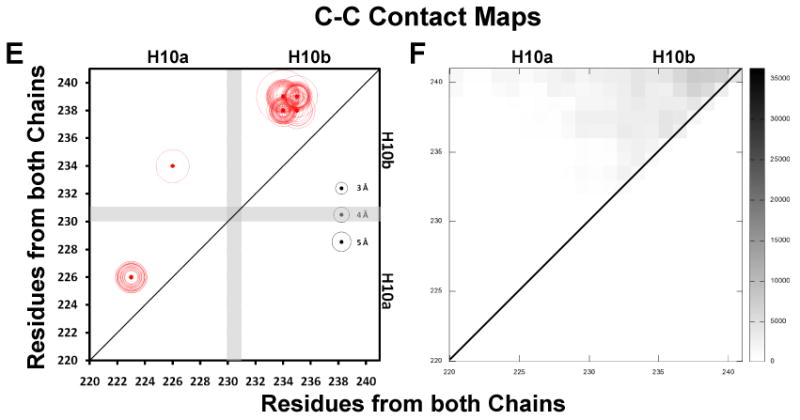
Intermolecular salt bridges are plotted as black dots, intramolecular salt bridges as red dots, other molecular contacts (fraction interhelical contacts close enough to allow cross-linking, defined as a Cα-Cα distance ≤ 25 Å (64), during the last 10 ns of all sixteen R2-216 MDSA trajectories summed over a total of 20,000 frames) are plotted as scaled black rectangles (scale to right of plot). Distances of salt bridges are indicated by circles (scaled to right of plot). A. N-N inter- and intramolecular salt bridges, residues 1-30. B. N-N intermolecular contacts, residues 1-30. C. C-C inter- and intramolecular salt bridges, residues 220-241. D. C-C intermolecular contacts, residues 220-241. E. C-N inter- and intramolecular salt bridges, residues 1-30 to 220-241. F. C-N intermolecular contacts, 1-30 to 220-241.
All salt bridges were plotted only above and molecular contacts were plotted above and below the diagonal. The position of each salt bridge is shown by a dot and the average length of that salt bridge in each of the sixteen simulations is proportional to the diameter of the circle surrounding the dot. The stronger the salt bridges, the smaller the diameters of the surrounding circles are (see scale on right side of each plot). The intrahelical salt bridges fall near the diagonal line of identity (red dots and red circles). Except for intermolecular interactions between N-N and C-C terminal domain pairs, the interhelical salt bridges (black dots and black circles) fall farther from the diagonal of identity passing through the middle of the sequence matrix box than the intramolecular ones.
“Sticky” N-terminal domains form extensive salt bridges with themselves and other portions of apoA-I
Figs. 7A-B are plots of the dynamics of salt bridge and molecular contacts between residues 1-30 in the N-terminal regions. The N-terminal 30 residues of human apoA-I, DEPPQSPWDRVKDLATVYVDVLKDSGRDYV, contain seven acidic (bold) and four basic (underlined) residues. Examination of Fig. 7A reveals the presence of a cluster of nine interhelical salt bridges (black dots) between the N-terminal domains: D1-R10, D1-K12, E2-R10, E2-K12, D1-K23, D1-R27, D9-R10, K12-D13 and R10-D13. These results suggest that salt bridges play a significant role in the persistent interhelical association of the two “sticky” N-terminal ends during MD simulations of the R2-216 ensemble. “Stickiness” is reflected in the overlap of intermolecular contacts and intermolecular salt bridges in the left quadrant of the sequence matrix plot.
Examination of the plots of salt bridge contacts between the N- and C-terminal domains, contacts between residues 1-30 and residues 220-241 (Figs. 7C-D), shows a series of salt bridges between essentially every acidic residue of the N-terminal domain and K238 and K239, and to a lesser extent between other basic and acidic residue combinations throughout the N-terminal and C-terminal domains. Thus not only are the proline-rich N-terminal domains self “sticky” but they also possess salt bridge-mediated “stickiness” to helix 10 (and other domains of apoA-I, see discussion).
“Promiscuous” C-terminal domains interact very little with each other
In contradistinction, examination of the sequence matrix plots of the dynamics of salt bridge and molecular contacts between residues 220-241 (helix 10) in the C-terminal regions in Figs. 7E -F reveals no dynamic intermolecular salt bridges and little intermolecular contact. These results are consistent with the generally wide separation between the C-terminal helix 10 pair.
Molecular details of “sticky” N-terminal interactions
Cross-eyed stereo images of three of the final structures of the R2-216 ensemble of sixteen MDSA simulations (Fig. 8) illustrate several of the intermolecular interactions between the first 10 residues of the “latched” N-terminal domains: salt bridges, E2←R10 and D9←R10 (Figs. 8A-B and Fig. 8 C, respectively) and proline stacking (Fig. 8B). Examination of all the final structures in the ensemble shows that the N-terminal domains are locked together in each particle of the R2-216 ensemble (Fig. S7 in Supporting Information). In six of the replicas, two E2-R10 salt bridges are formed between the “sticky” domains, in four of the replicas one E2-R10 salt bridge is formed, in one replica one D9-R10 salt bridge is formed, and in the five remaining replicas there are no salt bridges but the “sticky” domains are held together by close van der Waals interactions, predominantly through stacking of the prolines with themselves and with residues L14 and Y18. Additionally, in all sixteen replicas each N-terminal domain is attached to the lipid bilayer partially through the tryptophan residue W8 (shown in green in Fig. S7 in Supporting Information).
FIGURE 8. Cross-eyed stereo images in stick CPK representation of intermolecular contacts between overlapping N-N domains, residues 1-10, of three final structures from the R2-216 ensemble of sixteen MDSA simulations of the 160:24:2 (R2-2) particle.

Salt bridges are indicated by dashed green lines. Prolines are in yellow.
Testing of the “sticky” N-terminal domain hypothesis by T-jump-induced particle expansion
Our previous work suggested that the maximal diameter for the lipid core of the largest R2 apoA-I/HDL particle that can be reconstituted with POPC, R2-3 (Fig. 1B), would be approximately 91 Å and contain no more than 196 POPC molecules before fusing to form R3 or R4 particles (9). To test this hypothesis, we assembled a much larger all-atom R2 model, 240:36:2, and simulated it at 310K for 5 ns. In the 5 ns structure the N-terminal domains overlap by about 10 residues (Fig. S8A in Supporting Information, yellow arrowhead).
We then performed two 10 ns 500K MD T-jump simulations to expand the bilayer by 40-50% (27) thereby creating a force working to disengage the associated “sticky” N-terminal domains. After 1 ns, though the N-termini no longer overlapped, they remained engaged (Fig. S8B in Supporting Information, yellow arrowhead). However, the bilayer, rather than expanding to form a larger planar bilayer as expected, formed an invaginated cup-like shape (Figs. S8B-E in Supporting Information). In spite of this dramatic change in the shape of the lipid bilayer, it was not until 7 ns of simulation at 500K that the N-terminal domains disengaged (Fig. S8E in Supporting Information, yellow arrowhead).
Figs. 9A-B show detailed stereo images of the N- and C-terminal domains of the 500K T-jump simulations during the disengagement of the N-terminal domains between 6 and 7 ns. The final molecular interactions holding the N-termini together were salt bridges between D1 and R10 and E2 and the N-terminal NH3 (Figs. 9A). Upon repeating the T-jump, the “sticky” N-termini remained engaged until the simulation was stopped at 10 ns. These results indicate, in a qualitative manner, that the N-terminal domains are sufficiently self “sticky” to overcome the shear or bending modulus of the bilayer required to form a cup-shaped structure.
FIGURE 9. Select cross-eyed stereo images of a 240(POPC):36(UC): 2(apoA-I) particle subjected to an MD simulation at 310K for 5 ns and then a T-jump at 500K for 10 ns.
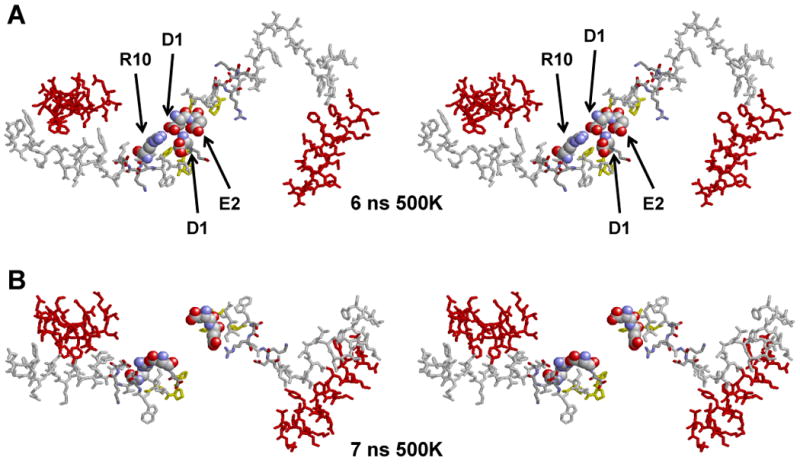
The yellow arrowheads indicate the position of the N-terminal proline-rich domains. A-E. Spacefilling representations of final structures of the following MD simulations: 5 ns at 310K and 1ns, 3 ns, 6 ns and 7 ns at 500K, respectively. Color code: Residues 1-43, white; helix 1 (residues 44-65), blue; helix 5 (residues 120-143), green; helix 8 (residues 187-208), skyblue; helix 10 (residues 220-243), red. F. Stick representations of the final molecular interactions holding the N-termini together in the T-jump simulation at 6 ns. G. Stick representations of the final molecular interactions after separation of the N-termini in the T-jump simulation at 7 ns. The salt bridges between D1 and R10 and E2 and the N-terminal NH3 are shown in spacefilling CPK representation. Color code: residues 1-43, white; helix 10, red; prolines, yellow.
Discussion
Proposed role of the “sticky” N-terminal domain in PL-rich HDL particle fusion
It is clear from Fig. 3 that PL-rich apoA-I/HDL particles containing FL-apoA-I exceeding a critical circumference (9) undergo particle-particle fusion. Further, it is clear that the N-terminal 43-residue domain is necessary for formation of the R2-2 particle (Fig. 1B): although fusion fails to occur following 40-50% expansion of R2-1 particles made from FL-apoA-I, equivalent expansion of R2-1 particles made from truncated (Δ43)apoA-I (25) results in fusion (Fig. 1A). Additional evidence supports the direct role of the N-terminal “sticky” domain in PL-rich HDL particle fusion; FL-apoA-I, but not Δ10apoA-I missing the proline-rich domain, is able to form larger R2-4 and R2-5 particles (Fig. 1B) with DMPC (44).
A recently published phylogenetic study of the sequence alignment of apoA-I also supports the “sticky” N-terminal domain model. In this study, residues 1, 2, 9 and 13 and residue 8 are conserved as acidic and tryptophan, respectively in all fifteen mammalian apoA-I sequences, while residues 10 and 12 are basic in essentially all of thirty-one species examined (2).
The average distance following MDSA between the ends of the N-terminal pair in the R2-2 PL-rich HDL particles is less than that for the smaller R2-0 and R2-1 particles, 15.5 Å versus 19.7 Å and 24.9 Å, respectively (Fig. S6 in Supporting Information). This suggests that the proline-rich N-terminal domain of apoA-I acts as a molecular latch that alone holds the terminal overlap ends of the R2-2 particle double belt together.
Bhat, et. al. (19, 20), using a combination of chemical cross-linking and mass spectrometry, suggested that the N- and C-terminal regions of each apoA-I chain in the double belt structure of PL-rich HDL are capable of forming intermolecular “hairpin” folds. They find a cross-link between R1-K118 demonstrating contact between the N-terminus and helix 4. Compatible with this report, coarse grained MD simulations that we performed recently showed “stickiness” of the N-terminal domain to helix 4 and helix 5 (56).
We propose that particle expansion beyond a critical threshold breaks the interaction between the “sticky” N-terminal repeats, exposing sufficient acyl chain surface area to solvent to create unstable particles that fuse upon collision (Fig. 10). We cannot at this time comprehensively define all of the molecular driving forces behind the “stickiness” of the N-terminal domain, both to itself and to other parts of apoA-I, other than to say that the “sticky” domain presents a number of potential salt bridges and van der Waals interactions that contribute to its “stickiness”. Certainly, the results of Figs. 6 and 9 and Figs. S7 and S8 in Supporting Information qualitatively suggest that, in spite of significant motion, N-terminal “stickiness” is substantial.
FIGURE 10. Schematic model of the polar lipid remodeling-switch hypothesis.
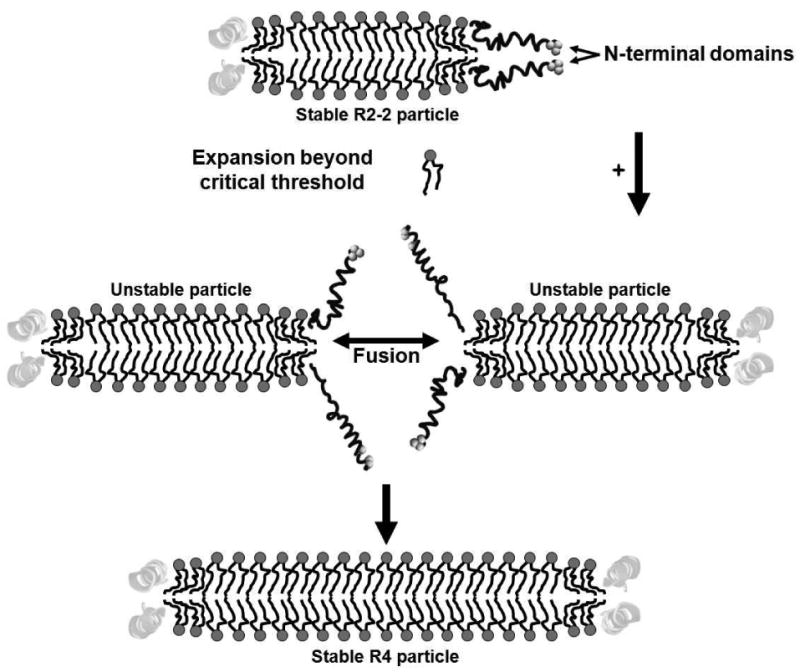
Postulated role of “sticky” N-terminal proline-rich domains in fusion between particles that have exceeded a critical polar lipid concentration. PL molecules are represented schematically in the usual ball (dark gray) and stick (black) motif. Particles are represented as bilayers in cross-section. Central domains (light gray ribbon helixes); terminal remodeling-switch domains (dark gray tube helixes).
Proposed role of the “promiscuous” C-terminal domain in PL-rich HDL particle polar lipid exchange
The experimental data in Fig. 2C clearly shows that polar lipid exchange flows down PL concentration gradients between PL-rich apoA-I/HDL particles. Equilibration of POPC between PL-rich apoA-I/HDL particles of different sizes occurs slowly, on a time scale of days at 37°C. From Fig. 2B, however, no measurable equilibration occurs between bulk POPC vesicles and PL-rich apoA-I/HDL particles. These results show that the presence of apoA-I on PL-rich apoA-I/HDL particles drives equilibration of POPC between particles. Although we as yet have no direct experimental data suggesting that the C-terminal helix 10 domains drive polar lipid exchange, helix 10 is known to have the highest lipid affinity of the many tandem amphipathic helical repeats in FL-apoA-I (57, 58) and it has been suggested that it is through helix 10 that apoA-I associates with cell membranes during ABCA1 PL-rich particle assembly (59). It seems reasonable to think that helix 10, because of its high lipid affinity and high mobility in PL-rich apoA-I/HDL particles, can interact with other PL-rich apoA-I/HDL particles in solution during collisions between particles to facilitate polar lipid exchange.
In conclusion, lipid exchange and equilibration and particle remodeling are critically important processes during metabolism of HDL particles (1, 6, 60-62); these processes, for example, are critical at every step in reverse cholesterol transport. Our Yin and Yang model for HDL may provide a potential conduit for additional biological processes—such as phospholipid transfer protein (63), CE transfer protein, and the CE transporter, SR-B1—to gain access to the lipid interior of HDL particles or the creation of hot spots with high chemical potential for binding of anti-inflammatory and anti-oxidant proteins.
For these reasons, our experimentally testable models suggesting the Yin and Yang model for the dynamic soft matter nature of the terminal domains of the double belt structure act as PL concentration-sensitive molecular switches for exchange, equilibration and remodeling of HDL particles, has substantial biological implications. Since more direct experimental approaches are unlikely to generate this level of detailed testable hypotheses, computational studies have emerged as being increasingly important in understanding the structure and dynamics of HDL. Site-directed mutagenesis studies, both experimental and computational, are currently underway in our laboratory to test the Yin and Yang hypothesis.
Supplementary Material
Acknowledgments
We would like to thank UAB Information Technology and Department of Mechanical Engineering for use of the IBM Blue Gene/L rack that they jointly maintain.
Abbreviations
- ABCA1
ATP-binding cassette transporter A1
- apo
apolipoprotein
- apoA-I/HDL
reconstituted high density lipoproteins
- CAD
coronary artery disease
- CE
cholesteryl ester
- DMPC
dimyristoylphosphatidylcholine
- FL
full length
- HDL
high density lipoproteins
- LCAT
lecithin:cholesterol acyl transferase
- MD
molecular dynamics
- MDSA
molecular dynamics simulated annealing
- MLV
multilamellar vesicles
- NDGGE
non-denaturing gradient gel electrophoresis
- PL
phospholipids
- POPC
palmitoyloleoylphosphatidylcholine
- RMSF
root mean square fluctuation
- R2 or R3 or R4
reconstituted high density lipoprotein particles containing 2, 3, or 4 molecules of apoA-I
- SR-BI
scavenger receptor B, type I
- T-jump
temperature-jump
- UC
unesterified cholesterol
- VMD
Visual Molecular Dynamics
Footnotes
This work was supported in part by the National Institutes of Health Grant P01 HL-34343 (to JPS).
Supporting Information Available: Figs. S1-S8 detail additional MD simulation results and analyses of the “sticky” and “promiscuous” domains. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Linsel-Nitschke P, Tall AR. HDL as a target in the treatment of atherosclerotic cardiovascular disease. Nat Rev Drug Discov. 2005;4:193–205. doi: 10.1038/nrd1658. [DOI] [PubMed] [Google Scholar]
- 2.Bashtovyy D, Jones MK, Anantharamaiah GM, Segrest JP. Sequence conservation of apolipoprotein A-I affords novel insights into HDL structure-function. J Lipid Res. 2010 doi: 10.1194/jlr.R012658. Epub Date 2010/12/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oram JF, Heinecke JW. ATP-binding cassette transporter A1: a cell cholesterol exporter that protects against cardiovascular disease. Physiol Rev. 2005;85:1343–1372. doi: 10.1152/physrev.00005.2005. [DOI] [PubMed] [Google Scholar]
- 4.Nichols AV, Blanche PJ, Gong EL, Shore VG, Forte TM. Molecular pathways in the transformation of model discoidal lipoprotein complexes induced by lecithin:cholesterol acyltransferase. Biochim Biophys Acta. 1985;834:285–300. doi: 10.1016/0005-2760(85)90001-3. [DOI] [PubMed] [Google Scholar]
- 5.Trigatti BL, Krieger M, Rigotti A. Influence of the HDL receptor SR-BI on lipoprotein metabolism and atherosclerosis. Arterioscler Thromb Vasc Biol. 2003;23:1732–1738. doi: 10.1161/01.ATV.0000091363.28501.84. [DOI] [PubMed] [Google Scholar]
- 6.Rye KA, Clay MA, Barter PJ. Remodelling of high density lipoproteins by plasma factors. Atherosclerosis. 1999;145:227–238. doi: 10.1016/s0021-9150(99)00150-1. [DOI] [PubMed] [Google Scholar]
- 7.Zlotnick A. Proceedings of the National Academy of Sciences of the United States of America. Vol. 101. 2004. Viruses and the physics of soft condensed matter; pp. 15549–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Segrest JP, Jones MK, Klon AE, Sheldahl CJ, Hellinger M, De Loof H, Harvey SC. A detailed molecular belt model for apolipoprotein A-I in discoidal high density lipoprotein. J Biol Chem. 1999;274:31755–31758. doi: 10.1074/jbc.274.45.31755. [DOI] [PubMed] [Google Scholar]
- 9.Li L, Chen J, Mishra VK, Kurtz JA, Cao D, Klon AE, Harvey SC, Anantharamaiah GM, Segrest JP. Double belt structure of discoidal high density lipoproteins: molecular basis for size heterogeneity. J Mol Biol. 2004;343:1293–1311. doi: 10.1016/j.jmb.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 10.Borhani DW, Rogers DP, Engler JA, Brouillette CG. Crystal structure of truncated human apolipoprotein A-I suggests a lipid-bound conformation. Proc Natl Acad Sci U S A. 1997;94:12291–12296. doi: 10.1073/pnas.94.23.12291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koppaka V, Silvestro L, Engler JA, Brouillette CG, Axelsen PH. The structure of human lipoprotein A-I. Evidence for the “belt” model. J Biol Chem. 1999;274:14541–14544. doi: 10.1074/jbc.274.21.14541. [DOI] [PubMed] [Google Scholar]
- 12.Maiorano JN, Davidson WS. The orientation of helix 4 in apolipoprotein A-I-containing reconstituted high density lipoproteins. J Biol Chem. 2000;275:17374–17380. doi: 10.1074/jbc.M000044200. [DOI] [PubMed] [Google Scholar]
- 13.Panagotopulos SE, Horace EM, Maiorano JN, Davidson WS. Apolipoprotein A-I Adopts a Belt-like Orientation in Reconstituted High Density Lipoproteins. J Biol Chem. 2001;276:42965–42970. doi: 10.1074/jbc.M106462200. [DOI] [PubMed] [Google Scholar]
- 14.Maiorano JN, Jandacek RJ, Horace EM, Davidson WS. Identification and structural ramifications of a hinge domain in apolipoprotein A-I discoidal high-density lipoproteins of different size. Biochemistry. 2004;43:11717–11726. doi: 10.1021/bi0496642. [DOI] [PubMed] [Google Scholar]
- 15.Li H, Lyles DS, Thomas MJ, Pan W, Sorci-Thomas MG. Structural determination of lipid-bound apo A-I using fluorescence resonance energy transfer. J Biol Chem. 2000;275:37048–37054. doi: 10.1074/jbc.M005336200. [DOI] [PubMed] [Google Scholar]
- 16.Li HH, Lyles DS, Pan W, Alexander E, Thomas MJ, Sorci-Thomas MG. ApoA-I structure on discs and spheres. Variable helix registry and conformational states. J Biol Chem. 2002;277:39093–39101. doi: 10.1074/jbc.M206770200. [DOI] [PubMed] [Google Scholar]
- 17.Davidson WS, Hilliard GM. The spatial organization of apolipoprotein A-I on the edge of discoidal high density lipoprotein particles: a mass specrometry study. J Biol Chem. 2003;278:27199–27207. doi: 10.1074/jbc.M302764200. [DOI] [PubMed] [Google Scholar]
- 18.Silva RA, Hilliard GM, Li L, Segrest JP, Davidson WS. A mass spectrometric determination of the conformation of dimeric apolipoprotein A-I in discoidal high density lipoproteins. Biochemistry. 2005;44:8600–8607. doi: 10.1021/bi050421z. [DOI] [PubMed] [Google Scholar]
- 19.Bhat S, Sorci-Thomas MG, Alexander ET, Samuel MP, Thomas MJ. Intermolecular contact between globular N-terminal fold and C-terminal domain of ApoA-I stabilizes its lipid-bound conformation: studies employing chemical cross-linking and mass spectrometry. J Biol Chem. 2005;280:33015–33025. doi: 10.1074/jbc.M505081200. [DOI] [PubMed] [Google Scholar]
- 20.Bhat S, Sorci-Thomas MG, Tuladhar R, Samuel MP, Thomas MJ. Conformational adaptation of apolipoprotein A-I to discretely sized phospholipid complexes. Biochemistry. 2007;46:7811–7821. doi: 10.1021/bi700384t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oda MN, Forte TM, Ryan RO, Voss JC. The C-terminal domain of apolipoprotein A-I contains a lipid-sensitive conformational trigger. Nature Structural Biology. 2003;10:455–460. doi: 10.1038/nsb931. [DOI] [PubMed] [Google Scholar]
- 22.Martin DD, Budamagunta MS, Ryan RO, Voss JC, Oda MN. Apolipoprotein A-I assumes a “looped belt” conformation on reconstituted high density lipoprotein. Journal of Biological Chemistry. 2006;281:20418–20426. doi: 10.1074/jbc.M602077200. [DOI] [PubMed] [Google Scholar]
- 23.Wu Z, Wagner MA, Zheng L, Parks JS, Shy JM, 3rd, Smith JD, Gogonea V, Hazen SL. The refined structure of nascent HDL reveals a key functional domain for particle maturation and dysfunction. Nat Struct Mol Biol. 2007;14:861–868. doi: 10.1038/nsmb1284. [DOI] [PubMed] [Google Scholar]
- 24.Wu Z, Gogonea V, Lee X, Wagner MA, Li XM, Huang Y, Undurti A, May RP, Haertlein M, Moulin M, Gutsche I, Zaccai G, Didonato JA, Hazen SL. The Double Super Helix model of high density lipoprotein. J Biol Chem. 2009 doi: 10.1074/jbc.M109.039537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones MK, Catte A, Patterson JC, Gu F, Chen J, Li L, Segrest JP. Thermal stability of apolipoprotein A-I in high-density lipoproteins by molecular dynamics. Biophys J. 2009;96:354–371. doi: 10.1016/j.bpj.2008.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klon AE, Segrest JP, Harvey SC. Molecular dynamics simulations on discoidal HDL particles suggest a mechanism for rotation in the apo A-I belt model. J Mol Biol. 2002;324:703–721. doi: 10.1016/s0022-2836(02)01143-9. [DOI] [PubMed] [Google Scholar]
- 27.Catte A, Patterson JC, Jones MK, Jerome WG, Bashtovyy D, Su Z, Gu F, Chen J, Aliste MP, Harvey SC, Li L, Weinstein G, Segrest JP. Novel changes in discoidal high density lipoprotein morphology: a molecular dynamics study. Biophys J. 2006;90:4345–4360. doi: 10.1529/biophysj.105.071456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Catte A, Patterson JC, Bashtovyy D, Jones MK, Gu F, Li L, Rampioni A, Sengupta D, Vuorela T, Niemela P, Karttunen M, Marrink SJ, Vattulainen I, Segrest JP. Structure of spheroidal HDL particles revealed by combined atomistic and coarse-grained simulations. Biophys J. 2008;94:2306–2319. doi: 10.1529/biophysj.107.115857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones MK, Catte A, Li L, Segrest JP. Dynamics of activation of lecithin:cholesterol acyltransferase by apolipoprotein A-I. Biochemistry. 2009;48:11196–11210. doi: 10.1021/bi901242k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Phillips JC, Wriggers W, Li Z, Jonas A, Schulten K. Predicting the structure of apolipoprotein A-I in reconstituted high density lipoprotein disks. Biophys J. 1997;73:2337–2346. doi: 10.1016/S0006-3495(97)78264-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Denis M, Haidar B, Marcil M, Bouvier M, Krimbou L, Genest J., Jr Molecular and cellular physiology of apolipoprotein A-I lipidation by the ATP-binding cassette transporter A1 (ABCA1) J Biol Chem. 2004;279:7384–7394. doi: 10.1074/jbc.M306963200. [DOI] [PubMed] [Google Scholar]
- 32.Rocco AG, Mollica L, Gianazza E, Calabresi L, Franceschini G, Sirtori CR, Eberini I. A model structure for the heterodimer apoA-IMilano-apoA-II supports its peculiar susceptibility to proteolysis. Biophys J. 2006;91:3043–3049. doi: 10.1529/biophysj.106.085886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shih AY, Freddolino PL, Arkhipov A, Schulten K. Assembly of lipoprotein particles revealed by coarse-grained molecular dynamics simulations. J Struct Biol. 2007;157:579–592. doi: 10.1016/j.jsb.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 34.Koivuniemi A, Heikela M, Kovanen PT, Vattulainen I, Hyvonen MT. Atomistic simulations of phosphatidylcholines and cholesteryl esters in high-density lipoprotein-sized lipid droplet and trilayer: clues to cholesteryl ester transport and storage. Biophys J. 2009;96:4099–4108. doi: 10.1016/j.bpj.2009.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rocco AG, Gianazza E, Calabresi L, Sensi C, Franceschini G, Sirtori CR, Eberini I. Structural features and dynamics properties of human apolipoprotein A-I in a model of synthetic HDL. J Mol Graph Model. 2009;28:305–312. doi: 10.1016/j.jmgm.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 36.Shih AY, Sligar SG, Schulten K. Maturation of high-density lipoproteins. J R Soc Interface. 2009 doi: 10.1098/rsif.2009.0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gogonea V, Wu Z, Lee X, Pipich V, Li XM, Ioffe AI, Didonato JA, Hazen SL. Congruency between Biophysical Data from Multiple Platforms and Molecular Dynamics Simulation of the Double-Super Helix Model of Nascent High-Density Lipoprotein. Biochemistry. 2010 doi: 10.1021/bi100588a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vuorela T, Catte A, Niemela PS, Hall A, Hyvonen MT, Marrink SJ, Karttunen M, Vattulainen I. Role of lipids in spheroidal high density lipoproteins. PLoS Comput Biol. 2010;6:e1000964. doi: 10.1371/journal.pcbi.1000964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yetukuri L, Soderlund S, Koivuniemi A, Seppanen-Laakso T, Niemela PS, Hyvonen M, Taskinen MR, Vattulainen I, Jauhiainen M, Oresic M. Composition and lipid spatial distribution of HDL particles in subjects with low and high HDL-cholesterol. Journal of Lipid Research. 2010;51:2341–2351. doi: 10.1194/jlr.M006494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kerr ID, Sankararamakrishnan R, Smart OS, Sansom MS. Parallel helix bundles and ion channels: molecular modeling via simulated annealing and restrained molecular dynamics. Biophys J. 1994;67:1501–1515. doi: 10.1016/S0006-3495(94)80624-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sankararamakrishnan R, Sansom MS. Structural features of isolated M2 helices of nicotinic receptors, Simulated annealing via molecular dynamics studies. Biophys Chem. 1995;55:215–230. doi: 10.1016/0301-4622(95)00006-j. [DOI] [PubMed] [Google Scholar]
- 42.Bassolino-Klimas D, Tejero R, Krystek SR, Metzler WJ, Montelione GT, Bruccoleri RE. Simulated annealing with restrained molecular dynamics using a flexible restraint potential: theory and evaluation with simulated NMR constraints. Protein Sci. 1996;5:593–603. doi: 10.1002/pro.5560050404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H, Tejero R, Monleon D, Bassolino-Klimas D, Abate-Shen C, Bruccoleri RE, Montelione GT. Homology modeling using simulated annealing of restrained molecular dynamics and conformational search calculations with CONGEN: application in predicting the three-dimensional structure of murine homeodomain Msx-1. Protein Sci. 1997;6:956–970. doi: 10.1002/pro.5560060502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gu F, Jones MK, Chen J, Patterson JC, Catte A, Jerome WG, Li L, Segrest JP. Structures of discoidal high density lipoproteins: a combined computational-experimental approach. J Biol Chem. 2010;285:4652–4665. doi: 10.1074/jbc.M109.069914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaleeacute L, Skeel R, Bhandarkar M, Brunner R, Gursoy A, Krawetz N, Phillips J, Shinozaki A, Varadarajan K, Schulten K. NAMD2: greater scalability for parallel molecular dynamics. J Comp Phys. 1999;151:283–312. [Google Scholar]
- 46.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 47.Jorgensen WL, Chandrasekhar J, Madura JD. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 48.MacKerell AD, Jr, Bashford D, Bellot M, Dunbrack RL, Jr, Evanseck J, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, III, Roux B, Schlenkrich M, Smith J, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 49.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. Journal of computational chemistry. 1983;4:187–217. [Google Scholar]
- 50.Feller SE, Pastor RW. Length scales of lipid dynamics and molecular dynamics. Pac Symp Biocomput. 1997:142–150. [PubMed] [Google Scholar]
- 51.Schlenkrich M, Brickmann J, MacKerell A, Jr, Karplus M. An empirical potential energy function for phospholipids: criteria for parameter optimization and applications. In: Merz KM, Roux B, editors. Biological Membranes: A Molecular Perspective from Computation and Experiment. Birkhauser; Boston: 1996. pp. 31–81. [Google Scholar]
- 52.Harvey SC, Tan RKZ, Cheatham TE. The flying ice cube: Velocity rescaling in molecular dynamics leads to violation of energy equipartition. Journal of Computational Chemistry. 1998;19:726–740. [Google Scholar]
- 53.Anantharamaiah GM, Garber DW. Chromatographic methods for quantitation of apolipoprotein A-I. Methods Enzymol. 1996;263:267–282. doi: 10.1016/s0076-6879(96)63019-5. [DOI] [PubMed] [Google Scholar]
- 54.Rogers DP, Brouillette CG, Engler JA, Tendian SW, Roberts L, Mishra VK, Anantharamaiah GM, Lund-Katz S, Phillips MC, Ray MJ. Truncation of the amino terminus of human apolipoprotein A-I substantially alters only the lipid-free conformation. Biochemistry. 1997;36:288–300. doi: 10.1021/bi961876e. [DOI] [PubMed] [Google Scholar]
- 55.Jonas A. Reconstitution of high-density lipoproteins. Methods Enzymol. 1986;128:553–582. doi: 10.1016/0076-6879(86)28092-1. [DOI] [PubMed] [Google Scholar]
- 56.Jones MK, Zhang L, Catte A, Li L, Oda M, Ren G, Segrest JP. Assessment of the Validity of the Double Super Helix Model for Reconstituted High Density Lipoproteins: A combined computational-experimental approach. J Biol Chem. 2010 doi: 10.1074/jbc.M110.187799. Epub date 10/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Palgunachari MN, Mishra VK, Lund-Katz S, Phillips MC, Adeyeye SO, Alluri S, Anantharamaiah GM, Segrest JP. Only the two end helixes of eight tandem amphipathic helical domains of human apo A-I have significant lipid affinity. Implications for HDL assembly. Arterioscler Thromb Vasc Biol. 1996;16:328–338. doi: 10.1161/01.atv.16.2.328. [DOI] [PubMed] [Google Scholar]
- 58.Mishra VK, Palgunachari MN, Datta G, Phillips MC, Lund-Katz S, Adeyeye SO, Segrest JP, Anantharamaiah GM. Studies of synthetic peptides of human apolipoprotein A-I containing tandem amphipathic alpha-helixes. Biochemistry. 1998;37:10313–10324. doi: 10.1021/bi980042o. [DOI] [PubMed] [Google Scholar]
- 59.Rogers DP, Roberts LM, Lebowitz J, Datta G, Anantharamaiah GM, Engler JA, Brouillette CG. The lipid-free structure of apolipoprotein A-I: effects of amino- terminal deletions. Biochemistry. 1998;37:11714–11725. doi: 10.1021/bi973112k. [DOI] [PubMed] [Google Scholar]
- 60.Rye KA, Wee K, Curtiss LK, Bonnet DJ, Barter PJ. Apolipoprotein A-II inhibits high density lipoprotein remodeling and lipid-poor apolipoprotein A-I formation. J Biol Chem. 2003;278:22530–22536. doi: 10.1074/jbc.M213250200. [DOI] [PubMed] [Google Scholar]
- 61.Liang HQ, Rye KA, Barter PJ. Remodelling of reconstituted high density lipoproteins by lecithin: cholesterol acyltransferase. J Lipid Res. 1996;37:1962–1970. [PubMed] [Google Scholar]
- 62.Rye KA, Duong M, Psaltis MK, Curtiss LK, Bonnet DJ, Stocker R, Barter PJ. Evidence that phospholipids play a key role in pre-beta apoA-I formation and high-density lipoprotein remodeling. Biochemistry. 2002;41:12538–12545. doi: 10.1021/bi025998k. [DOI] [PubMed] [Google Scholar]
- 63.Pussinen PJ, Jauhiainen M, Metso J, Pyle LE, Marcel YL, Fidge NH, Ehnholm C. Binding of phospholipid transfer protein (PLTP) to apolipoproteins A-I and A-II: location of a PLTP binding domain in the amino terminal region of apoA-I. J Lipid Res. 1998;39:152–161. [PubMed] [Google Scholar]
- 64.Young MM, Tang N, Hempel JC, Oshiro CM, Taylor EW, Kuntz ID, Gibson BW, Dollinger G. High throughput protein fold identification by using experimental constraints derived from intramolecular cross-links and mass spectrometry. Proc Natl Acad Sci U S A. 2000;97:5802–5806. doi: 10.1073/pnas.090099097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.