

Abstract
The mammalian cryptochromes mCRY1 and mCRY2 act as transcriptional repressors within the 24-h transcription-translational feedback loop of the circadian clock. The C-terminal tail and a preceding predicted coiled coil (CC) of the mCRYs as well as the C-terminal region of the transcription factor mBMAL1 are involved in transcriptional feedback repression. Here we show by fluorescence polarization and isothermal titration calorimetry that purified mCRY1/2CCtail proteins form stable heterodimeric complexes with two C-terminal mBMAL1 fragments. The longer mBMAL1 fragment (BMAL490) includes Lys-537, which is rhythmically acetylated by mCLOCK in vivo. mCRY1 (but not mCRY2) has a lower affinity to BMAL490 than to the shorter mBMAL1 fragment (BMAL577) and a K537Q mutant version of BMAL490. Using peptide scan analysis we identify two mBMAL1 binding epitopes within the coiled coil and tail regions of mCRY1/2 and document the importance of positively charged mCRY1 residues for mBMAL1 binding. A synthetic mCRY coiled coil peptide binds equally well to the short and to the long (wild-type and K537Q mutant) mBMAL1 fragments. In contrast, a peptide including the mCRY1 tail epitope shows a lower affinity to BMAL490 compared with BMAL577 and BMAL490(K537Q). We propose that Lys-537mBMAL1 acetylation enhances mCRY1 binding by affecting electrostatic interactions predominantly with the mCRY1 tail. Our data reveal different molecular interactions of the mCRY1/2 tails with mBMAL1, which may contribute to the non-redundant clock functions of mCRY1 and mCRY2. Moreover, our study suggests the design of peptidic inhibitors targeting the interaction of the mCRY1 tail with mBMAL1.
Keywords: Biophysics, Peptides, Protein Acylation, Protein-Protein Interactions, Transcription Regulation, Circadian Clock, Cryptochrome
Introduction
In mammals many physiological processes are regulated in a day-time-dependent manner. These circadian (24 h) rhythms are generated by circadian clocks, which are operated by transcriptional and translational feedback loops. In the central feedback loop, the bHLH-PAS (basic Helix-Loop-Helix-PER-ARNT-SIM) transcription factors mBMAL1 (brain and muscle ARNT-like protein) and mCLOCK (circadian locomotor output cycle kaput) activate the transcription of three period genes (mper1,2,3) and two cryptochromes (mcry1,2) (1). The mPER proteins and (even more potently) the mCRY proteins feedback-repress their own transcription by regulating the activity of mBMAL1 and mCLOCK (2, 3). Notably, the mBMAL1-mCLOCK transcription factor complex not only regulates the mper and mcry genes but also a large number of clock controlled genes, including genes involved in cell cycle regulation, cellular detoxification, and metabolism (4). Hence, the regulation of these transcription factors is of relevance for many body functions and associated diseases (e.g. sleep and depressive disorders, metabolic syndrome, cardiovascular diseases, and tumor formation) that are under the control of the circadian clock (5). The importance of mBMAL1 for clock function is clearly demonstrated by the fact that mBMAL1−/− knock-out mice show an immediate and complete loss of circadian rhythmicity at a behavioral and molecular level (6). Although mCRY1/mCRY2 double knock-out mice become totally arrhythmic, mCRY1−/− single knock-out mice exhibit a 1-h shorter period, and mCRY2−/− single knock-out exhibit mice a 1-h longer period (7–9). Hence, the two cryptochromes are partially redundant but also have nonredundant clock functions leading to the opposite effects of mCRY1 and mCRY2 disruption on the period length.
The cryptochromes are composed of an ∼500 amino acid photolyase homology region (PHR)6 (10) and variable C-terminal extensions, the tails (Fig. 1A). The mCRY tails together with a preceding predicted coiled coil (CC) region, which corresponds to the most C-terminal α-helix of the PHR (10, 11), are involved in the transcriptional repression of mCLOCK and mBMAL1 (12). In the following we will refer to the PHR lacking the C-terminal CC region as the photolyase homology core region (PHCR). Strikingly, the mCRY1CCtail fragment alone does not mediate transcriptional repression when fused to enhanced GFP (12). Moreover, mutations that are expected to destabilize the interface between the PHCR and the coiled coil region inhibit the transcriptional repression activity of both mCRY homologues and reduce the interaction of mCRY2 with mBMAL1, mPER1/2, and mCLOCK (13). Hence, the correct positioning of the coiled coil region with respect to the PHCR is critical for molecular interactions and transcriptional repression activities of the mammalian cryptochromes. Notably, both cryptochromes contain functional bipartite nuclear localization signals within their tails (12, 14). Furthermore, Ser-557 and Ser-553 in the mCRY2 tail are phosphorylated sequentially by DYRK1A (dual-specificity tyrosine-phosphorylated and -regulated kinase 1A) and glycogen synthase kinase-3β (15). Ser-553/557 phosphorylation triggers the proteasomal degradation of mCRY2 and thereby delays its accumulation and nuclear entry.
FIGURE 1.

Domain architecture and secondary structure prediction of mCRY1/2 and mBMAL1. A, domain architecture of full-length mCRY1/2 and mBMAL1 is shown. mCRY1 and mCRY2 are composed of a conserved PHR and nonconserved C-terminal tails. The PHR consists of an N-terminal αβ-domain and an α-helical domain, which includes a predicted CC region at its C-terminal end. In the PHR of photoreceptor-type cryptochromes, the chromophores methenyltetrahydrofolate and flavine adenine dinucleotide are non-covalently bound (34). In mBMAL1, the two PAS (PER-ARNT-SIM) domains (PAS-A and PAS-B) and the basic helix-loop-helix domain are shown. The C-terminal mCRYCCtail- and mBMAL1 fragments used in our biochemical studies are represented as black arrows. B, shown is a secondary structure prediction of the C-terminal mCRY1/2CCtail and mBMAL1 fragments. The amino acid sequences and secondary structure predictions (PSIPRED Version 3.0; (35)) of the mCRY1-(471–606), mCRY2-(489–592), mBMAL1-(577–625), and mBMAL1-(490–625) fragments studied herein are shown. Numbering of mBMAL1 residues refers to Isoform b/2, which contains 625 amino acids. mCRY1 peptides P1 and P2, which have been synthesized for interaction studies, are shown as yellow boxes. CC, predicted coiled coil; Ac, in vivo acetylated Lys-537. mCRY1/2CCT, mCRYCCtail fragments mCRY1-(471–606) and mCRY2-(489–592) including the coiled coil and tail regions; AA, amino acids.
In the C-terminal region of mBMAL1, the mutations A610S, A610T, and G611E were shown to reduce sensitivity to mCRY1/2 repression (16). Moreover, deletion of the last eight mBMAL1 residues reduces the mBMAL1-mCRY1 interaction in coimmunoprecipitation experiments, and insertions C-terminal to Ala-600 or Leu-606 severely affect circadian core oscillations and transcriptional activation (17). Interestingly, the most C-terminal 43 amino acids of mBMAL1 also mediate transcriptional activation by recruiting coactivators such as p300/CBP in a daily regulated manner, with a maximum efficiency around circadian time 6 (CT6) (18, 19). Furthermore, mCRY1 and mCRY2 inhibit the p300-induced transcriptional activation of mBMAL1-mCLOCK by ∼80% (19). Because mCRY1/2 protein levels in the suprachiasmatic nucleus are highest between CT12 and CT16 and are low at CT6 (3), it is conceivable that mCRY proteins displace transcriptional coactivators in a daily regulated manner. Collectively, the published data suggest that the C-terminal mBMAL1 region represents a regulatory switch that cycles in a day-time-dependent manner between an activating coactivator-bound “on” mode and a repressing mCRY-bound “off” mode.
Although literature reports about direct interactions of mCRY1 and mCRY2 with mCLOCK are inconsistent, there is no doubt that mCLOCK stabilizes the mBMAL1-mCRY interactions in a ternary mCRY-mBMAL1-mCLOCK complex (17, 20). Importantly, mCLOCK acetylates mBMAL1 in vivo specifically on Lys-537 (21) (Fig. 1B). Acetylation of Lys-537mBMAL1 occurs in a daily regulated manner with a peak at about CT15, i.e. during the repressive phase. Moreover, Lys-537 acetylation enhances the mCRY1-mBMAL1 interaction and thereby transcriptional repression (21).
To quantitatively analyze the mCRY-mBMAL1 interactions underlying the transcriptional regulation of the mBMAL1-mCLOCK complex, we have purified mCRY1/2CCtail proteins as well as two C-terminal mBMAL1 fragments of 5.5 and 14.3 kDa, the latter including the in vivo acetylated Lys-537. We show that mCRY1 (but not mCRY2) exhibits a lower affinity to the longer than to the shorter mBMAL1 fragment and compared with a mutant version of the longer mBMAL1 fragment, in which Lys-537 is exchanged to an acetyl mimetic glutamine. Using peptide scan analysis, we identify two mBMAL1 binding epitopes in mCRY1 and mCRY2 corresponding to the coiled coil region and a more C-terminal region within the tails. Isothermal titration calorimetry (ITC) experiments with mCRY coiled coil and tail epitope peptides revealed different mBMAL1 interactions of the mCRY1 and mCRY2 tails, which may contribute to the non-redundant clock functions suggested by mCRY1−/− and mCRY2−/− knock-out studies (7–9). Furthermore, we propose a molecular mechanism for the regulation of mCRY1 binding by Lys-537mBMAL1 acetylation, which involves electrostatic interactions predominantly with the mCRY1 tail. Our study also suggests the design of specific peptidic or small molecule ligands targeting the nonconserved interaction of the mCRY1/2 tails with mBMAL1.
EXPERIMENTAL PROCEDURES
Recombinant Expression and Purification of mCRY and mBMAL1 Proteins
C-terminal fragments of the mouse cryptochromes (mCRY1-(471–606) and mCRY2-(489–592)) and mouse mBMAL1 (mBMAL1-(577–625) and mBMAL1-(490–625)) were subcloned into a pGEX-6P2 expression vector using restriction sites 5′ BamHI (mCRY1, both mBMAL1 fragments) or SmaI (mCRY2) and 3′ NotI (all 4 fragments). The K537Q mutation was introduced into the mBMAL1-(490–625) construct using the QuikChange site-directed mutagenesis kit (Stratagene). The proteins were overexpressed as GST fusions in the Escherichia coli strain BL21(DE3) and purified via GSH affinity and size exclusion chromatography. For purification, 5–10 liters of mCRY or mBMAL1 expression cultures in TB (Terrific Broth) medium were induced with 0.1 mm isopropyl 1-thio-β-d-galactopyranoside at an A600 of ∼1. Expression was carried out for 5 h at 30 °C or overnight at 18 °C. Pellets were thawed on ice and homogeneously resuspended in lysis buffer containing 50 mm Tris-HCl, pH 7.8, 250 mm NaCl, 10 mm β-mercaptoethanol, 10% glycerol, 2 mm EDTA, and 1 mm phenylmethylsulfonyl fluoride. Cells were lysed by sonification or in a fluidizer, and insoluble material was removed by centrifugation. The supernatant was loaded onto a GSH affinity column. The GST tag was removed by cleavage with Prescission protease either on the GSH column (mBMAL1) or in batch (mCRY1/2) after elution of the GST-fused proteins with a buffer containing 50 mm Tris-HCl, pH 7.8, 150 mm NaCl, 10 mm β-mercaptoethanol, 5% glycerol, and 20–30 mm glutathione. Tag removal yielded recombinant mCRY and mBMAL1 proteins with the N-terminal extensions GPLGS (BamHI) or GPLGSPGIPG (SmaI, mCRY2) leading to the following molecular weights and isoelectric points (pIs) of the recombinant proteins: mCRY1-(471–606), 14,432.7 Da, pI = 8.91; mCRY2-(489–592), 12,087.0 Da, pI = 6.94; mBMAL1-(490–625), 14,263.8 Da, pI = 4.18; mBMAL1-(577–625), 5,482.0 Da, pI = 3.44. Fractions containing cleaved mCRY or mBMAL1 proteins were concentrated using an Amicon Ultra-15 filter device (Millipore, Bedford, MA) with a 3–10-kDa molecular weight cut off and loaded onto a Superdex HiLoad S75 16/60 size exclusion column (GE Healthcare) with a running buffer containing 25 mm Hepes, pH 7.8, 40 mm NaCl, 2 mm DTT, and 5% glycerol. The remaining GST was removed by applying mBMAL1- or mCRY-containing fractions onto a second GSH column. Fractions containing highly purified mCRY and mBMAL1 proteins were pooled, concentrated to typically 5 mg/ml (350–400 μm), and snap-frozen in liquid nitrogen. Samples were stored at −80 °C until measured.
Analytical Ultracentrifugation-Sedimentation Velocity Experiments
Sedimentation velocity experiments were performed with an Optima XL-I analytical centrifuge (Beckman Inc., Palo Alto, CA) using an An 60 Ti rotor with double-sector epon centerpieces or titanium centerpieces of 12-mm path length (Nanolytics, Germany) capped with sapphire windows. The proteins were kept in 25 mm Bis-tris propane, pH 7.8, 100 mm NaCl, 5 mm Tris(2-carboxyethyl)phosphine at concentrations of 0.07 mm for mCRY1-(471–606), 0.17 mm for mCRY2-(489–592), 0.2 mm for mBMAL1-(577–625), and 0.06 or 0.6 mm for mBMAL1-(490–625). The buffer density was measured with a DMA 5000 densitometer. All other auxiliary parameters were calculated from the buffer composition using SEDNTERP (22). The protein concentration distribution during sedimentation was monitored by absorption or interference detection. Sedimentation coefficient distributions were computed using the SEDFIT software package (23), resulting in a c(s)-distribution corrected for diffusion by means of a signal-average frictional coefficient f/f0, which was optimized during fitting. The combination of s and f/f0 allows an estimate of the molar mass Mf, and apparent sedimentation coefficients used for subsequent calculations were determined by integration of the area under the c(s) curve for the species of interest. Experimental hydrodynamic radii (Rh) were calculated with SEDNTERP. Expected Rh values for folded and unfolded proteins of equal molar mass were obtained using the empirical formulae for globular and guanidinium-HCl-unfolded proteins (Equations 1 and 2) (25).
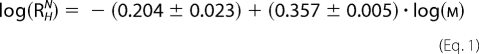 |
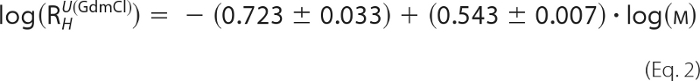 |
Circular Dichroism (CD) Spectroscopy
Purified protein samples were diluted to final concentrations between 13 and 60 μm in 25 mm NaH2PO4, pH 7.8, 5 mm Tris(2-carboxyethyl)phosphine buffer. CD spectra were measured by a Jasco J-715 spectropolarimeter using a 0.1-cm path length quartz cuvette and represent the mean molar ellipticity per amino acid residue of protein after buffer correction. Measurements were performed at 4 °C in a wavelength range from 190 to 250 nm with 0.1-nm intervals collecting data for 0.5 s at each point. For each measurement 10 spectra were used for accumulation. Analysis was performed using the CONTIN algorithm (26) with the reference dataset SMP56 (27, 28).
Fluorescence Polarization
mBMAL1 fragments were fluorescently labeled with FluorolinkTM Cy3.5 monoreactive Dye (GE Healthcare), which reacts with free amine groups (N-terminal amino groups and lysine side chain amino groups) of proteins. For labeling, a 15–20 mg/ml concentrated solution of purified mBMAL1 protein in a sodium carbonate buffer (0.1 m Na2CO3, pH 8.5) was incubated with the Cy3.5 dye for 2 h at 4 °C. For the fluorescence polarization measurement, the protein was transferred into a buffer containing 25 mm Hepes, pH 7.8, 50 mm NaCl, 2 mm dithioerythritol, 2.5% (v/v) glycerol with a desalting HiTrap column (GE Healthcare). Fluorescence polarization spectra were recorded with excitation of the Cy3.5 fluorophor at 581 nm and emission at 596 nm. A FluoroMax II spectrofluorimeter (Spex Industries, Edison, NJ) was used in the polarization mode at 10 °C. 500 nm Cy3.5-labeled mBMAL1 was titrated with increasing amounts of mCRY1 or mCRY2 proteins (concentrations 500 nm–300 μm) until saturation was reached. For each titration step, 30 measurements were accumulated and buffer-corrected. To obtain the dissociation constants (KD) for the mBMAL1-mCRY interactions, the concentration-dependent binding curve was fitted using a nonlinear regression function (Single Rectangular I, 3 Parameter, Hyperbola, SigmaPlot 10.0) provided by the program SigmaPlot.
ITC
The ITC experiments were performed using an ITC 200 MicroCalorimeter (MicroCal, Northampton, MA). All reagents were extensively dialyzed against a buffer containing 25 mm Bis-tris propane, pH 7.8, 100 mm NaCl, 5 mm Tris(2-carboxyethyl)phosphine, at 22 °C. The concentrations of the binding components in the reservoir solution were experimentally adjusted based on the preliminary knowledge of the interaction range. The concentration of the ligands was chosen between 0.5 and 0.9 mm, whereas the receptor was 10–15 times less. The typical titration consisted of 20 injections of 0.2–2-μl aliquots of the ligand into the receptor solution (250 μl in the cell), at time intervals of 180–360 s. The enthalpy changes ΔH upon binding, the association constant (KA), and the binding stoichiometry (N) were obtained directly, and the Gibbs energy (ΔG) − and entropy (ΔS) changes were calculated according to Equation 3. The dilution heat of the control titration, consisting of the identical titrant solution but with only buffer in the sample cell, was subtracted from each experimental titration. All steps of the data analysis were performed using the ORIGIN (Version 5.0) software provided by the manufacturer (Microcal).
SPOT Synthesis
Cellulose-bound peptide arrays were prepared according to standard SPOT synthesis protocols using a SPOT synthesizer (Intavis, Köln, Germany) as described in detail in Wenschuh et al. (29). The peptides were synthesized on amino-functionalized cellulose membranes (Whatman, Maidstone, UK) of the ester type prepared by modifying cellulose paper with Fmoc-b-alanine as the first spacer residue. In the second coupling step, the anchor position Fmoc-b-alanine-OPfp in dimethyl sulfoxide (DMSO) was used. Residual amino functions between the spots were capped by acetylation. The Fmoc group was cleaved using 20% piperidine in dimethylformamide. The cellulose-bound peptide arrays were assembled on these membranes by using 0.3 m solutions of Fmoc-amino acid-OPfp in 1-methyl-2- pyrrolidine. Side-chain protection of the Fmoc-amino acids used was as follows: Glu, Asp (OtBu); Ser, Thr, Tyr (tBu); His, Lys, Trp (Boc); Asn, Gln, Cys (Trt); Arg (Pbf). After the last coupling step, the acid-labile protection groups of the amino acid side chains were cleaved using 90% trifluoroacetic acid (TFA) for 30 min and 60% TFA for 3 h.
Binding Studies on Cellulose Membrane-bound Peptides
All primary incubation and washing steps were carried out under gentle shaking at room temperature. After washing the membrane with ethanol once for 10 min and 3 times for 10 min with Tris-buffered saline (TBS: 50 mm Tris-(hydroxymethyl)-aminomethane, 137 mm NaCl, 2.7 mm KCl, adjusted to pH 8 with 0.05% HCl), the membrane-bound peptide arrays were blocked for 3 h with blocking buffer (blocking buffer concentrate (Sigma), 1:10 in TBS containing 5% (w/v) sucrose) and then washed with TBS (1 × 10 min). Subsequently, the peptide arrays were incubated with 10 μm analyte solutions (mCRY2-(489–592) or Cy3.5 fluorescence-labeled mBMAL1-(577–625)) in TBS blocking buffer at 4 °C overnight. After washing 3 times for 10 min with TBS, analysis and quantification of peptide-bound mBMAL1 was carried out using a Lumi-Imager (Roche Applied Science). For mCRY2, a two-antibody system was used; anti-mCRY2/rabbit antibody (Alpha Diagnostics International, San Antonio, TX) in TBS was incubated at room temperature (1 h), and after washing 3 times with TBS, peroxidase-labeled anti-rabbit IgG was used as a secondary antibody and also incubated for 1 h. After washing 3 times 10 min with TBS, detection was done via chemiluminescence of the substrate.
Measurement of Spot Signal Intensities
Analysis and quantification of spot signal intensities were conducted with the Genespotter software package (Microdiscovery, Berlin, Germany). Genespotter has a fully automatic grid-finding routine, resulting in reproducible signal intensities. The spot signal is calculated from a circular region around the spot center detected in the image. The background signal for each spot is determined with a safety margin to the whole membrane background. The fluorescence of Cy3.5-labeled mBMAL1 was measured at 600 nm, and mCRY2 was detected via chemiluminescence.
Peptide Synthesis and Purification
Peptides P1 and P2 corresponding to the predicted coiled coil region of mCRY1 (P1, 472NHAEASRLNIERMKQIYQQLSRYRGLGLLASVPS505) and the C-terminal mBMAL1 binding epitope in the mCRY1 tail (P2, 564SQQTHSLKQGRSSAGTGLSSGKRPSQEE591) were synthesized using standard Fmoc chemistry on solid phase. Purification was performed on a C18 column using a gradient of water, ethanol, 0.08% TFA. The N termini of the peptides were protected by an acetyl group, and the C-termini were protected by amide.
RESULTS
Expression and Purification of mCRY and mBMAL1 Fragments
We have cloned, expressed, and purified C-terminal fragments of the mouse cryptochromes 1 and 2 (mCRY1-(471–606) and mCRY2-(489–592)), which contain the most C-terminal α-helix of the photolyase homology region (predicted CC) and the tail region (Fig. 1). Whereas the coiled coil region is well conserved between mCRY1 and mCRY2, their tails are clearly different. In addition, two C-terminal mBMAL1 fragments, mBMAL1-(490–625) and mBMAL1-(577–625), were constructed based on secondary structure predictions. The mCRYCCtail and mBMAL1 fragments were expressed in E. coli as GST fusion constructs and purified via affinity and size exclusion chromatography. The described purification scheme resulted in overall yields of ∼15 mg of highly purified mCRY or mBMAL1 proteins per liter of cell culture (supplemental Fig. S1). The identity of the purified proteins was confirmed by mass spectrometry.
Analysis of Self-oligomerization and Folding by Analytical Ultracentrifugation and CD Spectroscopy
To determine the oligomeric state of the mCRY and mBMAL1 fragments, we have performed analytical ultracentrifugation (AUC) sedimentation velocity experiments (Table 1, supplemental Fig. S2). These experiments showed that all fragments are monomeric at concentrations between 50 and 200 μm. The hydrodynamic (Stokes) radii determined by AUC analyses (Table 1) suggest that the mCRY and mBMAL1 proteins have somewhat elongated shapes and might be at least partially unstructured. Using CD spectroscopy, we have analyzed the secondary structure content of the purified mCRY and mBMAL1 fragments. The CD spectra (supplemental Fig. S3A) and their analysis (Table 2) indicate that all fragments are partially (between 30 and 40%) disordered. This may contribute to their enlarged hydrodynamic radii. Additionally our CD spectra confirmed the helicity of the synthetic peptide P1 comprising the predicted coiled coil region of the mCRY proteins (12).
TABLE 1.
Analysis of the oligomeric state and molecular shape of mBMAL1 and mCRYCCtail fragments by analytical ultracentrifugation
| Proteina | sb | RHc | RH globular | RH GuHCl-unfolded | MWd | Oligomeric state |
|---|---|---|---|---|---|---|
| S | nm | nm | nm | Da | ||
| mCRY1CCT | 1.146 | 3.26 | 1.91 | 3.43 | 14433 | Monomeric |
| mCRY2CCT | 1.176 | 2.50 | 1.79 | 3.12 | 12087 | Monomeric |
| BMAL490 | 1.322 | 2.62 | 1.90 | 3.41 | 14264 | Monomeric |
| BMAL577 | 0.667 | 1.98 | 1.35 | 2.03 | 5482 | Monomeric |
a The concentration of mCRY1/2 and BMAL490 was adjusted to A280 = 0.5 (corresponds to 0.07 mm mCRY1, 0.17 mm mCRY2, 0.06 mm BMAL490). The BMAL577 concentration was 0.2 mm. BMAL490 was also shown to be monomeric at A280 = 5 corresponding to 0.6 mm BMAL490. mCRY1/2CCT = mCRY1/2 coiled-coil-tail fragment. BMAL490/577 = mBMAL1-(490/577–625) fragment.
b s[S] = sedimentation coefficient in Svedberg units. The s values are normalized to 20 °C and water.
c RH = hydrodynamic (Stokes) radius.
d Sequence molecular weight, calculated as described under “Experimental Procedures.”
TABLE 2.
CD spectra suggest that the mBMAL1 and mCRY proteins are partially disordered and confirm the predicted helicity of the mCRY coiled coil region
| Fractionsa | Helix | Strand | Turn | Un-ordered | r.m.s.d.b | (N)r.m.s.d.b |
|---|---|---|---|---|---|---|
| % | % | % | % | |||
| Protein | ||||||
| mCRY1CCT | 0.19 | 0.25 | 0.23 | 0.33 | 0.048 | 0.022 |
| mCRY2CCT | 0.18 | 0.21 | 0.23 | 0.38 | 0.028 | 0.010 |
| BMAL490 | 0.09 | 0.26 | 0.24 | 0.41 | 0.018 | 0.008 |
| BMAL577 | 0.08 | 0.29 | 0.23 | 0.40 | 0.062 | 0.005 |
| Peptide P1c | 0.32 | 0.15 | 0.21 | 0.32 | 0.043 | 0.015 |
a For secondary structure analysis the CONTIN algorithm (26) was used with the reference dataset SMP56 (28). Helices include regular and distorted helices. Strands include regular and distorted β-strand.
b (N)r.m.s.d. = (normalized) root mean square deviation. Helices include regular and distorted helices. Strands include regular and distorted ß-strand (27).
c Peptide P1, mCRY peptide comprising the predicted coiled coil region.
Analysis of mCRY-mBMAL1 Interactions by Fluorescence Polarization
To find out if our purified C-terminal mCRYCCtail- and mBMAL1 fragments form stable heterodimeric complexes in solution and to determine their binding affinities, we have performed fluorescence polarization experiments. mBMAL1 fragments were labeled with Cy3.5, and mCRY fragments were titrated to a 500 nm solution of fluorescently labeled mBMAL1 (mCRY concentrations ranging from 500 nm to 300 μm). The shorter mBMAL1-(577–625) fragment bound to both mCRYCCtail fragments with a roughly 10 μm affinity (Fig. 2A). In contrast, the longer mBMAL1-(490–625) fragment bound to mCRY1 with an ∼40 μm affinity and to mCRY2 with an ∼10 μm affinity (Fig. 2B). The different affinities of mCRY1 and mCRY2 to the longer mBMAL1 fragment might be due to the fact that the Cy3.5 dye not only attaches to free N-terminal amino groups but also to side-chain amino groups of lysine residues. Whereas the mBMAL1-(577–625) fragment lacks lysine residues, the mBMAL1-(490–625) fragment contains three lysine residues (Lys-493, Lys-537, Lys-538). Notably, Lys-537 acetylation by mCLOCK enhances mCRY1 binding to mBMAL1 in a cellular environment (21). It is, therefore, conceivable that covalent modification of the mBMAL1-(490–625) fragment by the Cy3.5 dye or the lack of Lys-537 acetylation in the E. coli-expressed mBMAL1-(490–625) fragment specifically weakens mCRY1 binding in our assay.
FIGURE 2.

Fluorescence polarization spectra for mBMAL1-mCRY1/2 interactions. Polarization values are plotted against the concentration of the mCRY titrant. mCRY1-(471–606) and mCRY2-(489–592) proteins were titrated into 500 nm solutions of Cy3.5 fluorescently labeled mBMAL1-(577–625) (A) and mBMAL1-(490–625) (B) proteins. mCRY concentrations are stepwise increased from 500 nm to 300 μm. The increasing polarization values document the formation of mBMAL1-mCRY complexes. KD values are in the μm range (see the insets). The experiments were reproduced at least three times with similar results. mCRY1/2CCT, mCRYCCtail fragments. BMAL577 and BMAL490, mBMAL1-(577–625) and mBMAL1-(490–625) fragments, respectively.
Analysis of mCRY-mBMAL1 Interactions by Isothermal Titration Calorimetry
To assess the possible influence of a covalent modification of lysine residues or the N-terminal amino group by the Cy3.5 dye, in particular on the mCRY1-mBMAL1-(490–625) interaction, we also determined binding affinities by ITC using unlabeled mCRY and mBMAL1 proteins. In good agreement with the fluorescence polarization data, mCRY1 and mCRY2 bind to the mBMAL1-(577–625) fragment with a roughly 10 μm affinity (Fig. 3A and Table 3). Whereas mCRY2 shows a similar (∼10 μm) affinity to both mBMAL1 fragments, mCRY1 binds to the longer mBMAL1-(490–625) fragment with a roughly 20 μm affinity (Fig. 3B and Table 3). Values of 40 μm were never obtained for the mCRY1-mBMAL1-(490–625) interaction using ITC. This indicates that the lower affinity of mCRY1 to mBMAL1-(490–625) compared with mBMAL1-(577–625) is an intrinsic feature of the unlabeled proteins and the mCRY1-mBMAL1-(490–625) interaction is additionally weakened by the Cy3.5 dye in the fluorescence polarization experiments. Interestingly, the mutation of Lys-537 to Gln, which mimics the acetylation of Lys-537, increases the affinity of the mBMAL1-(490–625)-mCRY1 interaction to ∼10 μm (Table 3). This result suggests that the non-acetylated Lys-537 is indeed responsible for the lower (∼20 μm) affinity of mCRY1 to the wild-type mBMAL1-(490–625) fragment. According to our CD spectra, the K537Q mutation does not change the secondary structure content of the mBMAL1-(490–625) fragment (data not shown).
FIGURE 3.

Analysis of mCRY-mBMAL1 interactions by ITC. Representative ITC experiments for the interactions of mCRY2-(489–592) (receptor, 0.052 mm) with mBMAL1-(577–625) (ligand, 0.6 mm) (A) and mCRY1-(471–606) (receptor, 0.06) (B) with mBMAL1-(490–625) (ligand 0.8 mm) are shown. For both titrations the binding events are endothermic (heat is absorbed) and entropically favored. The top panels show the time response of the heat change upon the addition of the ligand. The best fits (lower panels) were obtained by using a single site binding model (best χ2 statistics) resulting in a 1:1 stoichiometry (N close to 1). At the used concentrations, receptor and ligand are monomeric according to our AUC measurements (Table 1).
TABLE 3.
ITC binding constants and thermodynamic parameters
ITC experiments were performed at 22 °C in 25 mm Bis-tris propane buffer, pH 7.8, 100 mm NaCl, 5 mm Tris(2-carboxyethyl)phosphine. At the concentrations used in the ITC experiments, all proteins were monomeric as shown by AUC (Table I). Binding was dominated by favorable entropy changes, and the contribution of TΔS to ΔG was significantly higher than ΔH. The BMAL490(K537Q)-P2 interaction was entropically and enthalpically favored. N, number of binding sites (N = ligand/receptor). Except for the mBMAL1-P1 complexes, the mBMAL1 fragments were used as ligands. Reported values and S.D. are the mean of at least three independent titrations.
| Complex | N | KD | ΔH | TΔS |
|---|---|---|---|---|
| μm | kcal/mol | kcal/mol | ||
| mCRY1CCT-BMAL577 | 0.9 | 10.5 ± 2.3 | 2.6 | 9.2 |
| mCRY1CCT-BMAL490 | 0.8 | 18.9 ± 5.0 | 1.8 | 8.1 |
| mCRY1CCT-BMAL490(K537Q) | 0.8 | 9.3 ± 2.2 | 1.4 | 8.2 |
| mCRY2CCT-BMAL577 | 1.1 | 7.8 ± 1.2 | 1.0 | 8.4 |
| mCRY2CCT-BMAL490 | 1.1 | 9.5 ± 2.2 | 4.2 | 11.1 |
| P1-BMAL577 | 1.0 | 10.7 ± 2.6 | 1.1 | 8.3 |
| P1-BMAL490 | 0.8 | 10.6 ± 3.0 | 1.3 | 8.1 |
| P1-BMAL490(K537Q) | 1.0 | 9.6 ± 2.0 | 0.7 | 7.5 |
| P2-BMAL577 | 0.9 | 3.3 ± 1.0 | 0.5 | 8.0 |
| P2-BMAL490 | 0.9 | 7.7 ± 1.9 | 7.3 | 14.2 |
| P2-BMAL490(K537Q) | 1.0 | 2.9 ± 0.9 | −0.6 | 6.8 |
Identification of mCRY-mBMAL1-interacting Epitopes by Peptide Scan Analysis
To map the mCRY-mBMAL1 interaction sites more accurately, we have performed peptide scan analyses. For determination of the mBMAL1 binding sites of mCRY1 and mCRY2, we used the Cy3.5-labeled mBMAL1-(577–625) fragment as analyte. Mapping of the mCRY2 binding site on the mBMAL1-(577–625) fragment was performed with an anti-mCRY2 antibody. The peptide scan analysis revealed two mBMAL1 binding sites in mCRY1 and mCRY2 (Fig. 4, A and B). One binding site corresponds to the predicted coiled coil region (Fig. 1). This epitope has been identified in both cryptochromes and comprises residues Ala-476 to Pro-504 in mCRY1 and residues Thr-494 to Pro-522 in mCRY2. The second epitope of both cryptochromes lies within their non-conserved tails and is interrupted. In mCRY1, the second epitope includes amino acids between Ser-564 and the C-terminal end and is interrupted at the acidic residues Glu-590, Glu-591, and Asp-592 (Fig. 4A). In mCRY2, the second epitope lies between Ala-540 and Thr-580 and includes a gap at residues Glu-566, Glu-567, and Pro-568 (Fig. 4B).
FIGURE 4.

Identification of mCRY-mBMAL1 interacting epitopes by peptide scan analysis. mCRY1mCRY1-(471–606) (A) and mCRY2-(489–592) (B) were incubated with Cy3.5-labeled mBMAL1-(577–625). mCRY1mCRY1-(471–606) and mCRY2-(489–592) were dissected into overlapping 10-mer sequences with an overlay of one amino acid (peptide scan). The resulting peptide array was synthesized using SPOT synthesis and probed against mBMAL1-(577–625). Signal intensities (SI) for each membrane spot are plotted against the first amino acid of the corresponding 10-mer peptide. The mCRY1 and mCRY2 membranes were incubated with Cy3.5-labeled mBMAL1-(577–625) (c = 10 μm). Binding to the mCRY peptides was detected by measuring the fluorescence emission of Cy3.5 on each membrane spot at 600 nm. Fluorescence emission of each spot was calculated from a circular region around the spot center detected in the membrane image. The presented results are global background-corrected. C, mBMAL1-(577–625) incubated with mCRY2-(489–592) is shown. Signal intensities for each membrane spot are plotted against the first amino acid of the corresponding 10-mer peptide. The membrane spots contain an array of 10-mer peptides covering the entire mBMAL1-(577–625) sequence with a shift of one amino acid. The mBMAL1 membrane was incubated with a 10 μm mCRY2 solution, and a two-antibody system was used to detect mCRY2 binding to the mBMAL1 peptides via chemiluminescence. The spot signal measured by means of chemiluminescence was calculated from a circular region around the spot center detected in the image. The presented results are global background-corrected. D, shown is a substitution analysis of the two mBMAL1 binding epitopes of mCRY1. Left, N-terminal epitope (473HAEASRLNIERMKQIYQQLSRYRGLGLLASVP504) corresponding to the predicted coiled coil region is shown. Significant effects of alanine mutations were only found in the depicted N-terminal peptide region. Right, C-terminal epitope within the mCRY1 tail region (565QQTHSLKQGRSSAGTGLSSGKRPSQEEDAQS595) is shown. Spots in the first row represent the wt mCRY1 sequences. Each spot of the second row corresponds to a mutated peptide in which one residue was replaced by alanine (mutated position as written to the left of the two spot columns). The mCRY1 membranes were incubated with Cy3.5-labeled mBMAL1-(577–625) (c = 10 μm). Signals were measured as described in A and B. Basic and acidic residues, whose substitution by alanine lead to reduced or enhanced mBMAL1-(577–625) binding, are highlighted in red and green, respectively.
Our peptide scan analysis of mBMAL1 revealed two binding sites for mCRY2 (Fig. 4C); that is, one epitope between residues Ala-598 and Ala-610, including a predicted α-helical mBMAL1 segment (Fig. 1B), and a second epitope between Leu-612 and the C-terminal end.
Substitutional Analysis of mBMAL1 Binding mCRY Epitopes
To determine which mCRY residues are most critical for the mBMAL1 interaction, we have SPOT-synthesized peptides including the conserved mCRY coiled coil epitope (mCRY1 sequence 473HAEASRLNIERMKQIYQQLSRYRGLGLLASVP504) and the major part of the mCRY1 tail epitope (565QQTHSLKQGRSSAGTGLSSGKRPSQEEDAQS595). We have exchanged each amino acid in the mCRY peptides to alanine and measured the binding of the modified peptides to Cy3.5 fluorescently labeled mBMAL1-(577–625) (Fig. 4D). This experiment showed that single alanine mutations of the negatively charged residues Glu-590, Glu-591, or Asp-592 in the mCRY1 tail peptide raise its affinity to mBMAL1 drastically, whereas the exchange of any single lysine or arginine to alanine lowers it. Similarly, the exchange of positively charged residues in the N-terminal part of the coiled coil epitope (Arg-478, Arg-483, and Lys-485 in mCRY1; Arg-496, Arg-501, and Lys-503 in mCRY2) to alanine significantly reduced the affinity to mBMAL1-(577–625). We conclude that the interaction with the overall acidic mBMAL1 fragments is driven predominantly by electrostatic interactions and the interruption of the mCRY1 tail epitope is likely due to electrostatic repulsion effects.
ITC Analysis of mBMAL1 Interactions with mCRY Peptides
To quantify the contributions of the two mBMAL1 binding epitopes of mCRY1 and to find out which epitope is responsible for the different binding affinities of mCRY1 to mBMAL1-(577–625), mBMAL1-(490–625), and mBMAL1-(490–625)K537Q, we have synthesized peptides comprising mCRY1 residues Asn-472 to Ser-505 corresponding to the predicted coiled coil region (peptide P1) as well as residues Ser-564 to Glu-591 within the mCRY1 tail region (peptide P2). CD spectroscopy showed that peptide P1 has a high α-helical content (as predicted), whereas peptide P2 is mostly disordered (Table 2 and supplemental Fig. S3B). Our ITC measurements revealed that peptide P1 binds to all three mBMAL1 fragments with an affinity of ∼10 μm (Table 3 and Fig. 5A). Peptide P2, however, binds to mBMAL1-(577–625) and to the mBMAL1-(490–625)K537Q mutant fragment with an affinity of ∼3 μm but to the wild-type mBMAL1-(490–625) fragment with a lower affinity of ∼8 μm (Table 3 and Fig. 5, B–D). In contrast to all other mCRY-mBMAL1 interactions that we have analyzed by ITC, the interaction of peptide P2 with mBMAL1-(490–625)K537Q is exothermic and enthalpically as well as entropically favored (Fig. 5D, Table 3). This indicates that the P2-mBMAL1-(490–625)K537Q complex involves a larger number of polar contacts (e.g. hydrogen bonds) than the other mCRY-BMAL1 interactions, which are entropically but not enthalpically favored (30).
FIGURE 5.

ITC analysis of the binding of mCRY peptides to mBMAL1. Shown are ITC experiments for binding of peptide P1 (ligand, 0.73 mm) to mBMAL1-(490–625) (receptor, 0.055 mm) (A), peptide P2 (receptor, 0.04 mm) to mBMAL1-(577–625) (ligand, 0.34 mm) (B), peptide P2 (receptor, 0.04 mm) to mBMAL1-(490–625) (ligand, 0.46 mm) (C), and peptide P2 (receptor, 0.025 mm) to mBMAL1-(490–625)K537Q (ligand, 0.34 mm) (D). Binding reactions are dominated by favorable entropy changes. A–C, binding events are endothermic and entropically favored. D, binding is exothermic and entropically and enthalpically favored. The top panels show the time response of the heat change upon addition of the ligand. The best fits (lower panels) were obtained by using a single site binding model (best χ2 statistics) resulting in a 1:1 stoichiometry (N close to 1). At the used concentrations, receptor and ligand are monomeric according to our AUC measurements (Table 1).
DISCUSSION
The C-terminal coiled coil and tail (CCtail) regions of the mammalian cryptochromes (mCRY1/2) and the C-terminal mBMAL1 region critically regulate the activity of the mBMAL1-mCLOCK transcription factor complex within the mammalian circadian clock (12, 16, 17). To provide mechanistic insights into the molecular interactions of the mCRYCCtail- and C-terminal mBMAL1 regions and their regulation by mBMAL1 acetylation on Lys-537 (21), we have purified mCRYCCtail proteins (mCRY1-(471–606) and mCRY2-(489–592)) and two C-terminal mBMAL1 fragments (mBMAL1-(577–625) and mBMAL1-(490–625)) and quantitatively analyzed their interactions by fluorescence polarization and ITC (Figs. 2 and 3 and Table 3). Although mCRY2-(489–592) bound equally well to both mBMAL1 fragments (KD = 8–10 μm), mCRY1 showed a roughly two times weaker interaction with the longer mBMAL1-(490–625) fragment that was additionally destabilized by the Cy3.5 dye used in the fluorescence polarization assay (KD ∼20 μm without and 40 μm with Cy3.5 dye). We conclude that the mBMAL1 region between residues 490 and 576 specifically weakens the binding of mCRY1 (but not mCRY2), presumably by masking mCRY1 binding sites located within the shorter mBMAL1-(577–625) fragment. Interestingly, the mutated mBMAL1-(490–625)K537Q fragment, in which Lys-537 acetylation is mimicked by a glutamine, binds to the mCRY1CCtail protein with a similar affinity (KD ∼10 μm) as mBMAL1-(577–625) (Table 3). The K537Q mutation, therefore, appears to unmask the mCRY1 binding sites in the mBMAL1-(577–625) fragment.
We have identified two mBMAL1 binding epitopes in the predicted coiled coil region and within the tails of mCRY1 and mCRY2 (Fig. 4, A and B). Whereas a synthetic peptide corresponding to the mCRY coiled coil region (P1) bound equally well to the short and to the long (wild-type and K537Q mutant) mBMAL1 fragments (KD ∼10 μm), peptide P2 including the mCRY1 tail epitope showed a roughly 2× lower affinity to mBMAL1-(490–625) than to mBMAL1-(577–625) (KD values ∼8 μm versus 3 μm) and bound to the mBMAL1-(490–625)K537Q mutant fragment with a similar affinity as to mBMAL1-(577–625) (KD ∼3 μm). Hence, the relative affinities of peptide P2 to our mBMAL1 fragments reflect those of the mCRY1CCtail fragment (Table 3). We conclude that the mCRY1 tail epitope accounts for the effects of the mBMAL1 region between residues 490 and 576 and of the Lys-537mBMAL1Gln mutation on the mBMAL1-mCRY1 interaction. Furthermore, the non-conserved mCRY tail epitopes are responsible for the different binding affinities of mCRY1 and mCRY2 to the mBMAL1-(490–625) fragment. The increased affinity of the mCRY1CCtail protein and the P2 tail peptide to the K537Q mutant version of mBMAL1-(490–625) likely mimics the effect of Lys-537 acetylation in vivo, which enhances mCRY1 binding to mBMAL1 and thereby down-regulation of mBMAL1-mCLOCK dependent transcription (21).
Our substitution analysis (Fig. 4D) revealed that alanine mutations of positively charged residues in both mCRY epitopes weaken the interaction with mBMAL1, whereas alanine mutations of the negatively charged residues Glu-590, Glu-591, and Asp-592, at which the mCRY1 tail epitope is interrupted, strengthen it. Because the mBMAL1 fragments used in this study are negatively charged (pI mBMAL1-(577–625) = 3.4; pI mBMAL1-(490–625) = 4.2), we suggest that binding of the mCRYCCtail fragments is driven by electrostatic interactions. We propose that in its non-acetylated state Lys-537 masks negative charges in mBMAL1 through intramolecular interactions and thereby interferes with mCRY1 binding. Lys-537 acetylation would weaken this masking effect and strengthen electrostatic interactions with positively charged mCRY1 residues predominantly in the tail (Fig. 6).
FIGURE 6.

Model for the regulation of mCRY1 binding by Lys-537BMAL1 acetylation. Top, in its non-acetylated state, Lys-537 masks negative charges in mBMAL1 through intramolecular interactions and thereby interferes with mCRY1 binding. Bottom, Lys-537 acetylation unmasks negative mBMAL1 charges and allows for electrostatic interactions with positively charged mCRY1 residues predominantly in its tail region.
Notably, Arg-501 and Lys-503 in the coiled coil region are important for the interaction of mCRY2 with mPER2 (31) as well as mBMAL1 (this study). Hence, binding of the mCRY coiled coil to mBMAL1 and mPER2 involves very similar molecular surfaces and is likely to be competitive. The functional importance of the coiled coil interaction with mBMAL1 is documented by the reduced efficiency of the R501E/K503R mCRY2 double mutant in transcriptional repression of the mBMAL1-mCLOCK complex (31). Yet the single mutations R501E or K503R weaken the binding of mCRY2 to full-length mPER2 but not to full-length mBMAL1. This is likely due the fact that mCRY interactions with mPER1 and mPER2 are predominantly (if not exclusively) mediated by the coiled coil region and do not require the mCRY tails or the PHCR (12, 31). In the repressive mBMAL1-mCRY complex, the additional and regulated interaction of mBMAL1 with the mCRY tails might facilitate the displacement of mPERs from the common coiled coil binding site.
It is striking that the P2 peptide binds to our mBMAL1 fragments with higher affinities than the mCRY1CCtail fragment (Table 3). This is probably due to the fact that this peptide ends at Glu-591 and, therefore, excludes one of the repulsive residues, Asp-592. Hence, our study suggests the design of tighter binding mCRY1-derived peptides by further elimination of negative charges or the addition of positive charges. Importantly, the K537Q mutation not only leads to an increased affinity of the P2 peptide to mBMAL1-(490–625) but also to an exothermic binding reaction, which is enthalpically and enthropically favored (Fig. 5D, Table 3). It is, therefore, conceivable that in the cell, peptide P2 would preferentially bind to mBMAL1, when it is acetylated on Lys-537 by mCLOCK. Peptide P2 and P2-derived potentially tighter binding mCRY1 tail peptides may, therefore, be used in a cell-based system to specifically inhibit the repressive mBMAL1(K543-Ac)-mCRY1 interaction. This could arrest the clock in a state where the mBMAL1-mCLOCK complex is transcriptionally active, possibly due to prolonged recruitment of p300/CBP transcriptional coactivators. It is conceivable that a peptide that interferes with the binding of the mCRY1 tail epitope would rather selectively target the mCRY1-mBMAL1 complex. Because our studies revealed different mBMAL1 interactions of the nonconserved mCRY1 and mCRY2 tails, an mCRY1-tail-derived peptide should not significantly affect mBMAL1-mCRY2 interactions. Furthermore, the tails are not required for the interactions of mCRY1 and mCRY2 with mPER2 and mPER1 (12, 31).
It is possible that the full-length mCRY or mBMAL1 proteins contain additional binding regions that further stabilize the mCRY-mBMAL1 complex. For the mCRYs, the correct alignment of the coiled coil region with the PHCR has been shown to be functionally important (12, 13). Because the isolated mCRYCCtail fragments are partially unstructured and peptide P2 is mostly disordered (supplemental Fig. S3, Table 2), the presence of the PHCR may enhance folding of the tail region. Indeed, a stabilizing interaction between the PHCR and the CCtail fragment of hCRY2 has been reported previously (32). Moreover, mBMAL1 interacts with the mCRY2 PHCR in a mammalian 2-hybrid system (31), and mCRY1 binds weakly to the PAS-B domain of mBMAL1 (20). Finally, mCRYs might bind more tightly to the mBMAL1-mCLOCK heterodimer, and in a repressive mPER-mCRY complex their binding might be enhanced by mPER interactions with the mBMAL1-mCLOCK PAS domains (33).
Additionally, posttranslational modifications such as acetylation (e.g. on Lys-537mBMAL1, see above) or phosphorylation (e.g. Ser-553/Ser-557 in the mCRY2 tail region (15)) may influence the mCRY-mBMAL1 interactions. Because Ser-553 and Ser-557 are located within the C-terminal mBMAL1 binding epitope of mCRY2 (Fig. 4B), their phosphorylation may influence or be affected by mBMAL1 binding. Furthermore, the tail epitopes of mCRY1 and mCRY2 both contain bipartite nuclear localization signals (585KRPX11KVQR602 in mCRY1, 559KRKX13KRAR578 in mCRY2) (12, 14) (Figs. 1B and 4, A and B), which might also be affected by the mBMAL1 interaction.
We have mapped the mCRY binding epitopes of mBMAL1 to the most C-terminal residues as well as a preceding predicted α-helix (Figs. 4C and 1B). Although it is tempting to speculate about a helical interaction between the mCRY coiled coil region and the predicted helical epitope of mBMAL1, deletions or mutations within the last 15 mBMAL1 residues have been reported to interfere with mCRY-mBMAL1 interactions and mCRY-dependent transcriptional repression (16, 17). Presumably, both epitopes are relevant to mCRY interactions, and it remains to be seen which part of the mCRYCCtail region binds to which mBMAL1 epitope.
With the presented work we have shown that the mCRY coiled coil and tail regions directly interact with the C-terminal 27 amino acids of the transcription factor mBMAL1. The mCRY1tail-mBMAL1 interaction is specifically affected by Lys-537, whose acetylation enhances mCRY1-mBMAL1 interactions in vivo. Our study suggests the design of peptide ligands targeting the interface between the mCRY1 tail region and mBMAL1. By inhibiting the repressive mBMAL1-mCRY1 interaction, such peptides may affect the transcriptional regulation of clock genes (and, hence, the circadian clock) and clock-controlled genes (and hence the circadian regulation of body functions).
Supplementary Material
Acknowledgments
We thank S. Uebel and E. Weyher-Stingl in the Core facility of the MPI of Biochemistry for help with biophysical experiments.
This work was supported by Deutsche Forschungsgemeinschaft Grants WO-695/3, WO-695/4, and WO-695/5 (to E. W.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
- PHR
- photolyase homology region
- CRY
- cryptochrome
- PER
- period
- PAS
- PER-ARNT-SIM
- PHCR
- photolyase homology core region
- CC
- coiled coil
- BMAL
- brain and muscle ARNT-like protein
- CLOCK
- circadian locomotor output cycle kaput
- m
- mouse/mammalian
- h
- human
- ITC
- isothermal titration calorimetry
- AUC
- analytical ultracentrifugation
- CT
- circadian time
- CBP
- cAMP-response element-binding protein (CREB)-binding protein
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl; 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
REFERENCES
- 1. Young M. W., Kay S. A. (2001) Nat. Rev. Genet. 2, 702–715 [DOI] [PubMed] [Google Scholar]
- 2. Griffin E. A., Jr., Staknis D., Weitz C. J. (1999) Science 286, 768–771 [DOI] [PubMed] [Google Scholar]
- 3. Kume K., Zylka M. J., Sriram S., Shearman L. P., Weaver D. R., Jin X., Maywood E. S., Hastings M. H., Reppert S. M. (1999) Cell 98, 193–205 [DOI] [PubMed] [Google Scholar]
- 4. Kondratov R. V., Antoch M. P. (2007) Trends Cell Biol. 17, 311–317 [DOI] [PubMed] [Google Scholar]
- 5. Bechtold D. A., Gibbs J. E., Loudon A. S. (2010) Trends Pharmacol. Sci. 31, 191–198 [DOI] [PubMed] [Google Scholar]
- 6. Bunger M. K., Wilsbacher L. D., Moran S. M., Clendenin C., Radcliffe L. A., Hogenesch J. B., Simon M. C., Takahashi J. S., Bradfield C. A. (2000) Cell 103, 1009–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thresher R. J., Vitaterna M. H., Miyamoto Y., Kazantsev A., Hsu D. S., Petit C., Selby C. P., Dawut L., Smithies O., Takahashi J. S., Sancar A. (1998) Science 282, 1490–1494 [DOI] [PubMed] [Google Scholar]
- 8. van der Horst G. T., Muijtjens M., Kobayashi K., Takano R., Kanno S., Takao M., de Wit J., Verkerk A., Eker A. P., van Leenen D., Buijs R., Bootsma D., Hoeijmakers J. H., Yasui A. (1999) Nature 398, 627–630 [DOI] [PubMed] [Google Scholar]
- 9. Vitaterna M. H., Selby C. P., Todo T., Niwa H., Thompson C., Fruechte E. M., Hitomi K., Thresher R. J., Ishikawa T., Miyazaki J., Takahashi J. S., Sancar A. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 12114–12119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Park H. W., Kim S. T., Sancar A., Deisenhofer J. (1995) Science 268, 1866–1872 [DOI] [PubMed] [Google Scholar]
- 11. Maul M. J., Barends T. R., Glas A. F., Cryle M. J., Domratcheva T., Schneider S., Schlichting I., Carell T. (2008) Angew. Chem. Int. Ed. Engl. 47, 10076–10080 [DOI] [PubMed] [Google Scholar]
- 12. Chaves I., Yagita K., Barnhoorn S., Okamura H., van der Horst G. T., Tamanini F. (2006) Mol. Cell. Biol. 26, 1743–1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McCarthy E. V., Baggs J. E., Geskes J. M., Hogenesch J. B., Green C. B. (2009) Mol. Cell. Biol. 29, 5465–5476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sakakida Y., Miyamoto Y., Nagoshi E., Akashi M., Nakamura T. J., Mamine T., Kasahara M., Minami Y., Yoneda Y., Takumi T. (2005) J. Biol. Chem. 280, 13272–13278 [DOI] [PubMed] [Google Scholar]
- 15. Kurabayashi N., Hirota T., Sakai M., Sanada K., Fukada Y. (2010) Mol. Cell. Biol. 30, 1757–1768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sato T. K., Yamada R. G., Ukai H., Baggs J. E., Miraglia L. J., Kobayashi T. J., Welsh D. K., Kay S. A., Ueda H. R., Hogenesch J. B. (2006) Nat. Genet. 38, 312–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kiyohara Y. B., Tagao S., Tamanini F., Morita A., Sugisawa Y., Yasuda M., Yamanaka I., Ueda H. R., van der Horst G. T., Kondo T., Yagita K. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 10074–10079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Takahata S., Ozaki T., Mimura J., Kikuchi Y., Sogawa K., Fujii-Kuriyama Y. (2000) Genes Cells 5, 739–747 [DOI] [PubMed] [Google Scholar]
- 19. Etchegaray J. P., Lee C., Wade P. A., Reppert S. M. (2003) Nature 421, 177–182 [DOI] [PubMed] [Google Scholar]
- 20. Langmesser S., Tallone T., Bordon A., Rusconi S., Albrecht U. (2008) BMC Mol. Biol. 9, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hirayama J., Sahar S., Grimaldi B., Tamaru T., Takamatsu K., Nakahata Y., Sassone-Corsi P. (2007) Nature 450, 1086–1090 [DOI] [PubMed] [Google Scholar]
- 22. Laue T. M. S. B. D., Ridgeway T. M., Pelletier S. L. (1992) Analytical Ultracentrifugation in Biochemistry and Polymer Science (Harding S., Rowe A. eds) pp. 90–125, Royal Society of Chemistry [Google Scholar]
- 23. Schuck P. (2000) Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Deleted in proof.
- 25. Uversky V. N. (2002) Eur. J. Biochem. 269, 2–12 [DOI] [PubMed] [Google Scholar]
- 26. Provencher S. W., Glöckner J. (1981) Biochemistry 20, 33–37 [DOI] [PubMed] [Google Scholar]
- 27. Sreerama N., Woody R. W. (2000) Anal. Biochem. 287, 252–260 [DOI] [PubMed] [Google Scholar]
- 28. Sreerama N., Woody R. W. (2004) Protein Sci. 13, 100–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wenschuh H., Volkmer-Engert R., Schmidt M., Schulz M., Schneider-Mergener J., Reineke U. (2000) Biopolymers 55, 188–206 [DOI] [PubMed] [Google Scholar]
- 30. Ladbury J. E., Klebe G., Freire E. (2010) Nat. Rev. Drug Discov. 9, 23–27 [DOI] [PubMed] [Google Scholar]
- 31. Ozber N., Baris I., Tatlici G., Gur I., Kilinc S., Unal E. B., Kavakli I. H. (2010) BMC Mol. Biol. 11, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Partch C. L., Clarkson M. W., Ozgür S., Lee A. L., Sancar A. (2005) Biochemistry 44, 3795–3805 [DOI] [PubMed] [Google Scholar]
- 33. Chen R., Schirmer A., Lee Y., Lee H., Kumar V., Yoo S. H., Takahashi J. S., Lee C. (2009) Mol. Cell 36, 417–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Berndt A., Kottke T., Breitkreuz H., Dvorsky R., Hennig S., Alexander M., Wolf E. (2007) J. Biol. Chem. 282, 13011–13021 [DOI] [PubMed] [Google Scholar]
- 35. Jones D. T. (1999) J. Mol. Biol. 292, 195–202 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.