

Abstract
Rhombencephalosynapsis (RES) is a rare congenital brain malformation typically identified by magnetic resonance imaging and characterized by fusion of the cerebellar hemispheres and dentate nuclei and vermian agenesis or hypogenesis. Although RES is frequently found in conjunction with other brain malformations and/or congenital anomalies, no specific molecular etiology has been discovered to date and no animal models exist. We identified two half sisters with alobar or semi-lobar holoprosencephaly (HPE) and partial RES, suggesting that genes linked to HPE may also contribute to RES. A deletion of seven base pairs in exon one of the ZIC2 gene (c.392_98del7) was identified in each of the two half sisters with HPE and partial RES. To identify genetic causes of RES and to assess whether genes identified in HPE have a role in RES, we tested 11 additional individuals with RES by high resolution chromosome analysis, chromosomal microarray analysis, and sequencing of four HPE genes. No mutations in ZIC2 or in other genes that cause HPE were identified, suggesting that mutation of ZIC2 is a rare cause of, or contributor to, rhombencephalosynapsis associated with HPE. In addition, an individual with a complex rearrangement of chromosome 22q13.3 and RES was identified, suggesting the presence of a dosage-sensitive gene that may contribute to RES in this region.
Keywords: cerebellum, chromosome microarray, holoprosencephaly, MTHFR, Phelan-McDermid syndrome, SHANK3, 22q13.3 deletion syndrome
Introduction
Rhombencephalosynapsis (RES) is a rare congenital brain malformation characterized by fusion of the cerebellar hemispheres and dentate nuclei and vermian agenesis or hypogenesis [Cohen, 2006; Pasquier et al., 2009; Sener, 2000; Toelle et al., 2002]. A thorough morphological study of 40 fetuses with RES ascertained by the presence of ventriculomegaly (90%) or other brain malformations on prenatal ultrasound at ≥ 22 weeks gestation demonstrated that RES in this circumstance was always associated with other brain abnormalities [Pasquier et al., 2009]. In addition to an abnormal ventricular system, the most common associated abnormalities included: fusion of the inferior and/or superior colliculi (55%), fusion of the thalami (13%), Purkinje cell heterotopias (20%), dysgenesis or agenesis of the corpus callosum (36%), lobar holoprosencephaly (5%), neural tube defects (8%), and VACTERL-H (vertebral anomalies, anal atresia, congenital heart defects, tracheo-esophageal fistula, renal abnormalities, and limb anomalies together with hydrocephalus; 15%) [Pasquier et al., 2009]. Gomez-Lopez-Hernandez syndrome (OMIM# 601853) is another sporadic clinical entity that also includes RES in addition to craniosynostosis, ataxia, trigeminal anesthesia, scalp alopecia, midface hypoplasia, corneal opacities, low-set ears, intellectual disability, and short stature; however, RES and alopecia may be the most consistent features of the syndrome [Sukhudyan et al., 2010]. Although RES is found frequently in conjunction with other congenital anomalies, no specific gene association or underlying molecular etiology has been discovered and no specific animal models exist [Pasquier et al., 2009; Toelle et al., 2002]. Based on observed chromosomal abnormalities, potential candidate gene regions have been identified in single individuals with RES harboring a 46,XX.ish der(10)t(2;10)(p25.3;q26.3)(Tel 2p+, Tel 10q−) [Lespinasse et al., 2004], an interstitial deletion of chromosome 2q [Truwit et al., 1991], a 1p microduplication [Pasquier et al., 2009], and a 7q microdeletion [Pasquier et al., 2009].
We initially identified two half sisters with alobar or semi-lobar holoprosencephaly (HPE) and partial RES who harbor a mutation in the ZIC2 gene (Fig 1), causing us to hypothesize that genes linked to HPE may also cause RES. HPE is a multifactorial and genetically heterogeneous brain malformation which in its most severe form is characterized by failure of the developing forebrain to divide into separate cerebral hemispheres and ventricles [Cohen, 2006]. Mutations in at least twelve distinct genes have been linked to HPE [Cohen, 2006; Solomon et al., 2010b]. To identify genetic causes of RES and to assess whether genes identified in HPE have a role in RES, we identified and tested additional individuals with RES by high resolution chromosome analysis, chromosomal microarray analysis, and sequencing of four known HPE genes.
Figure 1. Neuroimaging in patients with rhombencephalosynapsis.

MRI images for Patient 1 (top row): axial T2 images demonstrating partial superior rhombencephalosynapsis (left) and alobar holoprosencephaly (middle), and mid-line sagittal T1 image demonstrating a dysmorphic, hypoplastic, and abnormally positioned cerebellar vermis. MRI images for Patient 2 (bottom row): axial T1 images demonstrating partial central rhombencephalosynapsis (left) and semi-lobar holoprosencephaly (middle), and coronal T2 image again demonstrating rhombencephalosynapsis (right). White arrows point to RES.
Methods
Patient ascertainment
With approval of the Institutional Review Board for Baylor College of Medicine and Affiliated Hospitals, we used Texas Children’s Hospital (TCH) neuroradiology records from 2000–2006 and retrospectively identified an additional 15 children with RES. Eleven of these 15 patients were successfully located and were evaluated at the TCH Blue Bird Circle Clinic for Pediatric Neurology and enrolled in the study. Informed consent for genetic testing was obtained from parents, and when developmentally appropriate, assent was obtained from the child. Blood specimens were sent to the Baylor College of Medicine Medical Genetics Laboratories for chromosome analysis and chromosomal microarray analysis (CMA; versions 5 or 6) and to Gene Dx for the holoprosencephaly gene sequencing panel (includes SHH, TGIF, ZIC2, and SIX3). Radiology records and medical charts were also reviewed.
Array comparative genomic hybridization
Clinical chromosomal microarray analyses for all patients were performed with the use of the V5 or V6 BAC-based array designed by Baylor Medical Genetics laboratories as previously described [Lu et al., 2007]. Confirmatory FISH analyses were performed in the phytohemagglutinin-stimulated peripheral blood lymphocytes of patient 3 using standard procedures with the PAC and BAC clones RP3-402G11, RP5-925J7, RP11-66M5, RP11-164E23, and GS-99K24 specific for chromosome 22q13.3. In order to delineate the sizes and extents of the identified imbalances in patient 3, whole-genome high-resolution oligonucleotide microarray comparative genomic analysis was performed with the NimbleGen array 2.1M oligonucleotides (Roche NimbleGen Systems, Madison, WI, USA) in accordance with the manufacturer’s instructions.
Results
A deletion of seven base pairs in exon one of the ZIC2 gene (c.392_98del7) that is predicted to cause a frameshift at amino acid 131 (p.Pro131fsX84) was identified in each of the two half sisters with HPE and partial RES (Fig 1). Interestingly, Patient 1 had macrocephaly and scaphocephaly but Patient 2 had microcephaly; abnormal facial features were not appreciated. Both children have globally severe intellectual disabilities and motor deficits. Their shared mother did not carry the deletion in her peripheral blood. No additional mutations were identified in the gene regions covered by the Gene Dx sequencing panel in the other patients in our series.
G-banded chromosome analysis identified 46,Xinv(Y)(p11.3q11.23) in a boy with RES and encephalocele; CMA was not performed, and the father was not tested). G-banded chromosome analysis also revealed a complex, apparently balanced rearrangement 45,XX,dic(10;22)(q26.3;q13.3) in a girl with RES, diffusely thin corpus callosum with hypoplasia of the posterior body and splenium, enlarged ventricles, hypotonia, global developmental delay, and failure-to-thrive (Fig 2C; parents were not available for testing). However, CMA V5 revealed a gain in copy number on the subtelomeric region of chromosome 22 detected with two clones RP11-66M5 and RP5-925J7 and a loss in copy number detected by the next adjacent three clones RP3-402G11, RP11-164E23, and GS-99K24, and encompassing the SHANK3 gene (Fig 2A). FISH analyses verified these imbalances and showed a translocation of the long arms of chromosomes 10 and 22 (Fig 2D and E).
Figure 2. Summary of the results in Patient 3.
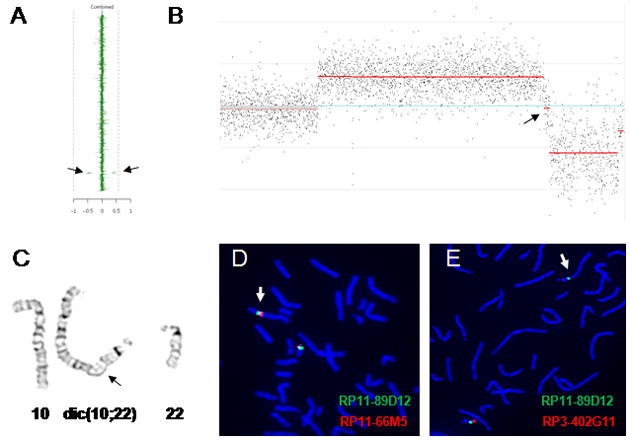
(A) Clinical aCGH plot showing a deletion and a duplication (denoted by arrows) on chromosome 22q13.3. (B) Array CGH plot (NimbleGen 2.1 M) shows the deletion and duplication in 22q13.3. The arrow points to the ~53 kb interspersed segment of normal copy-number. (C) G-banded partial karyotype of the patient showing derivative chromosome 10;22 (arrow indicates the breakpoint region on the derivative chromosome). (D and E) FISH verification of the rearrangement using BAC clones RP11-66M5 (duplicated on the dicentric chromosome; red; D) and RP3-402G11 (deleted on the dicentric chromosome; red; E) and using the control probe RP11-89D12 (green; D and E) demonstrated that the deletion and duplication arose at the translocation breakpoint.
Genome-wide oligonucleotide aCGH mapped the proximal duplication breakpoint between 46,511,147 and 46,512,467 and the distal duplication breakpoint between 48,780,779 and 48,782,683 (hg18). The proximal breakpoint of the terminal deletion was mapped between 48,835,877 and 48,837,261. In between these two CNVs, there is an ~53 kb segment of normal copy-number, suggesting a large DNA palindromic structure in formation [Tanaka et el. 2007] (Fig 2B). The other identified genome-wide changes represent known copy-number polymorphisms.
CMA also identified a paternally inherited microdeletion of 1p36.22 in a boy with RES, HPE, macrocephaly, intellectual disability, and epilepsy that most likely represents a familial copy number variant with no pathogenic significance since it was inherited from a phenotypically normal father. Based on the array design, the size of the deletion can be narrowed to between 154 kb and 199 kb with a maximum range between 11,753,084 and 11,951,933. The genes included in the deletion region are MTHFR, CLCN6, NPPA, NPPB, KIAA2013, and PLOD1.
We reviewed the neuroimages of all of the patients who were ascertained because they have RES and found that 76% of the patients have dysmorphic ventricles, and 54% of the patients had hydrocephalus requiring a ventriculoperitoneal shunt. Six of 17 patients (35%) have an absent, partially absent, or abnormally shaped corpus callosum. Neural tube defects were prominent (24%) in this series, occurring in two children with spina bifida and in two children with encephalocele. Of the 13 children who were examined, five of eight children older than age five years (63%) have intellectual disabilities (one child has moderate and four children have profound intellectual disabilities), and five of five children (100%) under the age of three years have global developmental delays. Ophthalmological findings were also common: two children have microphthalmia; one child has a small optic chiasm, absent septum pellucidum, and prior diagnosis of septo-optic dysplasia; and eight children (47%) have documented eye movement abnormalities (esotropia, exotropia, amblyopia, or nystagmus). Additional neuroimaging and clinical findings are summarized in Table I. None of the patients with RES of unknown etiology had family histories that appeared contributory except for the pertinent positives that are listed in the column entitled “Physical exam findings and clinical information”.
Table 1.
Clinical features of patients with rhombencephalosynapsis.
| Patient | Gender | Age at exam | Imaging Findings | Chromosome analyses | CMA | HPE Panel | Physical exam findings and clinical information |
|---|---|---|---|---|---|---|---|
| 1 | female | 6 y | RES, alobar HPE, absent CC, HC | normal | normal | ZIC2 c.392_98del7 exon1 | Weight < 3rd centile, hypotonia in extremities, profound ID, VPS, leg length discrepancy, bilateral hip dysplasia |
| 2 | female | 14 y | RES, semi-lobar HPE, partially absent CC | ND | ND | ZIC2 c.392_98del7 exon1 | Microcephaly, weight < 5th centile, hypertelorism, midface hypoplasia, brachydactyly, hypertonia, hyperreflexia, wheelchair-bound, profound ID |
| 3 | female | 2 y | RES, dysmorphic ventricles and CC | 45,XXder10,22; q26.3;22q13.3 | 22q13. 3 gain and loss | normal | Weight < 3rd centile, skin changes, gluteal cleft deviation, global hypotonia, decreased reflexes, GDD |
| 4 | male | 17 y | RES, HPE, left occipital porencephalic cyst, peri-ventricular white matter volume loss, HC | normal | 1p36.22 loss | normal | Macrocephaly, scoliosis, kyphosis, distal contractures, profound ID, localization-related epilepsy, VPS |
| 5 | male | 6 y | RES, monoventricle, left open-lip schizencephaly, calcifications, HC | normal | normal | normal | Macrocephaly, pronounced forehead, small mouth, wheelchair-bound, legs internally rotated, hypertonia, hyperreflexia, non-verbal, profound ID, localization-related epilepsy, VPS, 5 MM, gestational diabetes, breech presentation |
| 6 | female | 10 y | RES, small optic chiasm, dysmorphic ventricles, microphthalmia, thin brainstem and CC, decreased white matter | normal | normal | normal | Microcephaly, small eyes, midface hypoplasia, gum hyperplasia, nystagmus in all directions, hypertonia at ankles, legs rotated mildly externally, toe walks, moderate ID, autism, 24 week prematurity |
| 7 | female | 14 y | RES, dysmorphic ventricles, partially absent SP, frontal migrational abnormality | normal | normal | normal | L4 spina bifida, localization-related epilepsy |
| 8 | female | 5 y | RES, absent SP, small optic nerves, dysmorphic ventricles | normal | normal | normal | Nystagmus, disconjugate gaze, SOD |
| 9 | male | 10 y | RES, left peri-atrial gliosis | normal | normal | normal | Height and weight< 5th centile, mild slanting of palpebral fissures, learning and behavior problems, ADHD, esotropia, congenital vs. perinatal CMV exposure |
| 10 | male | 2 y | RES, dysmorphic ventricles and CC, Chiari II malformation, HC | normal | normal | normal | L5 spina bifida, GDD, VPS, gestational diabetes |
| 11 | male | 23 mo | RES, meningoencephaloce le, dysmorphic ventricles, HC | normal | ND | ND | Microcephaly, weight < 5th centile, bitemporal narrowing, downslanting palpebral fissures, epicanthal folds, axial hypotonia, hypertonia on right side, stereotyped behaviors, hyperreflexia on right, Babinski reflex on right, GDD, localization-related epilepsy, VPS, esotropia |
| 12 | female | 2 y | RES, Chiari I malformation with syrinx, dysmorphic ventricles and CC, polymicrogyria, HC, tectal lipoma, lipoma of filum terminale, tethered cord, extra vertebral segment | normal | normal | normal | Microcephaly, height and weight < 5th centile, hypertelorism, small eyes and palpebral fissures, hypertonia at ankles, external rotation of legs, GDD, VPS, exotropia, mother with type I diabetes mellitus |
| 13 | male | 2 y | RES, possible minimal forebrain fusion/lobar HPE | normal | ND | normal | Amblyopia, hypertonia at ankles, hyperreflexia in lower extremities, bilateral Babinski reflex, GDD, transposition of the great arteries |
| 14 | male | ND | RES, mild cerebral volume loss, subcutaneous scalp nodules | normal | ND | ND | Deceased, ID, in utero alcohol and illicit drug exposures |
| 15* | male | ND | RES, dysmorphic ventricles | Normal FISH for 22q11.2 deletion and telomeric FISH negative |
ND | ND | ADHD, duodenal atresia, velopharyngeal incompetence, exotropia, hearing loss |
| 16* | female | ND | RES | ND | ND | ND | Congenital CMV exposure |
| 17* | male | ND | RES, dysmorphic ventricles, encephalocele, mild Chiari II malformation, HC | 46,X.inv(Y) (p11.3q11.23) | ND | ND | GDD, VPS, bilateral esotropia, amblyopia |
Abbreviations: HC, hydrocephalus; RES, rhombencephalosynapsis; GDD, global developmental delay; MM, maternal miscarriage; ND, not done; CMA, chromosomal microarray; CC, corpus callosum; SOD, septo-optic dysplasia; HPE, holoprosencephaly; SP, septum pellucidum; ID, intellectual disability; VPS, ventriculoperitoneal shunt
The subjects marked with an asterisk were unable to be located, however, clinical information from chart review is provided.
Discussion
RES may occur in apparent isolation or in combination with other brain malformations, with neural tube defects, or with other congenital anomalies such as the VACTERL-H syndrome [Pasquier et al., 2009; Schwahn et al., 2004; Sener, 2000; Toelle et al., 2002; Truwit et al., 1991] or Gomez-Lopez-Hernandez syndrome [Sukhudyan et al., 2010]. RES has been reported previously in association with Chiari II malformations [Sener and Dzelzite 2003; Wan et al., 2005]; however, it also has been suggested that a small posterior fossa and compression of the cerebellar hemispheres can mimic the appearance of RES [Guntur Ramkumar et al., 2010]. Therefore, we cannot dismiss the possibility that the RES diagnosed in two individuals with Chiari II malformations in this study is not a real observation. There is likely an ascertainment bias towards the more severe and syndromic forms of RES as these individuals are more likely to undergo brain MRI examination. A prospective study of 3000 children undergoing brain MRI in Turkey found the frequency of RES to be 0.13% in this population [Sener 2000]. Partial RES is apparently an even more rare or under-ascertained malformation of the cerebellum [Demaerel et al., 2004; Wan et al., 2005].
The identification of a ZIC2 mutation in two half siblings with HPE and partial RES provides the first specific gene association with RES. Their shared mother does not carry the mutation, implicating germline mosaicism as the cause of recurrence. Familial recurrence of ZIC2 mutations due to presumed germline mosaicism has been reported previously, and germline mosaicism is more commonly observed with mutation of ZIC2 compared to other gene mutations that cause HPE [Solomon et al., 2010a]. Studies suggest that mutation of ZIC2 is identified in 3–8% of individuals with HPE [Grinberg and Millen, 2005; Solomon et al., 2010a], but association of ZIC2 mutation with RES has never been reported. However, variable expressivity is well documented within families and among affected individuals with ZIC2 mutations and HPE [Solomon et al., 2010a]. There are five ZIC genes (Zinc fingers in cerebellum) in humans and mice, and all five genes are expressed in the developing cerebellum [Grinberg and Millen, 2005]. Mice homozygous or heterozygous for Zic2 mutation or homozygous for Zic2 knockdown (Zic2Kd/Kd) develop holoprosencephaly, agenesis of the corpus callosum, neural tube defects, subtle cerebellar hypoplasia, dysmorphic ventricles, and skeletal abnormalities [Aruga et al., 2002; Elms et al., 2003; Nagai et al., 2000]. Zic1 null animals develop overt cerebellar hypoplasia and malformed folia with notable loss of granule cells in the anterior vermis [Aruga et al., 1998]. Interestingly, Zic1 and Zic2 are prominently expressed in the mid-line of the developing cerebellum, and Zic1+/−Zic2+/Kd double heterozygous mice lack the primary fissure in the cerebellar vermis, the posterior superior fissure in the cerebellar hemispheres, a lobule in the anterior vermis, and have a truncation of the most posterior lobule [Aruga et al., 2002]. Although not specifically mentioned by the authors, these data suggest that the pronounced deficits and abnormal cleavage patterns in the anterior cerebellum of Zic1+/−Zic2+/Kd animals represent a form of partial RES. These data strongly support our observation that mutation of ZIC2 contributes to RES or partial RES in humans, and suggest that patients with RES should also be screened for alterations in ZIC1 in addition to ZIC2. Notably, a single patient with mutation of ZIC2 was identified in a large series of patients with neural tube defects, suggesting a minor role for ZIC2 in human neural tube defects [Klootwijk et al., 2004]. Given the novel association of ZIC2 with RES, the theoretical concern exists that in rare cases, the presence of isolated RES may be a form fruste of HPE and genetic counseling should take this risk into consideration.
The identification of a second individual with RES, hypotonia, developmental delay, failure-to-thrive, and abnormality of 10q26.3 suggests that a causative or contributing gene or gene regulatory region may exist in this region; however, we did not detect any genomic imbalance in this region by aCGH. We did detect a complex rearrangement on chromosome 22 in this individual including microdeletion of the SHANK3 gene, suggesting that this child has 22q13.3 deletion syndrome (Phelan-McDermid syndrome, OMIM # 606232), which is consistent with her clinical presentation [Delahaye et al., 2009; Dhar et al., 2010; Phelan 2008; Wilson et al., 2003]. To date, RES has never been reported in the 22q13.3 deletion syndrome. We hypothesize that there is a dosage-sensitive gene in the deleted region on chromosome 22q which contributes to RES, however, we cannot dismiss the possibility that disruption of a gene in a regulatory region at 10q26.3 or that another distinct genetic alteration may have contributed to RES in this patient.
We also identified a subject with RES and HPE with a paternally-inherited microdeletion of 1p36.22. This deletion may represent a familial copy number variant with no pathogenic significance since it was inherited from a phenotypically normal father; however, this is not a CNV that is commonly observed in normal controls. Furthermore, the father has no known neurological deficits, but he has not had neuroimaging tests performed. Therefore, we cannot rule out incomplete penetrance or variable expressivity of phenotypes associated with this CNV. This microdeletion includes the MTHFR, CLCN6, NPPA, NPPB, KIAA2013, and PLOD1 genes. CLCN6 encodes a chloride channel that is expressed in brain tissue, and mutation of the gene is hypothesized to be a novel cause of neuronal ceroid lipofuscinosis; nevertheless, a role in the development of the cerebellum has not been reported [Pressey et al., 2010]. NPPA and NPPB encode atrial natriuretic polypeptide precursors; common variants in these genes have a role in inter-individual variation in blood pressure [Newton-Cheh et al., 2009]. PLOD1 encodes a collagen lysyl hydroxylase; recessive mutations in this gene cause Ehlers-Danlos syndrome type VI [Dembure et al., 1984]. Interestingly, mice homozygous for Mthfr gene deletion demonstrate cerebellar hypoplasia, depletion of external granule cells and Purkinje cell heterotopias primarily in the anterior lobules, and dysmorphic ventricles suggesting that MTHFR function is essential to normal cerebellar and ventricular development [Chen et al., 2005]. We do not know the mutation status of the proband’s remaining MTHFR allele, however, serum testing revealed a normal homocysteine level suggesting adequate enzyme activity. Given these observations, the association of RES with neural tube defects, and the well-documented association of MTHFR gene mutations and abnormal folate metabolism with neural tube defects, we propose that MTHFR haploinsufficiency is unlikely to be a sufficient cause of RES, however, MTHFR loss of function might be a genetic modifier that contributes to RES.
The genomic rearrangement 46,Xinv(Y)(p11.3q11.23) that was identified by G-banded chromosome analysis in a boy with RES and encephalocele is likely non-contributory, and therefore no further studies were performed.
Potential teratogenic causes of RES have been reported, and our case series suggests that congenital infection or gestational diabetes may also contribute to RES. Therefore, evidence to date suggests that the causes of RES are likely heterogeneous with complex interactions between predisposing genetic and environmental factors. The relative infrequent occurrence of RES compared with HPE or neural tube defects (despite an association that was confirmed by our study) may suggest lower expressivity of RES, potentially due to compensatory or redundant developmental mechanisms and a need for “multiple hits” for RES to occur. An important precedent for the multiple hit hypothesis exists with the example of Dandy-Walker malformation (DWM), another mid-line congenital cerebellar syndrome. Heterozygous loss of both ZIC1 and ZIC2 function in humans and mice is one cause of DWM identified to date, and while cerebellar abnormalities are completely penetrant in the mouse model, DWM is variably expressed [Grinberg et al., 2004]. Of note, heterozygous deletion of both the ZIC2 and ZIC5 genes in the 13q32 deletion critical region also has been associated with DWM [Mademont-Soler et al., 2010].
In summary, we identified a mutation of the ZIC2 gene as a cause of or predisposing factor to partial RES associated with HPE. When presented with a patient with RES, genetic counseling should consider the possibility that the individual and the parents may be at higher risk of having a child with HPE. The possibility that a gene within the deleted region on chromosome 22q13.3 may contribute to RES warrants further studies. The phenotypes of the patients with RES described herein add to the clinical knowledge of this relatively rare brain malformation.
Acknowledgments
M.B.R. is grateful for the support of NIH/NINDS grant 5K08NS062711-03. We thank Robert McNeil for image compilation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NINDS or the NIH. Some of the authors are based in the Department of Molecular and Human Genetics at Baylor College of Medicine which offers extensive genetic laboratory testing including use of arrays for genomic copy number analysis and derives revenue from this activity.
References
- Aruga J, Inoue T, Hoshino J, Mikoshiba K. Zic2 controls cerebellar development in cooperation with Zic1. J Neurosci. 2002;22:218–225. doi: 10.1523/JNEUROSCI.22-01-00218.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aruga J, Minowa O, Yaginuma H, Kuno J, Nagai T, Noda T, Mikoshiba K. Mouse Zic1 is involved in cerebellar development. J Neurosci. 1998;18:284–293. doi: 10.1523/JNEUROSCI.18-01-00284.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Schwahn BC, Wu Q, He X, Rozen R. Postnatal cerebellar defects in mice deficient in methylenetetrahydrofolate reductase. Int J Dev Neurosci. 2005;23:465–474. doi: 10.1016/j.ijdevneu.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Cohen MM., Jr Holoprosencephaly: clinical, anatomic, and molecular dimensions. Birth Defects Res A Clin Mol Teratol. 2006;76:658–673. doi: 10.1002/bdra.20295. [DOI] [PubMed] [Google Scholar]
- Delahaye A, Toutain A, Aboura A, Dupont C, Tabet AC, Benzacken B, Elion J, Verloes A, Pipiras E, Drunat S. Chromosome 22q13.3 deletion syndrome with a de novo interstitial 22q13.3 cryptic deletion disrupting SHANK3. Eur J Med Genet. 2009;52:328–332. doi: 10.1016/j.ejmg.2009.05.004. [DOI] [PubMed] [Google Scholar]
- Demaerel P, Morel C, Lagae L, Wilms G. Partial rhombencephalosynapsis. AJNR Am J Neuroradiol. 2004;25:29–31. [PMC free article] [PubMed] [Google Scholar]
- Dembure PP, Priest JH, Snoddy SC, Elsas LJ. Genotyping and prenatal assessment of collagen lysyl hydroxylase deficiency in a family with Ehlers-Danlos syndrome type VI. Am J Hum Genet. 1984;36:783–790. [PMC free article] [PubMed] [Google Scholar]
- Dhar SU, del Gaudio D, German JR, Peters SU, Ou Z, Bader PI, Berg JS, Blazo M, Brown CW, Graham BH, Grebe TA, Lalani S, Irons M, Sparagana S, Williams M, Phillips JA, 3rd, Beaudet AL, Stankiewicz P, Patel A, Cheung SW, Sahoo T. 22q13.3 deletion syndrome: clinical and molecular analysis using array CGH. Am J Med Genet A. 2010;152A:573–581. doi: 10.1002/ajmg.a.33253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elms P, Siggers P, Napper D, Greenfield A, Arkell R. Zic2 is required for neural crest formation and hindbrain patterning during mouse development. Dev Biol. 2003;264:391–406. doi: 10.1016/j.ydbio.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Grinberg I, Millen KJ. The ZIC gene family in development and disease. Clin Genet. 2005;67(4):290–296. doi: 10.1111/j.1399-0004.2005.00418.x. [DOI] [PubMed] [Google Scholar]
- Grinberg I, Northrup H, Ardinger H, Prasad C, Dobyns WB, Millen KJ. Heterozygous deletion of the linked genes ZIC1 and ZIC4 is involved in Dandy-Walker malformation. Nat Genet. 2004;36:1053–1055. doi: 10.1038/ng1420. [DOI] [PubMed] [Google Scholar]
- Guntur Ramkumar P, Kanodia AK, Ananthakrishnan G, Roberts R. Chiari II malformation mimicking partial rhombencephalosynapsis? A case report. Cerebellum. 2010;9:111–114. doi: 10.1007/s12311-009-0132-6. [DOI] [PubMed] [Google Scholar]
- Klootwijk R, Groenen P, Schijvenaars M, Hol F, Hamel B, Straatman H, Steegers-Theunissen R, Mariman E, Franke B. Genetic variants in ZIC1, ZIC2, and ZIC3 are not major risk factors for neural tube defects in humans. Am J Med Genet A. 2004;124A:40–47. doi: 10.1002/ajmg.a.20402. [DOI] [PubMed] [Google Scholar]
- Lespinasse J, Testard H, Nugues F, Till M, Cordier MP, Althuser M, Amblard F, Fert-Ferrer S, Durand C, Dalmon F, Pourcel C, Jouk PS. A submicroscopic unbalanced subtelomeric translocation t(2p;10q) identified by fluorescence in situ hybridization: fetus with increased nuchal translucency and normal standard karyotype with later growth and developmental delay, rhombencephalosynapsis (RES) Ann Genet. 2004;47:405–417. doi: 10.1016/j.anngen.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Lu X, Shaw CA, Patel A, Li J, Cooper ML, Wells WR, Sullivan CM, Sahoo T, Yatsenko SA, Bacino CA, Stankiewicz P, Ou Z, Chinault AC, Beaudet AL, Lupski JR, Cheung SW, Ward PA. Clinical implementation of chromosomal microarray analysis: summary of 2513 postnatal cases. PLoS One. 2007;2(3):e327. doi: 10.1371/journal.pone.0000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mademont-Soler I, Morales C, Armengol L, Soler A, Sanchez A. Description of the smallest critical region for Dandy-Walker malformation in chromosome 13 in a girl with a cryptic deletion related to t(6;13)(q23;q32) Am J Med Genet A. 2010;152A:2308–2312. doi: 10.1002/ajmg.a.33550. [DOI] [PubMed] [Google Scholar]
- Nagai T, Aruga J, Minowa O, Sugimoto T, Ohno Y, Noda T, Mikoshiba K. Zic2 regulates the kinetics of neurulation. Proc Natl Acad Sci U S A. 2000;97:1618–1623. doi: 10.1073/pnas.97.4.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton-Cheh C, Larson MG, Vasan RS, Levy D, Bloch KD, Surti A, Guiducci C, Kathiresan S, Benjamin EJ, Struck J, Morgenthaler NG, Bergmann A, Blankenberg S, Kee F, Nilsson P, Yin X, Peltonen L, Vartiainen E, Salomaa V, Hirschhorn JN, Melander O, Wang TJ. Association of common variants in NPPA and NPPB with circulating natriuretic peptides and blood pressure. Nat Genet. 2009;41:348–353. doi: 10.1038/ng.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquier L, Marcorelles P, Loget P, Pelluard F, Carles D, Perez MJ, Bendavid C, de La Rochebrochard C, Ferry M, David V, Odent S, Laquerriere A. Rhombencephalosynapsis and related anomalies: a neuropathological study of 40 fetal cases. Acta Neuropathol. 2009;117:185–200. doi: 10.1007/s00401-008-0469-9. [DOI] [PubMed] [Google Scholar]
- Phelan MC. Deletion 22q13.3 syndrome. Orphanet J Rare Dis. 2008;3:14. doi: 10.1186/1750-1172-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pressey SN, O’Donnell KJ, Stauber T, Fuhrmann JC, Tyynela J, Jentsch TJ, Cooper JD. Distinct neuropathologic phenotypes after disrupting the chloride transport proteins ClC-6 or ClC-7/Ostm1. J Neuropathol Exp Neurol. 2010;69:1228–1246. doi: 10.1097/NEN.0b013e3181ffe742. [DOI] [PubMed] [Google Scholar]
- Schwahn BC, Laryea MD, Chen Z, Melnyk S, Pogribny I, Garrow T, James SJ, Rozen R. Betaine rescue of an animal model with methylenetetrahydrofolate reductase deficiency. Biochem J. 2004;382(Pt 3):831–840. doi: 10.1042/BJ20030822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sener RN. Unusual MRI findings in rhombencephalosynapsis. Comput Med Imaging Graph. 2000;24:277–282. doi: 10.1016/s0895-6111(00)00025-2. [DOI] [PubMed] [Google Scholar]
- Sener RN, Dzelzite S. Rhombencephalosynapsis and a Chiari II malformation. J Comput Assist Tomogr. 2003;27:257–259. doi: 10.1097/00004728-200303000-00026. [DOI] [PubMed] [Google Scholar]
- Solomon BD, Lacbawan F, Mercier S, Clegg NJ, Delgado MR, Rosenbaum K, Dubourg C, David V, Olney AH, Wehner LE, Hehr U, Bale S, Paulussen A, Smeets HJ, Hardisty E, Tylki-Szymanska A, Pronicka E, Clemens M, McPherson E, Hennekam RC, Hahn J, Stashinko E, Levey E, Wieczorek D, Roeder E, Schell-Apacik CC, Booth CW, Thomas RL, Kenwrick S, Cummings DA, Bous SM, Keaton A, Balog JZ, Hadley D, Zhou N, Long R, Velez JI, Pineda-Alvarez DE, Odent S, Roessler E, Muenke M. Mutations in ZIC2 in human holoprosencephaly: description of a Novel ZIC2 specific phenotype and comprehensive analysis of 157 individuals. J Med Genet. 2010a;47:513–524. doi: 10.1136/jmg.2009.073049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon BD, Mercier S, Velez JI, Pineda-Alvarez DE, Wyllie A, Zhou N, Dubourg C, David V, Odent S, Roessler E, Muenke M. Analysis of genotype-phenotype correlations in human holoprosencephaly. Am J Med Genet C Semin Med Genet. 2010b;154C:133–141. doi: 10.1002/ajmg.c.30240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukhudyan B, Jaladyan V, Melikyan G, Schlump JU, Boltshauser E, Poretti A. Gomez-Lopez-Hernandez syndrome: reappraisal of the diagnostic criteria. Eur J Pediatr. 2010;169:1523–1528. doi: 10.1007/s00431-010-1259-7. [DOI] [PubMed] [Google Scholar]
- Toelle SP, Yalcinkaya C, Kocer N, Deonna T, Overweg-Plandsoen WC, Bast T, Kalmanchey R, Barsi P, Schneider JF, Capone Mori A, Boltshauser E. Rhombencephalosynapsis: clinical findings and neuroimaging in 9 children. Neuropediatrics. 2002;33:209–214. doi: 10.1055/s-2002-34498. [DOI] [PubMed] [Google Scholar]
- Truwit CL, Barkovich AJ, Shanahan R, Maroldo TV. MR imaging of rhombencephalosynapsis: report of three cases and review of the literature. AJNR Am J Neuroradiol. 1991;12:957–965. [PMC free article] [PubMed] [Google Scholar]
- Wan SM, Khong PL, Ip P, Ooi GC. Partial rhombencephalosynapsis and Chiari II malformation. Hong Kong Med J. 2005;11:299–302. [PubMed] [Google Scholar]
- Wilson HL, Wong AC, Shaw SR, Tse WY, Stapleton GA, Phelan MC, Hu S, Marshall J, McDermid HE. Molecular characterisation of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/PROSAP2 in the major neurological symptoms. J Med Genet. 2003;40:575–584. doi: 10.1136/jmg.40.8.575. [DOI] [PMC free article] [PubMed] [Google Scholar]