

Abstract
Membrane proteins are critical to cell physiology, playing roles in signaling, trafficking, transport, adhesion, and recognition. Despite their relative abundance in the proteome and their prevalence as targets of therapeutic drugs, structural information about membrane proteins is in short supply. This review describes the use of electron crystallography as a tool for determining membrane protein structures. Electron crystallography offers distinct advantages relative to the alternatives of X-ray crystallography and NMR spectroscopy. Namely, membrane proteins are placed in their native membranous environment, which is likely to favor a native conformation and allow changes in conformation in response to physiological ligands. Nevertheless, there are significant logistical challenges in finding appropriate conditions for inducing membrane proteins to form two-dimensional arrays within the membrane and in using electron cryo-microscopy to collect the data required for structure determination. A number of developments are described for high-throughput screening of crystallization trials and for automated imaging of crystals with the electron microscope. These tools are critical for exploring the necessary range of factors governing the crystallization process. There have also been recent software developments to facilitate the process of structure determination. However, further innovations in the algorithms used for processing images and electron diffraction are necessary to improve throughput and to make electron crystallography truly viable as a method for determining atomic structures of membrane proteins.
I. Introduction
Biological membranes surround all cells and mediate all their interactions with the outside world. Membrane proteins relay information or chemical substrates across the membrane and are key players in the biochemical events that take place either at the surface of cells or within membrane-bound organelles. Depending on the biological context, membrane proteins act as receptors, enzymes, channels, transporters, structural proteins and cell-cell adhesion molecules and, as such, contribute to an astounding variety of essential cellular functions, including transmembrane signaling, homeostasis, and energy conversion.
When considered on a genome-wide scale, membrane proteins comprise ~40% of all genes in eukaryotic, eubacterial and archaeal organisms (Wallin and von Heijne, 1998). Given their omnipresence and functional diversity, it is not surprising that membrane proteins play a pivotal role in numerous human pathologies. Important diseases resulting from defective membrane proteins include cystic fibrosis, several forms of cancer, Alzheimer’s disease, and various cardiomyopathies. In fact, ~60% of the therapeutic drugs currently used in the United States target membrane proteins (Drews, 2000).
Despite this tremendous relevance to basic cell biology and to therapeutic medicine, our understanding of membrane proteins from a structural perspective is limited, especially when it comes to visualizing membrane proteins in their natural lipid bilayer environment. To a large extent, this limitation is due to the prevalent tools for structure determination: X-ray crystallography and NMR spectroscopy. These tools have become increasingly successful with detergent-solubilized species, but are at a distinct disadvantage when membrane proteins are embedded in a lipid bilayer. In contrast, electron crystallography is particularly well suited to the study of membrane proteins within their native, membranous environment. Although electron crystallography provided the first 3D structure of a membrane protein - bacteriorhodopsin in 1975 - and has subsequently produced a handful of atomic structures, it has largely foundered in the fringes due to practical difficulties and to lack of a high-throughput approach. Here we review its current state-of-the-art and discuss the future developments that are necessary to allow electron crystallography to fulfill its promise.
II. The membrane environment
Lipid molecules are arranged as a continuous bimolecular layer of approximately 50–60 Å in thickness (Engelman, 1971; Mitra et al., 2004). The lipid bilayer is a structurally and chemically heterogeneous environment that complicates the surface chemistry of membrane proteins relative to their soluble cousins. Three distinct regions can be delineated in a cross-section of the bilayer: (i) the hydrophobic core populated by the lipid acyl chains, (ii) the hydrophilic layers flanking the core that are formed by lipid head groups and, and (iii) the aqueous regions at the outer margins (White and Wimley, 1999). The hydrophobic core is ~30 Å thick and largely impermeable to polar molecules and ions. This is a region with a low dielectric that favors long-range polar interactions and where the hydrophobic effect is absent. The length of the acyl chains and their degree of saturation influence the overall thickness and fluidity of the bilayer. The lipid head groups occupy 10–15 Å on either side of the hydrophobic core and serve to bind most of the water in these regions. As a result, the hydration of protein components and the magnitude of the hydrophobic effect are decreased compared with bulk aqueous solution. Lipids contain a variety of different head groups, which can include charge, dipoles, and carbohydrate groups. Finally, the surrounding aqueous environment in the vicinity of most membranes also displays distinct properties relative to bulk water. Due mainly to surface charge from the lipid head groups, this region typically displays a gradient in solutes, pH, and ions. As an additional complexity, the lipids composing biological membranes are heterogeneous (Brugger et al., 1997), differing between the two leaflets of the bilayer and between membrane compartments within a given cell (Pike et al., 2002). In this way, the cell can adjust, the thickness, surface charge and fluidity of membranes to meet the requirements of individual membrane proteins in different cellular compartments (Andersen and Koeppe, 2007; Dowhan, 1997; Lee, 2004; Nyholm et al., 2007; Yeagle, 1989).
In order to conform to their membrane environment, integral membrane proteins are amphiphilic in nature, with their transmembranous regions immersed in the hydrophobic core of a lipid bilayer and their extramembranous domains surrounded by water. The water-exposed domains adopt the diverse array of protein folds observed in soluble proteins, though their vicinity to the membrane surface likely influences their design. The structure of membrane domains are dictated by the physical and chemical constraints of the lipid bilayer (Popot and Engelman, 2000; Schulz, 2000; Ubarretxena-Belandia and Engelman, 2001; White and Wimley, 1999) and membrane proteins structures so far reveal membrane domains composed either of α-helical bundles or β-barrels. β-barrel architectures are largely constrained to the outer-membrane of bacteria and, in this review, we will focus on α-helical membrane proteins given their greater influence over the functioning of eukaryotic cells and of human tissues, in particular.
III. Why are there so few membrane protein structures?
This amphiphilic nature of membrane proteins represents a fundamental constraint on our ability to produce and to study these proteins. Specifically, there are serious hurdles associated with (i) over-expression of membrane proteins with native tertiary and quaternary structure; (ii) preservation of biological activity when membrane proteins are extracted from their native membrane environment with detergent; (iii) the large size of membrane protein/detergent complexes, which limits the application of solution NMR; and (iv) the difficulty in obtaining well-diffracting three-dimensional (3D) crystals for X-ray crystallography. These obstacles become more pronounced for large membrane protein complexes or for the less stabile membrane proteins that tend to come from eukaryotic sources. The limited capacity of the cell to accommodate over-expression is due either to the physiological consequences on membrane function or to the limited capacity to produce extra membrane surface area. Limited stability reflects the inability of detergent to duplicate the physical/chemical environment of the lipid bilayer; although the basic tripartite structure is present in a lipid micelle, the heterogeneity, lateral pressure, and charge distribution of a biological membrane is impossible to replicate. Finally, 3D crystallization relies primarily on intermolecular contacts between extramembranous regions of the protein and conditions for promoting these interactions while simultaneously stabilizing the intramembranous region of the protein are difficult to find.
Our inability to overcome these hurdles is clearly reflected in the Protein Data Bank (PDB): out of ~63,000 protein structures deposited as of September 2010, only ~691 structures come from 256 different membrane proteins (http://blanco.biomol.uci.edu/Membrane_Proteins_xtal.html). The β-barrel fold is greatly over-represented (25%) in the PDB, reflecting the enhanced stability of this fold and the ease of producing proteins from bacterial hosts. Nevertheless, there has been tremendous progress over the last several years and the number of membrane protein structures has started to increase exponentially (White, 2009). Recent success stories include a number of groundbreaking structures that have greatly illuminated their respective fields, creating an increased appetite for improving existing technologies and for finding new and better ways to study the structures of this vast class of biological macromolecules. By increasing our understanding of membrane protein structure and, specifically, by appreciating the influence of their native lipid environment on their function, we will be better equipped to understand the role of these proteins in human health and disease.
IV. Application of electron crystallography to membrane proteins
Electron crystallography is the only method capable of imaging membrane proteins in their lipid environment. This method was pioneered in the 1970s by Henderson and Unwin in their studies of bacteriorhodopsin (Henderson and Unwin, 1975) and relies on an ordered array of molecules within a bilayer in the form of two-dimensional (2D) sheets or tubes (Fig. 1). In the case of bacteriorhodopsin, ordering occurs in vivo on a specialized photosynthetic region of the plasma membrane, providing an ideal specimen to drive development of the necessary technologies. After significant advances in electron microscope design, imaging strategies and image reconstruction algorithms, the atomic structure of bacteriorhodopsin was published in 1990 (Henderson et al., 1990), just 6 years after the landmark X-ray crystallographic structure of photosynthetic reaction center (Deisenhofer et al., 1984). Based primarily on these early developments by Henderson and colleagues, electron crystallography has continued to be a powerful tool for studying 3D structure of membrane proteins (Table 1) at medium and high-resolution (Stahlberg et al., 2001; Subramaniam et al., 2002; Unger, 2001). In addition to bacteriorhodopsin, this methodology has yielded atomic structures of plant light-harvesting complex (Kühlbrandt et al., 1994), human red cell aquaporin-1 (Murata et al., 2000), eye lens aquaporin-0 (Gonen et al., 2005), rat aquaporin-4 (Hiroaki et al., 2006), glutathione transferase (Holm et al., 2006), prostaglandin E synthase (Jegerschold et al., 2008) and acetylcholine receptor (Unwin, 2005). In addition, 3D structures of ~25 other unique membrane proteins have been determined to medium-resolution (5–8 Å), and continuing efforts are expected to produce atomic models in the near future (e.g. (Hirai et al., 2002; Kukulski et al., 2005; Ubarretxena-Belandia et al., 2003). The recent structure of aquaporin-0 (AQP0) (Fig. 2) is noteworthy and deserves further mention, not only for its remarkable high-resolution (1.9 Å), but also for its unique ability to reveal essentially all of the lipid molecules that make up the membrane bilayer (Gonen et al., 2005). Thus, despite recent advances in the application of solid state NMR (Hong, 2006) and molecular dynamics simulations (Lindahl and Sansom, 2008), electron crystallography represents the best approach to understanding membrane protein structure in the context of a lipid bilayer.
Figure 1. Types of crystals used for membrane protein structure determination by electron crystallography.

(A) A planar bilayer (highlighted as a grey slab) with a coherent 2D array of proteins. These crystals must be tilted to collect data for a 3D analysis of their structure. Another related type of crystal (not shown) arises from flattened lipid vesicles that contain two overlapping 2D lattices. (B) A helical array of proteins in a cylindrical lipid vesicle. Because many different views are provides for the molecules, these helical crystals do not need to be tilted. In both cases, crystals are very thin (50 Å for single-layered bacteriorhodopsin crystals and 600 Å for tubular crystals of the nicotinic acetylcholine receptor) and, as a result, cryo-EM combined with image processing is the natural choice for solving their 3D structure.
Table 1.
3D structures of membrane proteins determined by electron crystallography.
| Membrane Proteina | Resol. (Å) |
Year | Reference |
|---|---|---|---|
| Eye lens Aquaporin 0 | 1.9 | 2005 | (Gonen, et al. 2005) |
| Aquaporin-4 | 2.8 | 2009 | (Tani et al., 2009) |
| Bacteriorhodopsin | 3.0 | 1997 | (Kimura et al., 1997) |
| Glutathione transferase | 3.2 | 2006 | (Holm et al., 2006) |
| Plant LHC-II | 3.4 | 1991 | (Kühlbrandt and Wang, 1991) |
| Bacteriorhodopsin | 3.5 | 1990 | (Henderson et al., 1990) |
| Prostaglandin E synthase | 3.5 | 2008 | (Jegerschold et al., 2008) |
| Aquaporin-1 | 3.8 | 2000 | (Murata et al., 2000) |
| Acetylcholine receptor | 4.0 | 2005 | (Unwin, 2005) |
| Human aquaporin 2 | 4.5 | 2005 | (Schenk et al., 2005) |
| Plant Aquaporin SoPIP2 | 5.0 | 2005 | (Kukulski et al., 2005) |
| Halorhodopsin | 5.0 | 2000 | (Kunji et al., 2000) |
| Bovine Rhodopsin | 5.5 | 2003 | (Krebs et al., 2003) |
| Porin PhoE | 6.0 | 1991 | (Jap et al., 1991) |
| Bacteriorhodopsin | 6.0 | 1975 | (Henderson and Unwin, 1975) |
| Glutathione transferase | 6.0 | 2002 | (Holm et al., 2002) |
| Bacteriorhodopsin | 6.5 | 1983 | (Leifer and Henderson, 1983) |
| Oxalate transporter OxlT | 6.5 | 2002 | (Hirai et al., 2002) |
| Frog Rhodopsin frog | 6.5 | 1997 | (Unger et al., 1997) |
| Ca2+-ATPase | 6.5 | 2002 | (Xu et al., 2002) |
| Glycerol channel GlpF | 6.9 | 2000 | (Stahlberg et al. 2000) |
| Gap junction channel | 7.0 | 2007 | (Oshima et al., 2007) |
| NhaA Na/ H+ antiporter | 7.0 | 2000 | (Williams, 2000) |
| EmrE multidrug transporter | 7.0 | 2003 | (Ubarretxena-Belandia et al. 2003) |
| hCTR1 Cu transporter | 7.0 | 2009 | (De Feo et al., 2009) |
| Gap junction channel | 7.5 | 1999 | (Unger et al., 1999) |
| Sec YEG complex | 8.0 | 2005 | (Bostina et al., 2005) |
| Plant photosystem II RC | 8.0 | 1998 | (Rhee et al., 1998) |
| Neurospora H+-ATPase | 8.0 | 1998 | (Auer et al., 1998) |
| Acetylcholine receptor | 9.0 | 1993 | (Unwin, 1993) |
Atomic-resolution structures in bold.
Auer, M., et al., 1998. Three-dimensional map of the plasma membrane H+-ATPase in the open conformation. Nature. 392, 840–843.
Bostina, M., et al., 2005. Atomic model of the E. coli membrane-bound protein translocation complex SecYEG. JMB. 352, 1035–1043.
Holm, P. J., et al., 2002. The 3-D structure of microsomal glutathione transferase 1 at 6 A resolution as determined by electron crystallography of p22(1)2(1) crystals. Biochim. Biophys. Acta. 1594, 276–285.
Jap, B. K., et al., 1991. Structural Architecture of an Outer Membrane Channel as Determined by Electron Crystallography. Nature. 350, 167–170.
Kimura, Y., et al., 1997. Surface of bacteriorhodopsin revealed by high-resolution electron crystallography. Nature. 389, 206–211.
Kunji, E. R., et al., 2000. The three-dimensional structure of halorhodopsin to 5 A by electron crystallography: A new unbending procedure for two-dimensional crystals by using a global reference structure. Proc Natl Acad Sci U S A. 97, 4637–42.
Leifer, D., Henderson, R., 1983. Three-dimensional Structure of Orthorhombic Purple Membrane at 6-5 A Resolution. J. Mol. Biol. 163, 451–466.
Rhee, K. H., et al., 1998. Three-dimensional structure of the plant photosystem II reaction centre at 8 A resolution. Nature. 396, 283–6.
Stahlberg, H., et al., 2000. The 6.9-A structure of GlpF: a basis for homology modeling of the glycerol channel from Escherichia coli. JSB. 132, 133–141.
Unger, V. M., et al., 1997. Arrangement of rhodopsin transmembrane alpha-helices. Nature. 389, 203–6.
Unger, V. M., et al., 1999. Three-dimensional structure of a recombinant gap junction membrane channel. Science. 283, 1176–1180.
Xu, C., et al., 2002. A structural model for the catalytic cycle of Ca2+-ATPase. JMB. 316, 201–211.
Figure 2. Selection of high-resolution membrane protein structures solved by electron crystallography.
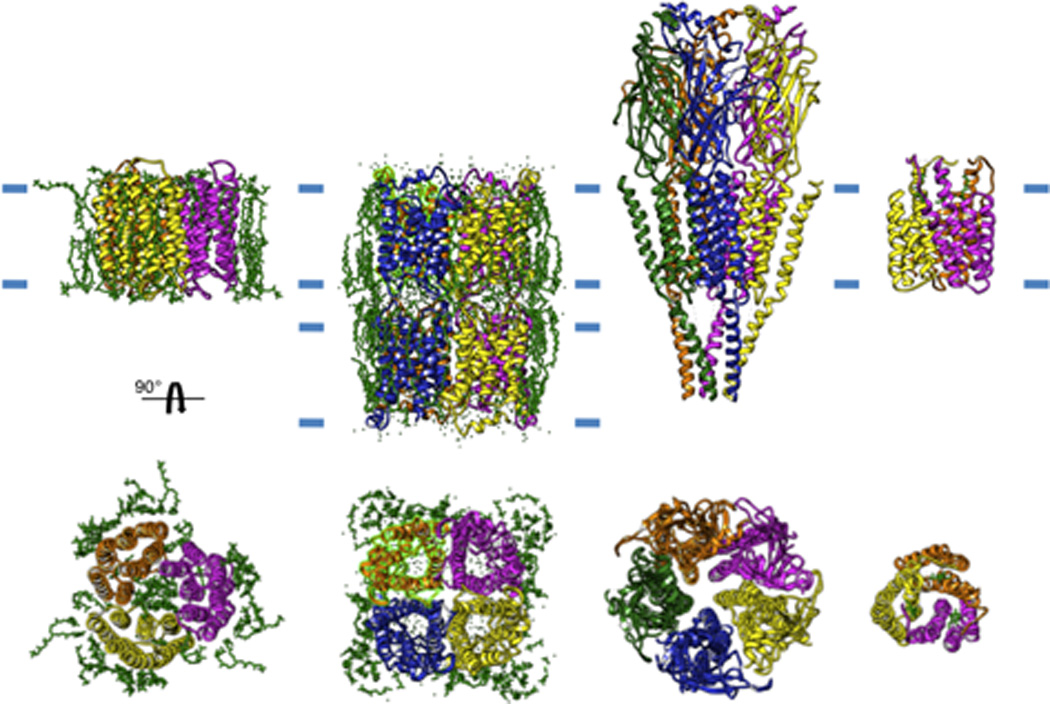
From left to right: a trimer of bacteriorhodopsin, a double-layer of the tetrameric AQP0, the heteropentameric acetylcholine receptor and a trimer of gluthathione transferase 1. The approximate boundaries of the bilayer is indicated by short blue lines and individual lipid molecules present in the structure are shown in green.
In special cases, crystallization within the lipid bilayer can be achieved directly within the native cellular membrane: e.g., bacteriorhodopsin from Halobacterium halobium, Ca2+-ATPase from mammalian sarcoplasmic reticulum (Zhang et al., 1998), and acetylcholine receptor from the electric organ of Torpedo marmorata (Unwin, 2005). More generally, these crystals are grown by reconstitution of purified, detergent-solubilized membrane proteins into lipid bilayers under defined conditions (for reviews see (Jap et al., 1992; Kühlbrandt, 1992; Mosser, 2001)). Reconstitution involves the controlled removal of detergent - by dialysis (Kühlbrandt, 1992), by controlled dilution (Remigy et al., 2003), by adsorption onto a hydrophobic resin (Rigaud et al., 1997) or by complexation with cyclodextrins (Signorell et al., 2006) - in presence of defined lipid species at an optimal lipid-to-protein ratio (LPR). By constraining a high density of a single protein species within a planar lipid bilayer, formation of a regular array within this bilayer becomes relatively favorable (Fig. 1). Given the physical constraint of molecules within the 2D plane of the bilayer, precipitants are not generally required for crystallization. Rather the most important factors appear to be the structural integrity and homogeneity of the protein, the choice of lipid species, the density of the protein within the bilayer, and the surface charge (as controlled by pH and lipid head group composition).
There are three predominant morphologies adopted by the resulting crystals: (i) flattened lipid vesicles with two, overlapping 2D lattices; (ii) tubular vesicles which retain a cylindrical shape and contain a helically organized array of membrane proteins; and (iii) a single, flat bilayer with a single, coherent 2D array of proteins (Fig. 1). Because these crystals are very thin (50 Å for single-layered bacteriorhodopsin crystals and 600 Å for tubular crystals of the nicotinic acetylcholine receptor), electron cryo-microscopy (cryo-EM) combined with image processing is the natural choice for solving their 3D structure.
V. Atomic structures by electron crystallography
1. Aquaporin
Water if the most prevalent molecule in biological tissues and life could not exist without its free circulation into and out of cells. Water can diffuse spontaneously across cell membranes but only at very low rates, and for this reason organisms from all three domains of life, from the simplest unicellular organisms to mammals, express membrane proteins called aquaporins that form specialized pores for water (Agre et al., 1993; Preston et al., 1992). There are many different types of aquaporins, at least 13 in mammals and at least 5 subfamilies in plants. Many of the mammalian aquaporins are expressed in the kidney, where water resorption is of critical physiological importance. Nevertheless, the original discovery of aquaporin was in the erythrocyte (AQP1), which set in motion a fierce competition to obtain the structure. Two different groups used electron crystallography to determine the first structures at 6–7 Å resolution (Cheng et al., 1997; Walz et al., 1997), which after several more years were improved to 3.8 Å (Murata et al., 2000), at which level the polypeptide chain could be traced. These structures revealed the protein fold for the first time and illustrated the path of water conduction across the membrane. Subsequent structures of aquaporins by electron crystallography include AQP2 (4.5 Å, (Schenk et al., 2005)) from apical membranes in the kidney collecting duct, AQP4 (2.8 Å, (Tani et al., 2009)) from basolateral membranes, plant aquaporin (SoPIP2 at 5.0 Å, (Kukulski et al., 2005)) and AQP0 from the eye lens at 2.5-1.9 Å resolution (Gonen et al., 2005; Hite et al., 2010).
The structures of AQP0 are notable for several reasons. First, this work represented the first time that methods of molecular replacement were applied in electron crystallography and the 1.9 Å resolution represented a new record for a mammalian membrane protein. Second, AQP0 formed double-layered crystals, which reflect the cell-cell junctions mediated by these channels in the lens (Fig. 2). Details of the structure showed how AQP0 closes its water channel upon formation of these junctions, a behavior that is important to maintaining proper hydration levels in the eye. Finally, these structures revealed a continuous lipid bilayer surrounding AQP0, elucidating specific interactions between the protein and its lipids. Furthermore, crystals formed from a different lipid species comprised a different oligomeric state of AQP0 (Hite et al., 2010), illustrating the strong effect that the lipid environment can have on membrane protein structure.
2. Acetylcholine receptor
Synaptic transmission at the neuromuscular junction is mediated by acetylcholine. The postsynaptic membrane is therefore loaded with the large heteropentameric acetylcholine receptor that recognizes acetylcholine and opens an ion channel. This opening depolarizes the membrane thus leading to an action potential and initiation of muscle contraction. A series of structures have been determined from tubular crystals of this nicotinic acetylcholine receptor from the electric organ of Torpedo marmorata (Fig. 2). Unlike the aquaporins, these crystals form spontaneously within the native biological membrane and a curvature is induced by the crystal contacts, which leads to a closed cylindrical shape with helical symmetry. Due to their limited size, these helical crystals harbor fewer molecules and produce a lower signal-to-noise ratio and the march to atomic resolution started modestly in 1981 with a structure at 30 Å resolution (Brisson and Unwin, 1984; Kistler and Stroud, 1981). Through a dogged improvement of imaging conditions and image processing algorithms (Beroukhim and Unwin, 1997), the resolution of the 3D structure gradually improved to 17 Å (Toyoshima and Unwin, 1990), 9 Å (Unwin, 1993), 4.6 Å (Miyazawa et al., 1999) and ultimately to 4 Å (Miyazawa et al., 2003), where an atomic model was built. High-resolution details have been modeled with reference to X-ray crystallographic structures of a soluble acetylcholine binding protein, which forms a pentameric structure related to the cytoplasmic domains (Brejc et al., 2001) and a bacterial homologue for the entire acetylcholine receptor (Hilf and Dutzler, 2008). However, the crystals of the nicotinic acetylcholine receptor have been used for studying of the mechanism of gating, by spraying acetylcholine onto the sample prior to rapid (msec time scale) freezing (Berriman and Unwin, 1994; Unwin, 1995). The fact that these crystals provide the native membrane environment and that the constituent acetylcholine receptor molecules represent the hetero-pentameric assembly present at mammalian neuromuscular junction mean that these studies provide invaluable insight that could not be obtained by X-ray crystallography.
3. Glutathione transferase
Aquaporin 0, the nicotinic acetylcholine receptor and bacteriorhodopsin are all abundant in their native cellular membranes, the latter forming crystals directly within the membrane of Halobacterium halobium. Thus, it is perhaps not surprising that these proteins can form large 2D arrays with long-range crystalline order which diffract to atomic resolution. But what about the vast majority of membrane proteins, which are present only at low to moderate concentration in their biological membranes? Can they also form membrane crystals that diffract to high-resolution? Microsomal glutathione transferase 1 is present at low levels in eukaryotic membranes, yet forms large membrane crystals that diffract to high-resolution This protein belongs to the superfamily of membrane-associated proteins in eicosanoid and glutathione metabolism. These proteins are key in the synthesis of mediators of fever, pain and inflammation as well as protection against reactive molecules and oxidative stress. The structure of the rat microsomal glutathione transferase 1 has been recently solved at 3.2 Å resolution in complex with glutathione by electron crystallography (Holm et al., 2006) and the related prostaglandin E synthase has been solved at 3.5 Å resolution (Jegerschold et al., 2008). These proteins form a homotrimer (Fig. 2) and the former structure revealed a binding site for glutathione that differed from the canonical soluble glutathione transferases.
VI. Advantages of membrane crystals
1. Crystallization within the membrane requires only moderate protein concentrations
Like soluble proteins, crystallization of detergent-solubilized membrane proteins for X-ray crystallography involves a phase transition that is facilitated by high protein concentrations (5–20 mg/ml). NMR also requires high concentrations of non-crystalline material to ensure suitable signal. In contrast, concentrations of 0.5–1 mg/ml are sufficient for reconstitution and crystallization of membrane proteins within the membrane bilayer. This is an important consideration for eukaryotic membrane proteins, which generally have low expression levels and a higher tendency to aggregate at higher concentrations. This tendency may be due to their exposure to very high detergent concentrations after concentration, or due to increased interactions between hydrophobic surfaces. A related benefit is that membrane crystals do not generally rely on a precipitating agent, such as high salt or polyethylene glycol. Such agents create non-physiological conditions in the aqueous phase that can lead to precipitation or unnatural conformations.
2. The membrane environment favors native protein conformation
A protein structure is far more informative if it represents a physiological conformation. In the case of membrane proteins, the inhomogeneous dielectric, charge distribution and lateral pressure of the bilayer can represent significant factors in determining the conformation and even the overall fold. In this regard, even lower resolution structures obtained from membrane crystals can yield valuable insights. For example, the X-ray structures of the EmrE multidrug transporter (Ma and Chang, 2004) caused considerable controversy which was resolved by electron crystallography (Ubarretxena-Belandia et al., 2003). EmrE is a multidrug transporter that catalyses the electrogenic efflux of various cationic aromatic hydrocarbons in exchange for two protons. The 3D structure of EmrE was first determined at 7 Å by electron crystallography (Fig. 3) and showed a bundle of eight transmembrane α-helices with one substrate molecule (tetrahenylphosphosphonium) bound near the centre (Ubarretxena-Belandia et al., 2003). The most remarkable finding was that EmrE formed an asymmetric homo-dimer with the two monomers related by a 180° rotation about an axis parallel to the membrane. This finding suggested that EmrE monomers are inserted with opposite topologies into the membrane. This antiparallel dimer represented a novel packing arrangement never before observed in a membrane protein. Subsequent X-ray structures of detergent-solubilized EmrE (Ma and Chang, 2004; Pornillos et al., 2005) revealed a completely different packing of transmembrane helices, physiological relevance of which was challenged in light of the electron crystallographic structure (Fleishman et al., 2006). As a result, the X-ray structures were ultimately revised to become more consistent with electron crystallographic data, showing the details of the antiparallel packing interaction at atomic resolution (Chen et al., 2007).
Figure 3. Membrane crystals preserve the native structure of membrane proteins.
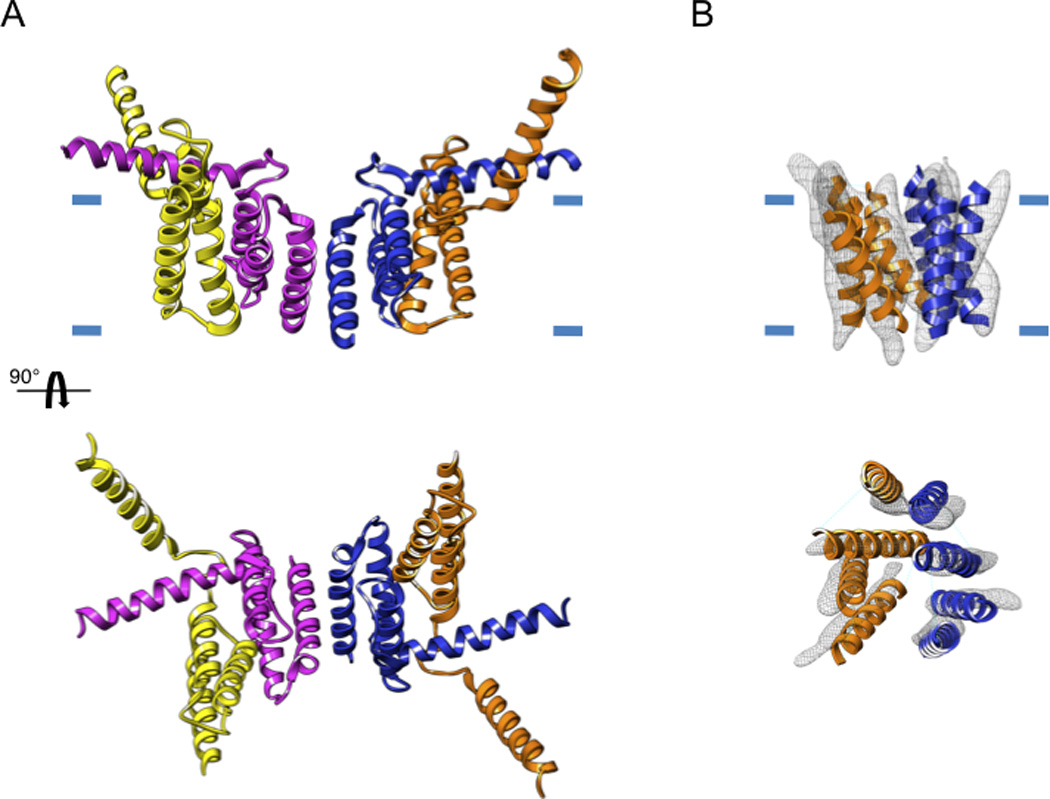
(A) The original X-ray structure showing a dimer of the multidrug-resistance antiporter from E. coli EmrE solved from 3D crystals formed with detergent-solubilized protein (pdb 1S7B). (B) The electron crystallographic map of monomeric EmrE (emdb emd-1087) solved at a resolution of 7 Å fitted with the model derived by Fleishman et al (Fleishman et al., 2006) (pdb 2I68). In the X-ray structure, two of the helices are protruding radically from the presumed bilayer plane, which is indicated by the short blue lines.
3. Conformational changes are more readily accommodated in membrane crystals
The physical constraints within membrane crystals are fewer than those for a 3D crystal, because the intermolecular interactions occur mainly in the 2D plane of the lipid bilayer rather than propagating isotropically in all three dimensions. Indeed, electron crystallographic studies of nicotinic acetylcholine receptor, rhodopsin and the Na+/H+ antiporter from E. coli (NhaA) have all included conformational changes induced by applying physiologically relevant stimuli to the membrane crystals. In the case of rhodopsin (reviewed in (Schertler, 2005)), light causes isomerisation of 11-cis retinal, thus initiating the photoactivation process, which involves an equilibrium between meta-I and meta-II conformations. Rhodopsin is also photoactive in 3D crystals, but X-ray diffraction from these crystals deteriorates dramatically after illumination, presumably due to a disordering caused by the corresponding conformational change. In contrast, membrane crystals accommodate these structural changes and have allowed the photocycle of this light-activated proton pump to be characterized by electron crystallography (Ruprecht et al., 2004). A similar was undertaken with the bacterial light-activated proton pump, bacteriorhodopsin, where electron crystallography has been instrumental in studying conformational changes not tolerated by 3D crystals (Hirai and Subramaniam, 2009).
In the case of NhaA, pH changes were used to study the transport cycle. The initial electron crystallographic map of NhaA at 7 Å (Williams, 2000) and the ensuing atomic structure by X-ray crystallography (Hunte et al., 2005) were both obtained at pH 4, i.e., with transport sites saturated with protons. To obtain mechanistic insight into other conformations of NhaA, NhaA membrane crystals were soaked in different buffers and electron crystallography revealed two pH activated states of the transporter (Appel et al., 2009), an approach that has not yet been possible with the 3D crystals.
4. Membrane crystals offer an optimal binding surface for aqueous ligands
The physiological topology of membrane proteins within membrane crystals exposes extramembranous surfaces to the aqueous medium and the high density of proteins within the crystal ameliorates “low occupancy” that can be obtained with ligands with limited affinity. This property has been exploited to study the conformational changes of the multidrug transporter EmrE in response to ligands of varying sizes (Korkhov and Tate, 2008; Tate et al., 2003; Ubarretxena-Belandia and Tate, 2004).
VII. Current hurdles in electron crystallography
Although it is clear from these examples that electron crystallography is well-suited for structure determination of membrane proteins, a number of significant practical considerations provide obstacles to the routine application of these methods, especially when it comes to resolutions ≤ 4 Å, where it becomes possible to decipher the chemical basis for the protein's function.
1. Screening of crystallization trials
The success of crystallographic methods relies on our ability to produce well-ordered crystals. The field of X-ray crystallography has made tremendous progress in developing high-throughput methods for 3D crystallization screening, using liquid handling robots for dispensing nanoliter scale droplets across thousands of different conditions (Luft et al., 2003) and sample loading robots for placing crystals in front of synchrotron X-ray beams. These methods have been particularly important for X-ray crystallography of membrane proteins, where a shotgun approach over a huge number of variables is generally required to produce high-resolution structures (Rees, 2001). These methods are facilitated by the macroscopic nature of 3D crystals, which can be rapidly and repeatedly imaged with a light microscope. In contrast, membrane crystals are microscopic (<10 µm across and 50–500 Å thick) and therefore require electron microscopy, which necessitates multiple pipetting steps for preparing an EM grid of each sample, insertion into the microscope through an airlock, followed by evaluation at various magnifications. Crystallization requires detergent removal, typically by dialysis, and strategies for high-throughput are only beginning to be developed. Such labor-intensive procedures have severely limited the number of parameters that can be investigated in an effort to discover or to optimize crystallization conditions.
2. Data collection
Once optimal conditions for crystallization and sample preparation are established, a 3D dataset includes images recorded from scores of well ordered crystals, which generally represent a small fraction of the total number of images recorded. This inefficiency results from a high rate of electron radiation damage, precludes prescreening of crystal quality if one wishes to record the highest resolution information in the image. This constraint makes reproducibility an essential element in the preparation of highly ordered membrane crystals. In contrast, X-ray crystallographers can screen tens to hundreds of crystals to find one that is well ordered, which can then used to provide a complete 3D dataset.
3. Flexibility of membrane crystals
The samples used for electron crystallography comprise a single bilayer studded with a 2D array of membrane proteins (Fig. 1). The corresponding lack physical constraints normal to this bilayer is advantageous from a physiological perspective, but means that they easily bend, curl and sometimes break into pieces. Such defects limit the usefulness of existing structure determination software, which assumes that all molecules lie within a given 2D plane. More specifically, maximal signal-to-noise ratio is obtained when all molecules in the crystal represent a single defined orientation and thus contribute coherently to the Fourier transform. Even the slightest deviation (e.g. 1° of bending) across the crystal degrades the signal, whereas larger amounts of bending or fragmentation render the data intractable with current software (Glaeser et al., 1991).
4. Anisotropic resolution
A 3D dataset is obtained by tilting crystals to a variety of angles within the electron microscope. However, there is an innate limit in the tilt angle, because crystal thickness increases dramatically above 60° and becomes essentially infinite at 90°. As a result, there is a missing cone of high-tilt data in the final dataset, which causes anisotropic resolution in the structure. This missing cone is exacerbated by the flexibility mentioned above, because the quality of data degrades at high tilt due to the fact that variations in the angle of view are amplified at high angle. This resolution anisotropy causes blurring of densities perpendicular to the membrane plane, complicating the interpretation of structures and the fitting of polypeptide chains.
VII. The future of electron crystallography
We are convinced that many of these obstacles can be overcome by a concerted effort to develop the appropriate methodologies. Indeed, several developments are already underway and, if pursued to a suitable endpoint, will facilitate a higher success rate in structure determination and thus will encourage more widespread use of electron crystallography in future studies.
1. High-throughput methods for crystallization
In a traditional manual screening the most important parameters affecting crystallization - i.e. phospholipid type, lipid-to-protein ratio, pH, temperature, detergent type, divalent cations, ionic strength, buffer, ligands, inhibitors, and additives - are surveyed in a very limited fashion. Thus, a large number of conditions must be screened in order to cover a sufficient range of relevant crystallization parameters. A key development will be to implement high-throughput screening of crystallization trials, first to establish general principles governing this process and, ultimately, to produce well-ordered membrane crystals (Fig. 4). As a start, two independent developments are underway, both involving liquid handling robots operating on a 96-well format. The first uses a dialysis block with 50 µl sample wells and an independent 2 ml reservoir for each sample (Kim et al., 2010; Vink et al., 2007). Using a commercial liquid-handling robot to refresh reservoir solutions frequently, detergent removal over a period of 4–14 days has been demonstrated, depending on the detergent, which is comparable to results obtained in more standard dialysis setups (e.g. buttons, capillaries or bags). The second approach to high-throughput relies on the ability of cyclodextrins to effectively remove detergent from ternary mixtures of detergent, lipid and protein, and thus effect membrane protein reconstitution (Signorell et al., 2007). A custom liquid-handling robot has been designed to titrate nanoliter amounts of cyclodextrin solutions to 10–50 µl of protein samples arrayed in 96 wells (Iacovache et al., 2010). Both approaches have been effective in producing membrane crystals and have potential to screen a broad array of parameters affecting the process. Both groups have also employed liquid-handling robots to prepare negatively stained grids, using magnetic platforms to hold down Ni grids during the staining process and liquid-handling robots to carry out the pipetting steps (Coudray et al., 2010; Hu et al., 2010). These methods built on an earlier negative staining robot that employed wells drilled into a block of Teflon (Cheng et al., 2007).
Figure 4. Pipeline for protein structure determination by electron crystallography.

Target membrane proteins are purified in detergent micelles in a stable and monodisperse form. Following the addition of lipids to form mixed micelles of protein, detergent and lipid, the crystallization process is studied by removing dialysis in a 96-well dialysis block. The 96 crystallization conditions are harvested, transferred to EM grids and negative stained with a liquid-handling robot. The EM grids are robotically inserted into the electron microscope, and images are recorded automatically and stored in a database. Thus, a broad range of parameters can be explored in an attempt to find large, well ordered crystals. Finally, image processing and structure determination can be carried out using, for example, the 2dx software.
2. High-throughput imaging of crystallization trials
These liquid-handling robots will generate large numbers of samples that must then be imaged by electron microscopy. This represents a huge bottleneck in the operation, given the logistics of operating the electron microscope. Fortunately, recent developments have resulted in several options for automated insertion and imaging of negatively stained samples. The first option involves an articulated 5-axis robotic arm that picks up individual EM grids with forceps, places them into the specimen holder, and then manipulate the holder through the airlock of a Tecnai F20 electron microscope (Potter et al., 2004). A second option divided the same procedure into two steps, employing a SCARA robot to manipulate the EM grids with a vacuum pickup, and a Cartesian robot to place the holder into a JEOL 1230 electron microscope (Hu et al., 2010). In both cases, specimen insertion was controlled by the program Leginon (Potter et al., 1999), which goes on to acquire a series of representative images from each sample and to place them in a database for later evaluation. In a very different approach, a so-called auto-loader was adapted for a Tecnai T12 microscope and its grid capacity was extended by placing the commercially available 12-grid cassettes onto an 8-position carousel, such that 96 grids can be accessed by custom control software (Coudray et al., 2010). This design was reminiscent of the Gating gun (Lefman et al., 2007), which accommodates 100 EM grids mounted in cartridges on a cylindrical drum within the vacuum of a Tecnai T12 microscope.
3. Preparing better ordered specimens
As discussed, membrane crystals are notoriously flexible and small forces distort the crystal during adsorption to a carbon support film, which itself is not perfectly flat. Two approaches to prepare flatter specimens include the back-injection method (Kuhlbrandt and Downing, 1989) and the carbon sandwich technique (Gyobu et al., 2004; Koning et al., 2003). The carbon sandwich technique appears to be the method of choice as it consistently yields improved resolution. Grids made of Mo are frequently used due to its lower coefficient of thermal expansion and tendency to minimize crinkling of the carbon support upon freezing. Still, we believe that further developments in the design of grids and support materials will be required in the quest for high-resolution. In addition, the 1.9 Å resolution obtained from AQP0 (Gonen et al., 2005) suggests that double-layered crystals have a greater stiffness than single layered ones, and crystallization conditions could be optimized for the growth of such double-layered membrane crystals.
4. Automated data collection
Automation of low dose imaging and collection of electron diffraction data promises to accelerate the throughput of structure determination and current developments in software for screening crystallization trials provide a solid foundation for this work (Cheng et al., 2007; Coudray et al., 2008; Coudray et al., 2010; Hu et al., 2010). Leginon (Potter et al., 1999) and SerialEM (Mastronarde, 2005) can automatically record low-dose images and if a strategy for locating large crystals at low magnification and evaluating crystal order through a quick peek at the electron diffraction were included, these programs would be highly effective for electron crystallography. Imaging of tilted specimens is particularly difficult, most likely due to charging phenomena. Thus, incorporating a spot-scan imaging mode, which reduces charge buildup (Downing and Glaeser, 1986), will optimize the success rate.
5. Correction of crystal lattice distortions
Lattice distortions produced from minor lattice faults can induce substantial shifts of large coherent areas of the crystal. Moreover, crystals can grow from multiple nuclei after proteins are integrated in the bilayer thereby leading to mosaic crystals. Such distortions can be addressed by image analysis and existing methods fall into two categories: unbending (Henderson et al., 1986) and correlation averaging (CA) (Saxton and Baumeister, 1982). While both methods have been shown capable of delivering high-resolution (3.5 Å) - purple membrane with unbending (Baldwin et al., 1988) and porin with CA (Sass et al., 1989) - there is a profound difference between them. Unbending strives to reconstruct a large, coherent 2D lattice which is then processed by Fourier methods, whereas CA aims to extract individual unit cells, which exhibit a high correlation with a selected reference, followed by alignment and averaging in real space. CA has the potential to correct for rotational disorder, both in-plane and out-of-plane, and it is thus best suited for the analysis of badly fragmented or bent lattices. This approach has already shown promise in the structure determination of a secondary transporter (Koeck et al., 2007), and we believe that future developments in this area will have significant impact.
6. Robust structure determination software
Improvements in data processing software are critical to the advancement of electron crystallography. Original developments in electron crystallography at the Medical Research Council (MRC) in the 1970's and 1980's produced a comprehensive set of programs that resulted in the atomic resolution structure of bacteriorhodopsin (Crowther et al., 1996). Since then, electron crystallographers have done relatively little to take advantage of the vast increase in computing power and programming infrastructure. In contrast, many refinements and new ideas have been implemented over the same time frame for single particle processing and electron tomography, developments that continue to be pursued today.
Nevertheless, several worthwhile programs have recently become available and facilitate image processing of membrane crystals. The 2dx initiative (Gipson et al., 2007) (www.2dx.unibas.ch/) provides a graphical user interface to the original MRC programs and features streamlined processing solutions with optional full automation that can potentially accelerate image processing and structure determination considerably. Similarly, the XDP software handles diffraction patterns by relying on the MRC code (Hirai et al., 1999). In contrast, IPLT is a new development for processing images and electron diffraction (www.iplt.org). This program takes advantage of a modern object oriented programming architecture and incorporates new strategies for correcting lattice distortions and untangling overlapping electron diffraction patterns (Philippsen et al., 2007; Philippsen et al., 2003). IPLT is innately extensible and appears to offer a good platform for implementing new algorithms.
An emphasis on electron diffraction represents an important avenue for future development. High resolution electron diffraction is relatively easy to collect from well ordered membrane crystals, whereas specimen instabilities and charging effects make the process of image collection laborious and time-consuming. Thus, if molecular replacement and phase extension methods could be routinely applied to electron diffraction data, similar to what is done by X-ray crystallographers, the inordinate difficulties in obtaining high resolution images could be bypassed and the rate of structure determination by electron crystallography could be greatly accelerated.
Bibliography
- Agre P, et al. Aquaporin CHIP: the archetypal molecular water channel. Am J Physiol. 1993;265:F463–F476. doi: 10.1152/ajprenal.1993.265.4.F463. [DOI] [PubMed] [Google Scholar]
- Andersen OS, Koeppe RE., 2nd Bilayer thickness and membrane protein function: an energetic perspective. Annu Rev Biophys Biomol Struct. 2007;36:107–130. doi: 10.1146/annurev.biophys.36.040306.132643. [DOI] [PubMed] [Google Scholar]
- Appel M, et al. Conformations of NhaA, the Na+/H+ exchanger from Escherichia coli, in the pH-activated and ion-translocating states. J Mol Biol. 2009;388:659–672. doi: 10.1016/j.jmb.2009.03.010. [DOI] [PubMed] [Google Scholar]
- Baldwin JM, et al. Images of purple membrane at 2.8 A resolution obtained by cryo-electron microscopy. J Mol Biol. 1988;202:585–591. doi: 10.1016/0022-2836(88)90288-4. [DOI] [PubMed] [Google Scholar]
- Beroukhim R, Unwin N. Distortion correction of tubular crystals: Improvements in the acetylcholine receptor structure. Ultramicroscopy. 1997;70:57–81. doi: 10.1016/s0304-3991(97)00070-3. [DOI] [PubMed] [Google Scholar]
- Berriman J, Unwin N. Analysis of transient structures by cryo-microscopy combinedwith rapid mixing of spray droplets. Ultramicroscopy. 1994;56:241–252. doi: 10.1016/0304-3991(94)90012-4. [DOI] [PubMed] [Google Scholar]
- Brejc K, et al. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature. 2001;411:269–276. doi: 10.1038/35077011. [DOI] [PubMed] [Google Scholar]
- Brisson A, Unwin PN. Tubular crystals of acetylcholine receptor. J Cell Biol. 1984;99:1202–1211. doi: 10.1083/jcb.99.4.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugger B, et al. Quantitative analysis of biological membrane lipids at the low picomole level by nano-electrospray ionization tandem mass spectrometry. Proc Natl Acad Sci U S A. 1997;94:2339–2344. doi: 10.1073/pnas.94.6.2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y-J, et al. X-ray structure of EmrE supports dual topology model. Proc Natl Acad Sci USA. 2007;104:18999–19004. doi: 10.1073/pnas.0709387104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A, et al. Towards automated screening of two-dimensional crystals. Journal of Structural Biology. 2007;160:324–331. doi: 10.1016/j.jsb.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A, et al. Three-dimensional organization of a human water channel. Nature. 1997;387:627–630. doi: 10.1038/42517. [DOI] [PubMed] [Google Scholar]
- Coudray N, et al. Automatic Acquisition and Image Analysis of 2D Crystals. Microscopy Today. 2008;16:48–49. [Google Scholar]
- Coudray N, et al. Automated Screening of 2D Crystallization Trials Using Transmission Electron Microscopy: A High-Throughput Tool-Chain for Sample Preparation and Microscopic Analysis. J Struct Biol. 2010 doi: 10.1016/j.jsb.2010.09.019. [DOI] [PubMed] [Google Scholar]
- Crowther RA, et al. MRC image processing programs. J Struct Biol. 1996;116:9–16. doi: 10.1006/jsbi.1996.0003. [DOI] [PubMed] [Google Scholar]
- Deisenhofer J, et al. X-ray structure analysis of a membrane protein complex. Electron density map at 3 A resolution and a model of the chromophores of the photosynthetic reaction center from Rhodopseudomonas viridis. J Mol Biol. 1984;180:385–398. doi: 10.1016/s0022-2836(84)80011-x. [DOI] [PubMed] [Google Scholar]
- Dowhan W. Molecular basis for membrane phospholipid diversity: why are there so many lipids? Annu Rev Biochem. 1997;66:199–232. doi: 10.1146/annurev.biochem.66.1.199. [DOI] [PubMed] [Google Scholar]
- Downing KH, Glaeser RM. Improvement in high resolution image quality of radiation-sensitive specimens achieved with reduced spot size of the electron beam. Ultramicroscopy. 1986;20:269–278. doi: 10.1016/0304-3991(86)90191-9. [DOI] [PubMed] [Google Scholar]
- Drews J. Drug discovery: a historical perspective. Science. 2000;287:1960–1964. doi: 10.1126/science.287.5460.1960. [DOI] [PubMed] [Google Scholar]
- Engelman DM. Lipid bilayer structure in the membrane of Mycoplasma laidlawii. J Mol Biol. 1971;58:153–165. doi: 10.1016/0022-2836(71)90238-5. [DOI] [PubMed] [Google Scholar]
- Fleishman SJ, et al. Quasi-symmetry in the cryo-EM structure of EmrE provides the key to modeling its transmembrane domain. J Mol Biol. 2006;364:54–67. doi: 10.1016/j.jmb.2006.08.072. [DOI] [PubMed] [Google Scholar]
- Gipson B, et al. 2dx—User-friendly image processing for 2D crystals. Journal of Structural Biology. 2007;157:64–72. doi: 10.1016/j.jsb.2006.07.020. [DOI] [PubMed] [Google Scholar]
- Glaeser RM, et al. Interfacial energies and surface-tension forces involved in the preparation of thin, flat crystals of biological macromolecules for high-resolution electron microscopy. J Microsc. 1991;161:21–45. doi: 10.1111/j.1365-2818.1991.tb03071.x. [DOI] [PubMed] [Google Scholar]
- Gonen T, et al. Lipid-protein interactions in double-layered two-dimensional AQP0 crystals. Nature. 2005;438:633–638. doi: 10.1038/nature04321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyobu N, et al. Improved specimen preparation for cryo-electron microscopy using a symmetric carbon sandwich technique. J Struct Biol. 2004;146:325–333. doi: 10.1016/j.jsb.2004.01.012. [DOI] [PubMed] [Google Scholar]
- Henderson R, et al. Model for the structure of bacteriorhodopsin based on high-resolution electron cryo-microscopy. J. Mol. Biol. 1990;213:899–929. doi: 10.1016/S0022-2836(05)80271-2. [DOI] [PubMed] [Google Scholar]
- Henderson R, et al. Structure of purple membrane from halobacterium halobium: recording, measurement and evaluation of electron micrographs at 3.5 Å resolution. Ultramicroscopy. 1986;19:147–178. [Google Scholar]
- Henderson R, Unwin PNT. Three-dimensional model of purple membrane obtained from electron microscopy. Nature. 1975;257:28–32. doi: 10.1038/257028a0. [DOI] [PubMed] [Google Scholar]
- Hilf RJ, Dutzler R. X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature. 2008;452:375–379. doi: 10.1038/nature06717. [DOI] [PubMed] [Google Scholar]
- Hirai T, et al. Three-dimensional structure of a bacterial oxalate transporter. Nat Struct Biol. 2002;9:597–600. doi: 10.1038/nsb821. [DOI] [PubMed] [Google Scholar]
- Hirai T, et al. Trehalose embedding technique for high-resolution electron crystallography: application to structural study on bacteriorhodopsin. J Electron Microsc (Tokyo) 1999;48:653–658. doi: 10.1093/oxfordjournals.jmicro.a023731. [DOI] [PubMed] [Google Scholar]
- Hirai T, Subramaniam S. Protein conformational changes in the bacteriorhodopsin photocycle: comparison of findings from electron and X-ray crystallographic analyses. PLoS One. 2009;4:e5769. doi: 10.1371/journal.pone.0005769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiroaki Y, et al. Implications of the aquaporin-4 structure on array formation and cell adhesion. J Mol Biol. 2006;355:628–639. doi: 10.1016/j.jmb.2005.10.081. [DOI] [PubMed] [Google Scholar]
- Hite RK, et al. Principles of membrane protein interactions with annular lipids deduced from aquaporin-0 2D crystals. EMBO J. 2010;29:1652–1658. doi: 10.1038/emboj.2010.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm P, et al. Structural Basis for Detoxification and Oxidative Stress Protection in Membranes. Journal of Molecular Biology. 2006;360:934–945. doi: 10.1016/j.jmb.2006.05.056. [DOI] [PubMed] [Google Scholar]
- Hong M. Oligomeric structure, dynamics, and orientation of membrane proteins from solid-state NMR. Structure. 2006;14:1731–1740. doi: 10.1016/j.str.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Hu M, et al. Automated electron microscopy for evaluating two-dimensional crystallization of membrane proteins. J Struct Biol. 2010 doi: 10.1016/j.jsb.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunte C, et al. Structure of a Na+/H+ antiporter and insights into mechanism of action and regulation by pH. Nature. 2005;435:1197–1202. doi: 10.1038/nature03692. [DOI] [PubMed] [Google Scholar]
- Iacovache I, et al. The 2DX robot: a membrane protein 2D crystallization Swiss Army knife. J Struct Biol. 2010;169:370–378. doi: 10.1016/j.jsb.2009.12.001. [DOI] [PubMed] [Google Scholar]
- Jap BK, et al. 2D crystallization: from art to science. Ultramicroscopy. 1992;46:45–84. doi: 10.1016/0304-3991(92)90007-7. [DOI] [PubMed] [Google Scholar]
- Jegerschold C, et al. Structural basis for induced formation of the inflammatory mediator prostaglandin E2. Proc Natl Acad Sci U S A. 2008;105:11110–11115. doi: 10.1073/pnas.0802894105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, et al. An automated pipeline to screen membrane protein 2D crystallization. J Struct Funct Genomics. 2010;11:155–166. doi: 10.1007/s10969-010-9088-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kistler J, Stroud RM. Crystalline arrays of membrane-bound acetylcholine receptor. Proc Natl Acad Sci U S A. 1981;78:3678–3682. doi: 10.1073/pnas.78.6.3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeck PJ, et al. Single particle refinement in electron crystallography: a pilot study. J Struct Biol. 2007;160:344–352. doi: 10.1016/j.jsb.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Koning RI, et al. Preparation of flat carbon support films. Ultramicroscopy. 2003;94:183–191. doi: 10.1016/s0304-3991(02)00263-2. [DOI] [PubMed] [Google Scholar]
- Korkhov VM, Tate CG. Electron crystallography reveals plasticity within the drug binding site of the small multidrug transporter EmrE. J Mol Biol. 2008;377:1094–1103. doi: 10.1016/j.jmb.2008.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühlbrandt W. Two-dimensional crystallization of membrane proteins. Q Rev Biophys. 1992;25:1–49. doi: 10.1017/s0033583500004716. [DOI] [PubMed] [Google Scholar]
- Kuhlbrandt W, Downing KH. Two-dimensional structure of plant light-harvesting complex at 3.7 A [corrected] resolution by electron crystallography. J Mol Biol. 1989;207:823–828. doi: 10.1016/0022-2836(89)90247-7. [DOI] [PubMed] [Google Scholar]
- Kühlbrandt W, et al. Atomic model of plant light-harvesting complex by electron crystallography. Nature. 1994;367:614–621. doi: 10.1038/367614a0. [DOI] [PubMed] [Google Scholar]
- Kukulski W, et al. The 5A structure of heterologously expressed plant aquaporin SoPIP2;1. J Mol Biol. 2005;350:611–616. doi: 10.1016/j.jmb.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Lee AG. How lipids affect the activities of integral membrane proteins. Biochim Biophys Acta. 2004;1666:62–87. doi: 10.1016/j.bbamem.2004.05.012. [DOI] [PubMed] [Google Scholar]
- Lefman J, et al. Automated 100-position specimen loader and image acquisition system for transmission electron microscopy. JSB. 2007;158:318–326. doi: 10.1016/j.jsb.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl E, Sansom MS. Membrane proteins: molecular dynamics simulations. Curr Opin Struct Biol. 2008;18:425–431. doi: 10.1016/j.sbi.2008.02.003. [DOI] [PubMed] [Google Scholar]
- Luft JR, et al. A deliberate approach to screening for initial crystallization conditions of biological macromolecules. J Struct Biol. 2003;142:170–179. doi: 10.1016/s1047-8477(03)00048-0. [DOI] [PubMed] [Google Scholar]
- Ma C, Chang G. Structure of the multidrug resistance efflux transporter EmrE from Escherichia coli. Proc Natl Acad Sci U S A. 2004;101:2852–2857. doi: 10.1073/pnas.0400137101. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Mastronarde DN. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 2005;152:36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Mitra K, et al. Modulation of the bilayer thickness of exocytic pathway membranes by membrane proteins rather than cholesterol. Proc Natl Acad Sci U S A. 2004;101:4083–4088. doi: 10.1073/pnas.0307332101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa A, et al. Nicotinic acetylcholine receptor at 4.6 A resolution: transverse tunnels in the channel wall. J Mol Biol. 1999;288:765–786. doi: 10.1006/jmbi.1999.2721. [DOI] [PubMed] [Google Scholar]
- Miyazawa A, et al. Structure and gating mechanism of the acetylcholine receptor pore. Nature. 2003;423:949–955. doi: 10.1038/nature01748. [DOI] [PubMed] [Google Scholar]
- Mosser G. Two-dimensional crystallogenesis of transmembrane proteins. Micron. 2001;32:517–540. doi: 10.1016/s0968-4328(00)00047-0. [DOI] [PubMed] [Google Scholar]
- Murata K, et al. Structural determinants of water permeation through aquaporin-1. Nature. 2000;407:599–605. doi: 10.1038/35036519. [DOI] [PubMed] [Google Scholar]
- Nyholm TK, et al. How protein transmembrane segments sense the lipid environment. Biochemistry. 2007;46:1457–1465. doi: 10.1021/bi061941c. [DOI] [PubMed] [Google Scholar]
- Philippsen A, et al. Collaborative EM image processing with the IPLT image processing library and toolbox. J Struct Biol. 2007;157:28–37. doi: 10.1016/j.jsb.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Philippsen A, et al. Iplt--image processing library and toolkit for the electron microscopy community. J Struct Biol. 2003;144:4–12. doi: 10.1016/j.jsb.2003.09.032. [DOI] [PubMed] [Google Scholar]
- Pike LJ, et al. Lipid rafts are enriched in arachidonic acid and plasmenylethanolamine and their composition is independent of caveolin-1 expression: a quantitative electrospray ionization/mass spectrometric analysis. Biochemistry. 2002;41:2075–2088. doi: 10.1021/bi0156557. [DOI] [PubMed] [Google Scholar]
- Popot JL, Engelman DM. Helical membrane protein folding, stability, and evolution. Annu Rev Biochem. 2000;69:881–922. doi: 10.1146/annurev.biochem.69.1.881. [DOI] [PubMed] [Google Scholar]
- Pornillos O, et al. X-ray structure of the EmrE multidrug transporter in complex with a substrate. Science. 2005;310:1950–1953. doi: 10.1126/science.1119776. [DOI] [PubMed] [Google Scholar]
- Potter CS, et al. Leginon: a system for fully automated acquisition of 1000 electron micrographs a day. Ultramicroscopy. 1999;77:153–161. doi: 10.1016/s0304-3991(99)00043-1. [DOI] [PubMed] [Google Scholar]
- Potter CS, et al. Robotic grid loading system for a transmission electron microscope. J Struct Biol. 2004;146:431–440. doi: 10.1016/j.jsb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Preston GM, et al. Appearance of water channels in Xenopus oocytes expressing red cell CHIP28 protein. Science. 1992;256:385–387. doi: 10.1126/science.256.5055.385. [DOI] [PubMed] [Google Scholar]
- Rees DC. Crystallographic analyses of hyperthermophilic proteins. Methods Enzymol. 2001;334:423–437. doi: 10.1016/s0076-6879(01)34484-1. [DOI] [PubMed] [Google Scholar]
- Remigy HW, et al. Membrane protein reconstitution and crystallization by controlled dilution. FEBS Lett. 2003;555:160–169. doi: 10.1016/s0014-5793(03)01105-0. [DOI] [PubMed] [Google Scholar]
- Rigaud JL, et al. Bio-Beads: an efficient strategy for two-dimensional crystallization of membrane proteins. J Struct Biol. 1997;118:226–235. doi: 10.1006/jsbi.1997.3848. [DOI] [PubMed] [Google Scholar]
- Ruprecht JJ, et al. Electron crystallography reveals the structure of metarhodopsin I. EMBO J. 2004;23:3609–3620. doi: 10.1038/sj.emboj.7600374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sass HJ, et al. Densely packed beta-structure at the protein-lipid interface of porin is revealed by high-resolution cryo-electron microscopy. Journal of Molecular Biology. 1989;209:171–175. doi: 10.1016/0022-2836(89)90180-0. [DOI] [PubMed] [Google Scholar]
- Saxton WO, Baumeister W. The correlation averaging of a regularly arranged bacterial envelope protein. Journal of Microscopy. 1982;127:127–138. doi: 10.1111/j.1365-2818.1982.tb00405.x. [DOI] [PubMed] [Google Scholar]
- Schenk AD, et al. The 4.5 A structure of human AQP2. J Mol Biol. 2005;350:278–289. doi: 10.1016/j.jmb.2005.04.030. [DOI] [PubMed] [Google Scholar]
- Schertler GF. Structure of rhodopsin and the metarhodopsin I photointermediate. Curr Opin Struct Biol. 2005;15:408–415. doi: 10.1016/j.sbi.2005.07.010. [DOI] [PubMed] [Google Scholar]
- Schulz GE. beta-Barrel membrane proteins. Curr Opin Struct Biol. 2000;10:443–447. doi: 10.1016/s0959-440x(00)00120-2. [DOI] [PubMed] [Google Scholar]
- Signorell GA, et al. Controlled 2D crystallization of membrane proteins using methyl-beta-cyclodextrin. J Struct Biol. 2006 doi: 10.1016/j.jsb.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Signorell GA, et al. Controlled 2D crystallization of membrane proteins using methyl-beta-cyclodextrin. J Struct Biol. 2007;157:321–328. doi: 10.1016/j.jsb.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Stahlberg H, et al. Two-dimensional crystals: a powerful approach to assess structure, function and dynamics of membrane proteins. FEBS Lett. 2001;504:166–172. doi: 10.1016/s0014-5793(01)02746-6. [DOI] [PubMed] [Google Scholar]
- Subramaniam S, et al. From structure to mechanism: electron crystallographic studies of bacteriorhodopsin. Philos Transact A Math Phys Eng Sci. 2002;360:859–874. doi: 10.1098/rsta.2001.0971. [DOI] [PubMed] [Google Scholar]
- Tani K, et al. Mechanism of aquaporin-4's fast and highly selective water conduction and proton exclusion. J Mol Biol. 2009;389:694–706. doi: 10.1016/j.jmb.2009.04.049. [DOI] [PubMed] [Google Scholar]
- Tate CG, et al. Conformational changes in the multidrug transporter EmrE associated with substrate binding. J Mol Biol. 2003;332:229–242. doi: 10.1016/s0022-2836(03)00895-7. [DOI] [PubMed] [Google Scholar]
- Toyoshima C, Unwin N. Three-dimensional structure of the acetylcholine receptor by cryoelectron microscopy and helical image reconstruction. The Journal of Cell Biology. 1990;111:2623–2635. doi: 10.1083/jcb.111.6.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubarretxena-Belandia I, et al. Three-dimensional structure of the bacterial multidrug transporter EmrE shows it is an asymmetric homodimer. Embo J. 2003;22:6175–6181. doi: 10.1093/emboj/cdg611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubarretxena-Belandia I, Engelman DM. Helical membrane proteins: diversity of functions in the context of simple architecture. Curr Opin Struct Biol. 2001;11:370–376. doi: 10.1016/s0959-440x(00)00217-7. [DOI] [PubMed] [Google Scholar]
- Ubarretxena-Belandia I, Tate CG. New insights into the structure and oligomeric state of the bacterial multidrug transporter EmrE: an unusual asymmetric homo-dimer. FEBS Lett. 2004;564:234–238. doi: 10.1016/S0014-5793(04)00228-5. [DOI] [PubMed] [Google Scholar]
- Unger VM. Electron cryomicroscopy methods. Curr Opin Struct Biol. 2001;11:548–554. doi: 10.1016/s0959-440x(00)00260-8. [DOI] [PubMed] [Google Scholar]
- Unwin N. Nicotinic acetylcholine receptor at 9 A resolution. J Mol Biol. 1993;229:1101–1124. doi: 10.1006/jmbi.1993.1107. [DOI] [PubMed] [Google Scholar]
- Unwin N. Acetylcholine receptor channel imaged in the open state. Nature. 1995;373:37–43. doi: 10.1038/373037a0. [DOI] [PubMed] [Google Scholar]
- Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J Mol Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- Vink M, et al. A high-throughput strategy to screen 2D crystallization trials of membrane proteins. J Struct Biol. 2007;160:295–304. doi: 10.1016/j.jsb.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallin E, von Heijne G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 1998;7:1029–1038. doi: 10.1002/pro.5560070420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walz T, et al. The three-dimensional structure of aquaporin-1. Nature. 1997;387:624–627. doi: 10.1038/42512. [DOI] [PubMed] [Google Scholar]
- White SH. Biophysical dissection of membrane proteins. Nature. 2009;459:344–346. doi: 10.1038/nature08142. [DOI] [PubMed] [Google Scholar]
- White SH, Wimley WC. Membrane protein folding and stability: physical principles. Annu Rev Biophys Biomol Struct. 1999;28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- Williams KA. Three-dimensional structure of the ion-coupled transport protein NhaA. Nature. 2000;403:112–115. doi: 10.1038/47534. [DOI] [PubMed] [Google Scholar]
- Yeagle PL. Lipid regulation of cell membrane structure and function. FASEB J. 1989;3:1833–1842. [PubMed] [Google Scholar]
- Zhang P, et al. Structure of the calcium pump from sarcoplasmic reticulum at 8-A resolution. Nature. 1998;392:835–839. doi: 10.1038/33959. [DOI] [PubMed] [Google Scholar]